
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable solvent extraction of gold and other metals with biomass chemicals†
Mark R. StJ.
Foreman
 *a,
Richard K.
Johansson
a,
Gloria
Mariotti
a,
Ingmar
Persson
b,
Behabitu E.
Tebikachew
a and
Mikhail S.
Tyumentsev
a
*a,
Richard K.
Johansson
a,
Gloria
Mariotti
a,
Ingmar
Persson
b,
Behabitu E.
Tebikachew
a and
Mikhail S.
Tyumentsev
a
aDepartment of Chemistry and Chemical Enginnering, Chalmers University of Technology, Goteborg, Sweden. E-mail: Foreman@chalmers.se
bDepartment of Molecular Sciences, Swedish University of Agricultural Sciences, Uppsala, Sweden
First published on 9th January 2024
Abstract
This paper presents the basis for an industrial gold purification process using an organic phase made from biomass derived organic chemicals is presented. The use of a sustainable kerosene (a biodiesel) as a diluent in a solvent extraction experiment is important as it allows the solvent extraction of metals to be done without petroleum kerosene. A chemical change in the organic phase which reduces the gold distribution ratio has been observed and rationalized with XANES/EXAFS, this change offers path towards more sustainable flowsheets for gold. A second process is also presented using biomass derived diluent, the parameters required to calculate the ratio of the activity functions of chloride and the tetrachloroaurate(III) complex in choline chloride media are within this paper. Additionally solvent extraction experiments using the new sustainable diluent for lithium and alkaline earth elements together with proof of principle experiments with stable lanthanides, 99mTc, 152Eu, 238Pu and 239Pu were performed.
Sustainability spotlightThis paper relates to goal 12 (sustainable consumption and production), a clear need exists for metal purifications during recycling and primary production. Solvent extraction is often used, industrial solvent extraction often uses unsustainable petroleum derived kerosene as a diluent and harmful extractants such as carcinogenic tributyl phosphate. In this paper we show how it is possible to selectively extract gold using an organic layer made from biomass derived diesel fuel with an extractant which can be formed from biomass. We also show how the biomass derived diluent has clear potential as a replacement for petroleum derived kerosene. With another sustainable diluent we enable the collection of data which will assist in process simulation and the rational design of separation processes. |
Introduction
Without good separations it will be impossible to obtain the pure materials needed for many applications. These separations are needed in both recycling and primary production. The ideal separation will produce pure materials, recover the vast majority of the target material, produce no additional waste and use little energy. For example while lithium ion cells can be recycled in a furnace, a large amount of energy is needed to operate the furnace1 and the metal recovery was inferior to that obtained with hydrometallurgy.2 Solvent extraction (liquid–liquid extraction) is a type of hydrometallurgical purification method when used in counter current extraction systems (such as mixer settlers) is able to produce high purity metals with very small losses of the wanted metal. In addition to solvent extraction as a purification method it is also used to determine thermodynamic constants,3 produce speciation data4 and is used in other scientific tasks.Some of the chemicals used in the solvent extraction of metals are profoundly harmful. For example, in academic research benzene has been used as a diluent in AKUFVE5 studies of lanthanide extraction.6 Diluent is the IUPAC preferred term for the liquid used to dissolve the extractants used to form the solvent phase. While the toluene7 used in high speed solvent extraction (SISAK8) used for research on superheavy elements is less carcinogenic than benzene in its acute toxicity is higher, at some radioactivity sites one is not allowed to handle the stock bottle in a fume hood for fire safety reasons thus requiring the chemist to handle it on the open bench.9 Within the solvent extraction community terms such “solvent” have different meanings to those used in other areas of chemistry, within this paper the IUPAC recommended nomenclature10 for solvent extraction is used. While tributyl phosphate has been used in industry for the purification of plutonium,11 rare earths12 and has been suggested as an extractant for gold13 it is a rodent carcinogen14 and the incineration of used solvent containing it will generate acidic materials such as P2O5.
At Chalmers and elsewhere an effort has been made to develop safer and more sustainable chemistry for the solvent extraction of metals. Often it has concentrated on the extractant, for example the phosphorus containing extractants used in the TRUEX15 process were replaced with malonamides to form the DIAMEX16 process. In industry a range of different fluids are used as diluents. It has been common to use kerosene type solvents for large scale solvent extraction processes, for example in the late 1980s the industrial scale solvent extraction of copper using hydroxyoximes used diluents such as Escaid100‡ and ordinary kerosene.17 By careful choice of diluent useful outcomes can be obtained. For example, Sellafield's reprocessing plants used a kerosene containing aromatic compounds to minimise tributyl phosphate degradation.18 As well as optimizing the performance of a process it is important to note that as the diluent is often the majority of the organic phase, by using a more sustainable diluent a greater fraction of the organic phase can be made from green chemicals. We wanted to test the hypothesis that gold (and other metals) could be extracted using a sustainable diluent in place of the petrochemical based diluents already used.
We present the use of an artificial kerosene type diluent. We are not alone in using artificial kerosenes, already AREVA's PUREX plants at La Hague (France) have successfully used an artificial kerosene (hydrogenated propylene tetramer) on the industrial scale.19 Within this paper we make the advance of using a sustainable artificial diluent. We present a metal extraction system for gold recycling in which all the organic chemicals used are sustainable. The new organic phase consists of a malonamide extractant diluted in a second generation biodiesel (HVO100). These are intended to be an advancement over the use of diethylene glycol dibutyl ether (dibutylcarbitol) by Vale and the use of methyl isobutyl ketone (MIBK) by Johnson Matthey/Rustenberg.20 We also reason that our HVO100 based organic phase will be less flammable than the 2,2,4-trimethyl-1,3-petanediol diisobutyrate advocated for the solvent extraction of gold.21 We also present more general results which indicate that HVO100 is a suitable replacement for other aliphatic kerosene grades in the solvent extraction of metals. For clarity the main organic phases for gold extraction are summarised in Table 1.
| Extractant | Diluent | Purpose | User |
|---|---|---|---|
| Dibutylcarbitol | Dibutylcarbitol | Gold production | Vale |
| MIBK | MIBK | Gold production | Johnson Matthey/Rustenberg |
| Tributyl phosphate | Kerosene | Gold recycling (suggestion) | Kim et al.13 |
| Tetra-2-ethylhexyl malonamide | HVO100 biodiesel | Gold recycling (suggestion) | This paper |
| Aliquat 336 | Dimethyloctanol | Gold recycling (suggestion) | This paper |
Experimental
HVO100 diesel fuel was purchased from a Tanka filling station in Vara (Sweden), it was made by Neste (Finland). Solvent 70 is a mixture of alkanes and cycloalkanes, this diluent (organic solvent) was made by Statoil (a Norwegian petroleum company, in 2018 the company changed name to Equinor). Tetrakis-(2-ethylhexyl) malonamide was made by the reaction of diethyl malonate and bis-(2-ethylhexyl) amine, this was vacuum distilled before use. Cyanex 923 was supplied by Cytec (since it was supplied Cytec was aquired by Solvay). Scrap gold was purchased from Pantbanken Sverige (a pawn broker), this was digested in aqua regia. The silver chloride was removed by centrifugation before the mixture of copper and gold chlorides was concentrated by evaporation. Alumina was supplied by Merck. Chlorine gas was generated by the reaction of potassium permanganate and concentrated hydrochloric acid.22 Argon gas was supplied by AirLiquid. All other chemicals were supplied by Aldrich. Further details of the materials, equipment and methods are given in the ESI.†A Inductively Coupled Plasma Mass Spectrometer (iCAP Q equipped with a ASX520) and Inductively Coupled Plasma Optical Emission Spectrometer (iCAP Pro equipped with a ASX560) equipment supplied by Thermoscientific were used to measure the concentrations of metals in aqueoussolutions. Gas chromatography mass spectroscopy experiments were conducted with a 7820A gas chromatography machine (Agilent) coupled with a 5977E MSD mass spectroscopy detector. Liquid scintillation counting of tritium and plutonium-238 (238Pu) was done using a Wallac Guardian 1414 liquid scintillation counter (PerkinElmer life sciences) using glass vials (7 ml). Other alpha counting was performed using a semiconductor detector supplied by ORTEC.
The majority of the metal distribution ratios were obtained in non-radioactive experiments. In these experiments metal solutions in aqueous (or mixed ethylene glycol/water) solutions of salts were shaken with equal volumes of organic phases in small (3.5 ml) glass vials with plastic lids. The distribution ratio is defined as the total concentration of an element in the organic layer divided by the total concentration of the element in the aqueous layer. If the organic phase contains 12 grams per litre of a metal in form A, while the aqueous layer contains 1 gram per litre of metal in form A and 2 grams per litre in form B then the distribution ratio will be four. Unlike equilibrium constants, which are strictly speaking defined using activites, the correct definition of distribution ratio is in chemical concentrations. Before shaking the tubes in a thermostated shaking system the lids were further secured with parafilm. After shaking the tubes were centrifuged before the lower layers were sampled with a pipette. To do this the pipette was adjusted to less than the volume of the lower phase (typically 400 μl samples were taken from 700 μl layers), a polyethene tip was attached to the pipette, the button on the pipette was depressed to the first stop before the tip touched the top layer in the vial. The tip was inserted into the lower phase. A small bubble of air was squeezed out of the tip by pressing harder on the control button before the control button was allowed to slowly rise until it was at the top of its travel. The pipette and tip were lifted out of the shaking vial, the outside of the tip wiped with paper towel before the sample was dispensed into a preweighed polyethene tube (15 ml) by depressing the control button to the first stop and then further down to the second and final stop. The samples were diluted before being examined with either ICPMS (iCAP Q) or ICPOES (iCAP Pro) supplied by Thermo Scientific. Further details of the experimental method and the individual experiments are presented in the ESI.†
All radiochemistry with tritium (3H), 99mTc, 152Eu and 238Pu (chemistry with radioactive substances) was performed inside a fumehood using a protocol which can be summed up as “clean hand/dirty hand”, in this protocol a hand is either gloved (and thus able to touch and manipulate objects which have radioactivity on their surfaces) or ungloved (thus able to operate the push button of the pipette). Always use a filter tip on the pipette to prevent radioactive aerosol droplets entering the pipette. The chemistry with 239/240Pu was performed in a negative pressure glovebox suitable for working with gram amounts of plutonium. The 239/240Pu samples were transferred from the glovebox to a fumehood where stainless steel planchets were prepared for alpha counting with a semiconductor detector. Both the 3H and 238Pu samples were measured with liquid scintillation counting. Gamma spectroscopy using germanium crystals cooled to liquid nitrogen temperature was used to measure 99mTc and 152Eu. The gamma lines at 140 and 122 keV were used to measure these radionuclides. The measurements were made using a detector within a lead castle lined with copper sheet (the copper sheet was to reduce the production and influence of secondary radiation on the counting process). The sampling of the liquids from the radioactive experiments was done in a different way, samples of the organic phases were collected by pipetting normally from upper layer with the push button pipette. The lower phase was sampled in the following manner (which requires more skill), start with one's thumb on the pipette button just before you touch the upper layer start to squeeze down. As you lower the tip through the upper layer a series of air bubbles will be blown out, continue to lower the tip while blowing bubbles. When your thumb reaches the first stop of the pipette stop pressing down and slowly relax the pressure on your thumb thus sucking up a sample. Take your thumb off the button and then raise the pipette tip out of the vial. Now transfer the sample either into a liquid scintillation counting vial (already containing the cocktail) for LSC dispense the liquid into a 6 ml high density polyethene tube with a screw cap for gamma counting. For transferring plutonium samples out of the glovebox for the creation of counting sources on a stainless steel disk place the sample in a polyethene tube. The sealed polyethene tube is transferred out of the glovebox and is taken into a radiochemical fumehood for further processing. As it is impossible for a nonradiochemist to learn how to safely perform this work by merely reading an instruction manual no further experimental details are given.
Gold L3-edge X-ray absorption data was collected in transmission mode at ambient temperature at the Balder synchrotron radiation beam-line at the MAX IV Laboratory, Lund University, Sweden.23,24 The solutions were contained in liquid cells made up by 2 mm Teflon spacers, Kapton foil as windows hold together with titanium frames. The Balder beamline is supplied by the MAX IV 3 GeV and 250 mA storage ring, which is maintained by top-up mode. For each sample three repeats were accumulated into an average spectrum. To minimize radiation damage, only a single EXAFS scan was recorded on each sample position. Immediately before the start of each repeat, the sample was shifted by 0.20 mm into a fresh position to reduce its radiolysis. The radiation was monochromatized by a Si[111] double crystal monochromator and mirrors were used to reject higher harmonics. The X-ray absorption spectra were energy calibrated by recording the XANES spectrum of a gold foil before and after the data collection of the samples with the first inflection point of the gold foil with the L3-edge defined as 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 919.0 eV.25 The experimental absorption spectra were treated by using standard procedures for pre-edge subtraction and spline removal and Fourier transformation by means of the program package EXAFSPAK.26Ab initio calculated EXAFS parameters, generated by the program FEFF v. 7.0,27 were used in the non-linear curve-fitting procedure.
919.0 eV.25 The experimental absorption spectra were treated by using standard procedures for pre-edge subtraction and spline removal and Fourier transformation by means of the program package EXAFSPAK.26Ab initio calculated EXAFS parameters, generated by the program FEFF v. 7.0,27 were used in the non-linear curve-fitting procedure.
Results and discussion
Malonamide in a sustainable alkane diluent (HVO100)
As the majority of the organic (solvent) phase's volume is often diluent, it was reasoned that the highest priority was to identify a sustainable diluent. We reasoned that the ideal diluent should be cheap, readily available, not toxic and its production should be sustainable. We reasoned that we should consider using a sustainable fuel which is made in large volumes as our diluent rather than attempting to devise a completely new diluent. We considered highly refined lamp oil and medical paraffin, but these were rejected for cost reasons and because the medicinal paraffin was far more viscous than the standard aliphatic kerosene type diluent used at Industrial Materials Recycling at Chalmers (Statoil's solvent 70). As spark ignition engine fuel (known as petrol in the UK) is volatile and has a low flash point, we choose to consider the heavier fuel oils used for compression ignition (Diesel) and turbofan (Jet) engines. While waste vegetable oil can be used without chemical modification in diesel engines its higher viscosity, low cetane number and other properties makes it less attractive as a fuel.28 While transesterification with methanol forms fatty acid methyl ester (FAME) diesel fuel which is more suitable for use in unmodified diesel engines for road vehicles than the unmodified vegetable oil, it is not a good diluent for solvent extraction. Its use caused the unwanted extraction of lanthanides by an organic phase containing Aliquat 336.29 More recently by hydrodeoxygenation vegetable oils30 have been converted into fuels31 which have been marketed by companies such as skyNRG and Neste. These fluids, which are devoid of aromatic compounds, are attractive as it has been shown in rats that dearomatized (catalytically hydrogenated) white spirit is less neurotoxic than a conventional white spirit containing 20% aromatics.32We purchased a sample of Neste's hydrotreated vegetable oil biodiesel which is sold as HVO100 in Sweden. Examination with GCMS indicated that this product was a mixture of alkanes with up to 18 carbons, the smallest major group of alkanes had 13 carbons. As HVO100 is a mixture of long chain alkanes, we decided to use it as alternative to an aliphatic petroleum-based diluent (solvent 70). The range of solvent extraction systems for gold33 include extraction from hydrochloric acid with tributyl phosphate34 or with trialkyl phosphine oxides (Cyanex 923).35 We decided that the use of a phosphorus containing extractant was undesirable. We reasoned that an amide extractant should be used instead-from our studies of activity issues in deep eutectic solvents we knew that gold can be extracted from chloride media using a malonamide diluted in solvent 70.36 As butanol can be obtained by fermentation,37 we reasoned that by chemical processing38–40 bis-(2-ethylhexyl) amine could be made from biomass. As E. Coli41 and Pichia Kudriavzevii42 can be used to produce malonic acid, we reason that N,N,N′,N′-tetrakis(2-ethylhexyl)malonamide43 can be made from biomass. While in this paper we have not used N,N,N′,N′-tetrakis(2-ethylhexyl)malonamide made from biomass. But as Wöhler demonstrated that biologically formed compounds have no “vital force”,44 we reason that an extractant formed using an unknown or even petroleum feedstock will have the same properties as one formed from biomass.
When a solution of gold(III) and copper(II) in chloride media was shaken with an equal volume of a solution of N,N,N′,N′-tetrakis(2-ethylhexyl) malonamide in HVO100 the vast majority of the gold was extracted (DAu = 13.7, 93% of the gold extracted in a single stage (1:1 phase ratio)). While gold(III) can be extracted with diethyl ether and diisopropyl ether from hydrobromic45 and hydrochloric46 acids respectively. We reason that our extraction chemistry is more sustainable due to the exceptional flammability of these dialkyl ethers and the tendency of diisopropyl ether to form explosive peroxides.47 Gold can be extracted from aqua regia with tributyl phosphate48 but this extractant is a rodent carcinogen so we choose to avoid it as much as possible, Cox reports that while INCO (now Vale) used dibutylcarbitol (diethylene glycol dibutyl ether) Matthey Rustenberg used MIBK (methyl isobutyl ketone) for the extraction of gold on the industrial scale.49 Cox also reports that in the Lonrho (now named Lonmin) flowsheet avoided solvent extraction of gold. The chloride leach liquor bearing gold, silver and platinum group metals in the Lonrho process is treated with sulfur dioxide to precipitate both gold and silver in metallic form.
