
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of chloride salts and microwaves on polyethylene terephthalate (PET) hydrolysis by iron chloride/acetic acid Lewis/Brønsted acidic deep eutectic solvent†
Marco
Rollo
 a,
Massimo A. G.
Perini
a,
Alessandro
Sanzone
b,
Lorenzo
Polastri
b,
Matteo
Tiecco
c,
Alejandro
Torregrosa-Chinillach
d,
Elisa
Martinelli
*a and
Gianluca
Ciancaleoni
*a
a,
Massimo A. G.
Perini
a,
Alessandro
Sanzone
b,
Lorenzo
Polastri
b,
Matteo
Tiecco
c,
Alejandro
Torregrosa-Chinillach
d,
Elisa
Martinelli
*a and
Gianluca
Ciancaleoni
*a
aDepartment of Chemistry and Industrial Chemistry, University of Pisa, Via Giuseppe Moruzzi 13, I-56124 Pisa, Italy. E-mail: elisa.martinelli@unipi.it; gianluca.ciancaleoni@unipi.it
bgr3n SA, Via Probello 19, 6963 Lugano, Switzerland
cChemistry Interdisciplinary Project (ChIP), School of Pharmacy, University of Camerino, Via Madonna delle Carceri, 62032 Camerino, MC, Italy
dDepartment of Organic Chemistry, Faculty of Sciences, Institute of Organic Synthesis (ISO), University of Alicante, Apdo. 99, 03080 Alicante, Spain
First published on 21st November 2023
Abstract
Chemical recycling offers a convenient solution for the disposal of plastic items made of polyethylene terephthalate (PET); however, there is still much room for improvement in terms of integration into the current waste treatment cycles. Recently, deep eutectic solvents (DESs) have exhibited interesting properties in PET glycolysis and hydrolysis, in some cases under mild conditions. In particular, we recently reported good results with Lewis/Brønsted acidic DESs (LBDESs) containing iron(III) chloride and sulfonic acids. However, the choice of weaker acids, such as acetic acid, is more cost effective and sustainable, with an associated reduced risk of corrosion and improved safety. In this study, we demonstrate that a simple post-reaction procedure significantly enhances the yield of terephthalic acid (TA) using FeCl3·6H2O/acetic acid (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) LBDES from 4% (literature value) to 54% under the same experimental conditions. Furthermore, we investigate the effect of chloride salts as additives and microwave irradiation on the reaction, achieving quantitative conversion and a high yield of TA in 10 minutes at 180 °C.
:1) LBDES from 4% (literature value) to 54% under the same experimental conditions. Furthermore, we investigate the effect of chloride salts as additives and microwave irradiation on the reaction, achieving quantitative conversion and a high yield of TA in 10 minutes at 180 °C.
Sustainability spotlightModern society needs to move from the current model of produce-use-discard to a circular economy, where no new polymers are produced and all the plastic is recycled. This is a very challenging task given that the quality of the recycled polymer is often negatively impacted by thermo-mechanical recycling processes. However, chemical recycling processes, such as depolymerization and repolymerization, can produce recycled polymers that are similar to virgin materials. Our goal is to develop new solvents for the depolymerization of polyethylene terephthalate, allowing the use of milder conditions with respect to existing industrial processes, in agreement with the following UN sustainable development goals: SDG 11 (sustainable cities and communities) and SDG 12 (responsible consumption and production). |
Introduction
Modern society cannot avoid the use of plastic items, some of which are disposable and have a very short life cycle (single-use plastic products, medical equipment, packaging materials, etc.). In many countries, recycling procedures exist to extend the life cycle of polymers; however, a large portion of plastic waste is landfilled, incinerated for energy recovery or mismanaged until it ends up in the ocean, with consequent pollution of soil, air and water.1 There are at least three different recycling levels: primary recycling involves reusing pre-consumer PET materials if they are still clean and uncontaminated; secondary processes (the most common, nowadays) involve the thermo-mechanical treatment of post-consumer items; and tertiary recycling involves the depolymerization of post-consumer plastic items to re-obtain the monomers (chemical recycling).2,3Tertiary recycling is still in its infancy; polymers, such as polyethylene,4 require harsh conditions5 and precious metal catalysts,6 and there is generally no selectivity.7 For condensation polymers, the potential is higher; indeed, research8–13 and industrial protocols already exist.2
In particular, polyethylene terephthalate (PET) is one of the preferred polymers for depolymerization studies; this research field is very active, and original ideas are continuously emerging.14–17 Generally, the depolymerization process of PET leads to bis(2-hydroxyethyl)-terephthalate (BHET) if ethylene glycol (EG) is the nucleophile,18 and to dimethyl terephthalate using methanol and terephthalic acid (TA) with water. In most cases, harsh conditions are necessary (T > 160 °C, many hours, sometimes high pressure) although exceptions can be found in recent studies.19–22
On the industrial side, many processes are used,23 including the pyrolysis of unsorted or sorted waste plastics and solvolysis, demonstrating widespread interest in the depolymerization process.
We recently contributed to this field by showing that the synergy between Lewis and Brønsted acids24–26 can be extremely beneficial in PET hydrolysis.27 Moreover, DESs formed by FeCl3·6H2O and organic sulfonic or carboxylic acids (mixed Lewis/Brønsted acidic DES, LBDESs) can efficiently hydrolyze PET under mild conditions (100 °C, 1 atm). The best performing LBDES contains methanesulfonic acid, but it is desirable to use different acids that are more environmentally friendly, cheaper, and easier to dispose of.
Acetic acid seems to be the ideal choice because it is cheaper and less corrosive than sulfonic acid (but still, significantly corrosive at high temperature28) and is generally synthesized by the carbonylation of methanol but can also be produced from lignocellulosic biomass via a bioconversion process.29,30 Acetic acid also has another advantage because it has a boiling point (Tb) of 118 °C, which is lower than that of MSA (Tb = 167 °C)27 and significantly different from that of EG (Tb = 197 °C), making the fractional distillation of the mixture to extract EG easier. However, our preliminary data showed that the liquid formed by FeCl3·6H2O/acetic acid (molar ratio 1:1, system LBDES1) is significantly less active than the FeCl3·6H2O/methanesulfonic acid (MSA) 1:1 LBDES, with PET conversion of around 20% and a terephthalic acid yield of around 4% in 30 minutes (0.3 g of PET in 4 g of LBDES, 100 °C, 1 atm).27
In this paper, we thoroughly characterize LBDES1, and we show that the performance of the system LBDES1 can be markedly improved from 4% of TA yield at 100 °C and 30 min to 56% under the same reaction conditions using a simple post-reaction treatment. We also demonstrate that the use of CaCl2 as an additive increases the yield up to 69% in 30 min, leading to quantitative conversion after 60 min of reaction at 100 °C. However, its use is not convenient because of the scale-up of the process.
The same reaction was also carried out by microwave (MW) irradiation,10,20,31 reducing the time to 10 min at 180 °C and greatly improving the PET/solvent ratio. Surprisingly, the results revealed that the beneficial effect of MW is that it is a fast and efficient heating strategy, without any significant specific effect.
Experimental details
PET plastics were collected directly from our department garbage containers, washed thoroughly with distilled water, dried and cut into flakes (approximate size 0.5 × 0.5 cm). Iron(III) chloride hexahydrate 97%, aluminum chloride hexahydrate 99% and glacial acetic acid > 99% were purchased from Alfa Aesar. Hydrochloric acid at 37%, sodium hydroxide at 98%, diethyl ether at 99.8%, acetone at 99.5%, DMSO-d6 at 99.8%, sulfuric acid at 99%, sodium chloride > 99.8% and calcium chloride > 93% were purchased from Sigma Aldrich. All the chemicals in this study were used without further purification. The water content of acetic acid was determined via Karl-Fischer titration (HI 904 Karl Fischer Coulometric Titration, Hanna Instruments), and it was 1600 ppm.LBDES preparation
Different mixtures were prepared by mixing FeCl3·6H2O with acetic acid (1:1 molar ratio, LBDES1, if not otherwise specified) and additives and/or water in suitable quantities under mild heating (approximately 80 °C) until homogeneous red dark liquid appeared.
