
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Temperature dependence of micelle shape transitions in copolymer solutions: the role of inter-block incompatibility†
M. J.
Greenall
 *a and
M. J.
Derry
b
*a and
M. J.
Derry
b
aSchool of Mathematics and Physics, University of Lincoln, Brayford Pool, Lincoln, LN6 7TS, UK. E-mail: mgreenall@lincoln.ac.uk
bAston Institute for Membrane Excellence, Aston University, Birmingham, B4 7ET, UK
First published on 9th April 2024
Abstract
The nature of the transition between worm-like and spherical micelles in block copolymer dispersions varies between systems. In some formulations, heating drives a transition from worms to spheres, while in other systems the same transition is induced by cooling. In addition, a sphere-worm interconversion can be accompanied either by an increase or a decrease in the core solvation, even if the direction of the temperature dependence is the same. Here, self-consistent field theory is used to provide a potential explanation of this range of behaviour. Specifically, we show that, within this model, the dependence of the transition on the incompatibility χBS of the solvophobic block B and the solvent S (the parameter most closely related to the temperature) is strongly influenced by the incompatibility χAB between B and the solvophilic block A. When χAB is small (χAB < 0.1), it is found that increasing χBS produces a transition from worm-like micelles to spheres (or, more generally, from less curved to more curved structures). When χAB is above 0.1, increasing χBS drives the system from spheres to worm-like micelles. Whether a transition is observed within a realistic range of χBS is also found to depend on the fraction of solvophilic material in the copolymer. The relevance of our calculations to experiments is discussed, and we suggest that the direction of the temperature dependence may be controlled not only by the solution behaviour of the solvophobic block (upper critical solution temperature-like versus lower critical solution temperature-like) but also by χAB.
Introduction
The morphologies of the self-assembled structures, such as micelles and vesicles, formed by amphiphilic block copolymers in solution can often be controlled by changing the temperature.1 These transitions have been investigated in a range of systems, with many studies focusing on structures formed by polymerisation-induced self-assembly (PISA).2–6 Recent work on thermoresponsive polymers produced by this method1 demonstrates that the full range of standard structures (spherical micelles, worm-like micelles, vesicles and lamellae) can be accessed in a single system by varying the temperature. Precise control over the self-assembled structures simply on changing the temperature raises the possibility of a range of applications, such as the release of proteins or enzymes encapsulated in a vesicle1 or the production of an oil that thickens at high temperatures.7Studies in this area are complicated by the fact that the nature of the transitions between structures varies significantly from one system to another. In many formulations, heating drives transitions towards more curved structures, for example, from worm-like micelles to spherical micelles8–13 or from vesicles to worm-like micelles.7,14
In other dispersions, the opposite temperature dependence is seen, and heating the system leads to transitions to less curved structures,15,16 so that sphere-to-worm17,18 and worm-to-vesicle19,20 transitions are observed as the temperature is increased.
In addition, systems that have the same direction of the temperature dependence can differ in how the core solvation changes during the transition, and recent studies on thermo-responsive block copolymers have found that transitions to less curved structures on heating can be accompanied either by increased or decreased solvation of the core.21,22
Self-consistent field theory (SCFT) calculations are a promising route for rationalising the varying temperature dependence observed in micellar systems. Although Monte Carlo methods23 provide a greater level of microscopic detail, and dissipative particle dynamics techniques24 give both more detail and information on the dynamics, self-consistent field theory allows a rapid scan of the parameter space.25 In addition, existing SCFT results25,26 show different dependence of the shape transitions on the solvent-solvophobic block chi parameter χBS, the quantity most usually associated with the temperature.25 Specifically, depending on the parameters chosen, increasing χBS is predicted by SCFT either to drive the system to form flatter structures25 or to form more curved structures.26 Our aim here is to find the factors, for example interaction strengths or block lengths, that lead to the difference in the direction of temperature dependence and to interpret our calculations in the light of experimental results. Although much of this experimental data comes from the PISA literature, the model does not depend on the dynamics of this process and should also be applicable to aggregates produced by other methods.
Method
Self-consistent field theory27 models polymer molecules as random walks in space. As a mean-field theory, SCFT does not take fluctuations into account and, as an equilibrium model, it finds states that correspond to a local minimum of the free energy. The system is treated as being incompressible, so that the volume fractions of the various species sum to 1 at all points, and the interactions between different molecular species i and j are assumed to be short ranged with a strength that can be quantified by a single parameter χij. These interactions are modelled by position-dependent fields, one associated with each species.To model micelle formation, a solution of AB diblock copolymers in solvent S is studied in the canonical ensemble, so that the overall volume fractions of all species remain fixed. The total degree of polymerisation N of the copolymer chains, the overall volume fraction ϕ of copolymer, the block volume fraction fA of the copolymer that consists of A monomers and the interaction parameters χAB, χAS and χBS are all specified at the beginning of the calculation, and the outputs are the position-dependent volume fractions ϕA(r), ϕB(r) and ϕS(r) and the free energy F. Since we are investigating broad trends rather than attempting to model a specific system, the volume of an A segment, the volume of a B segment and the volume of a solvent molecule are all set to the same value, which we write in terms of the segment density ρ0 as 1/ρ0.