For analytical separations MIBK has been used in conjunction with graphite furnace atomic adsorption spectroscopy (GF-AAS) to measure the gold content of aqua regia digests of gold ores,50 an alternative analytical method is to extract gold(III) from chloride media with DIBK (Diisobutyl ketone) on a polyacrylamide resin (Amberchrom CG71) followed by stripping from the resin with aqueous ammonia.51 MIBK is very flammable (flash point 17 °C) and has a high solubility in water (19 g dm−3)52 while dibutylcarbitol is expensive and it has a density (880 g dm−3) similar to water.53 We reason as the flash point of HVO100 is above 61 °C54 and as long chain alkanes are very insoluble in water that it is better than MIBK. As HVO100 is produced and used in bulk as a diesel fuel it will be far cheaper than dibutylcarbitol, it is also noteworthy that HVO100 has a low density (780 g dm−3).55 A density decrease from 880 to 780 grams per litre will increase the buoyancy of a droplet of diluent by a factor of 1.85 when the droplet is in water (density 997 g dm−3), so we reason that mixer settlers the phase separation will be better for an organic phase mainly made of HVO100 than for dibutylcarbitol.
Under the conditions of our first experiment 7% of the gold would be lost if a single extraction stage was used. But multiple stages of counter-current extraction are used then a higher recover of gold will occur. The fraction of a metal which will be rejected to the raffinate from a counter-current extraction system can be predicted from equation one56 where n is the number of stages and P is the mathematical product of the distribution ratio and the phase ratio (Vorg/Vaq). Within the ESI† a sheet is provided which indicates the meaning of all the symbols used in equations in this paper.
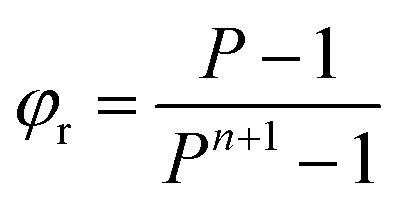 | (1) |
With three stages of counter-current extraction with a 1:1 phase ratio only 0.04% of the gold will be lost into the raffinate. As the copper distribution ratio was only 0.03, only circa 3% of the copper in the feed to the extraction plant will be extracted. This suggests that the simple process will decontaminate the gold by a factor of circa 33. The gold loaded organic phase was shaken with ethaline57,58 and the distribution ratio was 0.4 which suggests that with three counter-current stages of back extraction (stripping) that 96% of the gold would be stripped. While we had identified a system suitable for the purification of gold during our first attempt to deepen our understanding of the system additional experiments were performed (Fig. 1). A range of concentrations (0, 5, 10, 20 and 30% v/v)§ of N,N,N′,N′-tetrakis-2-ethylhexyl malonamide in HVO100 were shaken with the solution of copper(II), gold(III), lead(II), palladium(II) and zinc(II) chlorides in acidic sodium chloride, it was found that very little copper, lead and zinc is extracted (D ≤ 0.1) but the palladium and gold were well extracted.
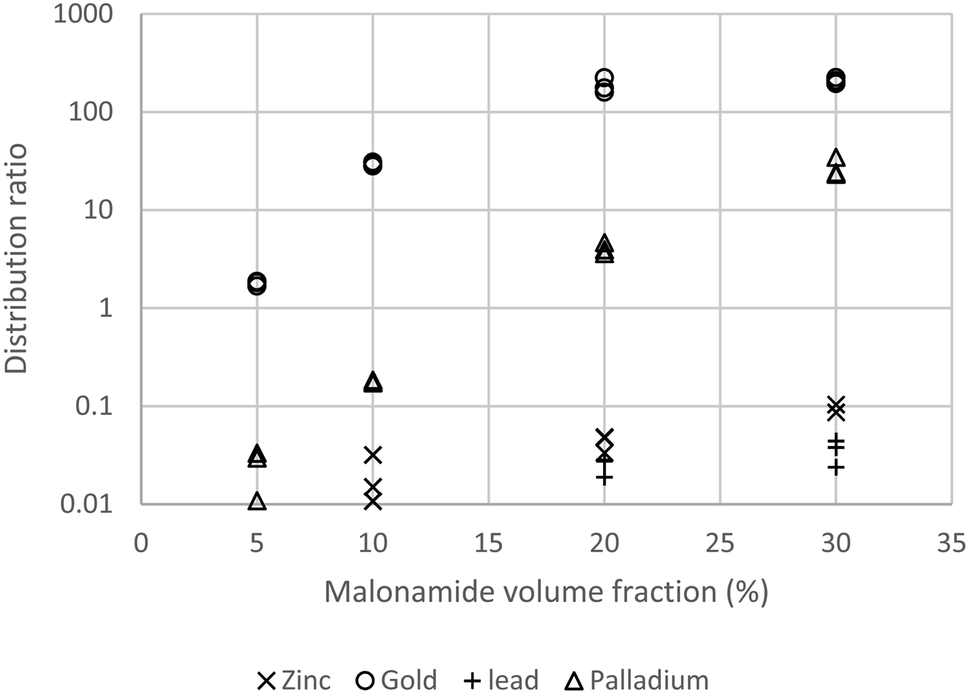 | ||
| Fig. 1 Distribution ratios of gold, lead, palladium and zinc obtained with different solutions of N,N,N′,N′-tetrakis-2-ethylhexyl malonamide in HVO100. The copper distribution ratio was below 0.02 for all conditions tested. The wavelengths used were 208.2 nm (Au), 221.8 nm (Cu), 220.4 nm (Pb), 229.7 nm (Pd) and 206.2 nm (Zn). | ||
These results suggest that it will be possible to remove the common impurities (silver, copper, zinc and palladium) from gold jewellery.59 Lead was included as sometimes 210Pb is found in jewellery60 as a result on improper recycling of gold encapsulated radiotherapy sources in the past.¶
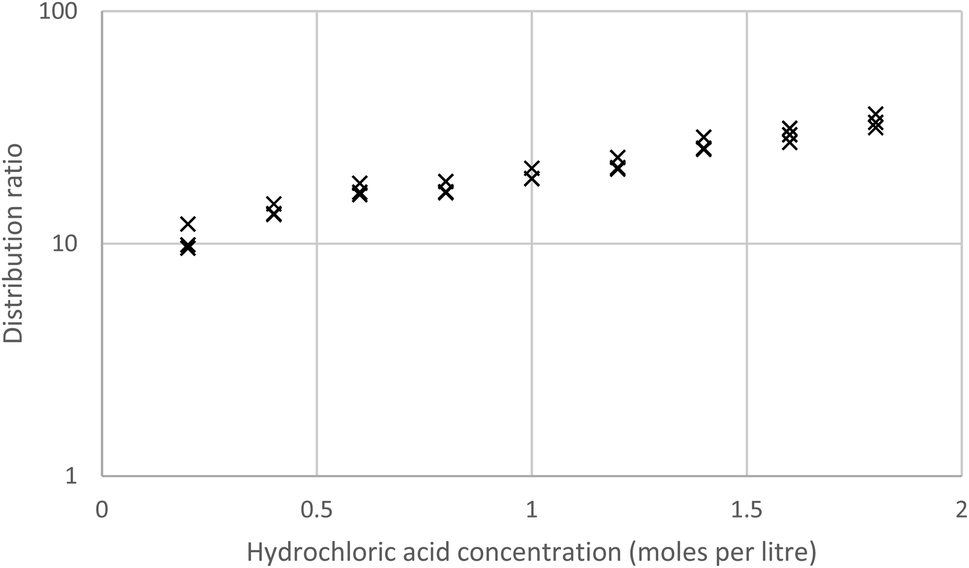
As MIBK extracts iron(III) as a MIBK complex of HFeCl4 (ref. 61) while malonamides can extract iron(III) as either complexes of FeCl3 or HFeCl4 (ref. 62) we compared the extraction of gold with that of iron from acidic chloride media with both MIBK and a 10% solution of the malonamide in HVO100. We made mixed metal solutions of aluminium, cadmium, cerium, cobalt, iron, gold, lanthanum, neodymium, praseodymium and zinc with a total chloride concentration of 2 moles per litre, where the hydrochloric acid content was allowed to vary. This was shaken with both MIBK and our malonamide solution. With the 10% solution of the malonamide solution and a contact time of two hours the gold distribution ratio tended to increase as the acid concentration of the aqueous was increased (Fig. 2). When one group of three tubes (1.8 M acid) were shaken for a far longer time an unexpectedly low distribution ratio was obtained. A jack-knife63 analysis indicates that these three points for 1.8 M acid are deviating from the line formed by the other points. Using all the data when the distribution ratio is given by D = m[HCl] + c, the values of m and c are 25.48 and 17.99 with standard deviations of 4.38 and 2.94. When the values were recalculated after excluding those nine points which were judged to contribute most to the random errors then the values were 30.55 and 16.86 with standard deviations of 1.93 and 1.94 respectively.
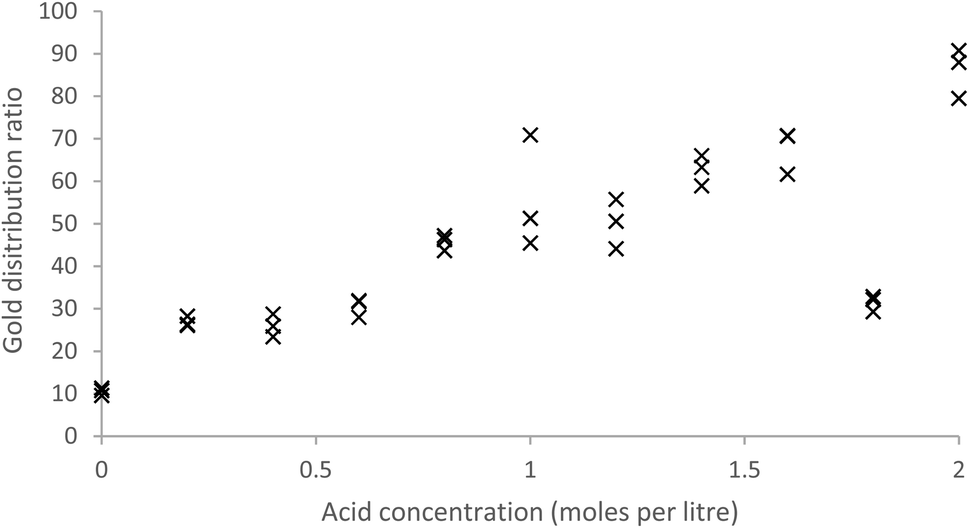 | ||
| Fig. 2 Distribution ratios obtained by shaking a 10% (v/v) solution of the malonamide (N,N,N′,N′-tetrakis-2-ethylhexyl malonamide) in HVO100 with gold containing aqueous solutions containing a total of 2 moles per litre of inorganic chloride, but different amounts of hydrochloric acid. | ||
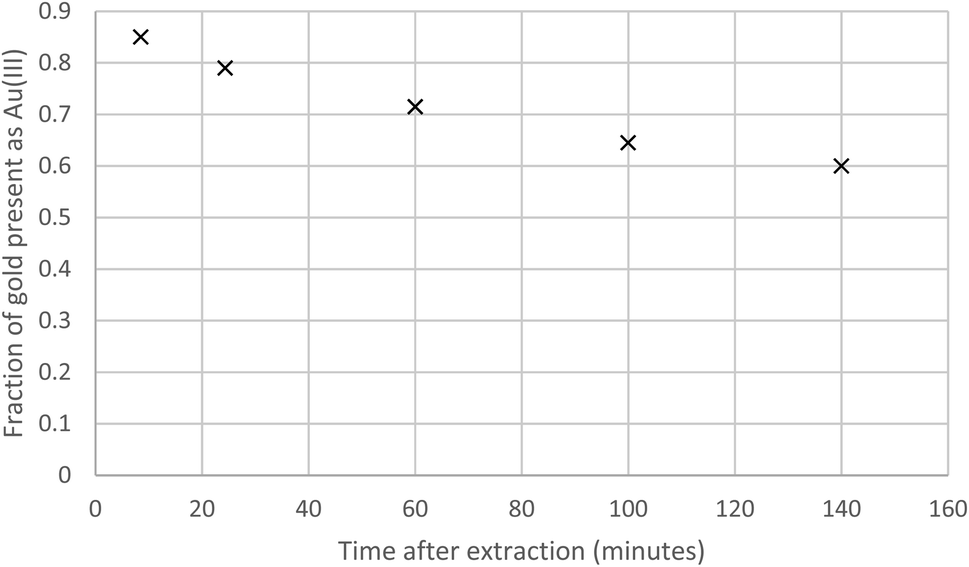
The association of the long shaking time with the lower gold distribution ratio, made us question our assumption that shaking experiment rapidly reaches equilibrium. A 10% (v/v) solution of the malonamide (N,N,N′,N′-tetrakis-2-ethylhexyl malonamide) in HVO100 was shaken with a solution of gold in a mixture of aqueous hydrochloric acid and sodium chloride. It was clear in Fig. 3 that when the shaking time was increased from six minutes to over 100 minutes that the distribution ratio decreased.
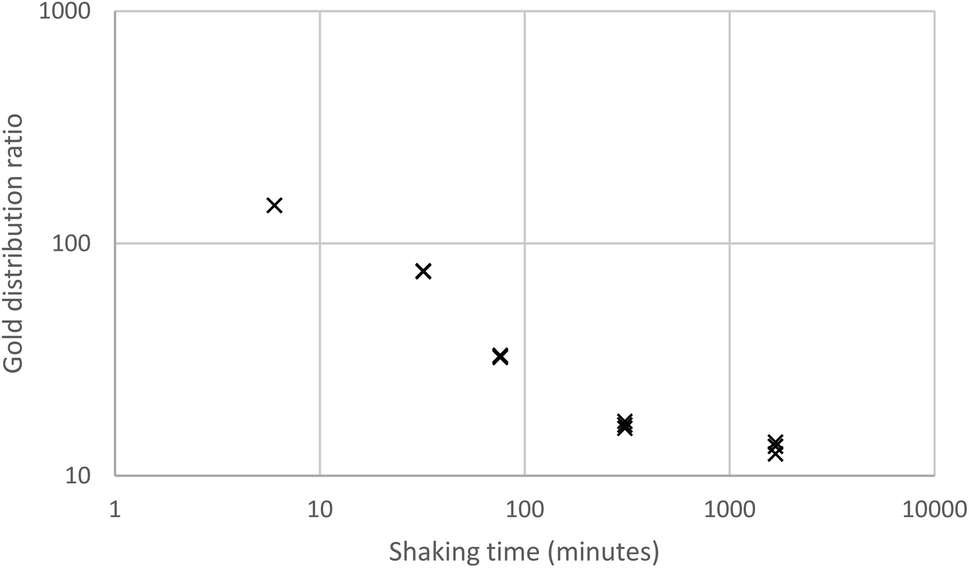 | ||
| Fig. 3 Variable shaking time experiment for the gold extraction using a 10% (v/v) solution of the malonamide (N,N,N′,N′-tetrakis-2-ethylhexyl malonamide) in HVO100. | ||
A reanalysis of the preliminary experiment published by ourselves in PCCP using a jackknife method64 for the experiment using 20% N,N,N′,N′-tetrakis-2-ethylhexyl malonamide in solvent 70 using either a two or four hour shaking time indicates that the distribution ratio is 1.288 ± 0.120 when all the data is included. When only the data for four hours is used then the distribution ratio was only 1.273 ± 0.246, when the data for a two hour contact time was used then the distribution ratio was 1.303 ± 0.115. Hence, we determined that it is likely that the evolution in the gold chemistry in that ICPMS based experiment is likely to have been masked by random errors. We reason that, due to chemical changes which occurs in the malonamide based system on a timescale of minutes to tens of minutes, that extraction with tetraalkyl malonamides in alkane dilutents, where possible, should not be used for the collection of the parameters for activity function equations or other thermodynamic data for gold. We reason that a chemical change which causes the gold distribution ratio to decline may be advantageous to the design of a sustainable flowsheet for gold purification. We reason if the gold can be extracted using a solvent extraction machine where the residence time of the organic phase is short, if the gold loaded organic phase is then allowed to age before stripping (back extracting) then if it can be stripped without the need for aqueous reagents which would later inhibit gold extraction. Then if it is possible to readily restore the gold to the well extracted form then we would have new opportunities to design better flowsheets. We tested the hypothesis that oxidation of the gold could be used to enable re-extraction after a redox (in situ reduction in the organic phase) had been used to assist the stripping of the gold. A solution of gold in a malonamide/HVO100 mixture which had been allowed to stand for four months was shaken with aqueous sodium chloride (3 M). The resulting gold loaded sodium chloride was combined with aqueous sodium chloride (3 M), hydrochloric acid (3 M) and water to form a mixture which contained gold in 1.8 M hydrochloric acid and 0.2 M sodium chloride. This stock was split in two. Half of it was bubbled with chlorine gas (three minutes) while the other half was not altered. Using a short shaking time (15 seconds) the distribution ratio (Table 2) was obtained with a 10% (v/v) solution of the malonamide in HVO100 (using triplicate shaking). The distribution ratio obtained with the chlorine treated gold solution was greater than that obtained with the untreated solution. A second group of three tubes were set up with the oxidized gold solution and the same organic phase. These were shaken and allowed to stand for 22 hours before being shaken again.
| Chlorine treated | Contact time | D Au |
|---|---|---|
| No | 15 seconds | 52.5 ± 5.0 |
| Yes | 15 seconds | 900 ± 187 |
| Yes | 22 hours | 35.9 ± 1.5 |
As the gold distribution ratio obtained with the long contact time for the chlorine treated aqueous was lower than that obtained with the nonchlorine treated aqueous we suspect that in acidic chloride media atmospheric oxygen|| is able to oxidize AuCl2− anions into AuCl4− anions. The AuCl4−/AuCl2− couple is reported to be equal to 0.9251 + 0.0296log10[AuCl4−] − 0.0296log10[AuCl2−] − 0.0591[Cl−] volt.65 Our experiment using chlorine gas indicates that it is possible to manipulate the gold distribution ratio using oxidation agents on the aqueous phase and to depend on the reduction of the gold in the organic phase. Strong gold solutions were shaken with N,N,N′,N′-tetrakis-2-ethylhexyl malonamide in dodecane before the organic phase was examined with EXAFS and XANES over a week after being formed. We used organic phases obtained by extracting gold from aqueous solutions with either high or low concentrations of hydrochloric acid. A solution of gold(III) chloride in sodium chloride was used as a structural standard, the vast majority of the gold(III) in this solution is in the form of AuCl4− anions. The gold complex is a square planar complex. This aqueous solution was used as a structural standard for the EXAFS experiment.