LBDES characterization
LBDES1 was characterized in terms of a comparison between the theoretical solid–liquid phase diagrams and the experimental melting points at different molar ratios. The melting points were measured with a thermometer via immersion of the samples in a Dewar with CO2/acetone mixture or liquid nitrogen. The melting points were taken in triplicate to avoid a kinetic effect on the melting of the mixtures. The solid–liquid theoretical curves were determined using eqn (1), which represents the solid–liquid equilibrium curve: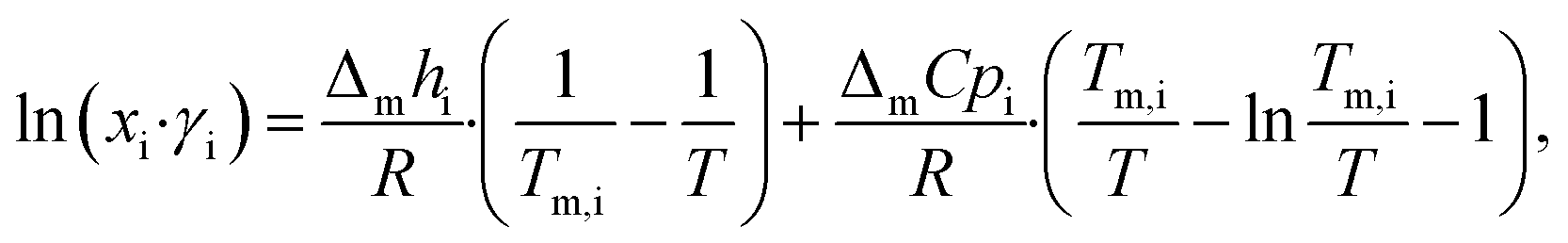 | (1) |
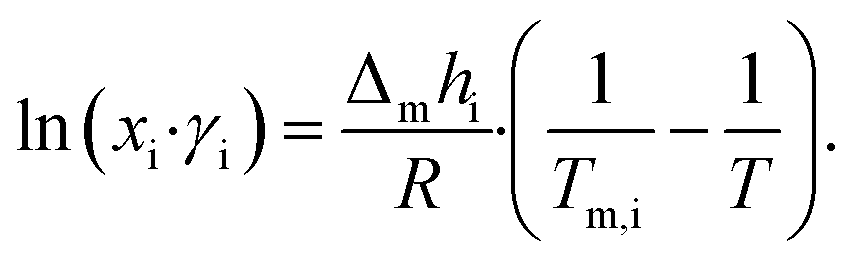 | (2) |
The theoretical melting temperatures were determined from the theoretical curves by considering the activity coefficients γi = 1. The eutectic points were determined as the minimum in the experimental curves, and they were compared to the theoretical ones. The experimental γi values were determined using eqn (3) with the experimentally observed melting temperatures:
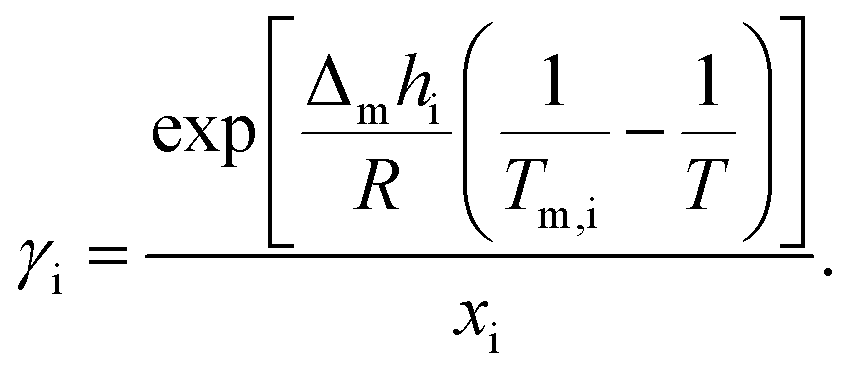 | (3) |
Characterization
1H and 13C NMR measurements were carried out using an FT NMR Joel JNM-ECZ500R MHz with a HFX probe at room temperature. The Fourier transform infrared-attenuated total reflectance (FTIR-ATR) spectra of terephthalic acid were recorded using an Agilent CARY 630 FTIR spectrometer. The conditions for ATR FT-IR measurements were a scan range of 400–4000 cm−1 with 32 scans, and a resolution of 4 cm−1. Conductivity measurements were carried out using a VIO series 7 conductometer with an integrated temperature probe. The measurements were conducted between 15 °C and 60 °C. Density analysis was carried out using an Anton Paar DMA 55 densimeter. The densimeter was calibrated using air and water with an error of ±1 × 10−5 g mL−1 for samples that have a density from 0.5 to 1.5 g mL−1. The measurements were conducted between 15 °C and 60 °C. Viscosity measurements were not carried out because of the corrosivity of the system toward steel. Comparing the flow rate with other DESs, the viscosity was evaluated to be lower than 100 cP.Improved post-reaction procedures for thermal hydrolysis of PET
For the catalytic experiments, a 15 mL screw-cap vial or a Carius tube equipped with a magnetic stirrer was filled with 300 mg of post-consumer PET and 4.0 g of LBDES (weight ratio PET/DES = 0.075). Hydrolysis reactions were carried out at temperatures ranging from 100 to 180 °C for 10–105 min. Warning: above 120 °C the autogenous pressure can reach high values (12 atm when T = 180 °C). Caution should be used for the glassware choice during all the operations. When the reaction was completed, about 20 mL of deionized water was added to the reaction solution to precipitate crude terephthalic acid (TA), oligomers and unreacted PET. The supernatant (straw yellow color) contains an aqueous solution of DES and EG from PET depolymerization. The solid fraction from the previous step was treated with an aqueous solution (20 mL) of NaOH (0.5 M), yielding a solution of di-sodium terephthalate (Na2TA) and solid Fe(OH)3 or Fe2O3. This solution was kept for 12 h in contact with the solid fraction to favor the quantitative neutralization of TA and eventually the conversion of oligomers (the presence of which was demonstrated in our previous study27) into Na2TA. After this time, unreacted PET flakes (if any) were removed manually, dried and weighted (w2 in eqn (4)). PET oligomers still insoluble in an alkaline medium were removed by filtration. Finally, TA was precipitated by adding 2 mL of a mineral acid (generally HCl 37% or H2SO4 40%). The obtained white powder of TA was then washed with water several times, dried at 80 °C overnight and weighed (wTA in eqn (5)).Conversion is calculated using the following equation:
 | (4) |
The yield of the purified TA is calculated using the following equation:
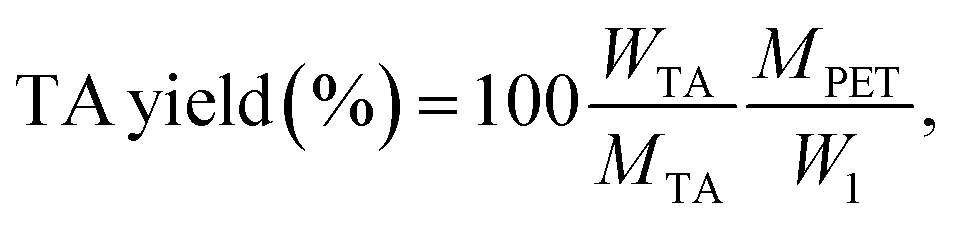 | (5) |
General procedures for the hydrolysis of PET via microwave irradiation
PET depolymerization via microwave heating was performed using an Anton Paar Microwave Reactor 400 equipped with a fiber-optic ruby thermometer. A 30 mL glass vial was filled with suitable quantities of LBDES1, water and PET flakes. PET/DES weight ratio was in the range of 0.075–0.4, while the water amount varied from 0 to 30 eq. compared to LBDES1. A silicon carbide (SiC) vessel was used to shield the content from microwave radiation, allowing for a comparison between the direct microwave heating and conventional heating of reaction mixtures.Microwave irradiation absorption
Microwave irradiation absorption was carried out at a fixed power (100 W) for 30 s, recording a temperature variation of 8 g of LBDES1 or deionized water. After the selected time, the irradiation power was turned off, and the time taken for the vial to return to room temperature was recorded.Solvent recycling
To test the solvent recycling, the reaction was conducted at 180 °C for 35 minutes, with a DES/PET ratio of 0.075 and in the presence of 20 equivalents of water with respect to iron(III). At the end of the reaction, no water was added. The reaction crude was filtered, and the solid fraction was treated as described above (see “Improved post-reaction procedures for thermal hydrolysis of PET”). The liquid fraction was reused for the next cycle without any treatment. After the last cycle, the remaining liquid was diluted with water and filtered, recovering the portion of TA that was soluble in the solvent.Environmental parameters
The sustainability of the process was evaluated using the environmental factor E,13 defined as follows: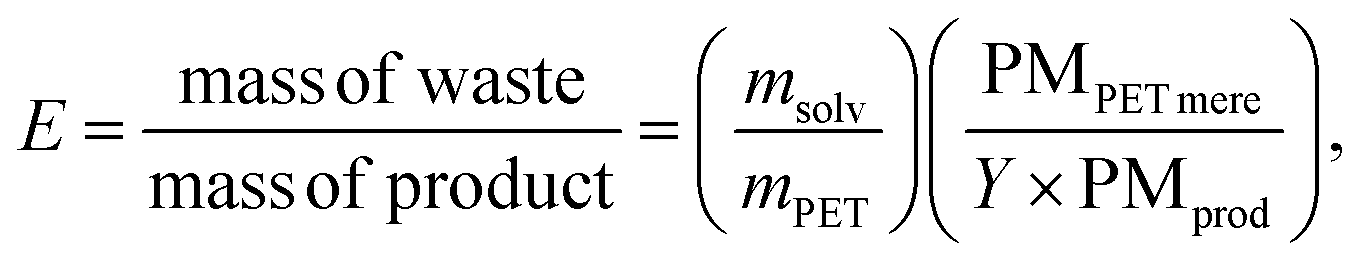 | (6) |
In addition to E, the energy economy coefficient (ε) is defined as
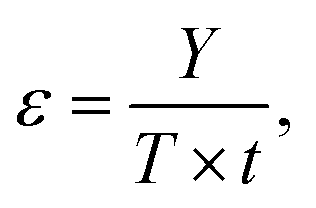
Finally, the energy impact factor (ξ) is the ratio between E and ε.
