
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Straightforward preparation of a tough and stretchable ion gel†
Aya
Saruwatari
 ab,
Yuji
Kamiyama
ab,
Akifumi
Kawamura
cd,
Takashi
Miyata
cd,
Ryota
Tamate
a and
Takeshi
Ueki
*ab
ab,
Yuji
Kamiyama
ab,
Akifumi
Kawamura
cd,
Takashi
Miyata
cd,
Ryota
Tamate
a and
Takeshi
Ueki
*ab
aResearch Center for Macromolecules and Biomaterials, National Institute for Materials Science, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: UEKI.Takeshi@nims.go.jp
bGraduate School of Life Science, Hokkaido University, Kita 10, Nishi 8, Kita-ku, Sapporo, Hokkaido 060-0810, Japan
cDepartment of Chemistry and Materials Engineering, Kansai University, 3-3-35, Yamate-cho, Suita, Osaka 564-8680, Japan
dOrganization for Research and Development of Innovative Science and Technology, Kansai University, 3-3-35, Yamate-cho, Suita, Osaka 564-8680, Japan
First published on 11th July 2024
Abstract
Ion gels, polymer networks swollen by ionic liquids, are expected to be applied to wearable devices that are tolerant to repeated stretching. High strength and excellent stretchability was achieved due to the numerous physical cross-links with abundant polymer chain entanglements in addition to a small number of immobile chemical cross-links, even though the ion gel was prepared by a facile methodology.
Ion gels are a class of gels swollen by ionic liquids (ILs). Because ILs are exclusively composed of ions, they have unique characteristics such as high ion conductivity, (electro)chemical stability, thermal stability, and low volatility. Owing to these features, ion gels have attracted attention as electrolyte membranes for batteries,1–3 transistors,4,5 and supercapacitors,6 gas absorption/separation membranes,7,8 catalyst-supported membranes.9,10 In addition to these previous studies, ion gels have recently been used as electrolyte membranes for artificial muscles and wearable devices attached to human skin,11–13 especially as strain sensors which measure body movements such as finger/wrist/elbow/knee bending, pronouncing, facial expressions, breathing, and pulse rate. Hydrogels are also candidates for strain sensors,14–19 but there may be serious problems such as the degradation of mechanical properties and the instability against long-term strains due to high volatility. To fulfil this purpose, many studies have focused on the design of stretchable ion gel. For example, Liu et al. applied a double network strategy, which has been widely explored in the research field of hydrogels. The ion gel composed of poly(2-acrylamido-2-methyl-1-propanesulfonic acid) (PAMPS) and 1-ethyl-3-methylimidazolium dicyanamide exhibited an ionic conductivity as high as 1.9 S m−1 with 66.4 wt% IL.20 They also found that the ion gel sensor had excellent stretchability over a wide temperature range between −70 and 100 °C. Sun et al. reported a tough ion gel composed of a poly(urethane–urea) polymer network and 1,2-dimethyl-3-ethoxyethyl-imidazolium trifluoromethylsulfonylimide.21 The ion gels showed no degradation in relative change of electrical resistance at 5% strain, even after severe cyclic tensile conditions of 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. Beyond the representative studies, a number of ion gels for wearable devices are available, including physically cross-linked ion gels based on intermolecular hydrogen bonding,22,23 self-assembled block copolymers,24 poly(ionic liquid)s,25,26 and so on.27,28 However, the preparation of mechanically superior ion gels generally requires complex polymer synthesis techniques, accompanied by advanced knowledge of organic chemistry. Stretchable ion gels that are easily prepared and showing suppressed residual strain for repeated stretching and stress relaxation—characteristics that are potentially applicable to wearable devices—remain rudimentary.
000 cycles. Beyond the representative studies, a number of ion gels for wearable devices are available, including physically cross-linked ion gels based on intermolecular hydrogen bonding,22,23 self-assembled block copolymers,24 poly(ionic liquid)s,25,26 and so on.27,28 However, the preparation of mechanically superior ion gels generally requires complex polymer synthesis techniques, accompanied by advanced knowledge of organic chemistry. Stretchable ion gels that are easily prepared and showing suppressed residual strain for repeated stretching and stress relaxation—characteristics that are potentially applicable to wearable devices—remain rudimentary.
Herein, we describe the preparation and characterization of a stretchable ion gel with excellent mechanical properties. The ion gel can be produced in a simple one-step procedure. As the monomer and cross-linker, methyl methacrylate (MMA) and ethylene glycol dimethacrylate (EGDMA), respectively, were combined with an extremely small concentration of photoinitiator ([I] = 0.02 mol% against MMA monomer in feed) and mixed with the IL, 1-ethyl-3-methylimidazolium trifluoromethylsulfonylimide ([C2mim][TFSI]). This mixture was irradiated with UV light.