Both organic gold solutions had identical EXAFS functions. The amplitude of the of the EXAFS function of the aqueous gold(III) solution is about twice that of the organic gold solutions, Fig. 4. This shows that it is expected that the number of chloride ligands is twice as many in the aqueous gold(III) complex than organic gold solution. It was possible for both aqueous and organic samples to fit the raw data to a model in which the gold complexes were homoleptic chloride complexes. From refinements of the raw EXAFS data it was possible to establish that the gold-chloride distance in the organic samples was 2.265 Å while for the aqueous sample it was 2.284 Å (Fig. 5). The homoleptic nature of the complexes and their consistency with the observed gold-chlorine distances in the [AuCl2]− and [AuCl4]− complexes in the solid state suggest that the gold underwent a reduction in the organic phase.66,67 However, when the organic phase was menthone, 30% Aliquat 336 in dimethyloctanol or 30% Aliquat 336 in eucalyptol no reduction of gold(III) to gold(I) was observed. In these three organic phases the gold was present as [AuCl4]− complexes.
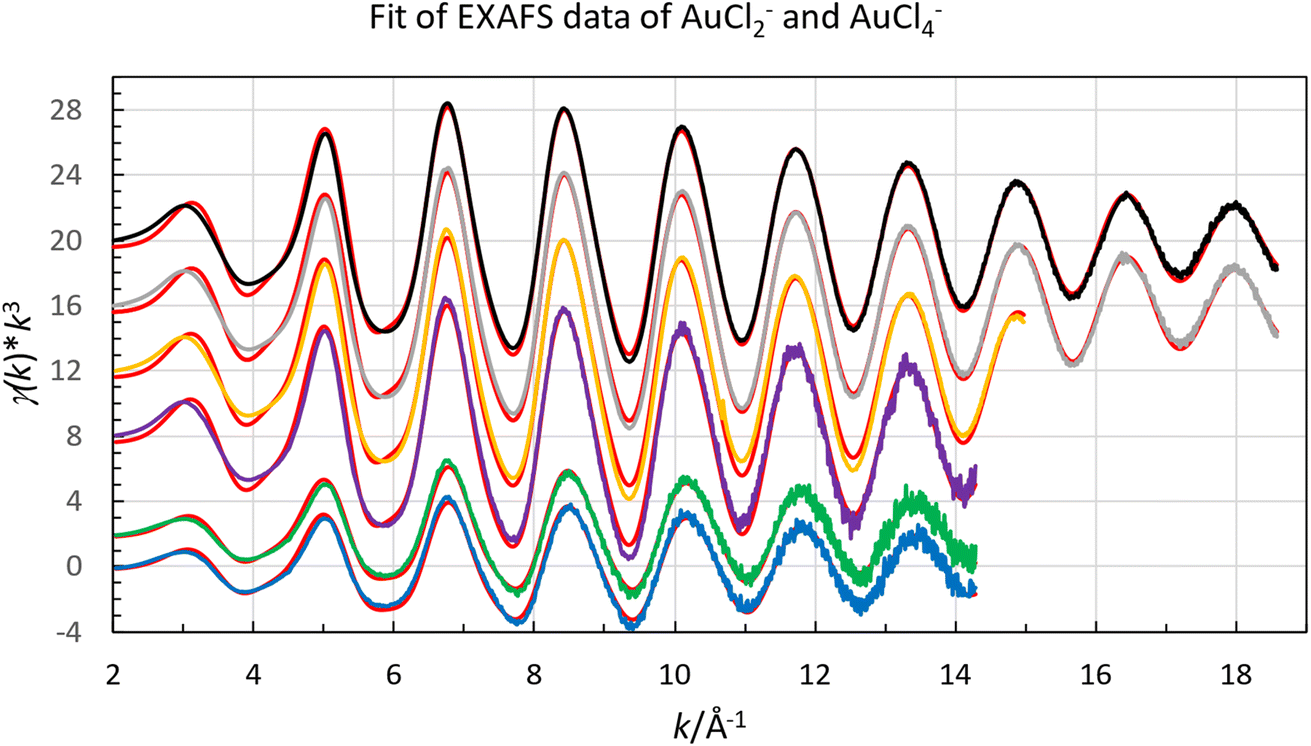 | ||
| Fig. 4 Fit of raw EXAFS data of gold(I) [AuCl2]−, in chloride containing organic solvents (extracted from low acid aqueous chloride media blue line, no offset, and extracted from hydrochloric acid green line offset +2), and gold(III), [AuCl4]−, in chloride containing aqueous solution (purple line, offset +8), in menthone (orange line offset 12), in Aliquat 336 in 3,7-dimethyloctanol (grey offset 16) and in Aliquat 336 in eucalyptol (black offset 20). With the calculated spectra, red lines, using the parameters reported in Table 3. | ||
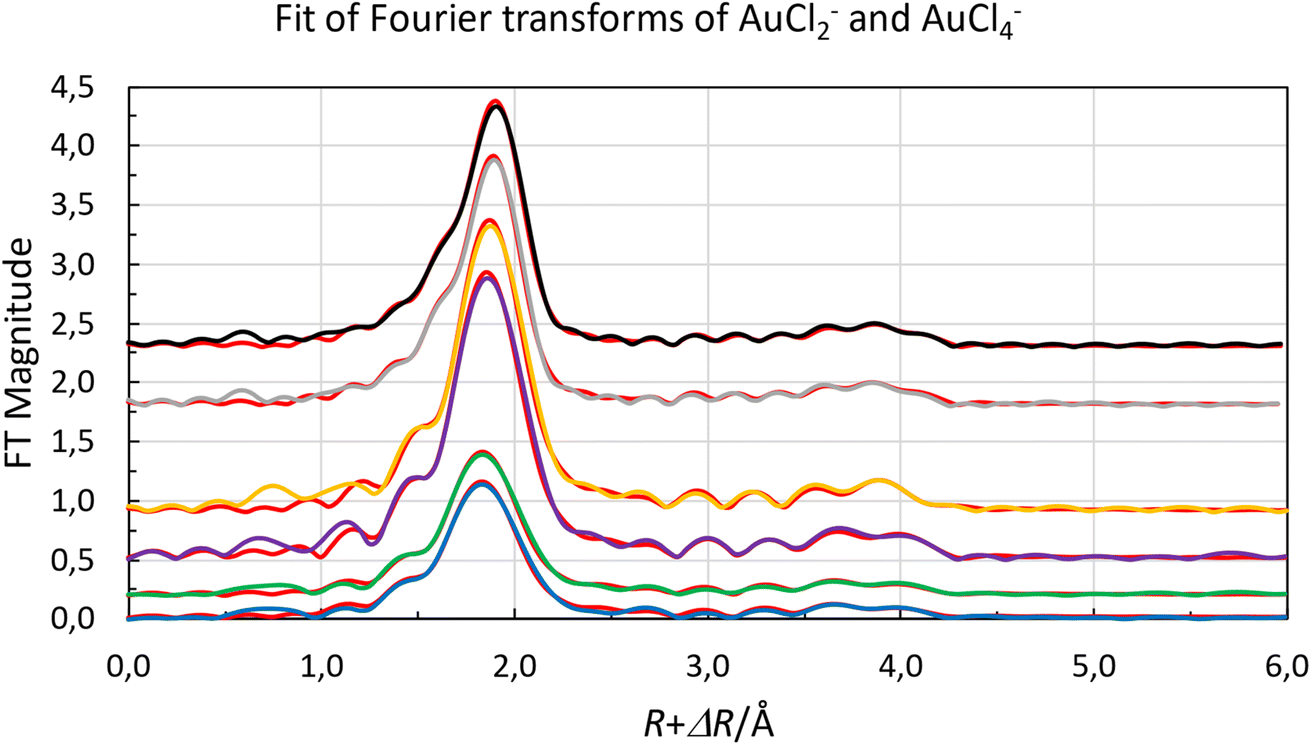 | ||
| Fig. 5 Fit of raw Fourier transforms of gold(I) [AuCl2]−, in chloride containing organic solvents (extracted from low acid aqueous chloride media blue line, no offset, and extracted from hydrochloric acid green line offset +0.5), and gold(III), [AuCl4]−, in chloride containing aqueous solution (purple line, offset +8), in menthone (orange line offset 0.9), in Aliquat 336 in 3,7-dimethyloctanol (grey offset 1.9) and in Aliquat 336 in eucalyptol (black offset 2.3). With the calculated spectra, red lines, using the parameters reported in Table 3. | ||
The XANES spectra of the stock solution on one hand and the extractant solutions are different. The XANES spectrum of the aqueous stock solution has a significant white line peaking at 11919.5 electron volts (eV) which is the signature of gold(III), Fig. 6. Neither metallic gold nor gold(I) complexes have any pronounced white line.68 The solutions of gold in the organic phase have XANES spectra which are typical for gold(I) complexes. The gold solutions all contained or were prepared from chloride rich media. Normally the XANES absorption edge can be expected to increase by 1.5 to 2.0 eV per oxidation state but in our case, it decreases with increasing oxidation number, 11919.0, 11917.4 and 11916.3 eV, for metallic gold, [AuCl2]− and [AuCl4]−, respectively, Fig. 6 and Table 3. This can be explained by the strong covalent bonding in the [AuCl2]− and [AuCl4]− complexes and a significant charge reduction on gold, and that increasing number of Au–Cl bonds reduces the charge density on gold to lower values than in metallic gold.
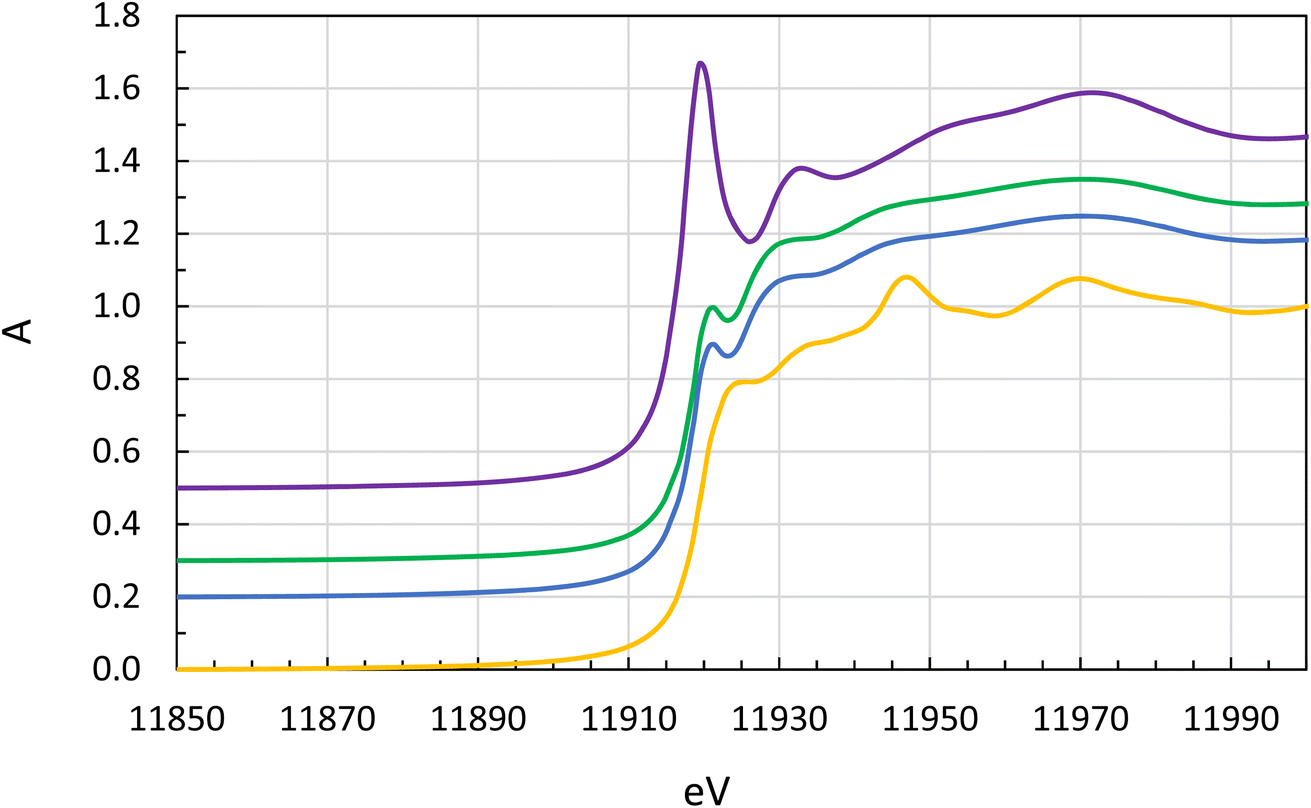 | ||
| Fig. 6 Normalized XANES spectra of metallic gold (yellow line, no offset), gold(I) [AuCl2]−, in chloride containing organic solvents (blue line, offset +0.2, no acid in the aqueous phase, and green line, offset +0.3, acid in the aqueous phase) and gold(III), [AuCl4]−, in chloride containing aqueous solution (purple line, offset +0.5). | ||
| Interaction | N | d | σ 2 | S o 2 | F |
|---|---|---|---|---|---|
| Aqueous stock solution, tetrachloroaurate( III ), [AuCl 4 ] − | |||||
| Au–Cl | 4 | 2.284(1) | 0.0030(1) | 1.05(1) | 11.5 |
| MS (AuCl4 sp,l) | 3 × 4 | 4.567 | 0.0043(2) | ||
| MS(Au–Cl–Cl nl) | 8 | 3.92(2) | 0.011(2) | ||
|
|||||
| Organic extraction solution from strongly acidic aqueous phase, dichloroaurate( III ), [AuCl 2 ] − | |||||
| Au–Cl | 2 | 2.265(1) | 0.0031(1) | 1.08(1) | 15.8 |
| MS (AuCl2 sp,l) | 3 × 2 | 4.531 | 0.0045(4) | ||
|
|||||
| Organic extraction solution from weakly acidic aqueous phase, dichloroaurate( III ), [AuCl 2 ] − | |||||
| Au–Cl | 2 | 2.264(1) | 0.0032(1) | 1.04(1) | 15.8 |
| MS (AuCl2 sp,l) | 3 × 2 | 4.528 | 0.0050(4) | ||
Further XANES/EXAFS work was performed using a solution of the malonamide in dodecane which had been recently shaken with a solution of gold in hydrochloric acid. XANES suggested that the gold was in the +3 oxidation state. Over a time span of 140 minutes much of the gold(III) was reduced into gold(I). It is of particular note that the kinetics of the reduction neither obeyed first or zero order kinetics, instead the gold reduction is second order with respect to gold. This indicates that with high concentrations of gold(III) in the organic phase that the reduction rate is higher than that when the concentration of Au(III) was low. An attempt was made to determine the order of the reaction. The data was fitted to a series of reactions of different ordered whose rates can be summed up by equation two (Fig. 7) (Table 4).
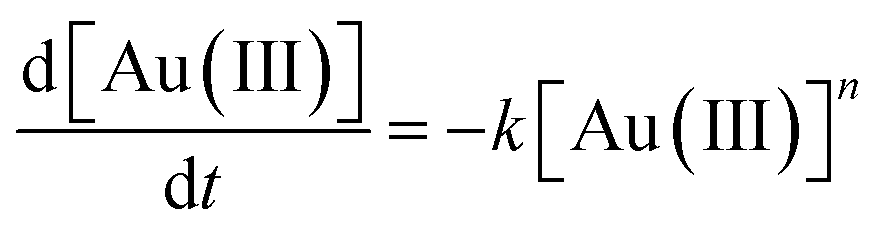 | (2) |
 | ||
| Fig. 7 The reduction of the gold(III) while it is in the organic phase made from malonamide and dodecane. | ||
| Reaction order | Integrated rate equation | Straight line equation | Σ(Fexp − Fmodel)2/Fexp |
|---|---|---|---|
| 0 | F = −k | F vs. t | 0.001797 |
| 1 | F = e−kt | LnF vs. t |
0.001002 |
| 2 | 1/F = (1/F0) + kt | (1/F) vs. t | 0.0004498 |
| 3 | F −2 = 1/F0−2 + 2kt | F −2 vs. t | 0.0001450 |
| 4 | F −3 = 1/F0−3 + 3kt | F −3 vs. t | 0.0000749 |
| 5 | F −4 = 1/F0−4 + 4kt | F −4 vs. t | 0.0002200 |
The goodness of the fit between the mathematical models for the different orders of reaction were considered by squaring the difference between the fraction of gold in the +3 oxidation state at each time, this value was then divided by the fraction of gold in the +3 oxidation state. The sum of these values for all five times were added. It was found that the reduction of the gold neither followed zero or first order kinetics with respect to the gold(III) concentration. With the data XANES data obtained at this time we do not feel confident in deciding the order of the reaction with respect to gold(III). But it is either second order or a still higher order.
If we assume that the gold(III) is extracted by an ion pairing mechanism where HAuCl4 is initially extracted by protonation of a malonamide molecule to form a lipophilic cation which enables the AuCl4− anion to enter the organic phase, then if the gold is converted into AuCl2− then the reduced lipophilicity of the anion will assist the stripping of the gold. As the AuCl4−/AuCl2− and Cl2/2Cl− couples are reported to be at 0.9251 and 1.3936 volts vs. the standard hydrogen electrode,69 it will be possible to regenerate AuCl4− anions using electrogenerated chlorine. We continued the work with the assumption that the malonamide diluted in the alkane mixture will extract gold, the smaller the ability of the organic phase to extract other metals such as iron the easier we reason it would be to use it to purify gold.