Results and discussion
LBDES characterization
In our previous publication, we demonstrated that the system FeCl3·6H2O/methanesulfonic acid is a DES because its melting point is lower than solid–liquid theoretical curves of the mixture at any composition. The same measurements are reported here for the FeCl3·6H2O/acetic acid mixture (LBDES1, Fig. 1).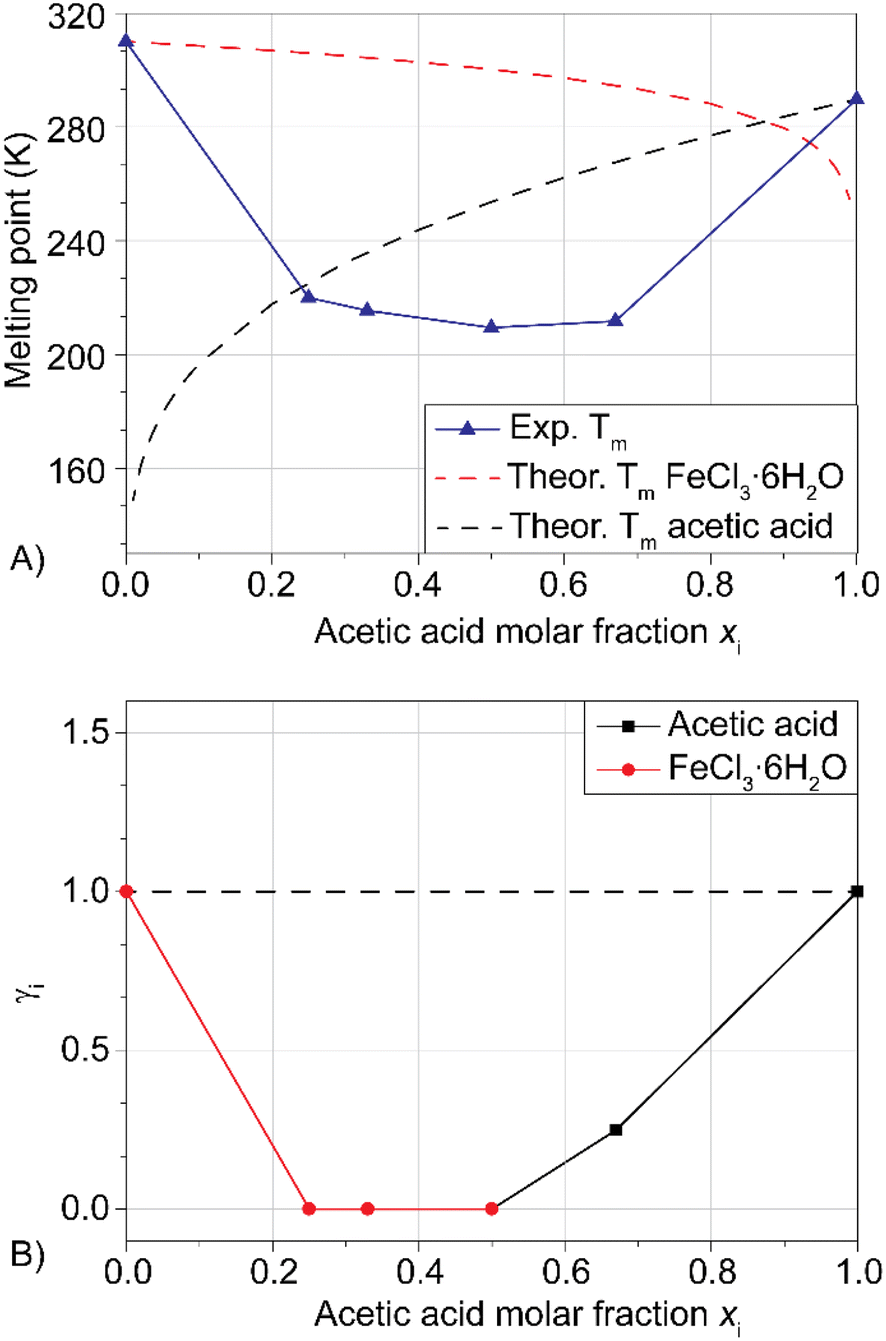 | ||
| Fig. 1 Panel A: experimental/theoretical solid–liquid phase diagrams for the FeCl3·6H2O/acetic acid LBDESs at various molar compositions; panel B: activity coefficients of the main component in the same liquids. Dashed lines indicate an ideal mixture. | ||
Additionally, in this case, the experimental Tm is significantly lower (ΔTm around 40 K) than that estimated by considering a simple eutectic without any specific interaction between the components. Coherently, the activity coefficients of the components are much lower than 1 in all compositions. Another consideration comes from the theoretical/experimental comparison: theoretical equations indicate the eutectic point at xacetic acid at around 0.9, while the experimental trend indicates the eutectic point at 0.5 (1:1 molar ratio). Interestingly, for the FeCl3·6H2O/methanesulfonic acid (MSA) mixture, the theoretical and experimental eutectic points are close to each other.27
To complete the LBDES characterization, the trends in the density and conductivity of LBDES1 with temperature were measured (Fig. S1, ESI†). This leads to an activation energy of conduction (Eσ) of 34 kJ mol−1. For comparison, the system FeCl3·6H2O/p-toluenesulfonic acid·H2O has a Eσ of 23 kJ mol−1.27 The difference is likely attributed to the lower acidity of acetic acid with respect to p-toluenesulfonic acid. A stronger acid generates more H3O+ ions, which are smaller and diffuse very efficiently (possibly also through the Grotthuss mechanism32). The other ions possibly present in LBDES1, apart from acetate, are FeCl2(H2O)4+ and Cl−, which are the ions expected from the crystal structure of FeCl3·6H2O.33
Improved post-reaction procedure
In our previous publication,27 the hydrolysis reaction of PET flakes (100 °C, 30 min) was followed by filtration of the reaction mixture, obtaining a solid composed of unreacted PET (manually separated, dried and weighted), oligomers and monomeric TA. The addition of NaOH 1 M separated TA from oligomers, forming an aqueous solution of sodium terephthalate (Na2TA). This procedure led, for LBDES1, to a conversion of 23% and a yield of 4%. The large difference between the two values is a direct consequence of the presence of oligomers, detected as a white powder dispersed in the alkaline liquid phase. Moreover, the surface of unreacted PET flakes appeared to be opaque and rough (Fig. 2b) possibly because of the reduced crystallinity (i.e. increased amorphous fraction) of the polymer at the surface.21,34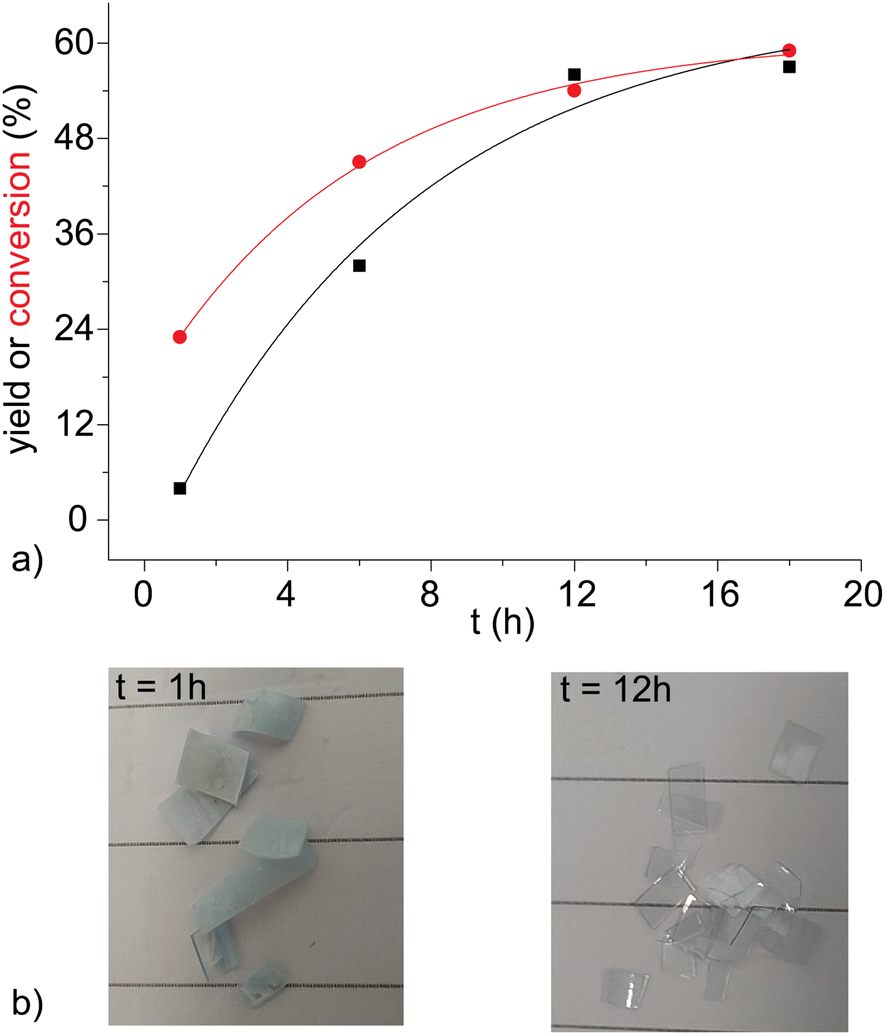 | ||
| Fig. 2 (a) Trend of TA yield vs. the duration of the post-reaction treatment with NaOH 0.5 M. Solid lines serve to guide only the eye. (b) Unreacted PET flakes were in contact with NaOH 0.5 M for 1 (left) or 12 (right) h. It is noteworthy that after 12 h, they are visibly thinner, more transparent and with a smooth surface than those after only 1 h. | ||
A mild post-reaction treatment maintaining all the recovered solid fractions (TA, oligomers and unreacted PET) in contact with 20 mL of NaOH 0.5 M for 12 h at room temperature under magnetic stirring was found to improve the conversion up to 54%. This suggests that the less crystalline polymer on the surface of the flakes was hydrolyzed and converted to Na2TA.21,34 The surface of the PET flakes was much smoother after this treatment (Fig. 2b). The white powder was also completely dissolved, with a consequent increase in yield to 56%. The percentages reported are the average of three reactions with deviations around ±5%. Fig. 2a shows that after 12 h of post-reaction treatment, the yield did not increase sensibly.