Details of SCFT applied to copolymers in a solvent can be found in, for example, ref.28. Specialising to the current case of AB diblock copolymers, the free-energy density measured with respect to that of a homogeneously mixed system can be written as28,29
 | (1) |
The derivation of the above free energy also gives a set of simultaneous equations that link the fields and densities.28 Each SCFT calculation is started by making initial guesses for the fields, which are then used to calculate the densities. These densities are then substituted back into the simultaneous equations to update the fields, and the process is repeated until convergence is achieved.
To compute the free energy of a spherical micelle, we work in spherical polar coordinates and assume spherical symmetry of the aggregates, making the problem one-dimensional.26 Reflecting boundary conditions are imposed at the centre and surface of the system. Once a solution corresponding to a micelle has been computed by the procedure outlined above, the micelle with the lowest free-energy density is found as follows. The volume of the calculation box is adjusted, the SCFT equations are solved again, and the new free-energy density is calculated. This process is continued until a minimum in the free-energy density is located. This is an approximate way of finding the equilibrium state of the whole system and can be justified as follows. The equilibrium state of the system is found by minimising the total free energy FT. For a system of constant total volume VT, this is equivalent to minimising the free-energy density FT/VT, since only FT changes. The macroscopic system minimises this quantity by varying the number of micelles.31 If the micelles are assumed all to be the same size and inter-micellar interactions are neglected, the system can be divided into identical boxes of volume V, each containing one micelle, and the number of micelles can be varied by changing V. If the free energy of each subsystem is F, the equilibrium state of the whole system can be found by minimising F/V (which, since all boxes are identical, is equal to FT/VT) with respect to V. Since the overall volume fraction of copolymer is kept constant, varying the box size means that the number of polymers in the box is changed at the same time. A more formal derivation of this method can be found in ref. 31. Similarly, worm-like micelles are modelled in cylindrical polar coordinates assuming cylindrical symmetry and no variation in density along the length of the cylinder. This latter assumption implies that the worms are treated as being inflexible and infinitely long, meaning that, for example, contributions to the free energy from the worm ends or from their curvature are neglected. Finally, Cartesian coordinates are used to investigate bilayers. Again, the problem is made one-dimensional, here by assuming that the density profile only varies perpendicular to the plane of the membrane. As in the cylindrical case, this treats the bilayers as being of infinite extent. This means that edge effects and curvature are not included in the model and no distinction is made between vesicles and lamellae. Given the assumed symmetries, the box volume V in eqn (1) must be replaced by an area A in the cylinder case and a length L in the bilayer calculation. The same procedure of varying the box size to find a minimum in the free-energy density is followed in the cylinder and bilayer cases, with the calculation now corresponding to finding an optimum number of cylinders per unit area or bilayers per unit length. To determine which morphology is predicted to be the equilibrium state for a given set of parameters, the free-energy densities are compared.
The diffusion equations are solved using a finite-difference method with a spatial step size of 0.005 in units of the root-mean-square end-to-end distance of the polymers. The dimensionless parameter s that specifies the distance along the polymer backbone varies from 0 to 1 and a step size of 0.0005 is used. The iterative procedure to solve the simultaneous equations involved in SCFT is implemented using simple mixing followed by damped Anderson mixing.32–35
The implementation of SCFT used here is equivalent to that used by Liaw et al.26 to study micelle and bilayer formation in block copolymer/solvent systems, and we have reproduced a representative sample of their results as a check on our method (Fig. S1, ESI†).