We compared the extraction of cadmium, gold, iron, palladium and zinc from 2 M hydrochloric acid by solutions of tetra-butyl malonamide, DEHBA (di-(2-ethyl-hexyl)butyramide), N,N,N′,N′-tetrakis-2-ethylhexyl malonamide, tributyl phosphate in HVO100 together with two pure ketones (acetophenone and menthone). The results are presented in Table 5. Of the organic phases tested only 10% N,N,N′,N′-tetrakis-2-ethylhexyl malonamide in HVO100 provided a gold distribution ratio greater than 50 together with having palladium, iron, cadmium and zinc distribution ratios below 0.1.
| Organic phase | D Au | D Pd | D Fe | D Zn | D Cd |
|---|---|---|---|---|---|
| 10% N,N,N′,N′-tetrakis-2-ethylhexyl malonamide in HVO100 | 113 | 0.058 | 0.048 | 0.028 | 0.038 |
| 30% tributyl phosphate in HVO100 | 451 | 0.015 | 3.29 | 0.64 | 0.14 |
| 100% tributyl phosphate | ∞ | 0.90 | 235 | 19.1 | 13.0 |
| 100% acetopheone | ∞ | 0.024 | 1.1 | 0.043 | 0.018 |
| 100% menthone | 29.0 | 0.017 | 0.011 | 0.0095 | 0.0045 |
| 100% tetra-butyl malonamide | ∞ | 17.0 | 499 | 70.7 | 394 |
| 30% DEHBA in HVO100 | ∞ | 0.53 | 0.33 | 0.12 | 0.20 |
| 30% bis-2-ethylhexyl hydrogen phosphate in HVO100 | 0.033 | 0.016 | 28.4 | 0.016 | 0.012 |
As modest distribution ratio was obtained using the terpene menthone, which would have enabled a process to be created which would be able to recover the vast majority of the gold using two counter current extraction stages with equal organic and aqueous flow rates (θ = 1), an experiment was performed in which the distribution of gold between menthone and aqueous mixtures of sodium chloride and hydrochloric acid (where the sum of the concentrations of sodium chloride and hydrochloric acid was 2 moles per litre) was measured at 30 °C (Fig. 8). The measurement was made at this temperature as it was made during an exceptionally hot day in Sweden.
 | ||
| Fig. 8 Distribution ratios obtained for gold with pure menthone as the organic phase, the aqueous phase was a series of combinations of sodium chloride and hydrochloric acid where the total concentration of these two electrolytes is 2 moles per litre. | ||
The work on gold chemistry at Chalmers includes the measurement of the gold content of shredded printed circuit boards. For health and safety reasons a visual search was made by a person with experience of building radio equipment in the 1990s (MRSJF), for cruciform transistors such as MRF234 and similar components on the boards. This is because they can contain beryllium oxide (beryllia),70 however none of the printed circuit boards examined were found to include transistors or power modules of the type associated with beryllia. Large samples of the waste were digested in aqua regia, after leaching for several days the liquid from the digestions was collected by filtration and diluted with water. The residue from the digestion was collected and dried. The filtered extracts were filtered and diluted with hydrochloric acid before being examined with ICPOES (Table 6).
| Element | Fraction of solid sample | Unit | ||
|---|---|---|---|---|
| Sample 1 | Sample 2 | Sample 3 | ||
| Al | 4.4 | 5.0 | 5.6 | % |
| Au | 268 | 352 | 446 | ppm |
| Ba | 0.56 | 0.61 | 0.70 | % |
| Ca | 0.66 | 0.69 | 0.75 | % |
| Cd | ND | ND | ND | — |
| Co | 232 | 156 | 264 | ppm |
| Cr | 825 | 870 | 1443 | ppm |
| Cu | 18.8 | 20.5 | 23.9 | % |
| Fe | 2.59 | 3.22 | 4.37 | % |
| Mg | 848 | 832 | 828 | ppm |
| Mn | 600 | 653 | 821 | ppm |
| Ni | 0.83 | 0.96 | 1.14 | % |
| Pb | 0.74 | 0.86 | 0.94 | % |
| Sr | 125 | 157 | 157 | ppm |
| Y | 33 | 38 | 43 | ppm |
| Zn | 1.11 | 1.31 | 1.65 | % |
A further batch of printed circuit boards were ground up after the removal of aluminium heat sinks. After sieving samples of the solids were digested in aqua regia before being examined with ICPOES. The presence of gold in the second batch of scrap was confirmed. As can be seen in Fig. 9 and 10 the metals were not uniformly distributed between the different size fractions.
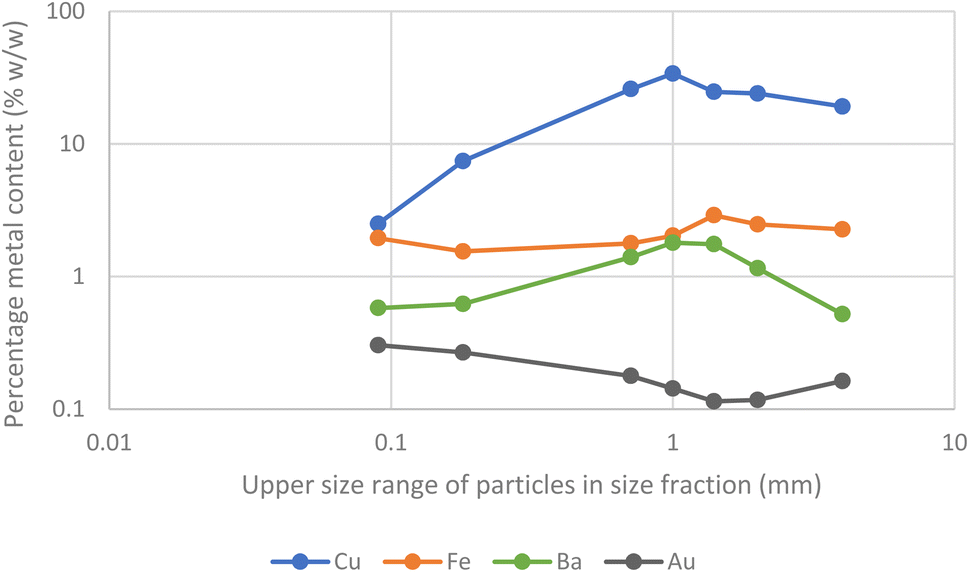 | ||
| Fig. 9 Barium, copper, iron and gold content of the different fractions of particle sizes obtained by grinding and then sieving. | ||
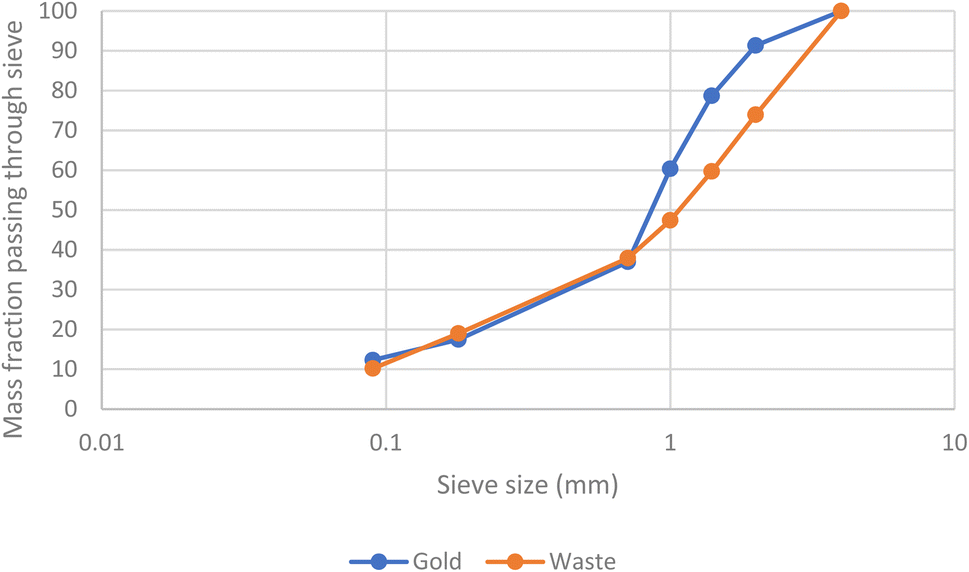 | ||
| Fig. 10 The fractions of the gold and the mass of all elements (the latter measured gravimetrically) passing a series of sieves. | ||
While the gold in the smallest particles is contaminated with the least copper, it is important to note that the majority of the gold is present in the larger particles. As a result, a chemical separation of gold from the other metals present in the waste printed circuit boards is needed.
We wanted to demonstrate the recovery of gold from the aqua regia digests of the waste ground in the first campaign. Part of each extract was spiked with a solution of gold in hydrochloric acid, these gold spiked solutions were then used in solvent extraction experiments with both MIBK and N,N,N′,N′-tetrakis-2-ethylhexyl malonamide (10% v/v) in HVO100. It was found that shaking with MIBK caused all the gold in the aqueous to be transferred into the organic phase, while we could extract gold with this ketone we choose to attempt a process based on the malonamide chemistry. In a separation funnel ten portions of an aqua regia extract containing gold (100 ml) were shaken with N,N,N′,N′-tetrakis-2-ethylhexyl malonamide (20% v/v) in HVO100 (50 ml) to form a gold loaded organic phase. This gold loaded organic phase was then shaken with hydrochloric acid before being stripped with ethaline to form a gold product. The shaking with the organic phase removed the majority of the gold from the portions of the aqua regia extract.
The gold product from this crude process was 66% gold (w/w) which was contaminated with aluminium, copper, iron and other metals. When the ratios of the impurity elements to gold were compared between the product and the feed solution, it was clear that the decontamination factors** were all greater than 1000. We suspect that the use of a separation funnel rather than a battery of mixer settlers operating in counter-current contributed to the contamination of the product with many of the elements. If we assume that the gold and copper distribution ratios are 14 and 0 respectively in both the extraction and scrubbing (selective back-extraction of impurities) stages of a process then with 2% entrainment of organic in aqueous and vica versa then the plant will behave as if the distribution ratios are 10.9 and 0.0204 respectively. It can be calculated that with three counter current stages for both extraction and scrubbing that a 91% recovery of gold will be possible with a decontamination factor of 5.3 × 106. It is likely that a better recovery of gold can be obtained by routing the aqueous effluent from the scrub into the extraction stage as was done in the counter-current centrifugal contactor experiments where TODGA††/TBP in kerosene was used to extract 241Am, 244Cm, 252Cf and 152Eu from nitric acid.71 In the future we may attempt to create and test a flowsheet for gold recovery in a mixer settler pilot plant.
While it would be possible to create a gold recycling process using Aliquat 336 in 3,7-dimethyloctanol, the malonamide in HVO100 system is better able to reject the zinc present in some gold jewellery alloys.
General suitability of HVO100 as a diluent for solvent extraction
| Element | Control | HVO100 | Solvent 70 | Solvesso 150 |
|---|---|---|---|---|
| Al | 1.4 | 1.8 | 1.9 | 2.1 |
| B | 2.5 | 2.4 | 2.7 | 2.7 |
| Ca | 0.59 | 0.64 | 0.75 | 0.72 |
| Na | 3.1 | 3.8 | 3.9 | 4.1 |
| Si | 1.7 | 1.7 | 1.9 | 1.8 |
| Diluent | Water content (ppm) |
|---|---|
| Solvent 70 (aliphatic kerosene) | 30.4 ± 11.5 |
| HVO100 | 34.2 ± 2.5 |
| Eucalyptol | 5581 ± 59 |
| Solvesso 150ND (aromatic kerosene) | 386 ± 95 |
WARNING: tritium (3H) is radioactive, open sources of tritium should only be used by a trained radiochemist working at a site suitably equipped for such work. Note that it is impossible to measure tritium with a Geiger–Muller tube detector as the low energy of the beta particles make them unable to pass through the mica window of a GM tube used for beta emitters.
| Nuclide | Aqueous phase | Organic phase | Distribution ratio |
|---|---|---|---|
| 99mTc | HCl (0.5 M) | HVO100 | 1.01 × 10−5(19) |
| 152Eu | HNO3 (28 mM) | HVO100 | 6.33 × 10−5(463) |
| 152Eu | NaNO3 (6 M) + HNO3 (28 mM) | HVO100 | 8.76 × 10−5(226) |
| 238Pu | HNO3 (0.5 M) | 30% TBP in HVO100 | 0.75 |
The plutonium distribution ratio obtained with nitric acid (0.5 M) is similar to those reported for 0.5 to 1.0 M nitric acid with 30% tributyl phosphate in dodecane.76 The results obtained using HVO100 indicate that the diluent is free of trace impurities which are able to strongly extract europium, plutonium and technetium.
The problem of metal extraction by impurities is compounded by the fact that the statement “a trace impurity which is able to extract one carrier free metal will be able to extract all metals” is capable of being false. Thus, we advise others considering working with carrier free radionuclides to perform their own extraction experiments with the carrier free metal that they will use. After showing that the HVO100 is unable to extract europium we conducted two extraction experiments with 152Eu. These serve two purposes, firstly in common with the protocol for the Ames test for mutagens77 we wished to include positive controls to demonstrate that 152Eu extraction from the sodium nitrate and dilute nitric acid are possible for solutions of solvating and acidic extractants respectively. We also tested the hypothesis that HVO100 contains an impurity which would inhibit the extraction of europium by Cyanex 923 under conditions related to those in the extraction stage of the Chinese TRPO process78 and the extraction of europium by DEHPA.79,80§§ In the TRPO process the actinides and lanthanides are extracted from high level liquid waste after the nitric acid content has been reduced to circa 1 M.81 As the extraction of the trivalent f-block elements is promoted by a high aqueous nitrate activity and suppressed by a high concentration of nitric acid82 in the aqueous phase we choose to extract carrier free europium from a concentrated sodium nitrate solution containing a low concentration of nitric acid. Under our conditions europium is extracted according to the following chemical equation (equation three).
| Eu(aq)3+ + 3NO3(aq)− + nR3PO(org) → [Eu(NO3)3(R3PO)n](org) | (3) |
We can write the following polynomial (equation four) where Q varies as a function of the parameters of the aqueous phase.
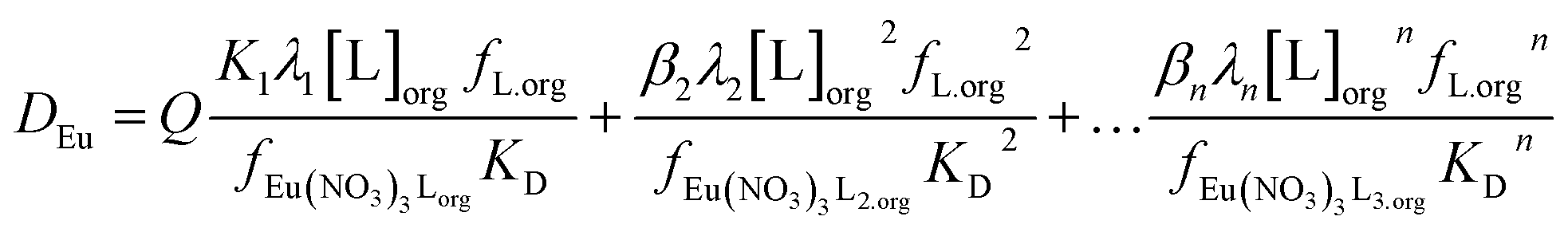 | (4) |
We shook a solution of sodium nitrate in dilute nitric acid with various concentrations of Cyanex 923 in HVO100. The aqueous solution was spiked with 152Eu before use. After withdrawing samples from both phases their radioactivity was determined by gamma spectroscopy, the results are given in Fig. 11. Here in common with the other radioactive extraction experiments the distribution ratio was obtained by dividing the activity (Bq ml−1) of the organic layer by that of the aqueous layer.
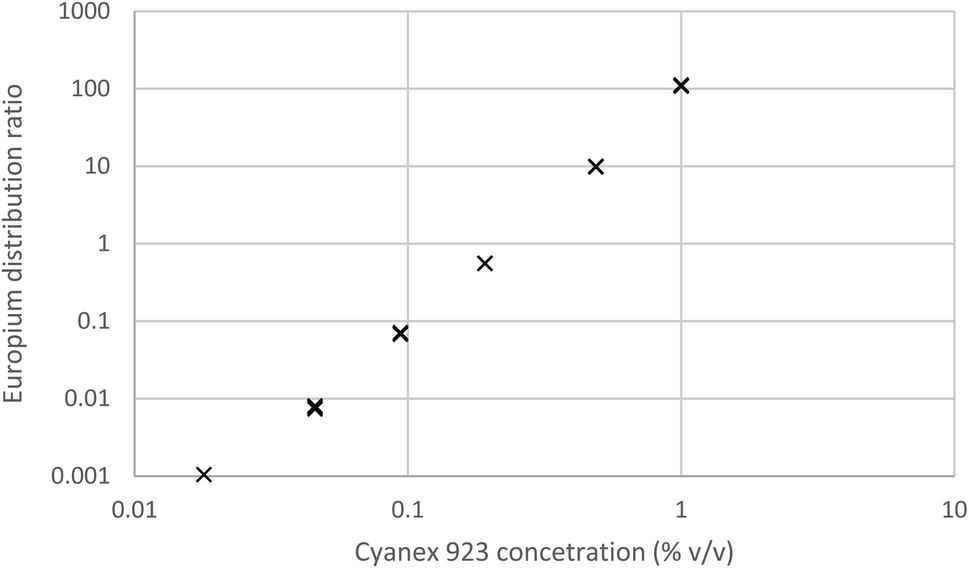 | ||
| Fig. 11 The variation of the europium distribution ratio as a function of the Cyanex 923 content of the organic phase. | ||
It was found that the europium distribution ratio was close to being proportional to the cube of the Cyanex 923 volume fraction of the organic phase, suggesting that only the tris-(trialkyl phosphine oxide) complex was extracted into the organic phase under the conditions used in the experiment. It is noteworthy that solvent extraction experiments (using aqueous nitrate salts and xylene as diluent) indicate that three trialkyl phosphine oxide molecules are required for the extraction of each lanthanide atom83 and with triisobutyl phosphine oxide that nine coordinate complexes of the formula [Ln(NO3)3(OPR3)3] have been reported for many of the lanthanides in the solid state.84 If we make the assumption that only neutral trinitrato tris-(trialkyl phosphine oxide) complexes are extracted then the equation for the europium extraction can be simplified to equation five.