The fact that the conversion and yield after the post-reaction treatment were similar implies that with this procedure, all the oligomers were hydrolyzed to Na2TA. It is noteworthy that keeping post-consumer 0.3 g of PET in 10 mL of NaOH 0.5 M for 12–18 h at room temperature did not lead to a measurable yield of Na2TA, therefore confirming that the hydrolysis reaction occurs in the LBDES.
For stronger acids, such as sulphonic ones, the conversion is so high that the improvement in the work-up is not relevant, but for weaker ones, such as acetic acid, it becomes crucial. The purity of obtained TA is verified by 1H (see Fig. 3), 13C NMR and IR spectroscopies (Fig. S3 and S4†). These impurities are mainly isomers of TA already present in the original PET blend, such as isophthalic acid (the singlet and triplet peaks are around 8.4 and 7.6 ppm, respectively, around 0.5%).
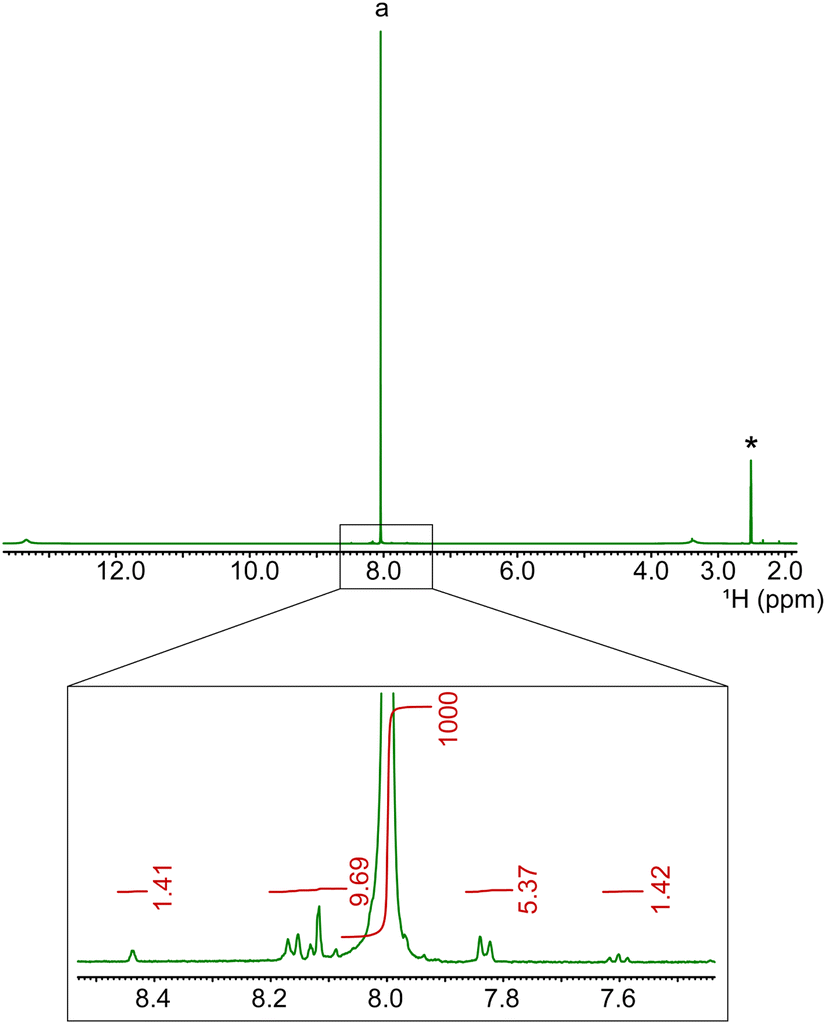 | ||
| Fig. 3
1H NMR spectrum (solvent DMSO-d6, room temperature) of TA resulting from the depolymerization of PET using FeCl3·6H2O/acetic acid (1:1) and improved work-up (see main text). The expansion of the aromatic region is shown to quantify the impurities. The asterisk denotes the residual solvent peak, and “a” is the aromatic signal of TA. | ||
Influence of additives
Another factor that can affect both conversion and yield is the presence of additives. We selected common chloride salts as additives because the DES already contains such anion, and the presence of other anions could interfere with liquid stability. For example, iron(III) sulfate does not form a stable liquid with acetic acid. Further, we opted only for cheap, non-toxic and readily available additives. We tested NaCl and CaCl2, which play a certain role in the depolymerization of PET35,36 and AlCl3, being a good Lewis acid and a known catalyst for trans-esterification reactions.37 DES is acidic and protic in nature; therefore, we used aluminum trichloride in its hexahydrate form because the anhydrous form reacts violently with water, liberating hydrogen chloride. Other chloride salts, such as barium, caesium, choline, ammonium, tetrabutylammonium, and benzyl tributyl, were insufficiently soluble to be tested.The speciation of chloride salts in DES is unclear because many equilibria could be active. For example, in the presence of a higher chloride concentration, FeCl2(H2O)4+ (the cation present in the solid state structure of FeCl6·6H2O33) could be converted into FeCl3(H2O)3 or FeCl4(H2O)2−. Even without these equilibria, chloride salts can enter the hydrogen bond network of the DES, interacting with acetic acid (CH3COOH⋯Cl−) or with water coordinated to iron. Hydrated AlCl3 has also more chances to interact owing to the water coordinated to aluminum, which can interact with bond hydrogen bond donors (such as –COO![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ) or acceptors (such as Cl− or –C
) or acceptors (such as Cl− or –C![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) OH). In all the cases, such interactions are insufficient to reach a high concentration of chloride salts.
OH). In all the cases, such interactions are insufficient to reach a high concentration of chloride salts.
In all the cases, we added from 1 to 4 equivalents of chloride salt with respect to the PET (0.3 g of PET = 1.60 mmol of monomeric units) based on the salt solubility in LBDES1. The results are summarized in Table 1. In all the cases, the liquid formed by FeCl3·6H2O and acetic acid (1:1) was used as solvent under the same experimental conditions (0.3 g of PET in 4 g of LBDES1, 100 °C, 30 min). The blank reaction (no additives added) gave a PET conversion of 54%.