Results and discussion
The technique outlined in the preceding section is now applied to a range of systems. Since our focus is on the effect of the properties of the polymer molecules and not their concentration, the polymer volume fraction ϕ is kept the same in all calculations. A value of ϕ = 0.05 is chosen, corresponding to a relatively dilute system. This choice is made because the edge of the calculation box would distort the aggregate at high concentrations. To reduce the number of parameters that are varied at once, the overall degree of polymerisation is also chosen to be the same in all computations. The value used, N = 250, is moderate in the context of studies of block copolymer self-assembly in solution.25,26 We follow ref. 16 by setting the solvent-solvophilic block chi parameter to 0.48, which is characteristic of a good solvent.26 The inter-block chi parameter χAB is changed from system to system, and is varied from 0.05, corresponding to moderately immiscible polymers, to 0.2, which gives strong segregation. The volume fraction fA of each copolymer that consists of solvophilic A monomers is also varied between systems, as this parameter has a strong influence on the shape of the aggregates.36 The solvent-solvophobic block chi parameter χBS is varied within each system to mimic the effect of temperature variation.25,26 Since both χAB and χAS are assumed not to vary with temperature, this approach assumes that χBS is much more responsive to temperature variations than the first two chi parameters. Evidence to support this focus on χBS in the current study of temperature dependence can be found in ref. 1 and 21, where variable-temperature NMR studies show that the solvation of the core block is much more sensitive to temperature than that of the shell block.Each system shown in Table 1 has a specified value of the inter-block chi parameter χAB and the solvophilic block degree of polymerisation NA. Since the total degree of polymerisation is fixed at 250, the degree of polymerisation of the solvophobic block is NB = 250 − NA and the solvophilic volume fraction of the copolymers is fA = NA/250. To focus the study, we concentrate on a range of parameters where spheres and cylinders are the most common morphologies. For each system, the solvent-solvophobic block chi parameter χBS is increased from 0.7 to 1.5, so that the core changes from strongly to weakly solvated.26 As χBS is changed, any shape transitions that occur are recorded, with the first letter in each cell in Table 1 representing the morphology (sphere, cylinder or bilayer) formed at low χBS and any subsequent letters representing the morphologies that form as χBS is increased.
| χ AB | |||||||
|---|---|---|---|---|---|---|---|
| N A|fA | 0.05 | 0.075 | 0.1 | 0.125 | 0.15 | 0.175 | 0.2 |
| 70|0.28 | B → C | B → C | C | C | C | S → C | S → C |
| 72|0.288 | B → C | B → C | C | C | C | S → C | S → C |
| 74|0.296 | B → C | C | C | C | S → C | S → C | S → C |
| 76|0.304 | B → C | C | C | C | S → C | S → C | S → C |
| 78|0.312 | B → C | C | C | S → C | S → C | S → C | S → C |
| 80|0.32 | B → C | C | C → S → C | S → C | S | S | S |
| 82|0.328 | C → S | C → S | C → S | S | S | S | S |
| 84|0.336 | C → S | C → S | S | S | S | S | S |
| 86|0.344 | C → S | C → S | S | S | S | S | S |
| 88|0.352 | C → S | C → S | S | S | S | S | S |
| 90|0.36 | C → S | C → S | S | S | S | S | S |
| 92|0.368 | C → S | S | S | S | S | S | S |
In two regions of Table 1, only one morphology (sphere or cylinder) forms over the range of χBS used. However, there are two further regions where a transition between cylinders and spheres is predicted. Specifically, for lower values of χAB and higher values of NA (bottom left of the table), a transition from cylinders to spheres is seen as χBS is increased. For higher values of χAB and lower values of NA (top right), the predicted transition runs in the opposite direction, from spheres to cylinders as χBS increases. In the top left of Table 1, where NA and χAB are both relatively small, there is a transition from bilayers to cylinders as χBS is increased.
These results suggest that the most important quantity in determining the direction of the dependence of the shape transitions on χBS could be the inter-block chi parameter χAB. However, it may be necessary to adjust the block lengths as well as χAB for the reversed transitions to be seen. For example, for NA ≥ 82, increasing χAB moves away from the cylinder–sphere transition to a region of parameter space where only one shape (the sphere) is seen. For the opposite (sphere–cylinder) transition to be observed, it is also necessary to lower NA to move into the top right of the table.
When NA = 80 and χAB = 0.1, there is a single system where two transitions occur: from cylinders to spheres and then from spheres back to cylinders. This system is on the border between the regions where cylinder–sphere and sphere–cylinder transitions occur, and the competition between the two morphologies is finely balanced. This re-entrant effect has also been predicted in mean-field calculations that use a single chi parameter,37 and the current calculations give some further information on the conditions for which this phenomenon might occur. We note that, If χBS is increased beyond 1.5, the re-entrant region can be extended to lower values of χAB. For example, at χBS = 1.8, the NA = 82, χAB = 0.075 system returns to the cylinder phase, giving a cylinder–sphere–cylinder sequence of morphologies overall. However, a strongly hydrophobic B-block with a low incompatibility with the hydrophilic A-block may be difficult to realise, and such strong hydrophobicity may lead to precipitation rather than self-assembly, meaning that it is likely that extremely fine tuning of the parameters to a region corresponding to the central area of Table 1 would be needed for a re-entrant transition to be seen in experiments.
To gain insight into the processes that lead to the transitions between spheres and cylinders predicted by the model, we plot cuts through the density profiles of the micelles that form as χBS is varied for three different systems. These are chosen to lie on a diagonal line starting on the left of the table in the region where cylinder–sphere transitions occur as χBS is increased and finishing on the top right where the transitions occur in the opposite (sphere–cylinder) direction. Since N is the same in all systems, distances may be measured in units of the root-mean-square end-to-end distance aN1/2 without introducing inconsistency between the figures. In each of Fig. 1–3, the local volume fractions of the A, B and S species are plotted. As described in the methods section, the volume of the calculation box is varied to find the structure with the lowest free-energy density, and the boundary of this box is marked to show that the aggregates are surrounded by solvent and not distorted by the edge of the system.
