
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charged hollow microgel capsules†
Nabanita
Hazra
 ,
Janik
Lammertz
,
Andrey
Babenyshev
,
Rebecca
Erkes
,
Fabian
Hagemans
,
Chandeshwar
Misra
,
Walter
Richtering
and
Jérôme J.
Crassous
*
,
Janik
Lammertz
,
Andrey
Babenyshev
,
Rebecca
Erkes
,
Fabian
Hagemans
,
Chandeshwar
Misra
,
Walter
Richtering
and
Jérôme J.
Crassous
*
Institute of Physical Chemistry, RWTH Aachen University, Landoltweg 2, 52074, Aachen, Germany. E-mail: crassous@pc.rwth-aachen.de
First published on 16th May 2024
Abstract
Responsive hollow microgels are a fascinating class of soft model systems at the crossover between polymer capsules and microgels. The presence of the cavity makes them promising materials for encapsulation and controlled release applications but also confers them an additional softness that is reflected by their peculiar behaviour in bulk and at interfaces. Their responsivity to external stimuli, such as temperature, pH, and ionic strength, can be designed from their synthesis conditions and the choice of functional moieties. So far most studies have focused on “small” hollow microgels that were mostly studied with scattering or atomic force microscopy techniques. In our previous study, we have shown that large fluorescent hollow poly(N-isopropylacrylamide) (PNIPAM) microgels could be synthesized using micrometer-sized silica particles as sacrificial templates allowing their investigation in situ via confocal microscopy. In this work, we extend this approach to charged large hollow microgels based on poly(N-isopropylacrylamide-co-itaconic acid) (P(NIPAM-co-IA)). Hereby, we compare the structure and responsivity of “neutral” (PNIPAM) and “charged” (P(NIPAM-co-IA)) hollow microgel systems synthesized under similar conditions with the same sacrificial template using confocal and atomic force microscopy and light scattering techniques. In particular, we could demonstrate the extremely soft character of the swollen charged hollow microgels and their responsivity to pH, ionic strength, and temperature. To conclude this study, the buckling behavior of the different capsules was investigated illustrating the potential of such systems to change its conformation by varying the osmotic pressure and pH conditions.
Microgels are cross-linked polymer networks typically swollen in a good solvent, such as water, and formed a three-dimensional macromolecular network with a colloidal size range.1,2 Microgels combine classic characteristics of polymers, colloids, and surfactants.3,4 That is, they are soft and porous, crystallize depending on concentration, and adsorb at interfaces since they lower the interfacial tension. The softness enables swelling, deformation, and interpenetration of microgels upon the influence of forces.1,5 In addition, microgels show swelling and deswelling behavior towards multiple external stimuli, such as temperature,2,6,7 pH,8–12 and ionic strength.13 Stimuli responsivity and softness make microgels appealing for industrial applications, such as catalysis14 or purification technology,15 but also for biomedical applications as biosensors,16,17 implant coatings18 or drug delivery systems.19–22 Advantageously, microgel properties can be adjusted to a great extent by changing their chemical composition or architecture.3,23,24
Hollow sensitive microgels provide promising characteristics and have been investigated in the past decades.25,26 They have a solvent-filled cavity that offers additional freedom for the expansion of the polymer chains and affects their mechanical behavior.27–30 Due to the cavity, hollow microgels are ideal as carriers with a high capacity for small species compared to conventional microgels.31–33 Particularly, (multi-)stimuli sensitivity enables controlled uptake and release of the guest species.34–37 Encapsulation and release may be triggered both by changing the network density38 and by changing the interactions between guest species and the microgel network.11,39 In previous studies, the preparation of poly(N-isopropylacrylamide) (PNIPAM) based nano/microcapsules has been achieved via various synthetic pathways, including semi-batch and temperature-programmed surfactant-free precipitation polymerization,40 inverse emulsion41,42 and miniemulsion43,44 polymerization, and layer-by-layer (LbL) assembly and click chemistry techniques.45 Among the different synthetic approaches, seed emulsion polymerization using a sacrificial polymeric core25,46 or inorganic silica core5,26,27,47 have been extensively applied for the creation of diverse PNIPAM based hollow microgels. Lapeyre et al.,47 Dubbert et al.48 and more recently Brugnoni et al.49 investigated hollow double-shell microgels with diameters of a few hundred nanometers. Lapeyre et al. demonstrated the multi-responsive character of their hollow microgels with an inner layer of PNIPMAM and an outer layer of PNIPAM copolymerized with either acrylic acid (AA) or phenylboronic acid (PBA) to implement an additional responsiveness toward pH or glucose, respectively.47 While Dubbert et al. utilized an inner shell of poly(N-isopropylacrylamide) (PNIPAM) and an outer shell of poly(N-isopropylmethacrylamide) (PNIPMAM), Brugnoni et al. employed an inner shell of poly(N-isopropylacrylamide-co-diacetone acrylamide) (P(NIPAM-co-DAAM)) and an outer shell of PNIPAM. In both cases, the inner and outer shells exhibit different temperature sensitivities, which may be used to tailor the temperature-triggered uptake and release of the guest species.