The presence of 2 equivalents of NaCl did not affect the PET conversion, which was basically the same as that of the blank (57%). Additionally, in this case (and in all the following ones), all the conversion values are the average of three independent measurements, with a standard deviation of around 5%. Increasing the number of salt equivalents led to undissolved material in the flask and was not investigated.
However, 2 equivalents of CaCl2 (0.17 equivalents with respect to the iron) boosted the conversion up to 69%, but a further increase in the concentration led to a loss of performance. Such an effect, even if significant, is not drastic, but it deserves further investigation.
The physical properties of the liquid FeCl3·6H2O/CaCl2/acetic acid (1:0.17:1, LBDES2) are slightly different from those of LBDES1. The density is higher (Table S2†), and more importantly, the conductivity is higher (Table S3†). This denotes that the solution contains more ions, demonstrating that CaCl2 dissociates (partially, at least) in its ions. More detailedly, Eσ is reduced to 27 kJ mol−1 (Fig. S2†), confirming that the ions are less paired together and, consequently, more mobile. It is also interesting to note that the melting point of LBDES2 is practically the same as that of LBDES1 (Table S4†).
Using LBDES2 as a solvent, conversion became quantitative after 60 min (Fig. 4), while yield reached 94% after 75 min. This result indicates a significant reduction in the reaction time, which is reported to be 180 min for a quantitative yield for the same DES but without CaCl2.27
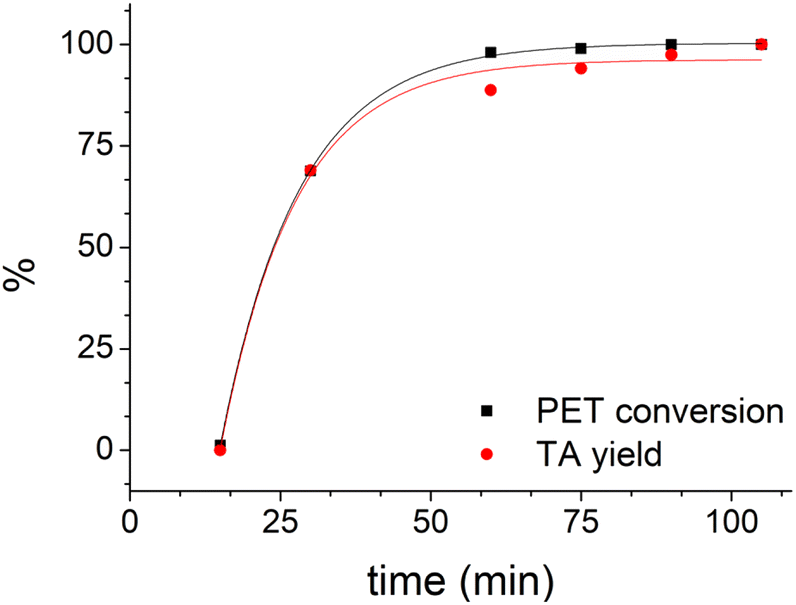 | ||
| Fig. 4 Trend of PET conversion and TA yield with time (0.3 g of PET in 4 g of LBDES2, 100 °C, conventional heating). Solid lines serve only to guide the eye. | ||
Finally, the addition of AlCl3·6H2O surprisingly did not affect the PET conversion (Table 1), as the latter did not change significantly by increasing the number of equivalents if we considered an experimental uncertainty of ±5%.
Proposing a single hypothesis to explain all these effects is not straightforward. In the absence of any additive, the catalytic effect of LBDES1 can be explained by a synergy between the Lewis and the Brønsted acids present in the DES. In particular, a likely mechanism is illustrated in Scheme 1: the Lewis acid can coordinate the carbonyl moiety, whereas the proton of the Brønsted acid can attack the ester oxygen.
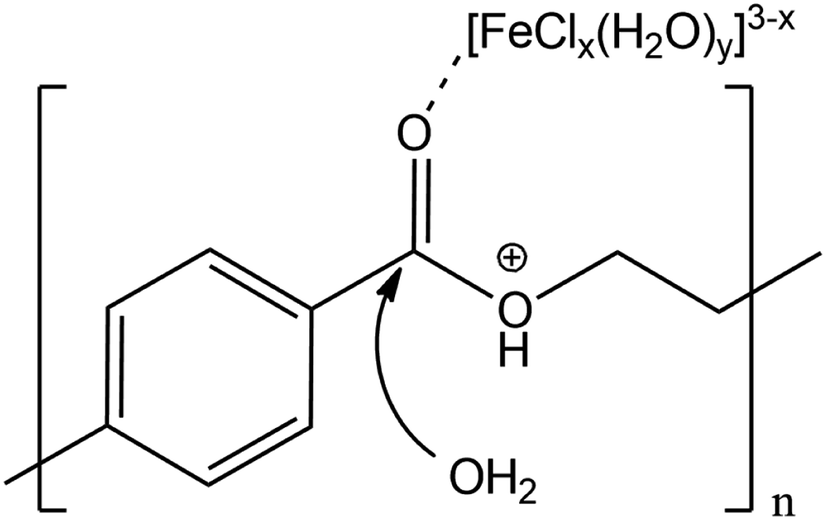 | ||
| Scheme 1 Proposed mechanism of double activation of the ester group by the Lewis and Brønsted acids. | ||
This double attack leads to a double activation of the ester group, making the ester carbon more prone to nucleophilic attack from water. This hypothesis can also explain the double role of water: it is necessary to carry on the hydrolysis reaction, but it also acts as a reaction inhibitor (see below and ref. 27). A trimolecular adduct is favored only at very high concentrations, and any dilution is detrimental to the reaction rate.
Under this assumption, it is possible to rationalize the role of the additives. We know from conductivity measurements that calcium chloride dissociates, at least partially, in its constituting ions, and it is also known that calcium ions strongly interact with water (calcium chloride is routinely used as a drying agent). Similarly, calcium coordinates some water molecules, favoring the reaction because it removes the inhibitor. If the calcium concentration exceeds its optimal value, too much water is sequestrated, and the hydrolysis is less efficient. However, sodium is known to be less effective in binding water because of its reduced charge density. This implies that sodium does not significantly reduce the amount of free water and that the conversion remains practically unaltered. Furthermore, NaCl has solubility issues in LBDES1 that limit its use at 2 equivalents.
Aluminum chloride brings its water molecules, increasing or maintaining stable inhibitor concentrations, depending on the number of water molecules that remain coordinated. It is expected to be a good Lewis acid; therefore, it is also possible that [AlClx(H2O)y]3−x replaces [FeClx(H2O)y]3−x for the activation of the carbonyl moiety (Scheme 1), leading to an unaltered mechanism and a similar PET conversion. Unfortunately, in this case, the solubility became an issue beyond 4 equivalents, not allowing further investigation. In addition, mixing AlCl3·6H2O and acetic acid did not lead to a stable liquid phase in the molar ratios 1:1, 1:2 and 2:1.
Microwave irradiation
The dilution of LBDES1 with water is highly desirable to reduce the corrosivity of the system and increase the amount of PET hydrolyzed in a single reaction. The maximum amount of PET processable depends on the available volume of liquid: if a PET flake is outside the liquid, it will not be depolymerized. In our case, the mass limit for 4 g of DES is 0.3 g of PET, which is not completely satisfactory. When water is added, the volume of the liquid phase increases, but as discussed above, excess water seriously slows down the reaction,20,22,27 and alternative strategies should be used. For instance, we previously demonstrated that 4 g of FeCl3·6H2O/MSA can depolymerize at least 1.2 g of PET divided into 4 aliquots (one per hour, together with 56 μL of water to replace those used for hydrolysis27) and that the DES can be reused by filtering unreacted PET and TA. Here, we decided to use MW irradiation to reach higher temperature values easily and rapidly to overcome the detrimental effects of water.First, we verified the ability of LBDES1 to absorb MWs by irradiating a sample of 8 g with a constant power of 100 W for 30 s (Fig. 5). Under these conditions, the sample increased its temperature to 53 K.