In the first system (Fig. 1), χAB = 0.05 and NA = 84. Here, the relatively large shell (A) block favours more curved structures. As χBS increases from 0.7 to 0.9, the radius of the cylinder increases, decreasing its curvature. This compresses the A-blocks in the shell into a progressively more unfavourable state. When χBS reaches 1, the system moves from the cylinder to the sphere phase, relaxing the A-blocks. Changing from cylinders to spheres increases the total interfacial area of the micelle cores. However, the system can pay the associated free-energy penalty due to the relatively low value of χAB. The core blocks also become more compressed when the micelles become spherical, but these are short compared to the other systems studied and the energy cost is again not sufficient to prevent the transition.
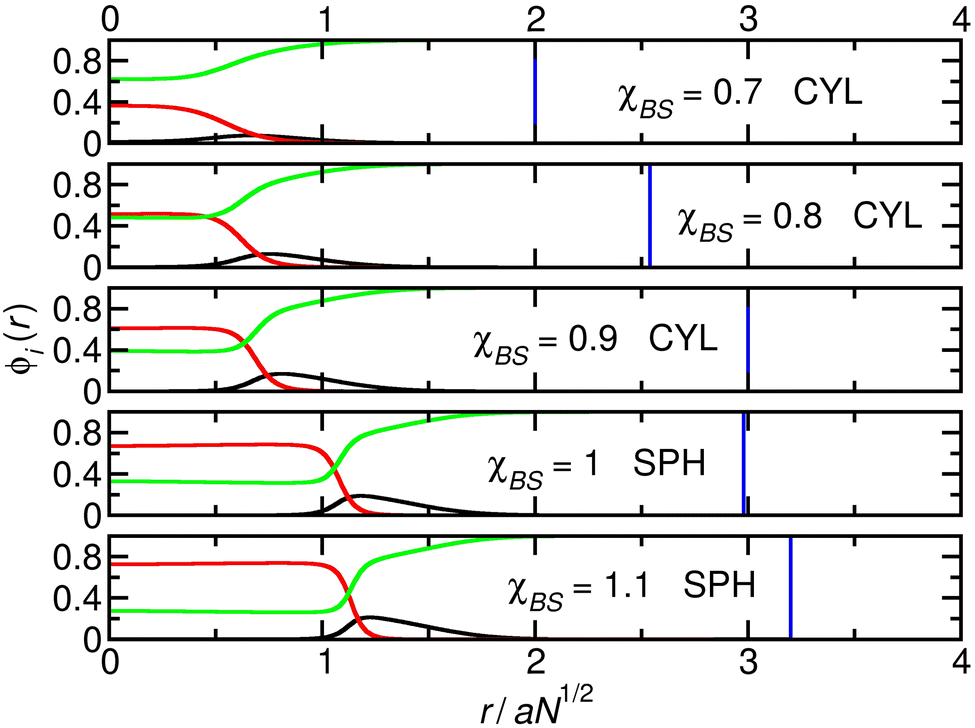 | ||
| Fig. 1 Cuts through the density profiles of the micelles that form when χAB = 0.05 and NA = 84. Black lines: A-blocks (shell); red lines: B-blocks (core); green lines: solvent. Vertical blue lines: boundary of the calculation box. | ||
We now turn our attention to the system (Fig. 2) at the far end of the diagonal cut through the table and contrast it with the previous case. Now, χAB = 0.2 and NA = 72, and the larger value of χAB means that the surface energy is a more important factor in determining the shape of the aggregates. This means that, as χBS increases, the system changes from the sphere to the cylinder phase to reduce the overall surface area of its solvophobic cores. This leads to a compression of the shell A-blocks, but these are relatively short, and the associated energy penalty is not enough to keep the system in the sphere phase. The relatively long core blocks also favour the transition to a less curved structure.