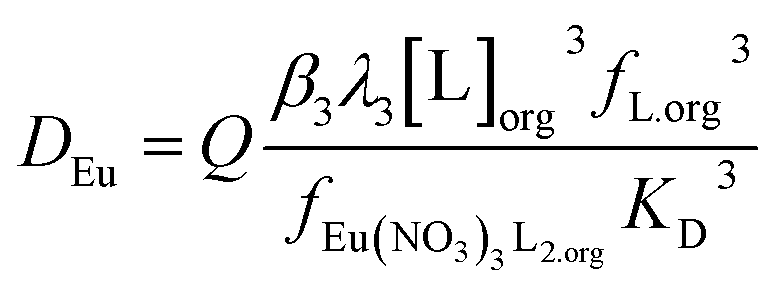 | (5) |
As the graph of the logarithm of the europium distribution ratio against the logarithm of the Cyanex 293 concentration (as a volume fraction) is 2.992 ± 0.093¶¶ we can be certain that over the experimental range used (1 to 0.018% (v/v)) that the cube of the activity function of the Cyanex 923 in the organic phase divided by the activity function of the [Eu(NO3)3(Cyanex 923)3] complex in the organic phase remains constant. Solvent extraction experiments were conducted with solutions of DEHPA (between 10 and 0.05% v/v) in HVO100 (Fig. 12). It was found that below 1% (v/v) of DEHPA that the europium distribution ratio was highly dependent on the concentration of the DEHPA. Above 1% the reported distribution ratio approached a plateau.
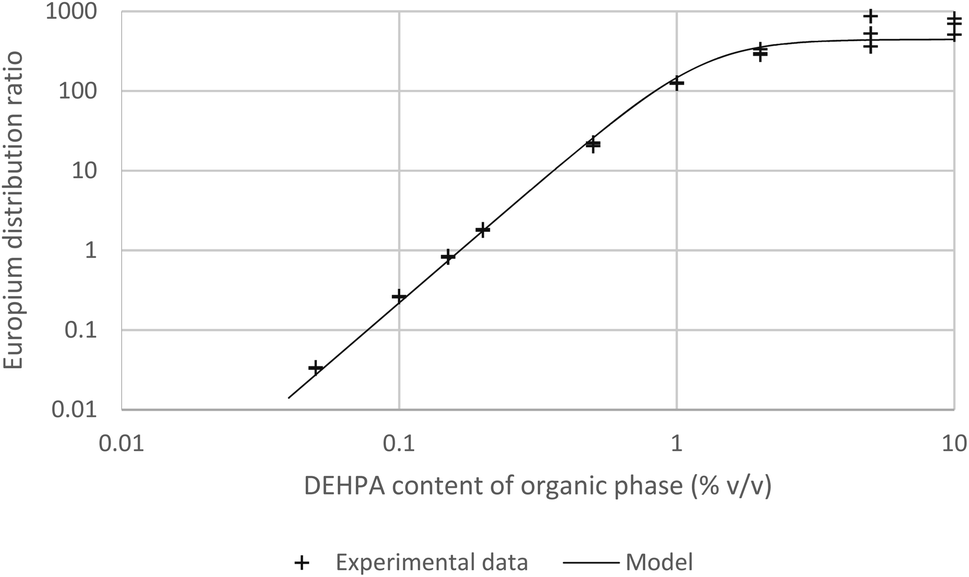 | ||
| Fig. 12 A graph of the europium distribution ratio against the DEHPA content of the organic phase. | ||
If we assume that all activity coefficients are equal to one and only a mononuclear neutral complex of europium with three conjugate anions of DEHPA is extracted then we can apply the mathematical method used to discuss the extraction of copper(II) and promethium(III) by acetylacetone85 to write the following equation which predicts the distribution ratio of the europium. In this equation λ3 is the partition coefficient of the neutral europium complex which is extracted, KA is the dissociation constant for aqueous DEHPA, KD is the partition coefficient of DEHPA and K1, β2, β3 and β4 are constants for the equilibria between europium and the complexes formed with the conjugate base of DEHPA (equation six). We are also ignoring oligomerisation of the DEHPA in the organic phase.
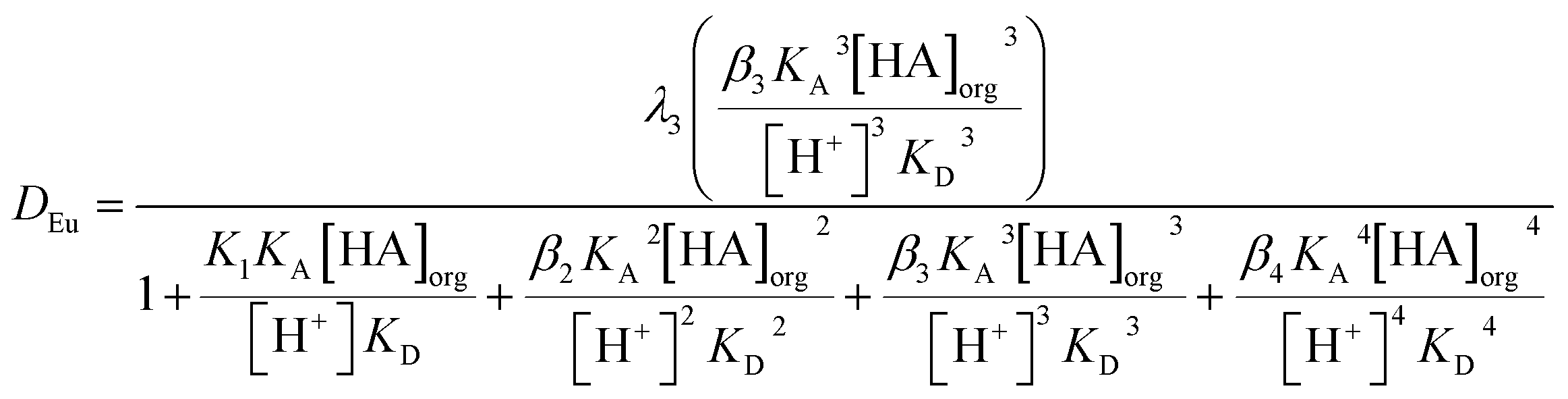 | (6) |
Through our use of very low chemical concentrations of europium we are able to perform the experiment at a constant acidity. We can simply the equation by combining many constants and concentrations into q constants to yield equation seven.||||
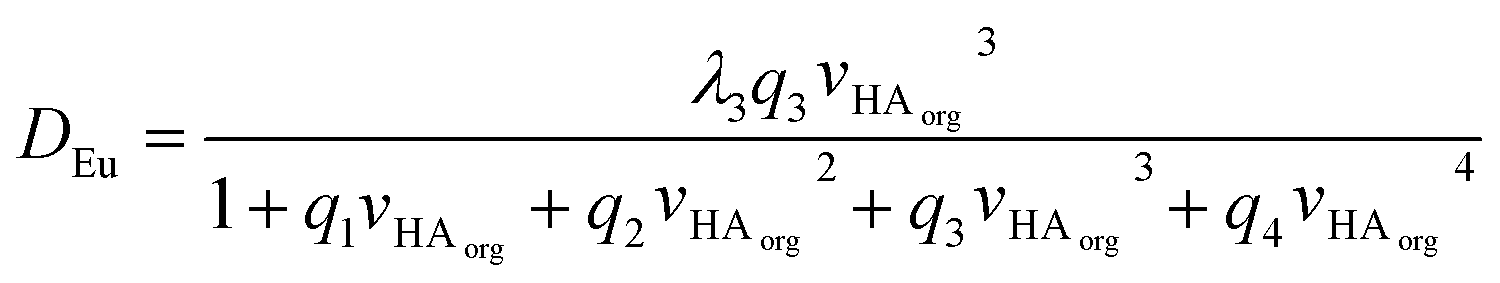 | (7) |
Using the solver function of excel q1, q2, q3, q4 and λ3 can be estimated to be 0.296%-1, 0.556%-2, 0.376%-3, zero and 681 respectively. A danger exists that when a large number of variables can be adjusted that a good but chemically spurious fit can be made of the experimental data. The equation was reduced in complexity to yield equation eight.
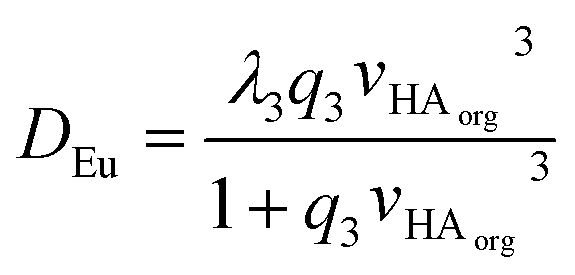 | (8) |
Using the reduced formula and the jackknife error estimation method λ3 and q3 were estimated to be 443.7 ± 50.3 and 0.496 ± 0.069 respectively. No further discussion will be made of the estimate for q3, it is important to note that when the term q3[HA]org3 is large that the distribution ratio will tend towards λ3. The value of λ3 obtained from fitting the data will be limited by both the partition coefficient of the neutral tris-DEHPA europium complex and the amount of entrainment. The degree of entrainment depends on the experimental conditions and also on the skill level of the worker sampling the two liquid layers. Distribution ratios in the range of 1000 have been reported for some experiments in which radioactive lanthanides are extracted with 1 M DHEPA from 0.1 M nitric acid using an aliphatic kerosene.86 Hence we can reason that it is possible to sample both liquid phases in a shaking tube experiment and the entrainment effect is insufficiently large to prevent the measurement of distribution ratios of up to 100. The lower limit of the distribution ratio which can be measured using HVO100 is likely to be further away (in terms of the magnitude of the log(D) value) as for a measurement of a very low distribution ratio the low activity organic phase can be sampled from the top without having to pass a pipette tip through another layer. On the other hand for the sampling of a low activity lower layer the tip must be passed through an upper phase with a much higher radioactivity level. The same arguments will apply to experiments using non-radioactive materials.
A further plutonium experiment was performed in a glovebox where equal volumes of a solution of plutonium (mainly 239/240Pu containing small amounts of 241Am arising from the decay of 241Pu and 238Pu arising from the neutron activation of 237Np) in nitric acid (6 M) was shaken with 30% tributyl phosphate in HVO100 for five minutes before being allowed to settle under the influence of gravity. During this experiment two clear layers were obtained, samples of these were collected by pipette. After the samples were posted out of the glovebox in a fume hood, small volumes were placed on stainless steel disks (planchets), after heating these were examined with alpha spectroscopy (Using a semiconductor detector supplied by ORTEC). Using the alpha emissions for 239Pu (5.245 MeV) and 240Pu (5.256 MeV) it was estimated that the plutonium distribution ratio was 7.3 while the americium distribution ratio was no greater than 0.088. Note that the alpha spectrometer is unable to distinguish between 241Am (5.638 MeV) and 238Pu (5.593 MeV) which increases the distribution ratio recorded by the experiment. This experiment indicates that it could be possible to use HVO100 as a diluent in actinide separation/purification processes such as the PUREX process.
WARNING: plutonium (238Pu, 239Pu, 240Pu) and americium (241Am) are radioactive and they are high specific activity alpha emitters which cause them to be exceptionally radiotoxic. It is important to avoid inhalation of alpha emitters, an excess of lung cancer has been observed in Mayak workers who were exposed to plutonium aerosols while working in the early Soviet nuclear sector.87 The europium used (152Eu) is a mixed beta/gamma emitter. The technetium used (99mTc) is the metastable excited state obtained by the beta decay of 99Mo. Work with radioactivity should only be undertaken by a trained radiochemist working at a site suitably equipped for such work. We strongly suggest that open sources (open containers) of radioactivity only be handled either in a fumehood or a glovebox.
We wished to explore the use of HVO100 in lanthanide chemistry, a series of solutions of lanthanum, neodymium and the alkaline earths in different concentrations of nitric acid were prepared. These were shaken with a solution of DEHPA in HVO100, it was found that the slopes of logarithms of distribution ratios against the aqueous acid concentration (computed at equilibrium) for calcium, lanthanum and neodymium were −1.997 ± 0.045, −2.949 ± 0.032 and −2.905 ± 0.050 respectively. These slopes are all close to the ideal slopes of −2, −3 and −3 which can be expected if all activity functions are equal to one in the aqueous phase.
| nR3PO(org) + Li(aq)+ + ClO4(aq)− → Li(ClO4)(R3PO)n(org) | (9) |
| nR3PO(org) + M(aq)2+ + 2ClO4(aq)− → M(ClO4)2(R3PO)n(org) | (10) |
In preliminary experiments solutions of most of the alkaline earths (Mg, Ca, Sr and Ba) together with lithium in sodium perchlorate solutions were shaken with solutions of Cyanex 923 in solvent 70, a terpene (eucalyptol) or a biokerosene (from skyNRG). These preliminary experiments indicated that these metals can be extracted by solutions of Cyanex 923 in these diluents. An experiment with three concentrations of Cyanex 923 in HVO100 was performed. Here dilute solutions of lithium together with all the alkaline earth metals from magnesium to barium in different concentrations of aqueous sodium perchlorate were shaken with 5, 10 and 30% (v/v) Cyanex 923 in HVO100 (Fig. 13). We demonstrated that the HVO100 was free of substances able to extract the alkaline earth metals by shaking the diluent with a strong solution of sodium perchlorate spiked with the metals, no metal extraction was observed in this experiment. Equally when a 30% (v/v) solution of Cyanex 923 in HVO100 was shaken with aqueous solutions of the metals without perchlorate no extraction was observed. The 30% (v/v) solution of Cyanex 923 in HVO100 other than sodium was able to extract all the s-block metals tested. For all concentrations of sodium perchlorate with 30% Cyanex 923 the calcium distribution ratio was too high to be measured reliably. When the higher concentrations of sodium perchlorate were used the magnesium and strontium distribution ratios were also too high for reliable measurement, but over the whole range it was possible to measure the distribution ratios of lithium and barium. For comparison we shook 30% DEHBA96 (N,N-di-2-ethylhexyl butamide) in HVO100 with the s-block metals in 6.68 M sodium perchlorate. The DEHBA was selected as ourselves97 and others98 have shown it to be suitable replacement for tributyl phosphate in some extraction systems. But sadly no extraction of barium, calcium, lithium, magnesium or strontium was observed with the solution of DEHBA.
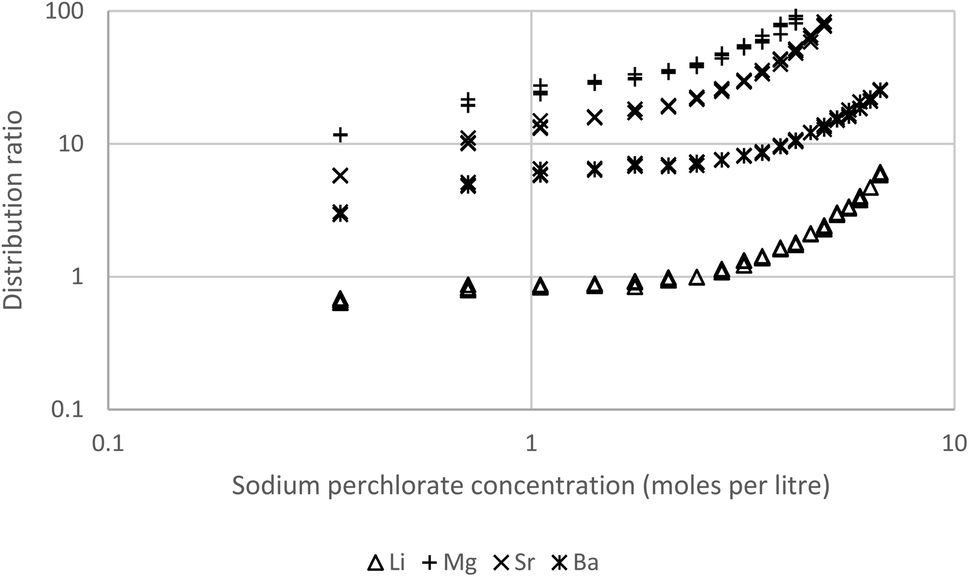 | ||
| Fig. 13 Lithium, magnesium, strontium and barium distribution ratios obtained by shaking a 30% solution of Cyanex 923 in HVO100 at 25 °C with sodium perchlorate solutions spiked with calcium, lithium, magnesium, strontium and barium. The calcium distribution ratios are too high to be measured. | ||
To lower the distribution ratios sodium perchlorate solutions of the s-block metals were shaken with 10% Cyanex 923 in the HVO100 diluent. As expected the distribution ratios of barium, lithium, magnesium and strontium were reduced. The experiment with 10% Cyanex 923 enabled us to record useful data for barium, magnesium and strontium (Fig. 14). Now the lithium distribution ratios were too small to be useful. As before the calcium distribution ratios were too high for reliable measurement. This high distribution ratio for calcium could be useful in the measurement of radium in calcium containing materials such as limestone,99 rather than using direct measurement of the sample by gamma spectroscopy we reason that the sample could be dissolved in perchloric acid, the calcium removed and then the radium isolated from the calcium depleted raffinate before being measured.