We previously reported a new class of ion gels cross-linked solely by polymer chain entanglements of ultrahigh-molecular weight polymers that did not undergo flow deformation and were stable over a wide range of temperatures and frequencies (UHMW ion gels). UHMW ion gels were prepared by mixing the monomer (MMA), solvent ([C2mim][TFSI]), and an extremely low concentration of thermal initiator (2,2′-azobis(isobutyronitrile) (AIBN)), and then heating the mixture to initiate free radical polymerization.29,30 Because the propagation reaction rate of free radical polymerization is significantly enhanced in ILs,31 almost 100% conversion of the monomer to UHMW was achieved. The synthetic strategy presented in this report is based on this procedure. Related to this ion gel preparation, in the research field of hydrogels, Miyata et al. and Suo et al. independently reported that a high monomer concentration and low cross-linker content yielded stretchable hydrogels in which polymer chain entanglements greatly outnumbered chemical cross-links.32–34 These pioneering works encouraged us to devise a synthetic strategy for an ion gel with enough toughness and stretchability, which would be applied to wearable devices. In addition, we attempted to use photoinitiated polymerization to prepare the ion gel instead of thermal initiation because rapid initiation and spatial control are required for processing thin films or flexible patterned shapes. More specifically, instead of the AIBN thermal initiator, an extremely small amount of the 2,2-diethoxyacetophenone (DEAP) photoinitiator, EGDMA as a cross-linker, MMA monomer, and [C2mim][TFSI] solvent were mixed and then irradiated with UV light (365 nm, 3 mW) to photoinitiate the polymerization. A transparent ion gel was obtained (Fig. 1(a)). The transmittance of 10 mm-thick ion gel exceeded 97.5% in the visible light region (400–800 nm) (Fig. 1(b)). The transparency was comparable to or higher than that of the previously reported ion gels even with a longer optical path length (Table S2, ESI†). Furthermore, the ion gel was more thermally stable compared to the hydrogel prepared with the same concentration of initiator, cross-linker and molar concentration of monomer (Fig. S2, ESI†). The hydrogel shrank to 70% of its initial size heated after 60 °C for 30 min and shrank to 80% kept after room temperature for 3 h due to solvent (water) evaporation. Contrary to this, the ion gel did not change in size and shape even after thermal aging. The ion gel is expected to maintain the performances longer than hydrogels owing to high thermal stability.
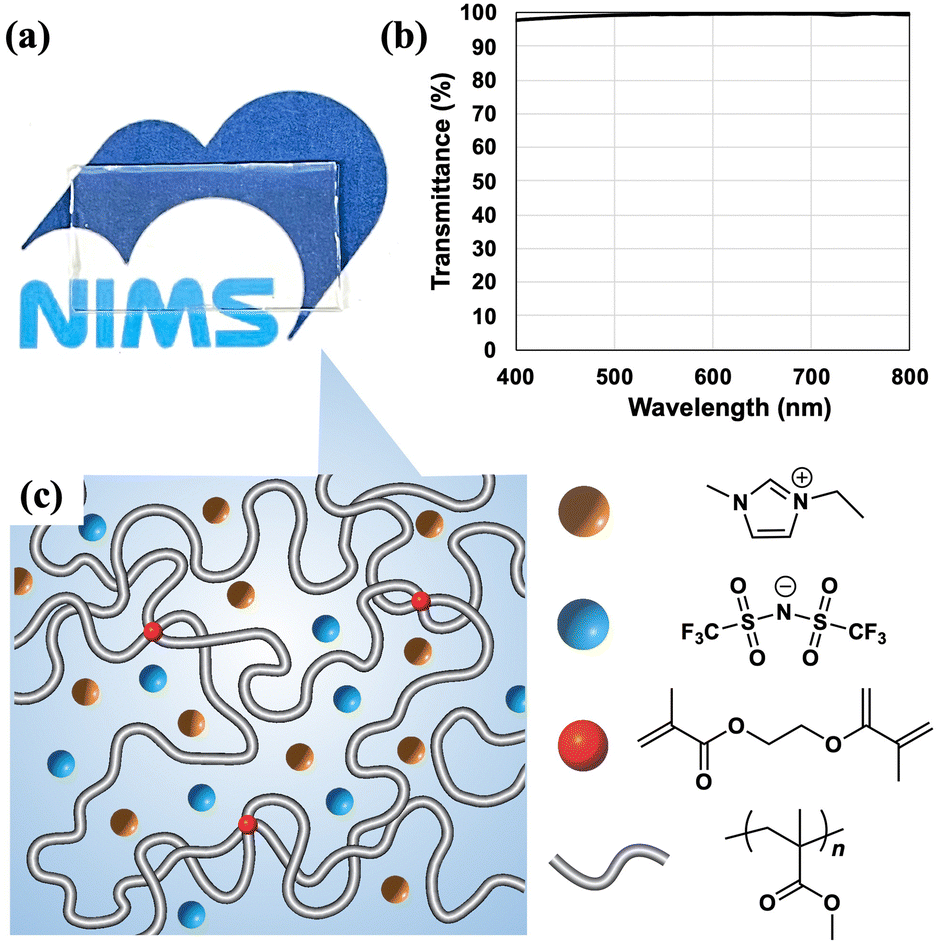 | ||
| Fig. 1 (a) A photograph, (b) a transmittance spectrum, and (c) a schematic illustration of the ion gel used in this study. The white bold lines represent PMMA with a small amount of cross-linker (red spheres). The orange and blue spheres represent the [C2mim]+, and [TFSI]−, respectively. | ||
First, the dependence of the mechanical properties on the cross-linker concentration ([CL] = 0.001–1 mol% vs. [MMA]) was evaluated. The concentrations of the monomer and initiator were fixed at 30 wt% and [I] = 0.02 mol% vs. [MMA], respectively, where topological polymer chain entanglements were expected to act as energy-dissipating components. A stress–strain curve was obtained from the tensile test, as shown in Fig. 2(a). The toughness (Fig. 2(b)), calculated from the area under the stress–strain curve, exhibits a maximum with respect to [CL]. Toughness