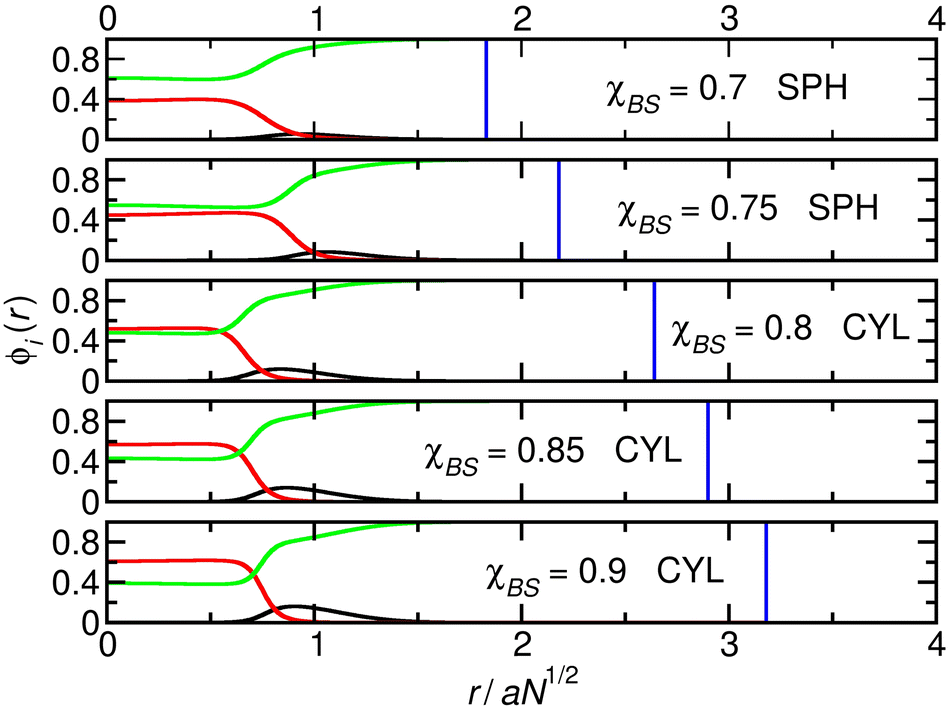 | ||
| Fig. 2 Cuts through the density profiles of the micelles that form when χAB = 0.2 and NA = 72. Black lines: A-blocks (shell); red lines: B-blocks (core); green lines: solvent. Vertical blue lines: boundary of the calculation box. | ||
The third system we focus on is that where the system changes from cylinders to spheres and then back again (Fig. 3). Here, χAB = 0.1 and NA = 80, intermediate between the values in the earlier systems, and there is a fine balance between the two phases. It is possible that a factor in the transition back to the cylinder phase at χBS = 1.5 is the unfavourable state of the B-blocks in the swollen core of the sphere phase as χBS increases. The fine balance between the various factors involved in determining the morphology is illustrated by the fact that the system moves from a phase with a lower micelle surface area per unit volume (cylinders), to one with a higher micelle surface area per unit volume (spheres), and back again. This is shown in Fig. S2 (ESI†), along with a corresponding plot of the interfacial width of the micelle surface.
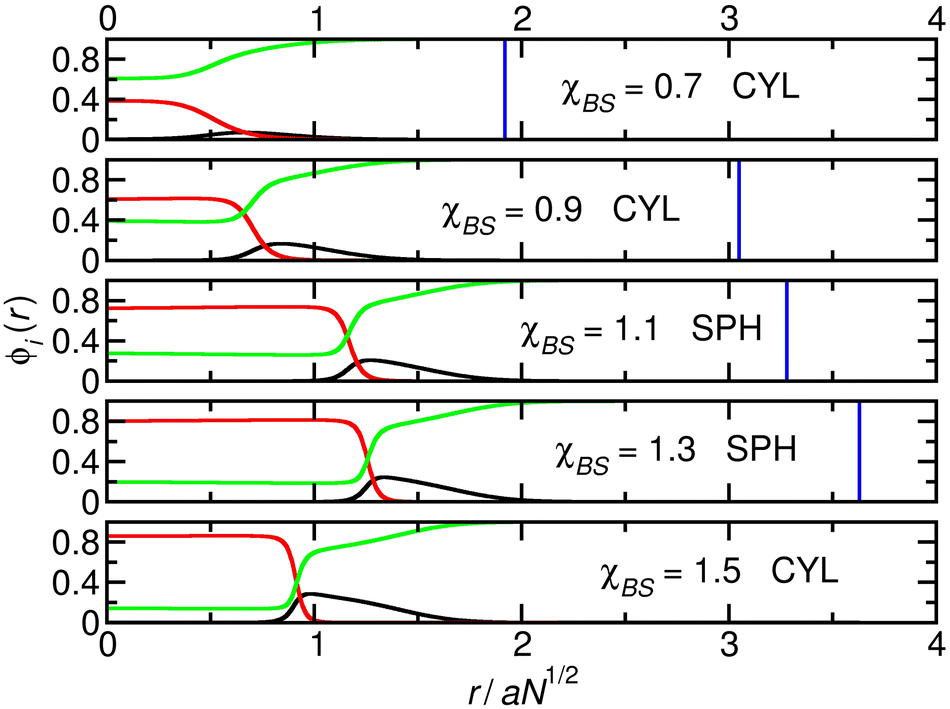 | ||
| Fig. 3 Cuts through the density profiles of the micelles that form when χAB = 0.1 and NA = 80. Black lines: A-blocks (shell); red lines: B-blocks (core), green lines: solvent. Vertical blue lines: boundary of the calculation box. In the final panel, the boundary line lies at r = 4.59 aN1/2 and is not plotted to make the range of r compatible with Fig. 1 and 2. | ||
We now discuss the potential connections between our calculations and experimental results. On the left-hand side of Table 1, where χAB < 0.1, the system moves from cylinders to spheres or from bilayers to cylinders, i.e., from less curved to more curved structures, as χBS is increased. If the value of χBS decreases on heating, that is, if the core block of the polymer displays upper critical solution temperature-like (UCST-like) behaviour, this corresponds to a transition to less curved structures as the temperature is increased (Fig. 4a). This is in line with the results of Liaw et al.,26 and we suggest that it may correspond to other systems where the core progressively and uniformly becomes more solvated as flatter structures are formed.1 This behaviour has been suggested21,22 to arise from ingress of the solvent into the centre of the core at higher temperatures, and the current calculations can be interpreted as being in line with this. Specifically, as the system changes from spheres to cylinders as χBS is lowered (which we are currently associating with a rise in temperature) in Fig. 1, the interfacial region becomes progressively less sharp, and the solvent penetrates further into the core of the micelle to the extent that, when χBS = 0.7, the separate and relatively less solvated core region seen in the spheres is no longer present. This effect is seen more clearly in calculations on the transition between cylinders and bilayers that occurs when χAB = 0.05 and NA = 70 (the top left corner of Table 1). These profiles are shown in Fig. 5, where there is a contrast between the relatively sharply defined core and interface regions in the cylindrical cases and the more diffuse core of the bilayers.
 | ||
| Fig. 4 Schematic diagrams of the association between aggregate curvature and the nature of the plasticisation of the core blocks. Part (a) shows the decrease in curvature that occurs on decreasing χBS for χAB< 0.1, associated with uniform plasticisation of the core blocks. Part (b) shows the increase in curvature on decreasing χBS for χAB > 0.1, associated with surface plasticisation of the core. | ||
 | ||
| Fig. 5 Cuts through the density profiles of the aggregates that form when χAB = 0.05 and NA = 70. Black lines: A-blocks (shell); red lines: B-blocks (core), green lines: solvent. Vertical blue lines: boundary of the calculation box. In the second panel, this boundary lies at x = 4.65 aN1/2 and is not plotted. | ||