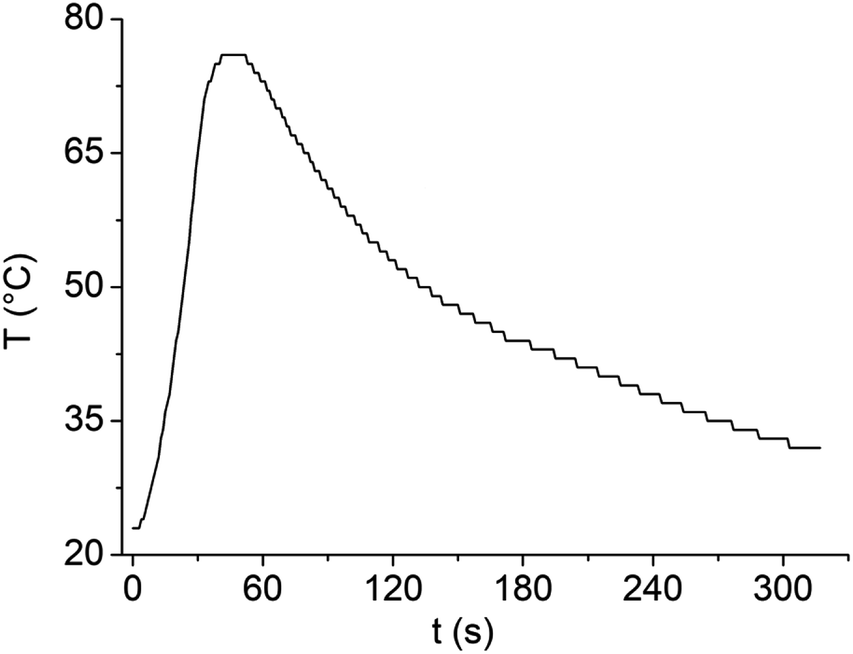 | ||
| Fig. 5 Temperature change of 8 g of LBDES1 under microwave irradiation of 100 W for 30 s. | ||
Therefore, the specific heat capacity of LBDES1 can be calculated using the following equation:
| P × t = Q = mcΔT | (7) |
The experimental results using MW irradiation are listed in Table 2. Run 1 was performed under experimental conditions similar to conventional heating (100 °C, 35 min, no water added, PET/solvent ratio = 0.075), showing slightly higher conversion and yield values, probably because the target temperature is reached faster. As expected, adding 10 eq. of water (w) with respect to iron(III) was strongly detrimental to the reaction, even after 60 min, both under MW (run 2) and conventional heating (run 3).
| Run | m (g) | T (°C) | t (min) | w (eq.) | PET conversion (%) | TA yield (%) |
|---|---|---|---|---|---|---|
| a Conventional heating. b Standard deviation calculated on 3 replicates: ±0.8. c Standard deviation calculated on 3 replicates: ±1.0. d Reaction heated by MW using a SiC reactor. | ||||||
| 1 | 1.125 | 100 | 35 | 0 | 74 | 73 |
| 2 | 1.125 | 100 | 60 | 10 | 0 | 0 |
| 3a | 1.125 | 100 | 60 | 10 | 0 | 0 |
| 4 | 1.125 | 140 | 10 | 0 | 100 | 87 |
| 5 | 1.125 | 140 | 35 | 10 | 30b | 26c |
| 6 | 1.125 | 140 | 60 | 20 | 4 | 2 |
| 7 | 1.125 | 180 | 10 | 0 | 100 | 92 |
| 8 | 1.125 | 180 | 10 | 10 | 100 | 91 |
| 9d | 1.125 | 180 | 10 | 10 | 100 | 88 |
| 10 | 1.125 | 180 | 35 | 20 | 100 | 96 |
| 11 | 4.000 | 180 | 10 | 10 | 100 | 95 |
| 12 | 6.000 | 180 | 10 | 10 | 100 | 94 |
By increasing T to 140 °C, the reaction was quantitative in 10 min (run 4), with a good yield (87%). Moreover, in this case, the addition of water led to a loss of conversion even when a longer time was used (run 5). Increasing the water addition (w) to 20 eq. (run 6) led to complete inactivity of the system, even with a longer reaction time (60 min).
At 180 °C, the reaction was always quantitative regardless of the amount of water added (from 0 to 20 eq., runs 7, 8 and 10), with TA yields always higher than 90%. As a confirmation that MW does not have a specific effect other than heating, run 9 was replicated with a SiC reactor, which efficiently absorbs MW.38 Comparable results were obtained, and the conversion and yield were 100% and 88% (run 9), respectively.
It is important to note that even at 180 °C and in the presence of ethylene glycol (coming from the depolymerization of PET), no trace of BHET was found in the 1H NMR spectrum (Fig. S6, ESI†). Moreover, BHET is soluble in basic water, but after some hours, it is hydrolyzed to Na2TA and EG.
Because the reaction was completed even in the presence of dilution, the amount of PET was increased to 4 g in run 11 and then up to 6 g in run 12 (ratio PET/DES = 0.4, PET/(DES + H2O) = 0.26) by maintaining the same amount of water (10 eq.) and DES (15 g) as well as the temperature (180 °C) and time (10 min) as in run 8. In both cases, the conversion was quantitative, and a high yield of pure TA was obtained. The result is particularly good compared with examples of acidic hydrolysis in the literature, in which the ratio PET/solvent typically varies from 0.09 to 0.4, reaction times from 140 to 300 min and temperatures from 98 to 140 °C.13
The experimental conditions of run 10 (Table 2) were used to test the solvent recycling. The reaction crude was filtered through a Whatman© filter paper, and the liquid fraction was reused without any treatment. At the end of the first cycle, the yield in TA was quite low (32%, Fig. 6) because TA was partially soluble in the LBDES. The yield increased in the second cycle by up to 82% because the DES solvent was saturated in TA. Finally, in the third cycle, the yield was found to exceed 100% (139%) because the TA lost in the first two cycles recovered after the complete work-up procedure. Globally, the conversion was always around 100%, and the yield over the three cycles was 78%. Furthermore, as the solid fraction and the filter remained impregnated with the solvent, some of the latter were lost during the filtration process.
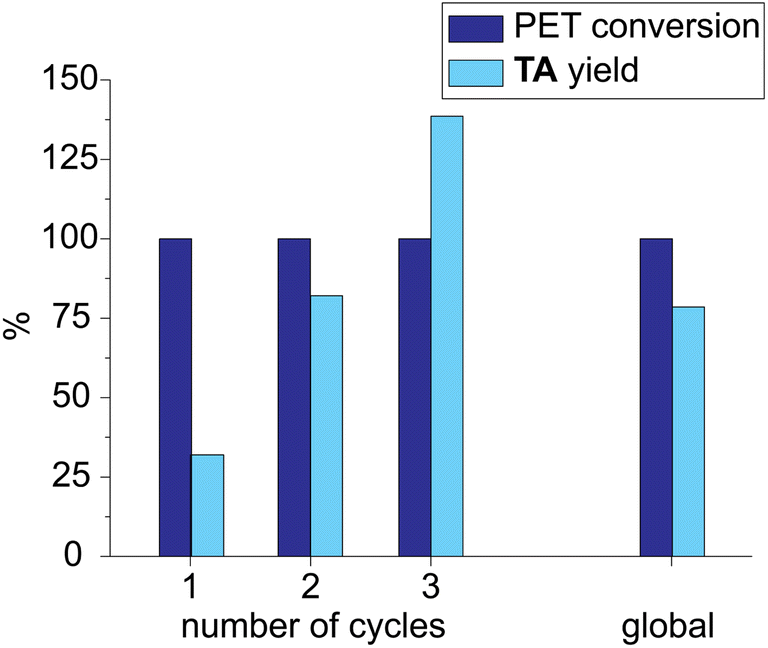 | ||
| Fig. 6 PET conversion and TA yield obtained by reusing the same LBDES for three cycles. | ||
MW irradiation can be compared to traditional heating through the environmental factor (E) defined as the mass of waste for the mass of the product. It should always be coupled with other factors, such as the energy economy (ε) and the combination of E and ε (environmental energy impact, ξ) (Table 3).13
| Run | E | ε (°C−1 min−1) | ξ (°C min) |
|---|---|---|---|
| 1 | 15.44 | 2.08 × 10−4 | 7.42 × 104 |
| 2 | — | — | — |
| 3 | — | — | — |
| 4 | 12.99 | 6.18 × 10−4 | 2.10 × 104 |
| 5 | 67.07 | 5.29 × 10−5 | 1.27 × 106 |
| 6 | 1119.14 | 2.50 × 10−6 | 4.48 × 108 |
| 7 | 12.16 | 5.13 × 10−4 | 2.37 × 104 |
| 8 | 19.09 | 5.06 × 10−4 | 3.78 × 104 |
| 9 | 19.74 | 4.89 × 10−4 | 4.04 × 104 |
| 10 | 24.40 | 5.35 × 10−4 | 4.56 × 104 |
| 11 | 5.15 | 5.27 × 10−4 | 9.79 × 103 |
| 12 | 3.46 | 5.23 × 10−4 | 6.61 × 103 |
The increase in T and decrease in t led to an improvement in the parameters (E and ξ should be minimized; ε should be maximized), and the strategy of diluting the system is particularly winning because it allows to processing of more PET concurrently. The environmental parameters are comparable to those obtained in our previous contribution27 using a much stronger acid, such as MSA (E = 2.88, ε = 4.07 × 10−5, ξ = 7.27 × 104). As depicted in Table 4, our method is compared with other methods proposed in recent literature, especially focusing on DES- and iron-based protocols.13
| Method | E | ε (°C−1 min−1) | ξ (°C min) | Ref. |
|---|---|---|---|---|
| FeCl3·6H2O/acetic acid/MW | 3.46 | 5.23 × 10−4 | 6.61 × 103 | This work |
| FeCl3·6H2O/MSA | 2.88 | 4.07 × 10−5 | 7.07 × 104 | 27 |
| NaOH/MW | 1.70 | 4.47 × 10−4 | 3.8 × 103 | 39 |
| H2SO4(aq.) | 8.87 | 2.22 × 10−5 | 3.99 × 105 | 40 |
| Urea/ZnCl2 | 0.407 | 1.63 × 10−4 | 2.5 × 103 | 41 |
| K2CO3/EG | 1.33 | 4.07 × 10−5 | 3.3 × 104 | 42 |
| FeCl3/2-ethylhexanol | 1.18 | 1.21 × 10−5 | 9.75 × 104 | 43 |
| FeCl3/ChCl/2-ethylhexanol | 0.57 | 2.65 × 10−5 | 2.15 × 104 | 43 |
To compare the sustainability of state-of-the-art PET management,23 a low temperature (100 °C) or a short time (10 min) when MWs are used makes our proposed technology competitive with other processes currently used for pyrolysis (T between 350 and 600 °C), glycolysis (T between 180 and 240 °C) or methanolysis (T between 180 and 280 °C, pressure between 20 and 40 atm).2
Conclusions
In this work, the use of the Lewis/Brønsted acidic deep eutectic solvent (LBDES) FeCl3·6H2O/acetic acid 1:1 (LBDES1) was explored for the depolymerization of PET through hydrolysis. Initial tests carried out at 100 °C for 30 minutes pointed out that LBDES1 depolymerized PET flakes with very low efficiency (PET conversion of 23% and TA yield of 4%) compared with recently investigated LBDES containing much stronger Brønsted acids.