increases as the [CL] increases from 0.001 to 0.01 mol%, but after passing through maximum at 1 MJ m−3 at 0.01 mol%, it decreases with an increase in the [CL] from 0.01 to 1 mol%. In other words, the toughness exhibits a nonlinear response to the concentration of the cross-linker when the concentrations of the monomer and initiator remain unchanged. As discussed later, the maximum toughness was due to the synergistic effect of a large number of physical cross-links based on polymer chain entanglements and a small number of covalent chemical cross-links. Fig. 2(b) also plots the toughness when the initiator concentration was increased to 0.04 mol% and 0.06 mol%, but the toughness values are significantly lower than those obtained at 0.02 mol%. This is because the polymer chain entanglements that contribute to toughness significantly decrease as the initiator concentration increases. By contrast, the Young's modulus (Eexp) increased slightly as the cross-linker concentration increased (Fig. 2(c)). Thus, in contrast to the conventional trend of gels becoming harder yet more brittle as [CL] increases, the ion gel developed in this study hardens and toughens at [CL] = 0.001–0.01 mol% when the monomer concentration is fixed as 30 wt% and an extremely low concentration of photo-initiator (0.02 mol% vs. monomer) is used.
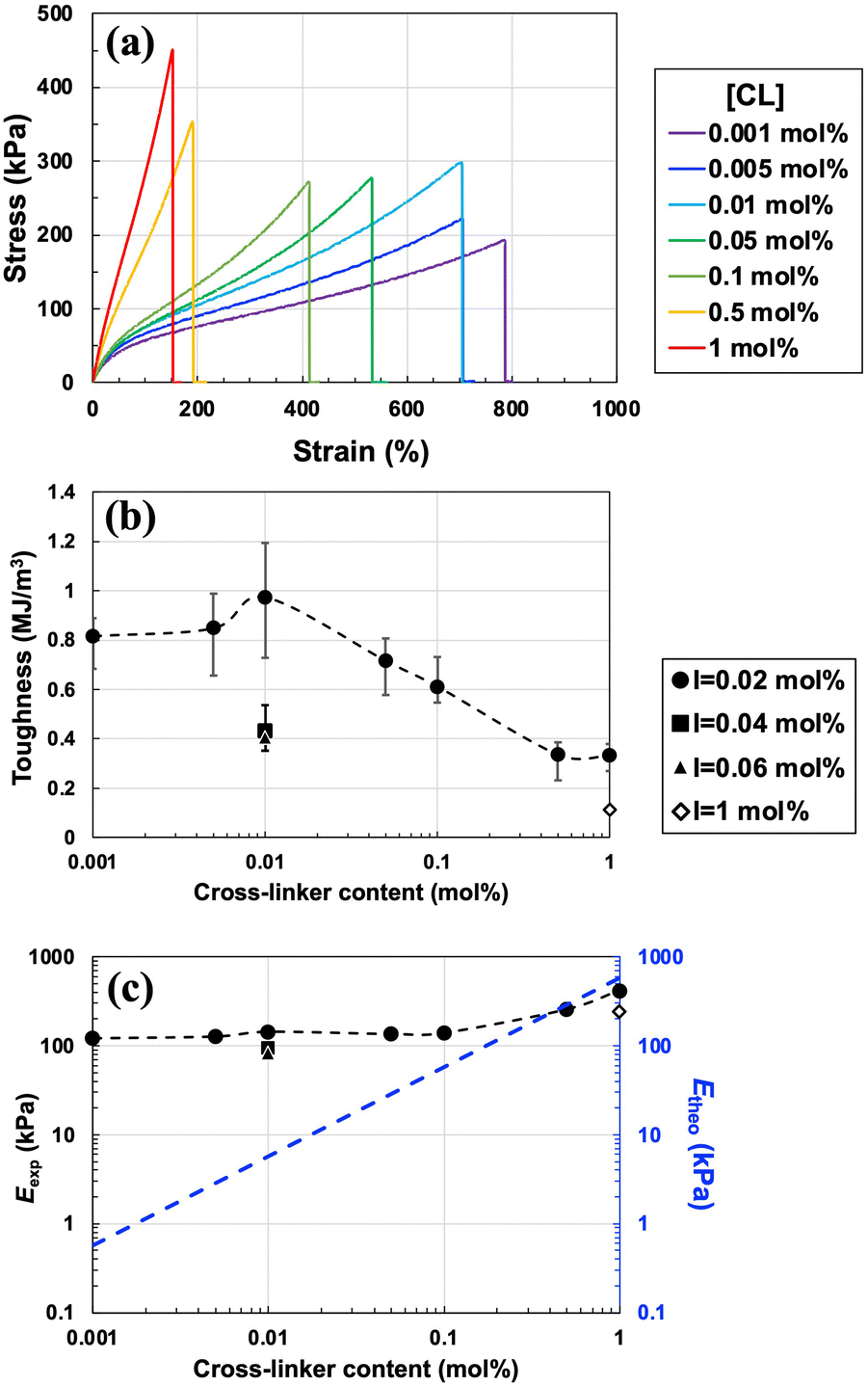 | ||
| Fig. 2 (a) Tensile stress–strain curves of various ion gels with varying cross-linker concentrations and an initiator content of 0.02 mol%. Toughness (b) and Young's modulus (c) of the ion gels in relation to cross-linker and initiator contents of 0.02 (circle), 0.04 (triangle), 0.06 (square), and 1 mol% (diamond). (c) The primary axis plots Young's modulus (Eexp), as determined from the stress–strain curve (a), while the secondary axis is the theoretical value of the Young's modulus (Etheo), calculated based on the cross-linker concentration at I = 0.02 mol%. | ||
We speculated that physical cross-links based on polymer chain entanglements inside the gel were the cause of such nonlinear behaviour and roughly investigated the effect of physical cross-links by calculating the Young's modulus. Assuming that the chemical cross-links are uniformly dispersed in the polymer network and that all of them contribute to the stress, the theoretical value of the Young's modulus (Etheo) was calculated from νtheo35,36 based on the affine network model (eqn (1) and (2)).
| νtheo = Cf/2 = nCLf/2 V | (1) |
| Etheo = 3νtheokT | (2) |
| νexp = Eexp/3kT | (3) |
 | ||
| Fig. 3 Relationship between the cross-linker content in the pre-gel solution and the ratio of experimental to theoretical cross-linking densities (νexp/νtheo) of the ion gels. | ||
If polymer chain entanglements are significantly correlated with the mechanical properties of the gel, particularly in the lower region of the chemical cross-linking density, the molecular weight of the polymer chain appears to directly contribute to the probability of polymer chain entanglement inside the polymer network. Note that the molecular weight in this discussion indicates the degree of monomer linkage of the linear polymer, assuming that no chemical cross-links are introduced, rather than the degree of polymerization (molecular weight) between two cross-links along the polymer chain. In the free-radical polymerization of linear polymers, the molecular weight of the polymer produced is inversely proportional to the root of the initiator concentration in the feed if no