If the core blocks of the polymers display UCST-like behaviour but χAB is such that the system lies on the right-hand side of Table 1, then SCFT predicts that the temperature dependence of the shape transitions will be reversed, i.e., that increasing temperature will cause progressively more curved structures to form. This form of temperature dependence has been linked with surface plasticisation, i.e., the penetration of solvent into the surface of the aggregate core,7,11,14 which makes the core surface layer behave like an extension of the shell, causing a transition to more curved structures (Fig. 4b). Again, the density profiles calculated from SCFT can be interpreted as corroborating this. In Fig. 2, where χAB = 0.2 and NA = 72, the system moves from cylinders to spheres as χBS is lowered, which is still being associated with a rise in temperature. As in Fig. 1, the interfacial region becomes broader as χBS is reduced, but here a separate core region remains, even at χBS = 0.7.
In a third class of systems,16 the system transitions to less curved aggregates as the temperature is increased, but the core becomes less solvated as this happens. We suggest that these systems lie on the right of Table 1, but that, as noted elsewhere,21,22 the dependence of χBS on temperature is lower critical solution temperature (LCST)-like so that χBS increases on heating. This behaviour corresponds to Fig. 2, but with increasing χBS now associated with increasing temperature. The association between surface plasticisation and the formation of more curved structures (Fig. 4b) has also been made for these systems,21,22 and (as noted above) the solvophobic cores of the spheres in Fig. 2 show a broad surface region that remains distinct from the centre of the core.
The above calculations of the density profiles are encouraging, but it should be noted that the solvation of the core and the surface are strongly connected in SCFT, with the broadening of the solvated layer at the surface of the core that we have tentatively associated with surface plasticisation being accompanied by increased solvation of the centre of the core in all results presented here. Furthermore, although the change from the interfacial to the core region is smoothed out in the cases (such as the top panel of Fig. 5) where the solvent penetrates strongly into the centre of the core, the core solvation does not have the uniform profile suggested in, for example,ref. 1 and 21. These considerations mean that the distinction between plasticisation of the surface versus uniform plasticisation of the core made in discussions of experimental data is not very marked in the model. In addition, the fact that a separate core region is present in (for example) the cylinders in Fig. 5 but not in the bilayers could simply result from the fact that the bilayer core thickness is less than the core diameter of the cylinders.38
Conclusions
We have shown that self-consistent field theory predicts that the shapes of the aggregates that form in AB block copolymer solutions are strongly dependent on the inter-block incompatibility χAB of the copolymers. The importance of the solvophobic/solvophilic block interactions in driving transitions between spheres and worms has been noted in recent experiments39 and simulations,40 and our results show the utility of SCFT in investigating this effect over a wide range of parameters.The current work has also demonstrated that the dependence of the shape transitions on the solvophobicity χBS of the core-forming B-block can be reversed within SCFT by varying χAB, with increasing χBS driving transitions from less to more curved structures for lower χAB and from more to less curved structures for higher χAB. At an intermediate value of χAB, a re-entrant transition37 (cylinders–spheres–cylinders) as a function of χBS has been found. From a technical point of view, these results demonstrate the importance of setting χAB carefully in SCFT calculations. It is also possible to rationalise a range of experimental observations in terms of our results by also considering the solution behaviour (UCST-like versus LCST-like) of the core blocks of the copolymers.