Thus, the improvement in LBDES1 efficiency was achieved by adding a simple post-reaction procedure, specifically by keeping unreacted PET, dispersed oligomers and the crude TA for 12 h in contact with an aqueous solution of NaOH (0.5 M) at room temperature under stirring. This step significantly improved both PET conversion and yield in TA, thus making all the depolymerization processes more effective. Furthermore, the effect of chloride salts as additives was investigated, and it was found that cheap and non-toxic CaCl2 positively affected the solvent performance, thus increasing the conversion up to 69%. This effect is explained by the known ability of calcium ions to coordinate water molecules, whose excess acts as an inhibitor. Moreover, the increase in conversion and yield is not so marked as to justify the use of an additive, especially because of a scale-up of the process.
Results obtained for the same reaction carried out under microwave (MW) irradiation demonstrated that if the temperature was increased to 180 °C, the reaction was quantitative and very high TA yields were obtained at low reaction time (10 minutes), even in the presence of 10 equivalents of excess water. The presence of the latter is an important goal because it allows processing in a single reaction to a significantly higher amount of PET (PET/solvent ratio = 0.26 in mass).
Under these conditions, the environmental parameters were comparable with those of much stronger acids, with an evident improvement in corrosion, costs, eco-sustainability and the ease of disposing of exhaust solvent.
Data availability
Data will be made available on request.Author contributions
Marco Rollo: investigation, data curation, writing – original draft. Massimo A. G. Perini, Alejandro Torregrosa-Chinillach: investigation. Alessandro Sanzone, Lorenzo Polastri: supervision. Matteo Tiecco, Elisa Martinelli, Gianluca Ciancaleoni: supervision, conceptualization, formal analysis, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by the Ministry of University and Research (MUR) as part of the PON 2014–2020 “Research and Innovation” resources – Green/Innovation Action – DM MUR 1061/2022. University of Pisa is acknowledged for financial support (Fondi di Ateneo 2022 and PRA_2022_34).References
- R. Geyer, J. R. Jambeck and K. L. Law, Production, Use, and Fate of All Plastics Ever Made, Sci. Adv., 2017, 3(7), e1700782, DOI:10.1126/sciadv.1700782.
- M. Crippa and B. Morico, PET Depolymerization: A Novel Process for Plastic Waste Chemical Recycling, Stud. Surf. Sci. Catal., 2019, 179, 215–229, DOI:10.1016/B978-0-444-64337-7.00012-4.
- H. Sardon and A. P. Dove, Plastics Recycling with a Difference: A Novel Plastic with Useful Properties Can Easily Be Recycled Again and Again, Science, 2018, 360, 380–381, DOI:10.1126/science.aat4997.
- P. Johar, E. L. Rylott, C. R. McElroy, A. S. Matharu and J. H. Clark, Biologically Bound Nickel Accelerated De-Polymerization of Polyethylene to High Value Hydrocarbons and Hydrogen, RSC Sustainability, 2023, 1, 117–127, 10.1039/d2su00001f.
- Y. Liu, K. Chandra Akula, K. Phani Raj Dandamudi, Y. Liu, M. Xu, A. Sanchez, D. Zhu and S. Deng, Effective Depolymerization of Polyethylene Plastic Wastes under Hydrothermal and Solvothermal Liquefaction Conditions, Chem. Eng. J., 2022, 446(4), 137238, DOI:10.1016/j.cej.2022.137238.
- X. Wu, A. Tennakoon, R. Yappert, M. Esveld, M. S. Ferrandon, R. A. Hackler, A. M. Lapointe, A. Heyden, M. Delferro and B. Peters, et al., Size-Controlled Nanoparticles Embedded in a Mesoporous Architecture Leading to Efficient and Selective Hydrogenolysis of Polyolefins, J. Am. Chem. Soc., 2022, 144, 5323–5334, DOI:10.1021/jacs.1c11694.
- L. D. Ellis, S. V. Orski, G. A. Kenlaw, A. G. Norman, K. L. Beers, Y. Román-Leshkov and G. T. Beckham, Tandem Heterogeneous Catalysis for Polyethylene Depolymerization via an Olefin-Intermediate Process, ACS Sustain. Chem. Eng., 2021, 9, 623–628, DOI:10.1021/acssuschemeng.0c07612.
- Y. Yang, J. Liu, F. S. Kamounah, G. Ciancaleoni and J. W. Lee, A CO2-Catalyzed Transamidation Reaction, J. Org. Chem., 2021, 86, 16867–16881, DOI:10.1021/ACS.JOC.1C02077.
- A. Kamimura, Y. Shiramatsu and T. Kawamoto, Depolymerization of Polyamide 6 in Hydrophilic Ionic Liquids, Green Energy Environ., 2019, 4, 166–170, DOI:10.1016/j.gee.2019.01.002.
- O. A. Attallah, A. Janssens, M. Azeem and M. B. Fournet, Fast, High Monomer Yield from Post-Consumer Polyethylene Terephthalate via Combined Microwave and Deep Eutectic Solvent Hydrolytic Depolymerization, ACS Sustain. Chem. Eng., 2021, 9, 17174–17185, DOI:10.1021/acssuschemeng.1c07159.
- Y. Wang, Y. Zhang, H. Song, Y. Wang, T. Deng and X. Hou, Zinc-Catalyzed Ester Bond Cleavage: Chemical Degradation of Polyethylene Terephthalate, J. Cleaner Prod., 2019, 208, 1469–1475, DOI:10.1016/j.jclepro.2018.10.117.
- L. Biermann, E. Brepohl, C. Eichert, M. Paschetag, M. Watts and S. Scholl, Development of a Continuous PET Depolymerization Process as a Basis for a Back-to-Monomer Recycling Method, Green Process. Synth., 2021, 10, 361–373, DOI:10.1515/gps-2021-0036.
- E. Barnard, J. J. Rubio Arias and W. Thielemans, Chemolytic Depolymerisation of PET: A Review, Green Chem., 2021, 23, 3765–3789, 10.1039/d1gc00887k.
- L. G. Schaerer, R. Wu, L. I. Putman, J. M. Pearce, T. Lu, D. R. Shonnard, R. G. Ong and S. M. Techtmann, Killing Two Birds with One Stone: Chemical and Biological Upcycling of Polyethylene Terephthalate Plastics into Food, Trends Biotechnol., 2023, 41, 184–196, DOI:10.1016/j.tibtech.2022.06.012.
- S. Bhattacharjee, M. Rahaman, V. Andrei, M. Miller, S. Rodríguez-Jiménez, E. Lam, C. Pornrungroj and E. Reisner, Photoelectrochemical CO2-to-Fuel Conversion with Simultaneous Plastic Reforming, Nat. Synth., 2023, 2, 182–192, DOI:10.1038/s44160-022-00196-0.