In addition, Wypysek et al.50 used hollow microgels based on poly(N-isopropylacrylamide-co-itaconic acid) (P(NIPAM-co-IA)) with a diameter of a few hundred nanometers. The microgels are neutral at low pH values and negatively charged at high pH values due to deprotonation of the acidic group. The neutral microgels are mainly sensitive to temperature changes whereas the charged microgels are mainly sensitive to changes in ionic strength. Hollow microgels possess exceptional architecture and morphology, making them particularly valuable for encapsulating sensitive molecules like enzymes and proteins, as well as entrapping toxins and bioactive molecules. In a recent work of Wypysek et al.33 they synthesized hollow polyelectrolyte anionic P(N-isopropylacrylamide-co-itaconic acid) microgels and investigated their encapsulation behaviour as nanocontainers for the model protein cytochrome c as a function of pH. They employed fluorescence-lifetime imaging (FLIM) to determine the spatial distribution of the protein within the hollow microgels. Their findings elucidated how pH fluctuations, from low to high levels, affected the protein's electrostatic interactions within the microgels, alternating between no interaction, attraction, and repulsion.33
Hollow microgels are distinguished by the relative size of shell thickness and radius. Additionally, hollow microgels with a thin shell in comparison to the cavity are comparable to microcapsules and might exhibit different mechanical behavior.51 We note however, that most of the hollow microgels based on a silica sacrificial template produced so far use a core of only a few hundred nanometers in diameter. In our previous work, we described the synthesis and characterization of highly monodisperse micron-sized hollow PNIPAM microgels with relatively thin shells and temperature sensitivity.52 This capsule size is optimal to determine accurately the system dimensions using light scattering techniques and for in situ imaging by using confocal fluorescence microscopy when the polymer network is labelled. The capsules were found to deform when exposed to an external osmotic pressure induced by the addition of free polymers in the bulk solution. It was demonstrated that high molecular weight dextran macromolecules were apparently hindered from entering the cavity. Exceeding a critical osmotic pressure, the capsules buckle. According to the theory of elasticity,53 the Youngs modulus of the capsules was determined from the critical buckling osmotic pressure.
In the present study, we extend the synthesis of such micrometric hollow microgel capsules to charged microgel capsules. Starting with the same silica core particles as templates, two synthesis routes are described leading to well-defined and monodisperse “neutral” PNIPAM hollow microgel capsules and “charged” P(NIPAM-co-IA) hollow microgel capsules. We present the characterization of these two systems based on different scattering and microscopy techniques. The pH dependence of the P(NIPAM-co-IA) hollow microgel capsules was further tested confirming the extended swelling of the network under low pH conditions. Such particles were found to be much softer than their neutral analogues, which was confirmed by their high deformation when adsorbed at a solid substrate, using atomic force microscopy. Finally, the buckling of PNIPAM and P(NIPAM-co-IA) microgel capsules at high osmotic pressure set by the addition of high molecular weight dextran was investigated under different pH conditions.