cross-linker is added to the polymerization system. For this reason, we explored the dependence of the toughness of the ion gels on the initiator concentration ([I] = 0.02–2 mol%) fixing [MMA] and [CL] at 30 wt% and 0.01 mol% vs. [MMA], respectively, exhibiting maximum toughness (Fig. 2(b)). We also prepared ion gels with [CL] = 0 and 1 mol% vs. [MMA] as a reference. For [CL] = 1 mol%, in the case of the typical cross-linker concentrations for the preparation of polymer gels, ion gels possessing sufficient mechanical strength for handling were obtained at all initiator concentrations ([I] = 0.02–2 mol%). However, when a cross-linker concentration is reduced to 1/100th of the typical initiator concentration ([CL] = 0.01 mol%), no gelation occurs at [I] ≥ 0.2 mol%, although the polymerization reaction appeared to proceed (Table S1, ESI†). Although self-standing gels were obtained at low initiator concentrations of [I] = 0.04 and 0.06 mol%, the toughness and Young's modulus was less than [I] = 0.02 mol%, indicating poor polymer chain entanglement (Fig. 2(b) and (c) and Fig. S4, ESI†). To clarify the effect of the molecular weight of the polymers synthesized in the IL, the molecular weight of the polymers inside the gels was evaluated by changing [I] from 0.02 to 0.04 and 0.06 mol% without adding the cross-linker ([CL] = 0 mol%) (Table 1). At [I] = 0.02 mol%, the molecular weight of poly(methyl methacrylate) (PMMA) exceeded 1000 kDa, whereas at [I] = 0.04 and 0.06 mol%, the molecular weight was less than 1000 kDa, reducing the Young's modulus and toughness. To obtain ion gels with high moduli and toughness, it is important to induce abundant polymer chain entanglements, which can be cross-links and dissipate energy, by lowering the amount of initiator. We have previously reported that in the ion gels formed by UHMW polymers (Mn ≥ 1000 kDa) as a matrix, in the absence of cross-linkers, the abundant polymer chain entanglements act as physical cross-links, resulting in stable elastic gels without flow deformation in a wide range of temperature and frequency regions. Although a self-standing ion gel was obtained in the absence of UHMW polymers (Mn = 175 kDa), flow deformation was observed at high temperatures and in the low-frequency range.29 The effect of molecular weight was also observed in the ion gels loaded with a small amount of chemical cross-linkers. Note that we have already observed that the monomer conversion is significantly lowered when the initiator concentration in the feed is less than 0.02 mol%.29 This suggests that the remaining low-Tg component of the unreacted monomer inside the resultant gel acts as a plasticizer, which reduces the robustness of the materials. Therefore, the amount of initiator should be reduced, but the conversion should be kept nearly 100%, making the residual monomer concentration negligible. To determine monomer conversion, 1H-NMR was measured for the ion gels without cross-linker instead of the cross-linked ion gels that are not soluble in deuterated solvent, and the conversion was found to be almost 100% (Table 1).