Within SCFT, several extensions of the current work are possible. Firstly, instead of varying the block ratio while keeping the overall degree of polymerisation constant, it could be easier to make a detailed comparison with experiments, especially those involving polymerisation-induced self-assembly20 if the length of one block (typically, the solvophilic stabiliser) were to be held constant with the other being varied. The parameters in the model would also need to be set more carefully for comparison with a given experiment. As it stands, the three χ parameters have been treated as independent of each other, and the temperature dependence of only one of these parameters (χBS) has been considered. Including experimental information on the flexibility of the polymer molecules and allowing this to differ between the two blocks would be desirable. In the current work, the volumes occupied by an A segment, a B segment and an individual solvent molecule are all assumed to be equal, which means that our results are more applicable to systems where the solvent molecules are closer in volume to the monomer molecules, such as those involving n-dodecane.12 However, within the continuum SCFT used here, it is possible to relax this assumption (see, for example, ref. 31), and this would be needed for comparison with formulations where the solvent molecules are further in volume from the monomers.
Since many recent investigations into micelle and vesicle formation in copolymer solutions involve polymerisation-induced self-assembly, it would also be useful to model the dynamics of this process, which can cause shifts in the phase boundaries.41 Such more detailed studies could be guided by the parameters identified in the present work and could allow experimentalists to identify formulations with the thermal response required by a specific application in advance. The trends identified in the current model could also be used to construct a simpler theory of micelle formation42 in which the interfacial region43 is modelled in a way that takes the interaction of all three species into account.40
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank S. J. Hunter and P. D. Topham for useful discussions and comments on the manuscript. M. J. D. wishes to thank EPSRC for funding (grant number EP/Y005309/1).Notes and references
- S. J. Byard, C. T. O’Brien, M. J. Derry, M. Williams, O. O. Mykhaylyk, A. Blanazs and S. P. Armes, Chem. Sci., 2020, 11, 396–402 RSC.
- V. Baddam, L. Välinen, L. Kuckling and H. Tenhu, Polym. Chem., 2022, 13, 3790–3799 RSC.
- F. H. Sobotta, M. T. Kuchenbrod, C. Grune, D. Fischer, S. Hoeppener and J. C. Brendel, Polym. Chem., 2021, 12, 1668–1680 RSC.
- H. Phan, V. Taresco, J. Penelle and B. Couturaud, Biomater. Sci., 2021, 9, 38–50 RSC.
- J. Wan, B. Fan and S. H. Thang, Chem. Sci., 2022, 13, 4192–4224 RSC.
- Z. Zhao, S. Lei, M. Zeng and M. Huo, Aggregate, 2023, 5, e418 CrossRef.
- M. J. Derry, O. O. Mykhaylyk and S. P. Armes, Angew. Chem., Int. Ed., 2017, 56, 1746–1750 CrossRef CAS PubMed.
- Y. Pei, N. C. Dharsana, J. A. van Hensbergen, R. P. Burford, P. J. Roth and A. B. Lowe, Soft Matter, 2014, 10, 5787–5796 RSC.
- Y. Pei, O. R. Sugita, L. Thurairajah and A. B. Lowe, RSC Adv., 2015, 5, 17636–17646 RSC.
- N. Busatto, V. Stolojan, M. Shaw, J. L. Keddie and P. J. Roth, Macromol. Rapid Commun., 2019, 40, 1800346 CrossRef PubMed.
- K. L. Thompson, L. A. Fielding, O. O. Mykhaylyk, J. A. Lane, M. J. Derry and S. P. Armes, Chem. Sci., 2015, 6, 4207–4214 RSC.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS PubMed.
- N. Audureau, F. Coumes, J.-M. Guigner, P. T. N. Thi, C. Ménager, F. Stoffelbach and J. Rieger, Polym. Chem., 2020, 11, 5998–6008 RSC.