- Y. Yang, S. Sharma, C. Di Bernardo, E. Rossi, R. Lima, F. S. Kamounah, M. Poderyte, K. Enemark-Rasmussen, G. Ciancaleoni and J. W. Lee, Catalytic Fabric Recycling: Glycolysis of Blended PET with Carbon Dioxide and Ammonia, ACS Sustain. Chem. Eng., 2023, 11, 11294–11304, DOI:10.1021/acssuschemeng.3c03114.
- I. Agostini, B. Ciuffi, R. Gallorini, R. A. Maria, D. Chiaramonti and L. Rosi, Recovery of Terephthalic Acid from Densified Post-Consumer Plastic Mix by HTL Process, Molecules, 2022, 27, 7112, DOI:10.3390/molecules27207112.
- A. Sheel and D. Pant, Chemical Depolymerization of PET Bottles via Glycolysis, Recycl. Polyethylene Terephthalate Bottles, 2019, 61–84, DOI:10.1016/b978-0-12-811361-5.00004-3.
- X. L. Wang, W. L. An, R. Du, F. Tian, Y. Yang, X. Zhao, S. Xu and Y. Z. Wang, Rapid Hydrolysis of PET in High-Concentration Alcohol Aqueous Solution by Pore Formation and Spontaneous Separation of Terephthalate, J. Environ. Chem. Eng., 2023, 11(2), 109434, DOI:10.1016/j.jece.2023.109434.
- J. J. Rubio Arias and W. Thielemans, Instantaneous Hydrolysis of PET Bottles: An Efficient Pathway for the Chemical Recycling of Condensation Polymers, Green Chem., 2021, 23, 9945–9956, 10.1039/d1gc02896k.
- S. Giraldo-Narcizo, N. Guenani, A. M. Sánchez-Pérez and A. Guerrero, Accelerated Polyethylene Terephthalate (PET) Enzymatic Degradation by Room Temperature Alkali Pre-Treatment for Reduced Polymer Crystallinity, ChemBioChem, 2023, 24(1), e202200503, DOI:10.1002/cbic.202200503.
- D. D. Pham and J. Cho, Low-Energy Catalytic Methanolysis of Poly(Ethyleneterephthalate), Green Chem., 2021, 23, 511–525, 10.1039/d0gc03536j.
- A. Maisels, A. Hiller and F. G. Simon, Chemical Recycling for Plastic Waste: Status and Perspectives, ChemBioEng Rev., 2022, 9, 541–555, DOI:10.1002/cben.202200024.
- H. Qin, X. Hu, J. Wang, H. Cheng, L. Chen and Z. Qi, Overview of Acidic Deep Eutectic Solvents on Synthesis, Properties and Applications, Green Energy Environ., 2020, 5, 8–21, DOI:10.1016/j.gee.2019.03.002.
- Y. Bai, X. F. Zhang, Z. Wang, T. Zheng and J. Yao, Deep Eutectic Solvent with Bifunctional Brønsted-Lewis Acids for Highly Efficient Lignocellulose Fractionation, Bioresour. Technol., 2022, 347, 126723, DOI:10.1016/j.biortech.2022.126723.
- Y. B. Jiang, F. L. Yu, B. Yuan, C. X. Xie and S. T. Yu, Preparation of High-Quality Alkylated Gasoline with Low Alkane-to-Alkene Ratio Catalyzed by Polyether-Based Brønsted-Lewis Acidic Deep Eutectic Solvent, Fuel, 2023, 340, 127565, DOI:10.1016/j.fuel.2023.127565.
- M. Rollo, F. Raffi, E. Rossi, M. Tiecco, E. Martinelli and G. Ciancaleoni, Depolymerization of Polyethylene Terephthalate (PET) under Mild Conditions by Lewis/Brønsted Acidic Deep Eutectic Solvents, Chem. Eng. J., 2023, 456, 141092, DOI:10.1016/j.cej.2022.141092.
- S. S. Chadwick, Ullmann's Encyclopedia of Industrial Chemistry, ed. B. Elvers, S. Hawklins and G. Schulz, Wiley-VCH Verlag GmbH & Co. KGaA, Hoboken, NJ, USA, 1988, vol. 16, DOI:10.1108/eb049034.
- J. R. Andreesen, A. Schaupp, C. Neurauter, A. Brown and L. G. Ljungdahl, Fermentation of Glucose, Fructose, and Xylose by Clostridium Thermoaceticum: Effect of Metals on Growth Yield, Enzymes, and the Synthesis of Acetate from CO 2, J. Bacteriol., 1973, 114, 743–751, DOI:10.1128/jb.114.2.743-751.1973.
- E. Budsberg, R. Morales-Vera, J. T. Crawford, R. Bura and R. Gustafson, Production Routes to Bio-Acetic Acid: Life Cycle Assessment, Biotechnol. Biofuels, 2020, 13, 154, DOI:10.1186/s13068-020-01784-y.
- Y. Luo, E. Selvam, D. G. Vlachos and M. Ierapetritou, Economic and Environmental Benefits of Modular Microwave-Assisted Polyethylene Terephthalate Depolymerization, ACS Sustain. Chem. Eng., 2022, 11, 4209–4218, DOI:10.1021/acssuschemeng.2c07203.
- N. Agmon, The Grotthuss Mechanism, Chem. Phys. Lett., 1995, 244, 456–462, DOI:10.1016/0009-2614(95)00905-J.
- M. D. Lind, Crystal Structure of Ferric Chloride Hexahydrate, J. Chem. Phys., 1967, 47, 990–993, DOI:10.1063/1.1712067.
- T. Chen, W. Zhang and J. Zhang, Alkali Resistance of Poly(Ethylene Terephthalate) (PET) and Poly(Ethylene Glycol-Co-1,4-Cyclohexanedimethanol Terephthalate) (PETG) Copolyesters: The Role of Composition, Polym. Degrad. Stab., 2015, 120, 232–243, DOI:10.1016/j.polymdegradstab.2015.07.008.
- D. Stanica-Ezeanu and D. Matei, Natural Depolymerization of Waste Poly(Ethylene Terephthalate) by Neutral Hydrolysis in Marine Water, Sci. Rep., 2021, 11, 4431, DOI:10.1038/s41598-021-83659-2.
- N. Trejo-Carbajal, K. I. Ambriz-Luna and A. M. Herrera-González, Efficient Method and Mechanism of Depolymerization of PET under Conventional Heating and Microwave Radiation Using T-BuNH2/Lewis Acids, Eur. Polym. J., 2022, 175, 111388, DOI:10.1016/j.eurpolymj.2022.111388.
- J. A. Abedi and N. M. Roscher, Transesterification with Hydrated Aluminum Chloride: A Convenient Method for the Transesterification of Long Chain Alcohols and Acid Sensitive Carboxylic Acids, Synth. Commun., 1989, 19, 1539–1549, DOI:10.1080/00397918908051049.
- Q. Li, X. Yin, W. Duan, L. Kong, B. Hao and F. Ye, Electrical, Dielectric and Microwave-Absorption Properties of Polymer Derived SiC Ceramics in X Band, J. Alloys Compd., 2013, 565, 66–72, DOI:10.1016/j.jallcom.2013.02.176.
- A. Căta, M. N. Ştefănuţ, I. M. C. Ienaşcu, C. Tănasie and M. Miclău, Alkaline Hydrolysis of Polyethylene Terephthalate under Microwave Irradiation, Rev. Roum. Chim., 2017, 62, 531–538 Search PubMed.
- S. D. Mancini and M. Zanin, Post Consumer Pet Depolymerization by Acid Hydrolysis, Polym.-Plast. Technol. Eng., 2007, 46, 135–144, DOI:10.1080/03602550601152945.
- Q. Wang, X. Yao, Y. Geng, Q. Zhou, X. Lu and S. Zhang, Deep Eutectic Solvents as Highly Active Catalysts for the Fast and Mild Glycolysis of Poly(Ethylene Terephthalate)(PET), Green Chem., 2015, 17, 2473–2479, 10.1039/c4gc02401j.
- E. Sert, E. Yılmaz and F. S. Atalay, Chemical Recycling of Polyethlylene Terephthalate by Glycolysis Using Deep Eutectic Solvents, J. Polym. Environ., 2019, 27, 2956–2962, DOI:10.1007/s10924-019-01578-w.
- L. Zhou, X. Lu, Z. Ju, B. Liu, H. Yao, J. Xu, Q. Zhou, Y. Hu and S. Zhang, Alcoholysis of Polyethylene Terephthalate to Produce Dioctyl Terephthalate Using Choline Chloride-Based Deep Eutectic Solvents as Efficient Catalysts, Green Chem., 2019, 21, 897–906, 10.1039/c8gc03791d.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00205e |
| This journal is © The Royal Society of Chemistry 2024 |