1 Materials and methods
Materials
3-Aminopropyl triethoxysilane (APTES, Fluka), tetraethyl orthosilicate (TEOS, Sigma-Aldrich), ethanol absolute (absolute EtOH, VWR Chemicals), ammonium hydroxide solution (Amonia, Merck) fluorescein isothiocyanate isomer I (FITC, Sigma Aldrich), 3-(Trimethoxysilyl)propylmethacrylate (MPS, AppliChem), N-isopropylacrylamide (NIPAM, Acros Organics), N,N′-methylenebisacrylamide (BIS, Sigma-Aldrich), potassium peroxodisulfate (KPS, Fluka), dimethyl itaconate (DMI, Sigma-Aldrich), methacryloxyethyl thiocarbamoyl rhodamine B (MRB, Polysciences, Inc.) and sodium hydroxide (NaOH, Alfa Aesar) were used as received.Synthesis
The synthesis of micro-sized responsive capsules was executed in four steps as schematically depicted in Fig. 1. In the first step, silica templates were generated through a semi-continuous Stöber method. In doing so, seed concentration and other factors were varied to approximate the appropriate particle size and polydispersity. In the second step, the surface of the silica template was modified. In the third step, a precipitation polymerization with either NIPAM and BIS or DMI, NIPAM and BIS, together with the fluorescent dye MRB, was performed in order to produce labelled core–shell particles. The neutral monomer DMI was chosen because it does not reduce the colloidal stability and can be saponified to itaconic acid (IA) in contrast to charged monomers. In the fourth step, the silica cores were dissolved in NaOH solution and the charged microcapsules were obtained. | ||
| Fig. 1 Schematic four-step synthesis of microgel capsules, including preparation of silica templates, modification of silica surface, microgel shell polymerization, and silica core dissolution. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) as a free polymer. The osmotic pressure of the dextran solution π can be calculated from the weight percentage of the solution β as: πDextran150000 = 286β + 87β2 + 5β3 for 0.2 < β < 15.55 Stock solutions of Dextran 150000 of concentration 20 wt% were prepared in Milli-Q water and buffer solutions of pH = 3 and 9. All the buckling experiments were performed at a Dextran concentration of 10 wt% which corresponds to an osmotic pressure of ≈16.5 kPa52,55 and the microgel concentrations of 2.3 × 10−3 and 2.6 × 10−3 wt% for the PNIPAM and P(NIPAM-co-DMI) hollow microgels, respectively. To quench the charged hollow microgels at different pH, a solution of hollow microgels, 10 wt% Dextran, and 1 mM NaOH (pH = 11) was prepared. After 1 hour, HCl (2 mM) was added to the same solution to reduce the pH up to 3. Finally, again after 1 hour, 2 mM NaOH was added to increase the pH to 11. The fraction of buckled particles fB was estimated by counting the number of individual buckled and spherical particles based on more than 100 microgels.
000 g mol−1) as a free polymer. The osmotic pressure of the dextran solution π can be calculated from the weight percentage of the solution β as: πDextran150000 = 286β + 87β2 + 5β3 for 0.2 < β < 15.55 Stock solutions of Dextran 150000 of concentration 20 wt% were prepared in Milli-Q water and buffer solutions of pH = 3 and 9. All the buckling experiments were performed at a Dextran concentration of 10 wt% which corresponds to an osmotic pressure of ≈16.5 kPa52,55 and the microgel concentrations of 2.3 × 10−3 and 2.6 × 10−3 wt% for the PNIPAM and P(NIPAM-co-DMI) hollow microgels, respectively. To quench the charged hollow microgels at different pH, a solution of hollow microgels, 10 wt% Dextran, and 1 mM NaOH (pH = 11) was prepared. After 1 hour, HCl (2 mM) was added to the same solution to reduce the pH up to 3. Finally, again after 1 hour, 2 mM NaOH was added to increase the pH to 11. The fraction of buckled particles fB was estimated by counting the number of individual buckled and spherical particles based on more than 100 microgels.
1.1 Methods
000 frames and a frame rate of 500 Hz were recorded. Diluted samples were temperature-controlled using an Okolab incubation unit, adjusting temperatures from 20 °C to 40 °C in 5 °C intervals with a temperature sensor near the sample. The data were evaluated with the software DDMSoft.§52 We refer readers to the ESI† for further details on the data evaluation.