- I. R. Dorsman, M. J. Derry, V. J. Cunningham, S. L. Brown, C. N. Williams and S. P. Armes, Polym. Chem., 2021, 12, 1224–1235 RSC.
- X. Wang, J. Zhou, X. Lv, B. Zhang and Z. An, Macromolecules, 2017, 50, 7222–7232 CrossRef CAS.
- L. P. D. Ratcliffe, M. J. Derry, A. Ianiro, R. Tuinier and S. P. Armes, Angew. Chem., Int. Ed., 2019, 58, 18964–18970 CrossRef CAS PubMed.
- Q. Yue, S.-P. Wen and L. A. Fielding, Soft Matter, 2022, 18, 2422–2433 RSC.
- N. J. Warren, M. J. Derry, O. O. Mykhaylyk, J. R. Lovett, L. P. D. Ratcliffe, V. Ladmiral, A. Blanazs, L. A. Fielding and S. P. Armes, Macromolecules, 2018, 51, 8357–8371 CrossRef CAS PubMed.
- C. György, S. J. Hunter, C. Girou, M. J. Derry and S. P. Armes, Polym. Chem., 2020, 11, 4579–4590 RSC.
- P. J. Docherty, C. Girou, M. J. Derry and S. P. Armes, Polym. Chem., 2020, 11, 3332–3339 RSC.
- O. J. Deane, J. Jennings and S. P. Armes, Chem. Sci., 2021, 12, 13719–13729 RSC.
- S. J. Hunter and S. P. Armes, J. Colloid Interface Sci., 2023, 634, 906–920 CrossRef CAS PubMed.
- Y. Termonia, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 890–895 CrossRef CAS.
- Y.-D. Yan, Y.-H. Xue, H.-Y. Zhao, H. Liu, Z.-Y. Lu and F.-L. Gu, Macromolecules, 2019, 52, 6169–6180 CrossRef CAS.
- I. Ianiro, S. P. Armes and R. Tuinier, Soft Matter, 2020, 16, 2342–2349 RSC.
- C. Y. Liaw, K. J. Henderson, W. R. Burghardt, J. Wang and K. R. Shull, Macromolecules, 2015, 48, 173–183 CrossRef CAS.
- S. F. Edwards, Proc. Phys. Soc., 1965, 85, 613–624 CrossRef CAS.
- R. Wang, P. Tang, F. Qiu and Y. Yang, J. Phys. Chem. B, 2005, 109, 17120–17127 CrossRef CAS PubMed.
- M. W. Matsen, Phys. Rev. Lett., 1995, 74, 4225–4228 CrossRef CAS PubMed.
- M. W. Matsen, Soft Matter, Wiley-VCH, Weinheim, 2006, ch. 2 Search PubMed.
- M. J. Greenall, D. M. A. Buzza and T. C. B. McLeish, J. Chem. Phys., 2009, 131, 034904 CrossRef CAS PubMed.
- D. G. Anderson, J. Assoc. Comput. Mach., 1965, 12, 547–560 CrossRef.
- K.-C. Ng, J. Chem. Phys., 1974, 61, 2680–2689 CrossRef CAS.
- R. B. Thompson, K. O. Rasmussen and T. Lookman, J. Chem. Phys., 2004, 120, 31–34 CrossRef CAS PubMed.
- P. Stasiak and M. W. Matsen, Eur. Phys. J. E: Soft Matter Biol. Phys., 2011, 34, 110 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- A. M. Rumyantsev, F. A. M. Leermakers, E. B. Zhulina, I. I. Potemkin and O. V. Borisov, Langmuir, 2019, 35, 2680–2691 CrossRef CAS PubMed.
- G. Battaglia and A. J. Ryan, J. Am. Chem. Soc., 2005, 127, 8757–8764 CrossRef CAS PubMed.
- L. Hahn, T. Zorn, J. Kehrein, T. Kielholz, A.-L. Ziegler, S. Forster, B. Sochor, E. Lisitsyna, N. A. Durandin, T. Laaksonen, V. Aseyev, C. Sotriffer, K. Saalwächter, M. Windbergs, A.-C. Pöppler and R. Luxenhofer, ACS Nano, 2023, 17, 6932–6942 CrossRef CAS PubMed.
- V. S. Kravchenko, I. V. Portnov and I. I. Potemkin, Macromolecules, 2023, 56, 7626–7635 CrossRef CAS.
- A. A. Gavrilov, R. M. Shupanov and A. V. Chertovich, Polymers, 2020, 12, 2599 CrossRef CAS PubMed.
- L. Leibler, H. Orland and J. C. Wheeler, J. Chem. Phys., 1983, 79, 3550–3557 CrossRef CAS.
- E. Helfand and Y. Tagami, J. Polym. Sci., Part B: Polym. Lett., 1971, 9, 741–746 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00331d |
| This journal is © The Royal Society of Chemistry 2024 |