Unlike chemical gels, which are formed by covalent bonds with a permanent lifetime, physically cross-linked gels usually exhibit a reduction in stress when strain is applied, owing to the short lifetime of the cross-links, that is, stress relaxation. For example, UHMW ion gel29,30 exhibits excellent mechanical properties over a wide range of frequencies and temperatures, but the stress relaxation originating from the loop-out of the polymer chain can be a serious drawback in the realization of material applications such as strain sensors, which are intended to be subjected to strain for a long period of time. We expected that our optimized ion gel would exhibit less stress relaxation because it not only possesses physical cross-links based on polymer chain entanglements but also a small amount of immobile covalent chemical cross-links. Thus, we attempted to compare the stress relaxation behaviour of the toughest ion gel ([CL] = 0.01 mol%, [I] = 0.02 mol%) with an ion gel prepared without cross-linker ([CL] = 0 mol%, [I] = 0.02 mol%) (Fig. 4). Although the initial stresses were comparable as well as these samples exhibited similar stresses at the initial strain (Fig. S3, ESI†), the relaxation time of the ion gel incorporating chemical cross-linkers was more than 10 times longer than that of the ion gel without cross-linkers, indicating that the introduction of a small amount of cross-linker (one chemical cross-linker per 1000 kDa of MMA) improved the long-term durability under constant strain.
 | ||
| Fig. 4 Stress relaxation tests at γ = 10% for ion gels with a cross-linker content of 0.01 mol% (blue plot) and 0 mol% (black plot). In both samples, the initiator content was 0.02 mol%. | ||
In addition, to evaluate the capability of recovery from repetitive strain, an important mechanical property for wearable devices such as strain sensors and flexible batteries, the ion gels were subjected to 80 successive loading–unloading cycles at a strain of 0–300% with 10 min intervals between cycles (Fig. 5). In this study, we compared the toughest ion gel ([CL] = 0.01 mol%, [I] = 0.02 mol%) with the ion gel prepared without cross-linker ([CL] = 0 mol%, [I] = 0.02 mol%). Hysteresis was observed for both ion gels, which was attributed to energy dissipation owing to the relaxation of polymer chain entanglements. At [CL] = 0 mol%, the slope of the stress–strain curve in the low-strain region decreased, and the residual strain gradually accumulated after the cycle. After 80 cycles, the residual strain finally reached ≈20%. By contrast, the ion gel loaded with [CL] = 0.01 mol% showed almost no slope change, and the final residual strain was less than only 10%. Given that the residual strain indicates disentanglement of the polymer chains, the introduction of a small amount of cross-linker suppressed the disentanglement of the polymer chains, resulting in a significant improvement in the cyclic strain resistance of the ion gels. When physically cross-linked gels formed by non-covalent bonds such as ionic bonds are stretched, the initial bonds break and temporary bonds are formed with other partners. After unloading, the temporary bonds are repetitively formed and broken to return to the initial bonds by the driving force of rubber elasticity.39,40 In present system, physical cross-linking is based on polymer chain entanglement, however; similar “exchangeable cross-linking” must play important role on mechanical property of material. The introduction of chemical cross-linkers, which are permanent bonds, is expected to inhibit the formation of temporary bonds, resulting in a higher recovery to the initial state. To further investigate the kinetics of disentanglement and entanglement for the ion gels with and without cross-linkers, we are planning to measure the hysteresis and residual strain of the ion gel depending on tensile speed, on deformation range and on waiting time.