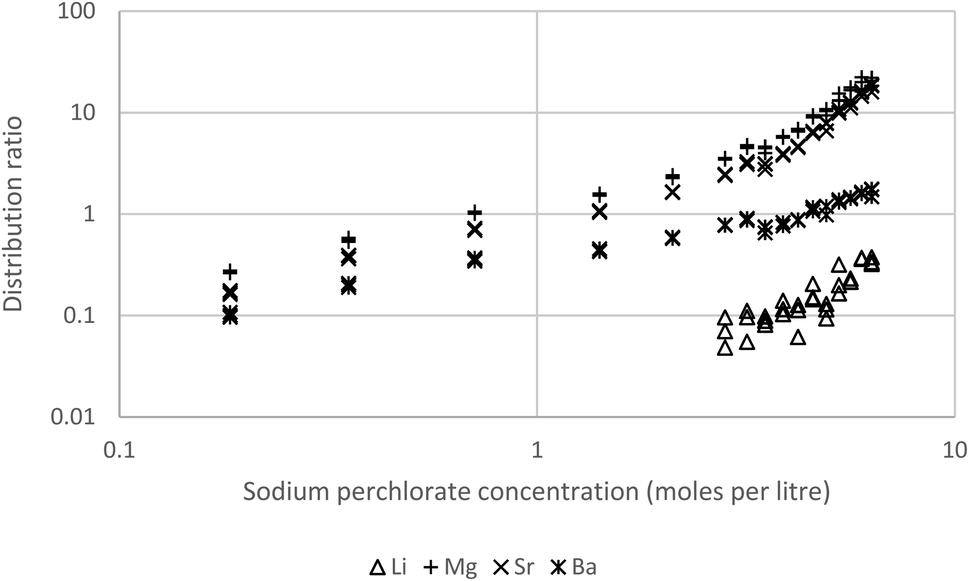 | ||
| Fig. 14 Lithium, magnesium, strontium and barium distribution ratios obtained by shaking a 10% solution of Cyanex 923 in HVO100 at 25 °C with sodium perchlorate solutions spiked with calcium, lithium, magnesium, strontium and barium. The calcium distribution ratios are too high to be measured. | ||
In order to be able to obtain useful data for calcium we further decreased the concentration of the Cyanex 923 to 5% (v/v) in the HVO100. As hoped we were able to obtain calcium distribution ratios in our preferred interval (Fig. 15). These result obtained for s block elements further support the hypothesis that HVO100 is a suitable replacement for aliphatic petroleum kerosene as a diluent in solvent extraction.
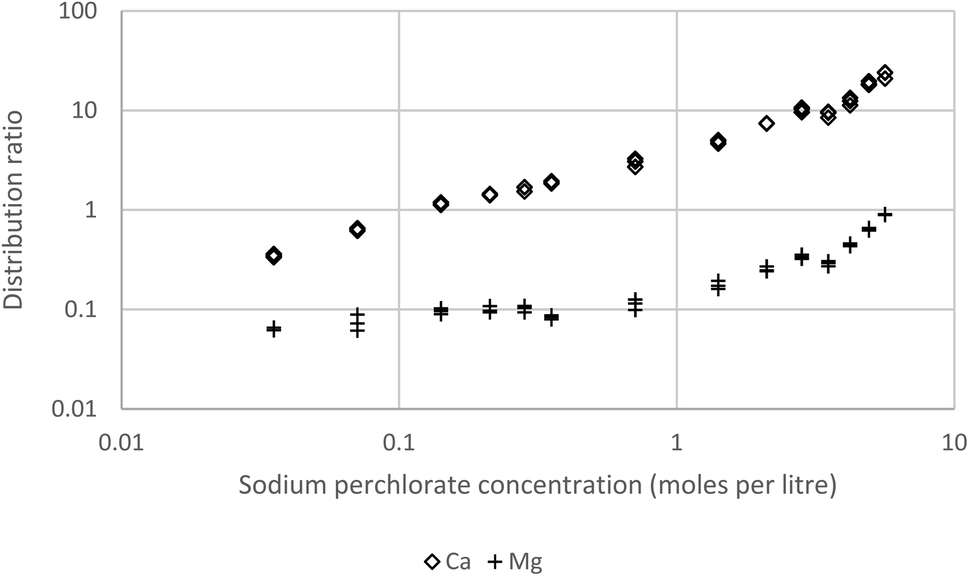 | ||
| Fig. 15 Calcium and magnesium ratios obtained by shaking a 10% solution of Cyanex 923 in HVO100 at 25 °C with sodium perchlorate solutions spiked with calcium, lithium, magnesium, strontium and barium. The barium and lithium distribution ratios are too low to be measured. The strontium distribution ratios are very similar to those of magnesium, for clarity they are not shown on the graph. | ||
As a final solvent extraction experiment solutions of the s-block elements in weakly acidic sodium nitrate solutions were shaken with a mixture of Cyanex 923 (30% v/v) and HVO100, it was found that no extraction of barium, lithium or strontium occurred. However extraction of calcium and magnesium was observed, the extraction of these two elements was greater when the concentration of the sodium nitrate was higher (Fig. 16). This is consistent with the formation and extraction of neutral dinitrate complexes by the Cyanex 923. It may be possible by making a careful analysis of the solvent extraction of the s-block elements to obtain an insight into the chemical activities of these elements in concentrated nitrate and perchlorate media. However the in depth consideration of these issues is outside the scope of this paper.
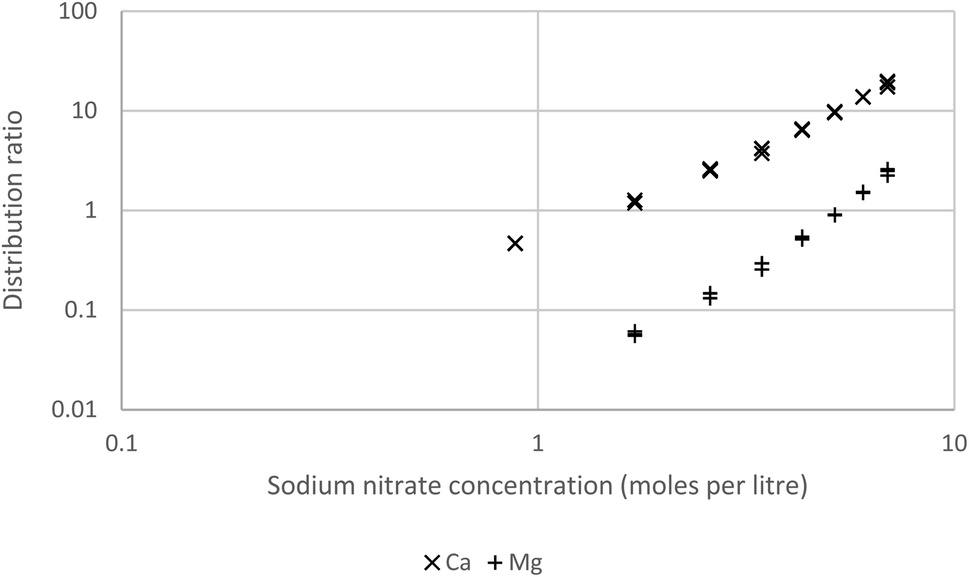 | ||
| Fig. 16 A graph of the distribution ratios of calcium and magnesium as a function of the sodium nitrate. | ||
WARNING: Beryllium is exceptionally toxic. It is carcinogenic and can cause a serious lung disease. All beryllium samples were handled inside a fumehood as if they were radioactive samples.
To compliment our understanding of HVO100 the extraction of all the non-radioactive alkaline earth elements by solutions of DEHPA in HVO100 was investigated by shaking different concentrations (5 to 50% v/v) of DEHPA in HVO100 with an acetate buffer solution containing the elements. It was found that the vast majority of the magnesium, calcium, strontium and barium was extracted by the organic phases. However the beryllium was more interesting, while the distribution ratios for beryllium being extracted from non-coordinating aqueous media by DEHPA in benzene has been reported to be very high.100 In our case modest distribution ratios were obtained. The shape of the graph in Fig. 17 suggests that even without DEHPA that extraction of beryllium is possible. This was ascribed to the fact that beryllium forms a stable complex with acetate anions. In this complex four beryllium atoms form a tetrahedron with an oxygen atom at its centre,101 since the 1930s it has been known that the Be4O core of basic beryllium acetate is surrounded by six acetate groups.102 The lipophilic exterior of the Be4O(OAc)6 complex will enable it to be slightly extracted by the diluent but in the same experiment the strong coordination of beryllium to acetate has changed the order of extractability of the alkaline earths from that observed by McDowell and Coleman. It is possible to inhibit the extraction of a metal by the formation of stable hydrophilic complexes.103 If we assume that the only forms of beryllium in the aqueous phase are Be2+, Be4O(OAc)6 and BeA2 (Where A is the conjugate base of DEHPA) then when we assume all activity functions are equal to one we could write eqn (11).
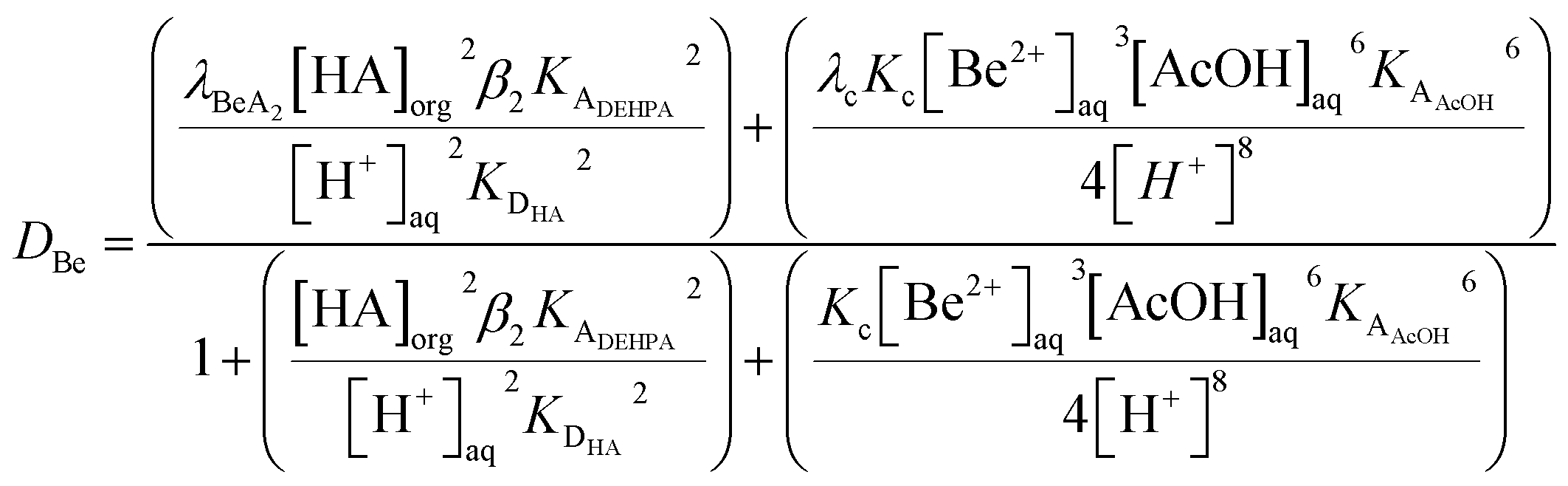 | (11) |
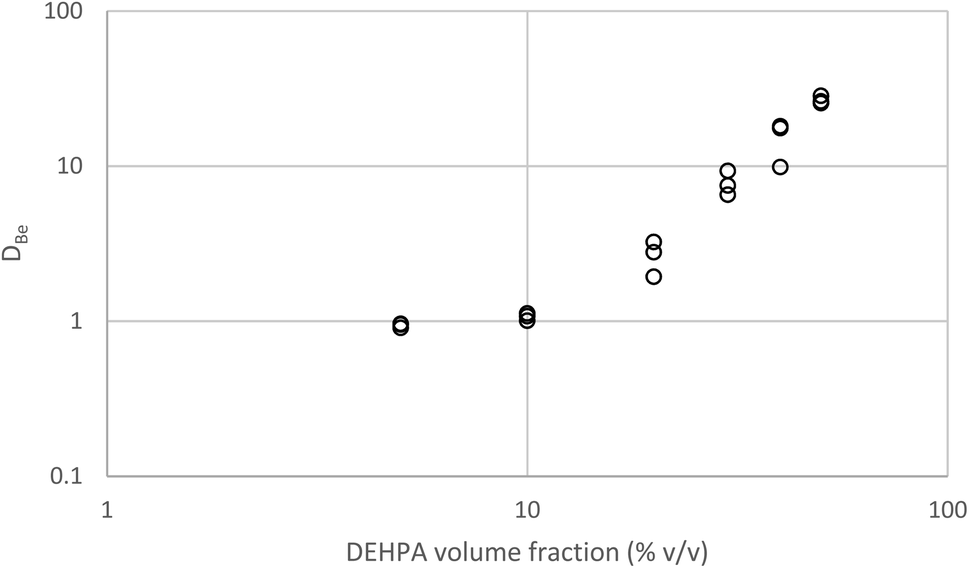 | ||
| Fig. 17 A graph of beryllium distribution ratio against the DEHPA fraction in the organic phase (Diluent HVO100). | ||
If eqn (11) is true then it is clear that the beryllium distribution ratio will change as a function of the total beryllium concentration in the system due to the formation of the polynuclear species. The formation of polymeric beryllium species has been reported elsewhere.104 In common with Anthony Hill's disinclination to prepare more bis-(3-methylbut-1-yn-1-yl)mercury105 we feel unmotivated to work with higher concentrations of beryllium for health and safety reasons. However at a future time using radioactive 7Be in an AKUFVE it could be possible to measure the stability constants for the beryllium acetate complexes using a procedure similar to that used to investigate the binding of thorium to isosaccharinic acid.106
It can be reasoned that rather than dismissing a diluent as “dangerous” based on extreme exposures such as high pressure injection of fuel during diesel engine (compression ignition engine) servicing115 and aspiration into the lungs after swallowing large volumes of liquid,116 we should consider the chronic exposures which are more likely for adult workers in a solvent extraction plant.
The length of time a 70 kg worker whose hands have a surface area of 840 cm2 can safely cover both hands each day with JP8 jet fuel before adsorbing the daily reference oral dose for hydrcarbons (RfD)117*** has been considered in the literature, part of this assessment was the measurement of the permeability of fuel components through skin.118 We have applied this methodology for the assessment of systemic effects to a series of hydrocarbons. It was found that the exposure time for aromatic diluents was short (1 to 4 min) while for large alkanes (C12 and C13) this time was long (>40 min). It is noteworthy that with porcine skin (pig's ear) that tetradecane and hexadecane adsorption was not observed while the adsorption of smaller alkanes (Nonane to Trdecane) was observed.119 This literature result suggests that the larger alkanes (such as hexadeane and octadecane) in the HVO100 are less likely to cause systemic effects than the shorter chain alkanes. It has also been shown that as the length of alkanes are increased from C11 to C16 that their ability to damage cells and irritate the skin peaks at C13 before decreasing again.120 As the HVO100 is mostly comprised of alkanes with more than 13 carbons it may well be less irritating to skin than solvent 70 (Statoil) which is a mixture of alkanes and cycloalkanes with between 11 and 14 carbon atoms.121
It has been shown that the more carbons are present in an alkane that the less adsorption occurs within the digestive system.122 The same authors reported that no difference in the adsorption rate exists for n-, iso- and cycloalkanes with the same number of carbons. As the alkanes in HVO100 have larger numbers of carbons than those in our standard aliphatic kerosene (solvent 70) we judge that for oral exposure the HVO100 alkanes are less harmful. Albler et al. and others121,123 have pointed out that for high molecular weight solvents, the threat due to inhalation of the vapour becomes smaller as the boiling point of the solvent increases. The Swedish workplace air limit (based on an eight hour day) for long chain alkanes (decanes and heavier alkanes) is 350 mg m−3, while the limits for jet fuel (Jet A-1) and diesel fuel are 250 and 350 mg m−3.124 As the equilibrium concentrations in air above pure tridecane, tetradecane, and pentadecane are 206, 59 and 16 mg m−3. Because HVO100 comprises of alkanes with 13 or more carbons it is not possible at 20 °C for the vapour concentration in a sealed room containing a puddle of HVO100 to exceed the strict Swedish limits. In the absence of mechanical processes which generate aerosols we reason that HVO100 has a good workplace safety profile.
While we have shown that it is possible to purify gold using our very sustainable organic phase, it was reasoned that the malonamide/HVO100 mixture was unsuitable for the determination of the data needed for the computation of activity functions as the gold changes chemical form in the organic phase. We sought out an alternative organic phase in which the gold would be more chemically stable in the organic phase.
Aliquat 336 based organic phases for gold extraction
It was decided that we should compare this extraction with a series of less sustainable organic phases which can be made from commercially available reagents. We considered the use of Aliquat 336 which is very close to the ionic liquid N8881Cl. It is known that the gold distribution ratios obtained when ethyl benzene solutions of Aliquat 336 are shaken with aqueous solutions of gold(III) in chloride media are very high. While a high distribution ratio might appear to be attractive, if it is not possible to reduce the distribution ratio below one then back extraction of metals in a process can be impossible. Also, due to entrainment, when the distribution ratio becomes very high it becomes impossible to measure the distribution ratio.125 We sought a diluent which would allow us to decrease the gold distribution ratio obtained with moderate concentrations of Aliquat 336. As it is known that the inclusion of alcohols in kerosenes used as diluents with Aliquat 336 tend to decrease metal distribution ratios,126 we considered the use of 3,7-dimethyloctanol as a diluent for Aliquat 336. It was found that the gold distribution ratios obtained when 30, 20 or 10% (v/v) Aliquat 336 solutions in dimethyloctanol was shaken with gold(III) in acidic sodium chloride solutions were exceptionally high. Even when dilute solutions (5, 2.5 or 1%) of Aliquat 336 were used the distribution ratios were high. But when the experiment was repeated with choline chloride as the main electrolyte more modest distribution ratios were obtained. The distribution ratios varied between 47 and 0.09 suggesting that it would be possible to strip (back extract) gold by changing the choline chloride concentration. While we were attempting to devise gold recycling methods we recognised the profligacy of not attempting to obtain activity function parameters from the data. If we assume that all of the gold is present as AuCl4− ions then the extraction of the gold can be described with the equation twelve.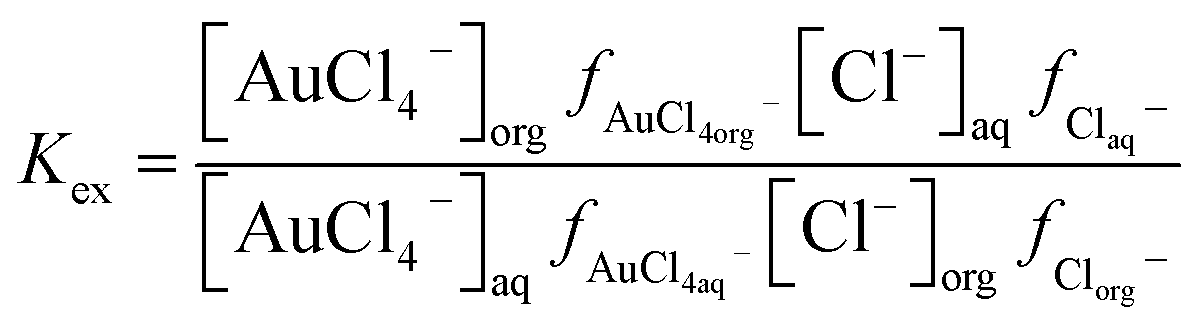 | (12) |
By combination with a Pitzer equation, we can obtain eqn (13).†††
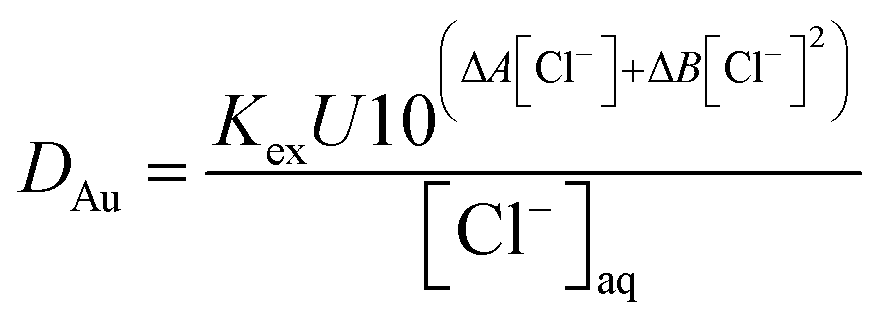 | (13) |
It was possible with the solver feature of excel to estimate the values of KexU, ΔA and ΔB. The standard deviations on KexU, ΔA and ΔB were estimated using the jack-knife method. We found with either 30% (v/v) Aliquat 336 in eucalyptol or 30% (v/v) Cyanex 923 (a trialkyl phosphine oxide) in HVO100 that the distribution ratios of the impurity elements are higher than that observed with 30% (v/v) malonamide in HVO100. Hence, if a need exists to extract the impurity elements in an industrial plant, we suggest that the gold is first extracted with the malonamide before the other elements are recovered with the other organic phase. This would be an example of an early separation of gold from the impurity element, a literature example of such an early separation would be the uranium/plutonium separation in the UREX process.127
Measurements were made of the gold distribution ratio obtained with a series of different solutions of Aliquat 336 in 3,7-dimethyloctanol. In this experiment, the denser phase was varied between the deep eutectic solvent and an aqueous choline chloride solution with the same concentration of choline chloride per litre. It was found for all three concentrations of Aliquat 336 that as the ethaline content of the lower phase increased that the gold distribution ratio decreased. We considered the hypothesis that the system can be described by eqn (14).
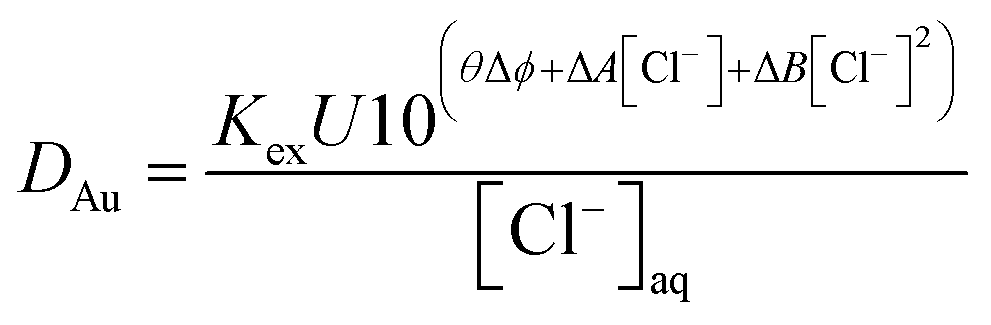 | (14) |
As the chloride concentration remained constant we are able to write a simpler eqn (15).
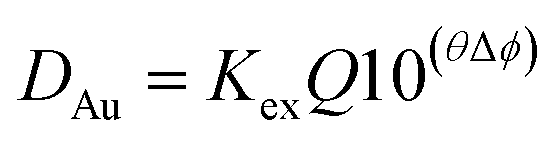 | (15) |
When all the collected data was used the standard deviations on the parameters (KexQ and Δϕ) were large, the jack-knife jawas used to identify the data points which contributed most to the standard deviations. By excluding a few points, new estimates of the parameters were obtained with smaller standard deviations. Using the 1% aliquat solution the best estimates of KexQ and Δϕ were 1.20 ± 0.09 and −3.19 ± 0.12. While with the 5% (v/v) solution of Aliquat 336 these parameters were 11.9 ± 2.5 and −3.26 ± 0.16. These two estimates of Δϕ differ by less than one estimated standard deviation, so they are assumed to be the same value. The estimate of Δϕ made using the 30% Aliquat 336 solution was −2.70 ± 0.11 which differs more from the other estimates. The difference between the estimates made using 1 and 30% Aliquat 336 is 0.49 while the sum of the estimated standard deviations is 0.23. In the paper in PCCP, a different formula was used to model the distribution ratio of gold at 30 °C using the malonamide a value of −3 was obtained,‡‡‡ the kinetics and EXAFS/XANES experiments cast a shadow of doubt on those gold results published in 2020. However it should be noted that the rate of reduction of gold(III) in the organic phase is proportional to the gold(III) concentration raised to a high power, as a result very dilute solutions of gold(III) in the malonamide/alkane mixtures are likely to be more stable.
A limit of the area of chemical space must be accepted when writing a paper, as we have only determined gold distribution ratios for water/choline chloride/ethylene glycol systems with a single ionic strength it is impossible to know how or if Δϕ (Δψ) will change as a function of the ionic strength. We suggest that before anyone attempts to operate a commercial scale gold recovery system using either the malonamide or aliquat chemistry that they determine Δϕ under a wider range of conditions. We also urge anyone attempting to perform gold extraction at a larger scale to study the hydrodynamic behaviour of the system,128 and the kinetics of both the extraction129 and stripping of gold and the other metals present.
Conclusions
A selective and reversible extraction of gold using a biomass derived organic phase is thermodynamically feasible. Within this paper we have demonstrated the extraction of gold and plutonium using sustainable organic reagents at TRL§§§ 3. In the event that the malonamide is unavailable then a lipophilic chloride ionic liquid (Aliquat 336) can be used instead for gold extraction. A series of purity tests using radionuclides with high specific activities (large activity in terms of disintegrations per second {Bq} per mole of element) suggest that the sustainable diesel fuel has a very low concentration of substances able to extract metals while positive control experiments with europium indicate that the diluent is free of substances which inhibit the extraction of the metal.When exposure to diluent vapor, oral exposure to small amounts and skin contact is considered, the HVO100 has a favourable workplace safety profile. Due to a lack of toxicology and industrial hygiene information about 3,7-dimethyloctanol in the literature it is not possible to make a similar assessment of this substance, hence we suggest that when a choice exists that the malonamide in HVO100 system should be used for industrial scale gold purification. However for the collection of activity coefficient data we recommend the use of Aliquat 336 in 3,7-dimethyloctanol.
Author contributions
Mark R. StJ. Foreman all chemistry unless otherwise stated, all radiochemistry, devising the study, and writing of the paper and supervision of GM, RKJ and MST. Richard Johansson gold extraction experiments in dimethyloctanol. Gloria Mariotti the second grinding campaign and the analysis of the solid samples. Behabitu E. Tebikachew and Mikhail S. Tyumentsev synthesis of the monoamide (DEHBA) and the malonamide reagents respectively. Ingmar Persson was responsible for the collection of XANES and EXAFS data before interpretation of the data.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work was supported under a Horizon 2020 project (Codename TARANTULA, grant agreement number 821159) an ERA-MIN project (Codename LIMEX, ERA-MIN reference number ERA-MIN-2018_57, VINNOVA grant number 2019-03432) and an EIT RawMaterials project (Codename RareGreen, project ID 21130). We would like to thank RagnSells Recycling AB for funding and supplying gold containing printed circuit boards. We would like to thank Christian Ekberg for discussions on statistical issues relating to the estimation of random errors. The plutonium chemistry was performed during a training event which was part of the A-CINCH project (Augmented Cooperation in Education and Training in Nuclear and Radiochemistry). The A-CINCH project received funding from the Euratom research and training programme 2019–2020 under grant agreement No. 945301. We would like to thank Stellan Holgersson, Johan Eriksson and Marcus Hedberg for their assistance in the alpha lab at Chalmers during the training event. We also wish to thank Teodora Retegan Vollmer for her role in attracting the funding required for the A-CINCH project. We wish to thank Chelsea Foreman for proofreading the paper. We acknowledge MAX IV Laboratory for time on Beamline Balder under Proposal 20220171. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496.Notes and references
- Z. A. Kader, A. Marshall and J. Kennedy, Emergent Mater., 2021, 4, 725 CrossRef CAS.
- J. Quan, S. Zhao, D. Song, T. Wang, W. He and G. Li, Sci. Total Environ., 2022, 819, 153105 CrossRef CAS PubMed.
- S. Allard and C. Ekberg, J. Solution Chem., 2006, 35, 1173 CrossRef CAS.
- J. A. Schramke, D. Rai, R. W. Fulton and G. R. Choppin, J. Radioanal. Nucl. Chem., 1989, 130, 333 CrossRef CAS.
- J. Rydberg, Acta Chem. Scand., 1969, 23, 647 CrossRef CAS.
- J. Rydberg and Y. Albinsson, Solvent Extr. Ion Exch., 1989, 7, 577 CrossRef CAS.
- J. P. Omtvedt, J. Alstad, T. Bjørnstad, Ch. E. Düllmann, K. E. Gregorich, D. C. Hoffman, H. Nitsche, K. Opel, D. Polakova, F. Samadani, F. Schulz, G. Skarnemark, L. Stavsetra, R. Sudowe and L. Zheng, Eur. Phys. J. D, 2007, 45, 91 CrossRef CAS.
- H. Persson, G. Skarnemark, M. Skalberg, J. Alstad, J. O. Liljenzin, G. Bauer, F. Haberberger, N. Kaffrell, J. Rogowski and N. Trautmann, Radiochim. Acta, 1989, 48, 177 CrossRef CAS.
- Personal Communication from G. Skarnemark.
- N. M. Rice, H. M. N. H. Irving and M. A. Leonard, Pure Appl. Chem., 1993, 65, 2373 CrossRef CAS.
- L. Leay, K. Tucker, A. Del Regno, S. L. M. Schroeder, C. A. Sharrad and A. J. Masters, Mol. Phys., 2014, 112, 2203 CrossRef CAS.
- F. Xie, T. A. Zhang, D. Dreisinger and F. Doyle, Miner. Eng., 2014, 56, 10 CrossRef CAS.
- M. Bae, J. C. Lee, H. Lee and S. Kim, Sep. Purif. Technol., 2020, 235, 116154 CrossRef CAS.
- L. L. Arnold, W. R. Christenson, M. Cano, M. K. StJohn, B. S. Wahle and S. M. Cohen, Fundam. Appl. Toxicol., 1997, 40, 247–255 CrossRef CAS PubMed.
- J. D. Law, T. G. Garn, D. H. Meikrantz and J. Warburton, Sep. Sci. Technol., 2010, 45, 1769 CrossRef CAS.
- O. Courson, M. Lebrun, R. Malmbeck, G. Pagliosa, K. Römer, B. Sätmark and J.-P. Glatz, Radiochim. Acta, 2000, 88, 857 CrossRef CAS.
- M. Cox, in Science and Practice of Liquid-Liquid Extraction, ed. J. D. Thornton, Oxford Science Publications, Oxford, 1992, ch. 1, vol. 2, pp. 45–46 Search PubMed.
- W. Batey, in Science and Practice of Liquid-Liquid Extraction, ed. J. D. Thornton, Oxford Science Publications, Oxford, 1992, ch. 2, vol. 2, p. 146 Search PubMed.
- S. De Sio, I. Klurb, E. Tison, C. Bouyer, D. Lebeau, F. Goutelard, L. Séjourné, C. Eysseric and N. Vigier, Procedia Chem., 2016, 21, 17 CrossRef.
- M. Cox, in Science and Practice of Liquid-Liquid Extraction, ed. J. D. Thornton, Oxford Science Publications, Oxford, 1992, ch. 1, vol. 2, pp. 56–61 Search PubMed.
- E. Paatero and M. Haapalainen, US Pat., 8926730, 2015.
- M. Casey, J. Leonard, B. Lygo and G. Procter, Advanced Practical Organic Chemistry, Blackie, Glasgow and London, 1990 Search PubMed.
- K. Klementiev, K. Norén, S. Carlson, K. G. V. Sigfridsson Clauss and I. Persson, The BALDER beam-line at the MAX IV Laboratory, J. Phys.: Conf. Ser., 2016, 712, 1–5 Search PubMed.
- I. Persson, D. Lundberg, É. G. Bajnóczi, K. Klementiev, J. Just and K. G. V. Sigfridsson Clauss, EXAFS study on the coordination chemistry of the solvated copper(II) ion in a series of oxygen donor solvents, Inorg. Chem., 2020, 59, 9538 CrossRef CAS PubMed.
- H. Thompson, A. Attwood, D. Gullikson, E. Howells, M. Kim, K.-J. Kirz, J. Lindau, I. Liu, Y. Pianetta, P. Robinson, A. Scofield, J. Underwood, J. Vaughan, D. Williams and G. Winick, X-Ray Data Booklet 3rd Rev, Lawrence Berkeley National Laboratory, Berkeley, 2009 Search PubMed.
- G. N. George and I. J. Pickering, EXAFSPAK – A Suite of Computer Programs for Analysis of X-Ray Absorption Spectra, SSRL, Stanford, CA, 1993 Search PubMed.
- S. I. Zabinsky, J. J. Rehr, A. Ankudinov, R. C. Albers and M. Eller, M. Multiple-Scattering Calculations of X-ray Absorption Spectra, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 2995–3009 CrossRef CAS PubMed.
- D. Capuano, M. Costa, S. Di Fraia, N. Massarotti and L. Vanoli, Renewable Sustainable Energy Rev., 2017, 69, 759 CrossRef CAS.
- M. R. StJ. Foreman, Cogent Chem., 2016, 2, 1139289 CrossRef.
- M. Parravicni, C. Barro and K. Boulouchos, Fuel, 2021, 292, 120177 CrossRef.
- M. Ameen, M. Tazli, S. Yusup, A. Ramli and M. Yasir, Renewable Sustainable Energy Rev., 2017, 80, 1072 CrossRef CAS.
- H. R. Lam, P. Plenge and O. S. Jorgensen, Neurotoxicol. Teratol., 2001, 23, 603 CrossRef CAS PubMed.
- N. R. Das and S. N. Bhattacharyya, Talanta, 1976, 23, 535 CrossRef CAS PubMed.
- A. A. Yadav and S. M. Khopkar, Sep. Sci., 1970, 5, 637 CAS.
- F. J. Alguacil and I. Martin, Sep. Sci. Technol., 2003, 38, 2055 CrossRef CAS.
- P. Cen, K. Spahiu, M. S. Tyumentsev and M. R. S. Foreman, Phys. Chem. Chem. Phys., 2020, 22, 11012 RSC.
- P. H. Pfromm, V. Amanor-Boadu, R. Nelson, P. Vadlani and R. Madl, Biomass Bioenergy, 2010, 34, 515 CrossRef CAS.
- H. Tummes, H. Noeske and W. Kascha, Process for the Improved Manufacture of 2-Ethylhexanol, US Pat., 4318588, 1979.
- M. E. Janka, R. T. Hembre, S. D. Barnicki, R. S. Huss, X. Shan, S. R. Testerman, T. A. Upshaw and D. W. Fuller JR, Transfer Hydrogenation Process, US Pat., 9988329, 2018.
- J. W. Clark and A. L. Wilson, Process for Preparing branched Chain Aliphatic Secondary Amines, US Pat., 2319848, 1943.
- C. W. Song, J. W. Kim, I. J. Cho and S. Y. Lee, ACS Synth. Biol., 2016, 5, 1256 CrossRef CAS PubMed.
- J. A. Dietrich, Recombinant host cells for the production of malonate, US Pat., 10385368, 2019.
- A. Rout and K. Binnemans, Dalton Trans., 2013, 43, 1862 RSC.
- F. Wöhler, Pogg. Ann., 1828, 12, 253 Search PubMed.
- Y. Yokoyama, T. Yokota, S. Hayashi and E. Izawa, Geochem. J., 1996, 30, 175 CrossRef.
- D. Maljkovic, D. Maljkovic D and A. Paulin, Solvent Extr. Ion Exch., 1992, 10, 477 CrossRef CAS.
- W. Hunter and J. Downing, J. Soc. Chem. Ind., 1949, 68, 362 CrossRef CAS.
- M. Bae, J.-C. Lee, H. Lee and S. Kim, Sep. Purif. Technol., 2020, 235, 116154 CrossRef CAS.
- M. Cox, in Science and Practice of Liquid-Liquid Extraction, ed. J. D. Thornton, Oxford Science Publications, Oxford, 1992, ch. 1, vol. 2, pp. 56–61 Search PubMed.
- S. Terashima, S. Itoh and A. Ando, Geostand. Newsl., 1992, 16, 9 CrossRef CAS.