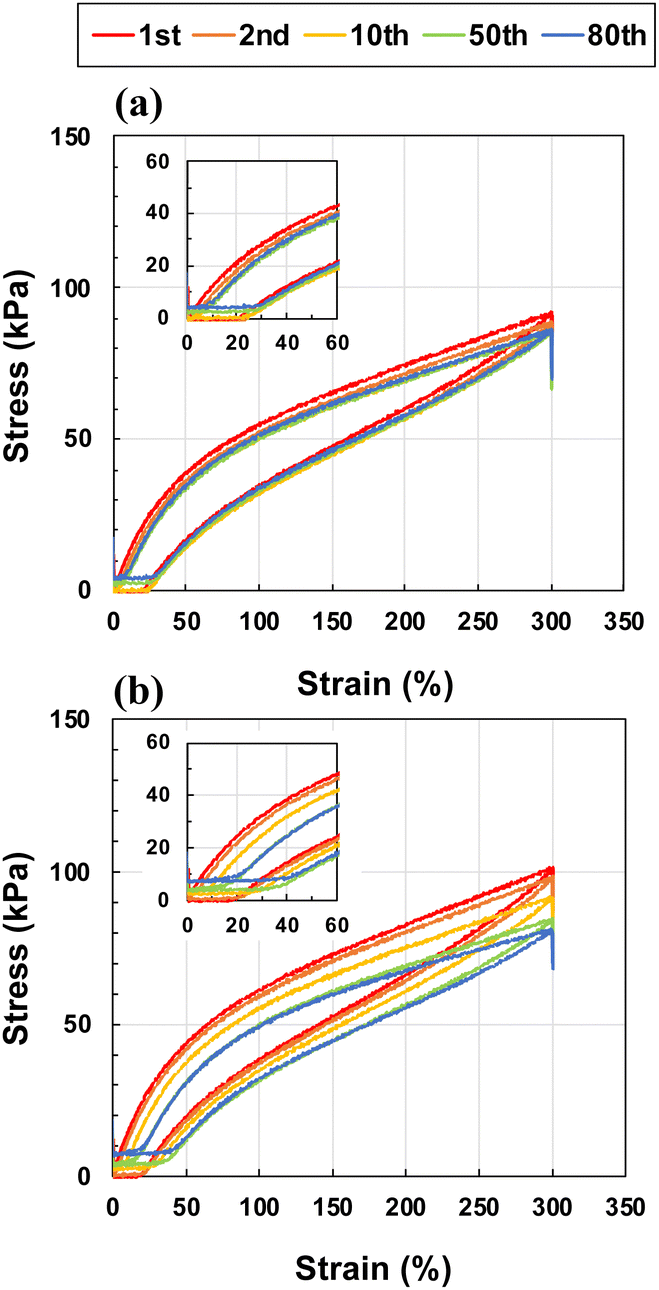 | ||
| Fig. 5 Cyclic stress–strain curves at 300% strain at a tensile rate of 10 cm min−1 for the ion gels with cross-linker contents of (a) 0.01 mol% and (b) 0 mol% at the same initiator content of 0.02 mol%. Each cycle was separated by a waiting time of 600 s. The inset shows magnified images. | ||
In general, there is a trade-off relationship between the ionic conductivity guaranteed by the amount of IL and the mechanical properties of the gels.41,42 Sun et al. reported a cyclic strain-tolerant ion gel containing 50 wt% IL as a solvent. The ion gel showed ≈20% residual strain after 10 cycles with a 200% loading strain.21 Song et al. also reported that an ion gel swollen by 40 wt% ILs showed no residual strain after 50 cycles with a 200% loading strain.25 In this study, we prepared an excellent expansion-strain-tolerant ion gel swelling up to 70 wt% using ILs as solvents. Compared to the previously reported ion gels containing nearly 70 wt% IL (Table S2, ESI†), the ion gel in this study showed low residual strain, considering that the tensile fatigue tests were conducted under relatively severe conditions with a 300% strain. We expected that the gel would exhibit both good electrochemical properties and durability against cyclic strain.
For demonstration as strain sensors, the ion gel ([CL] = 0.01 mol%, [I] = 0.02 mol%) sandwiched between copper foils at both ends were manually stretched while the current was measured at a constant voltage. The relative resistance responded reversibly within five cycles and increased as the strain was increased to 10%, 25%, and 50% (Fig. S5, ESI†), indicating that the ion gel has the ability to sense different movements of the human body. To investigate the stability against more repetitive stretching, the ion gel was subjected to 100 and 50 consecutive loading–unloading cycles at 10 and 50% strain, respectively (Fig. S6, ESI†). The changes of the relative resistance were stable throughout the overall cycles. The ion gel detected different mechanical strains with reversible and stable signal output.