- I. K. Pitcairn, P. E. Warwick, J. A. Milton and D. A. H. Teagle, Anal. Chem., 2006, 78, 1290 CrossRef CAS PubMed.
- T. Oshima and K. Miyake, AIChE J., 2021, 67, e17214 CrossRef CAS.
- W. Batey, in Science and Practice of Liquid-Liquid Extraction, ed. J. D. Thornton, Oxford Science Publications, Oxford, 1992, ch. 2, vol. 2, p. 138 Search PubMed.
- SAFETY DATA SHEET NEXBTL Renewable Diesel; Neste 100% NEXBTL diesel; Neste Green 100 diesel, https://www.neste.com/sites/default/files/attachments/nexbtl_renewable_diesel_version3.0_safetysheet.pdf accessed 4 October 2022 Search PubMed.
- Neste Renewable Diesel Handbook, https://www.neste.com/sites/default/files/attachments/neste_renewable_diesel_handbook.pdf, accessed 4 October 2022 Search PubMed.
- G. Choppin, J.-O. Liljenzin and J. Rydberg, Radiochemistry and Nuclear Chemistry, Butterworth-Heinemann, Woburn, 2002 Search PubMed.
- A. P. Abbott, G. Capper, K. J. McKenzie and K. S. Ryder, J. Electroanal. Chem., 2007, 599, 288 CrossRef CAS.
- V. Agieienko and R. Buchner, Phys. Chem. Chem. Phys., 2022, 24, 5265 RSC.
- W. Stankiewicz, B. Bolibrzuch and M. Marczak, Gold Bull., 1998, 31, 119 CrossRef CAS.
- D. Jones, B. Bjarngard, N. Simon and J. Harley, Br. J. Radiol., 1968, 41, 944 CrossRef CAS PubMed.
- B. R. Reddy and P. V. R. Bhaskara Sarma, Hydrometallurgy, 1996, 43, 299 CrossRef CAS.
- M. C. Costa, M. Martins and A. P. Paiva, Sep. Sci. Technol., 2004, 39, 3573 CrossRef CAS.
- G. Meinrath, C. Ekberg, A. Landgren and J. O. Liljenzin, Talanta, 2000, 51, 231 CrossRef CAS PubMed.
- G. Meinrath, C. Ekberg, A. Landgren and J. O. Liljenzin, Talanta, 2000, 51, 231 CrossRef CAS PubMed.
- M. A. Diaz, G. H. Kelsall and N. J. Welham, J. Electroanal. Chem., 1993, 361, 25 CrossRef CAS.
- F. H. Allen, The Cambridge Structural Database: a quarter of a million crystal structures and rising, Acta Crystallogr., Sect. B, 2002, 58, 380 CrossRef PubMed.
- Inorganic Crystal Structure Database 1.4.6, Release 2022-2, FIZ Karlsruhe, Eggenstein-Leopoldshafen, Germany, 2022 Search PubMed.
- J. G. Parsons, M. V. Aldrich and J. L. Gardea-Torresdey, Environmental and biological applications of extended X-ray absorption fine structure (EXAFS) and X-ray absorption near edge structure (XANES) spectroscopies, Appl. Spectrosc. Rev., 2002, 37, 187–222, DOI:10.1081/ASR-120006044.
- M. A. Diaz, G. H. Kelsall and N. J. Welham, J. Electroanal. Chem., 1993, 361, 25 CrossRef CAS.
- G. R. Jessop, VHF/UHF Manual, Radio Society of Great Britain, Potters Bar, 4th edn, 1983 Search PubMed.
- G. Modolo, H. Asp, H. Vijgen, R. Malmbeck, D. Magnusson and C. Sorel, Solvent Extr. Ion Exch., 2007, 26, 62 CrossRef.
- N. D. Zinina, A. L. Timashova, M. V. Pavlovskaya and D. F. Grishin, Pet. Chem., 2014, 34, 392 CrossRef.
- M. J. C. Rezende, C. R. Perruso, D. de A. Azevedo and A. C. Pinto, J. Chromatogr. A, 2005, 1063, 211 CrossRef CAS PubMed.
- C. Hill, C. Madic, P. Baron, M. Ozawa and Y. Tanaka, J. Alloys Compd., 1998, 271–273, 159 CrossRef CAS.
- Y. J. Zhu, Radiochim. Acta, 1995, 68, 95 CrossRef CAS.
- S. De Sio, C. Sorel, E. Bosse and P. Moisy, Radiochim. Acta, 2013, 101, 373 CrossRef CAS.
- D. M. Maron and B. N. Ames, Mutat. Res., 1983, 113, 173 CAS.
- J. C. Wang and C. L. Song, Solvent Extr. Ion Exch., 2001, 19, 231 CrossRef.
- D. F. Peppard, G. W. Mason, J. L. Maier and W. J. Driscoll, J. Inorg. Nucl. Chem., 1957, 4, 334 CrossRef CAS.
- C. Tunsu, Y. Menard, D. O. Eriksen, C. Ekberg and M. Petranikova, J. Cleaner Prod., 2019, 218, 425 CrossRef CAS.
- Y. Zhu and R. Jiao, Nucl. Technol., 1994, 108, 361 CrossRef CAS.
- Y. A. El-Nadi, N. E. El-Hefny and J. A. Daoud, Solvent Extr. Ion Exch., 2007, 25, 225 CrossRef CAS.
- M. L. P. Reddy, R. L. Varma, T. R. Ramamohan, S. K. Sahu and V. Charavortty, Solvent Extr. Ion Exch., 1998, 16, 795 CrossRef CAS.
- A. Bowden, P. N. Norton and A. W. G. Platt, Inorg. Chem., 2011, 50, 2553 CrossRef CAS PubMed.
- J. Rydberg, G. R. Choppin, C. Musikas and T. Sekine, in Solvent Extraction Principles and Practice, ed. J. Rydberg, Marcel Decker, New York, 2nd edn, 1992, ch. 4, pp. 139–154 Search PubMed.
- I. Svantesson, G. Persson, I. Hagström and J. O. Liljenzin, J. Inorg. Nucl. Chem., 1980, 42, 1037 CrossRef CAS.
- N. A. Koshurnikova, M. G. Bolotnikova, L. A. Ilyin, I. B. Keirim-Markus, Z. S. Menshikh, P. V. Okatenko, S. A. Romanov, V. I. Tsvetkov and N. S. Shilnikova, Radiat. Res., 1998, 149, 366 CrossRef CAS PubMed.
- Anonymous, C. O. M.(2020) 474 – Critical Raw Materials Resilience: Charting a Path towards Greater Security and Sustainability, European Commission, Brussels, 2020 Search PubMed.
- K. Brandt, Historical Development of Secondary Lithium Batteries, Solid State Ionics, 1994, 69, 173 CrossRef CAS.
- T. Kulsartov, Z. Zaurbekova, I. Tazhibayeva, Y. Ponkratov, V. Gnyry, G. Kizane, E. Nesterovc and N. Varlamovac, Study of tritium and helium generation and release from lead-lithium eutectics Li15.7Pb under neutron irradiation, Fusion Eng. Des., 2019, 146, 1317 CrossRef CAS.
- M. R. StJ. Foreman, Reactor Accident Chemistry an Update, Cogent Chem., 2018, 4, 1450944 CrossRef.
- M. Ubeyli, Effect of Lithium Enrichment on the Tritium Breeding Characteristics of Various Breeders in a Fusion Driven Hybrid Reactor, J. Fusion Energy, 2009, 28, 300 CrossRef.
- K. Nishizawa, T. Takano, I. Ikeda and M. Okahara, Extractive Separation of Lithium Isotopes by Crown Ethers, Sep. Sci. Technol., 1988, 23, 333 CrossRef CAS.
- L. Zhang, L. Li, D. Shi, J. Li, X. Peng and F. Nie, Selective extraction of lithium from alkaline brine using HBTA-TOPO synergistic extraction system, Sep. Purif. Technol., 2017, 188, 167 CrossRef CAS.
- S. Umetani and M. Matsui, Solvent-Extraction of Alkaline-Earth Metals with 4-Acyl-5-Pyrazolones and Polydentate Phosphine Oxides, Anal. Chem., 1992, 64, 2288 CrossRef CAS.
- G. Thiollet and C. Musikas, Synthesis and uses of the amides extractants, Solvent Extr. Ion Exch., 1989, 7, 813 CrossRef CAS.
- E. Aneheim, C. Ekberg, M. R. S. Foreman, E. Lofstrom-Engdahl and N. Mabile, Studies of a Solvent for GANEX Applications Containing CyMe4-BTBP and DEHBA in Cyclohexanone, Sep. Sci. Technol., 2012, 47, 663 CrossRef CAS.
- D. R. Prabhu, G. R. Mahajan and G. M. Nair, Di(2-ethyl hexyl)butyramide and di(2-ethyl hexyl)isobutyramide as extractants for uranium(VI) and plutonium(IV), J. Radioanal. Nucl. Chem., 1997, 224, 113 CrossRef CAS.
- L. A. Najam, F. M. Al-Jomaily and E. M. Al-Farha, Natural radioactivity levels of limestone rocks in northern Iraq using gamma spectroscopy and nuclear track detector, J. Radioanal. Nucl. Chem., 2011, 289, 709 CrossRef CAS.
- W. J. McDowell and C. F. Coleman, J. Inorg. Nucl. Chem., 1966, 28, 1083 CrossRef CAS.
- W. H. Bragg, Nature, 1923, 111, 532 CrossRef CAS.
- L. Pauling and J. Sherman, Proc. Natl. Acad. Sci. U. S. A., 1934, 20, 340 CrossRef CAS PubMed.
- S. Andersson, K. Eberhardt, C. Ekberg, J. O. Lijenzin, M. Nilsson and G. Skarnemark, Radiochim. Acta, 2006, 94, 469 CrossRef CAS.
- I. S. El-Yamani, M. Y. Farah and E. N. Abd El-Messieh, J. Radioanal. Chem., 1978, 45, 357 CrossRef CAS.
- R. D. Dewhurst, A. F. Hill and M. K. Smith, Organometallics, 2006, 25, 2388 CrossRef CAS.
- S. Allard and C. Ekberg, J. Solution Chem., 2006, 35, 1173 CrossRef CAS.
- C. I. Bliss, Ann. Entomol. Soc. Am., 1940, 33, 721 CrossRef CAS.
- J. B. Allison, Studies on the toxicity of hydrocyanic acid, Iowa State Coll. J. Sci., 1928, 2(4), 243–252 CAS.
- R. G. Dale, Br. J. Radiol., 1985, 58, 515 CrossRef CAS PubMed.
- M. Morin, R. Masse and J. Lafuma, C. R. Acad. Sci. III., 1990, 311, 459 CAS.
- Y. Tsareva, I. Deltour, M. Sokolnikov, P. Okatenko, V. V. Vostrotin, S. J. Schonfeld and J. Schüz, Radiat. Environ. Biophys., 2016, 55, 291 CrossRef CAS PubMed.
- K. J. Johnson, B. H. Alexander, M. M. Doody, A. J. Sigurdson, M. S. Linet, L. G. Spector, R. W. Hoffbeck, S. L. Simon, R. M. Weinstock and J. A. Ross, Br. J. Cancer, 2008, 99, 545 CrossRef CAS PubMed.
- A. Stewart, J. Webb, D. Giles and D. Hewitt, Lancet, 1956, 271, 447 CrossRef PubMed.
- T. Estlander, O. Kari, R. Jolanki and L. Kanerva, Contact Dermatitis, 1998, 38, 1 CrossRef PubMed.
- H. Bekler, A. Gokce and F. Parmaksizoglu, J. Hand Surg. Eur. Vol., 2007, 32E, 394 CrossRef PubMed.
- E. A. Makrygianni, F. Palamidou and A. G. Kaditis, Pediatr. Pulmonol., 2016, 51, 560 CrossRef PubMed.
- D. A. Edwards, M. D. Andriot, M. A. Amoruso, A. C. Tummey, C. J. Bevan, A. Tveit, L. A. Hayes, S. H. Youngren and D. V. Nakles, Development of Fraction Specific Reference Doses (RfD) and Reference Concentrations (RfC) for Total Petroleum Hydrocarbons (TPH), Amherst Scientific Publishers, Amherst (Massachusetts), 1997 Search PubMed.
- J. N. McDougal, D. L. Pollard, W. Weisman, C. M. Garrett and T. E. Miller, Toxicol. Sci., 2000, 55, 247 CrossRef CAS PubMed.
- F. Muhammad, N. A. Monteiro-Riviere, R. E. Baynes and J. E. Riviere, J. Toxicol. Environ. Health, Part A, 2005, 68, 719 CrossRef CAS PubMed.
- F. Muhammad, N. A. Monteiro-Riviere and J. E. Riviere, Toxicol. Pathol., 2005, 33, 258 CrossRef CAS PubMed.
- F.-J. Albler, K. Bica, M. R. S. J. Foreman, S. Holgersson and M. S. Tyumentsev, J. Cleaner Prod., 2017, 167, 806 CrossRef CAS.
- P. W. Albro and L. Fishbein, Biochim. Biophys. Acta, 1970, 219, 437 CrossRef CAS PubMed.
- R. H. McKee, R. Tibaldi, M. D. Adenuga, J.-C. Carrillo and A. Margary, Regul. Toxicol. Pharmacol., 2018, 92, 439 CrossRef CAS PubMed.
- Occupational Exposure Limit Values (AFS2011:18).
- J.-O. Liljenzin, Solvent Extr. Ion Exch., 2019, 37, 226 CrossRef CAS.
- F. D. M. Fabrega and M. B. Mansur, Hydrometallurgy, 2007, 87, 83 CrossRef CAS.
- J. Mitchell, R. M. Counce, J. S. Watson, B. B. Spencer and G. D. Del Cul, Nucl. Technol., 2010, 170, 422 CrossRef CAS.
- A. H. Thaker, M. Darekar, K. K. Singh and V. V. Buwa, Chem. Eng. Res. Des., 2018, 139, 174 CrossRef CAS.
- S. Javanshir, M. Abdollahy, H. Abolghasemi and A. K. Darban, Int. J. Miner. Process., 2011, 98, 42 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00078h |
| ‡ Escaid100 is an ExxonMobil product containing at least 18% aromatics with a boiling point between 195 and 245 °C. While Escaid110 has no more than 0.5% aromatics. |
| § In hydrometallurgy and industrial solvent extraction it is normal to express the concentration of an extractant in the organic phase using volume percentages. |
| ¶ In the ESI we present a flowsheet for a process for 210Pb contaminated gold. |
| || E° for O2 + 4H+ + 4e− → 2H2O is +1.23 volts. |
| ** DFAu/Cu = ([Au]product[Cu]feed)/([Au]feed[Cu]product) |
| †† Tetra octyl diglycolamide. |
| ‡‡ 152Eu is produced by the neutron bombardment of stable 151Eu in a nuclear reactor, as a result the 152Eu stock solution will contain some stable europium atoms. |
| §§ Note that DEHPA (di(2-ethylhexyl)phosphoric acid) is also known by several other names in the solvent extraction literature which include D2EHPA, bis(2-ethylhexyl) hydrogen phosphate and di(2-ethyl hexyl) orthophosphoric acid. |
| ¶¶ Standard deviation estimated by the jackknife method. |
| |||| The ratio of the highest to the lowest distribution ratios in the radioactive solvent extraction experiments are limited by entrainment, the need for the pipette tip to pass through the upper layer when samples of the lower layer are taken, by the non infinite nature of the partition coefficient of the extracted metal complex and by contamination of the polyethene vials used for gamma counting. To simplify the experimental procedure only gravity was used for phase disengagement and to reduce the effects of contamination of the outsides of vials with radioactivity the counting tubes were never handled with a glove which had previously touched a shaking tube. Also the tubes were held within an uncontaminated plastic beaker, all additions to the tubes were made using a pipette where the tip of the pipette was inserted into the mouth of the counting tube. But the pipette tip was not allowed to touch the inside of the counting tube. |
| *** The RfD is of the daily human exposure limit designed to prevent adverse outcomes in humans if chronic exposure was to occur. The RfDs used do not consider carcinogenic effects. |
| ††† The derivation of the equation is shown in the ESI. |
‡‡‡ In the PCCP paper it was quoted as 3, as the equation 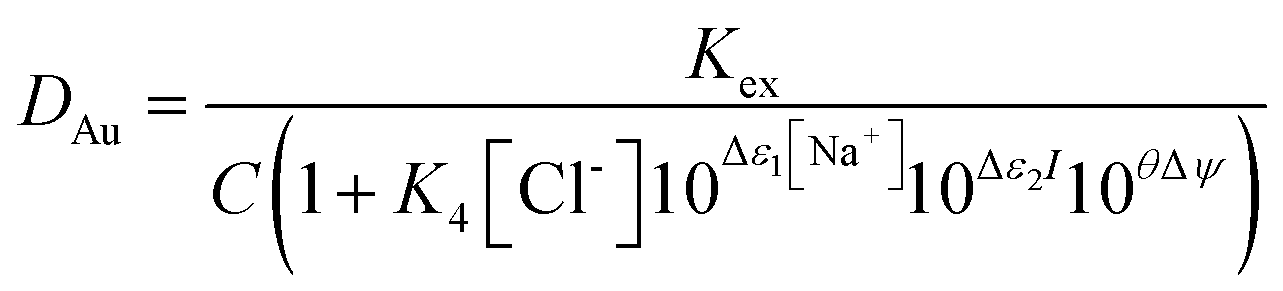 is used if we use Q instead of C then if we write DAu = KexQ10(θΔϕ) then the sign of Δϕ will be opposite to of the Δψ in the PCCP paper but the magnitude shall be the same. is used if we use Q instead of C then if we write DAu = KexQ10(θΔϕ) then the sign of Δϕ will be opposite to of the Δψ in the PCCP paper but the magnitude shall be the same. |
| §§§ TRL means Technology Readiness Level, it is a scale from first laboratory experiments (one) to successful deployment in industry (nine). |
| This journal is © The Royal Society of Chemistry 2024 |