Conclusions
In this paper, we report an ion gel consisting of a [C2mim][TFSI] solvent and MMA as a matrix polymer cross-linked with a small amount of EGDMA. The ion gel was obtained by a facile one-step procedure and exhibited a high toughness of up to ≈1 MJ m−3 and excellent recovery capability against loading–unloading cycles when the composition of the pre-gel solution was appropriately optimized. In contrast to the conventional mechanical behaviour of gels, which become stiff and brittle as the [CL] increases, the Young's modulus and toughness of our ion gels change non-linearly as a function of [CL] at 0.001–0.01 mol% [CL]. The comparison between Etheo and Eexp at a [CL] lower than 0.1 mol% indicates that the numerous physical cross-links derived from the polymer chain entanglements and the small amount of permanent chemical cross-links synergistically contribute to the toughness and stiffness of the gel.The initiator concentration ([I]) in the pre-gel solution also plays an important role in maintaining the mechanical properties of the resultant ion gel. As the toughness and self-standing capability decreased when [I] was significantly increased, an increase in the degree of monomer linkage (i.e., increasing the molecular weight of the polymer to UHMW when the chemical cross-links were ignored) was found to induce polymer chain entanglements and ensure that the ion gel was mechanically strong. Furthermore, because unreacted MMA monomers accidentally plasticize and reduce the Tg of the ion gel, [I] should be controlled to the extent that conversion reaches ≈100%. The ion gel with the highest toughness, despite the low number of chemical cross-linkers (one chemical cross-linker per every 10000 repeating units of MMA), demonstrated a 10-fold longer relaxation time and less than half its the residual strain after 80 successive loading–unloading cycles than the ion gel without a cross-linker, indicating an improvement in long-term durability.
This study would underpin the strategic design of ion gels useful for wearable electronics, soft robotics, and other applications owing to its simple material preparation procedure, high strength, and high resilience to repetitive strains.
Author contributions
Investigation: A. S. supervision: T. U. writing – original draft: A. S. and T. U. writing – review and editing: Y. K., A. K., T. M., R. T., and T. U.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by a Grant-in-Aid for JSPS Fellows (22KJ010202 to A. S.) and JSPS KAKENHI grants (23H02030 to T. U.).References
- M. Watanabe, M. L. Thomas, S. G. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS.
- N. Chen, H. Zhang, L. Li, R. Chen and S. Guo, Adv. Energy Mater., 2018, 8, 1702675 CrossRef.
- L. C. Tome, L. Porcarelli, J. E. Bara, M. Forsyth and D. Mecerreyes, Mater. Horiz., 2021, 8, 3239–3265 RSC.
- S. H. Kim, K. Hong, W. Xie, K. H. Lee, S. Zhang, T. P. Lodge and C. D. Frisbie, Adv. Mater., 2013, 25, 1822–1846 CrossRef CAS.
- J. Lee, M. J. Panzer, Y. Y. He, T. P. Lodge and C. D. Frisbie, J. Am. Chem. Soc., 2007, 129, 4532–4533 CrossRef CAS.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- L. C. Tomé and I. M. Marrucho, Chem. Soc. Rev., 2016, 45, 2785–2824 RSC.
- M. G. Cowan, D. L. Gin and R. D. Noble, Acc. Chem. Res., 2016, 49, 724–732 CrossRef CAS PubMed.
- P. Snedden, A. I. Cooper, K. Scott and N. Winterton, Macromolecules, 2003, 36, 4549–4556 CrossRef CAS.
- R. T. Carlin and J. Fuller, Chem. Commun., 1997 Search PubMed.
- X. Fan, S. Liu, Z. Jia, J. J. Koh, J. C. C. Yeo, C. G. Wang, N. E. Surat'man, X. J. Loh, J. Le Bideau, C. He, Z. Li and T. P. Loh, Chem. Soc. Rev., 2023, 52, 2497–2527 RSC.
- N. Gao and C. Pan, SmartMat, 2023, 5, e1215 CrossRef.
- M. Wang, J. Hu and M. D. Dickey, JACS Au, 2022, 2, 2645–2657 CrossRef CAS.
- Z. Wang, Y. Cong and J. Fu, J. Mater. Chem. B, 2020, 8, 3437–3459 RSC.
- G. Li, C. Li, G. Li, D. Yu, Z. Song, H. Wang, X. Liu, H. Liu and W. Liu, Small, 2022, 18, 2101518 CrossRef CAS.
- Q. Wu, A. Chen, Y. Xu, S. Han, J. Zhang, Y. Chen, J. Hang, X. Yang and L. Guan, Soft Matter, 2024, 20, 3666–3675 RSC.
- R. Li, J. Ren, M. Li, M. Zhang, Y. Li and W. Yang, Soft Matter, 2023, 19, 5723–5736 RSC.
- Y. Niu, H. Liu, R. He, M. Luo, M. Shu and F. Xu, Small, 2021, 17, 2101151 CrossRef CAS.
- J. Yang, Z. Xu, J. Wang, L. Gai, X. Ji, H. Jiang and L. Liu, Adv. Funct. Mater., 2021, 31, 2009438 CrossRef CAS.
- Y. Ding, J. Zhang, L. Chang, X. Zhang, H. Liu and L. Jiang, Adv. Mater., 2017, 29, 1704253 CrossRef.
- T. Li, Y. Wang, S. Li, X. Liu and J. Sun, Adv. Mater., 2020, 32, 2002706 CrossRef PubMed.
- Z. Tang, D. Liu, X. Lyu, Y. Liu, Y. Liu, W. Yang, Z. Shen and X. Fan, J. Mater. Chem. C, 2022, 10, 10926–10934 RSC.
- Y. Wang, Y. Liu, R. Plamthottam, M. Tebyetekerwa, J. Xu, J. Zhu, C. Zhang and T. Liu, Macromolecules, 2021, 54, 3832–3844 CrossRef CAS.
- K. G. Cho, S. An, D. H. Cho, J. H. Kim, J. Nam, M. Kim and K. H. Lee, Adv. Funct. Mater., 2021, 31, 2102386 CrossRef CAS.
- T. Li, F. Liu, X. Yang, S. Hao, Y. Cheng, S. Li, H. Zhu and H. Song, ACS Appl. Mater. Interfaces, 2022, 14, 29261–29272 CrossRef CAS.
- Z. Yu and P. Wu, Adv. Mater., 2021, 33, 2008479 CrossRef CAS.
- Y. M. Kim and H. C. Moon, Adv. Funct. Mater., 2020, 30, 1907290 CrossRef CAS.
- W. Li, L. Li, S. Zheng, Z. Liu, X. Zou, Z. Sun, J. Guo and F. Yan, Adv. Mater., 2022, 34, 2203049 CrossRef CAS PubMed.
- Y. Kamiyama, R. Tamate, T. Hiroi, S. Samitsu, K. Fujii and T. Ueki, Sci. Adv., 2022, 8, eadd0226 CrossRef CAS.
- Y. Kamiyama, R. Tamate, K. Fujii and T. Ueki, Soft Matter, 2022, 18, 8582–8590 RSC.
- P. Kubisa, Eur. Polym. J., 2020, 133, 109778 CrossRef CAS.
- C. Norioka, Y. Inamoto, C. Hajime, A. Kawamura and T. Miyata, NPG Asia Mater., 2021, 13, 34 CrossRef CAS.
- J. Kim, G. G. Zhang, M. X. Shi and Z. G. Suo, Science, 2021, 374, 212–216 CrossRef CAS PubMed.
- M. Shi, J. Kim, G. Nian and Z. Suo, Extreme Mech. Lett., 2023, 59, 101953 CrossRef.
- W. Xue, M. B. Huglin and T. G. J. Jones, Eur. Polym. J., 2005, 41, 239–248 CrossRef CAS.
- D. Calvet, J. Y. Wong and S. Giasson, Macromolecules, 2004, 37, 7762–7771 CrossRef CAS.
- J. Baselga, I. Hernandez-Fuentes, I. F. Pierola and M. A. Llorente, Macromolecules, 1987, 20, 3060–3065 CrossRef CAS.
- J. Baselga, M. A. Llorente, I. Hernández-Fuentes and I. F. Piérola, Eur. Polym. J., 1989, 25, 477–480 CrossRef CAS.
- R. Shi, T. L. Sun, F. Luo, T. Nakajima, T. Kurokawa, Y. Z. Bin, M. Rubinstein and J. P. Gong, Macromolecules, 2018, 51, 8887–8898 CrossRef CAS PubMed.
- R. Long, K. Mayumi, C. Creton, T. Narita and C.-Y. Hui, Macromolecules, 2014, 47, 7243–7250 CrossRef CAS.
- M. A. Susan, T. Kaneko, A. Noda and M. Watanabe, J. Am. Chem. Soc., 2005, 127, 4976–4983 CrossRef CAS.
- M. M. Mok, X. Liu, Z. Bai, Y. Lei and T. P. Lodge, Macromolecules, 2011, 44, 1016–1025 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details for materials; preparation procedures of ion gels; measurement procedures; 1H NMR spectrum; observation results of gelation behaviour; and additional characterization results of the ion gels. See DOI: https://doi.org/10.1039/d4sm00628c |
| This journal is © The Royal Society of Chemistry 2024 |