2 Results and discussion
The radius of fluorescent-labelled silica seeds was determined via SLS and DLS (refer to Fig. S1, ESI†) at 179 nm and 178 nm, respectively.60 These small, labelled silica seeds were further used for the synthesis of the micro-sized silica-core particles. The SEM analysis confirmed the low size polydispersity (4%) of the core-particles with a mean radius determined from statistical analysis of 50 particles at 530 ± 3 nm as shown in Fig. 2A. After surface functionalization, either a PNIPAM or P(NIPAM-co-IA) microgel shell was synthesized. | ||
| Fig. 2 (A) Scanning electron micrographs of the silica core-particles. (B) Confocal fluorescence micrographs of the core–shell PNIPAM and (C) P(NIPAM-co-IA) particles in dispersion, Scale bars: 5 μm. | ||
Confocal fluorescence images of the core–shell silica/P(NIPAM) and silica/P(NIPAM-co-IA) microgels in dispersion are presented in Fig. 2B and C confirming the successful synthesis procedure due to the clear identification of core and shell. AFM measurements performed on the two systems in the dried state (see Fig. S2, ESI†) further confirm the presence of the core with a maximal height slightly higher than 1000 nm for both systems in the order of the silica core diameter. Both systems are also characterized by limited spreading at the surface evidenced by an average diameter in the order of 1200 and 1300 nm for the neutral and charged core–shell microgels, respectively. In this case the evaluation is limited by the topography of the particles which prevents clear imaging of the adsorbed corona close to the silica core.
The cores of the two core–shell systems were subsequently dissolved in NaOH solution to yield hollow microgel capsules, as outlined in the synthesis section. We first investigate the dried capsules via confocal and AFM imaging. A dramatic change in the microstructure could be observed in comparison to the core–shell systems. Although individual objects can be observed, the resulting particles at the glass surface are highly deformed and present a buckled and wrinkled conformation characteristic of collapsed soft capsules as shown from the AFM height imaging in the dried state of drop casted samples in Fig. 3. The microgel capsule contact areas, which are mostly circular, exhibit large variations in diameter ranging from approximately 1 to 3.5 μm, and are by far exceeding the diameters of the core–shell systems. The neutral hollow microgels spread and flatten at the substrate adopting either a wrinkled conformation characterized by a lower diameter or a fully adsorbed conformation (Fig. 3A–C). Their average size is almost two times smaller than their charged counterparts, which is also reflected by their height profiles. In comparison, for the charged hollow microgels we could even observe very large and thin and sometimes partially disrupted systems that could correspond to ruptured capsules (Fig. 3D–F). From the difference between height profiles, we could assume that after adsorption and drying neutral microgels maintain capsule integrity. The large spreading of the charged microgel capsules can further be rationalized by their softness and the strong repulsive electrostatic interactions of their shell with the negatively charged ozone treated substrate. Two sample preparations, i.e., drop casting and spin-coating, performed on hydrophilic glass substrates were compared. Spin-coating the samples results in a lower distribution of diameters, however much fewer objects could be detected on a single micrograph, resulting so far in rather poor statistics based on seven capsules. Characteristic AFM height micrographs of spin-coated neutral and charged capsules are shown in Fig. S3A and B (ESI†) together with their average height profiles in Fig. S3C (ESI†). The mean diameter estimated from this analysis was 1820 ± 320 nm and 3400 ± 300 nm for the neutral and charged microgels, respectively. The corresponding average heights were measured at 21 ± 5 nm and 6.0 ± 1.3 nm. Comparing the average height and diameter of spin-coated and drop casted microgel capsules in Fig. S3D (ESI†), we observed that the squared radius scales with height, which would indicate the dried microgel volume (assuming a disk geometry) is almost the same for all capsules, i.e., that both the neutral and charged capsules are monodisperse and have the same mass. We further investigate their swollen conformation when adsorbed at untreated glass using CLSM (Fig. 4). The majority of the systems appear as hollow capsules with the presence of a large cavity confirmed by 3D reconstruction from z-stack imaging (Fig. 4E–H). These systems coexist with microgels strongly deformed at the glass. In addition, due to their extreme softness and their adhesion to the glass surface, some of the hollow charged microgels become strongly deformed along the shear created during the sample preparation as shown in Fig. 4B, D and F.
 | ||
| Fig. 3 AFM height micrographs of the neutral PNIPAM (50 × 50 μm2, 20 × 20 μm2, 10 × 10 μm2) (A)–(C) and charged P(NIPAM-co-IA, pH = 9) (50 × 50 μm2, 20 × 20 μm2, 10 × 10 μm2, zoom in) (D)–(F) capsules respectively. Measurements were performed under dry conditions after drop casting using the tapping mode. | ||
 | ||
| Fig. 4 CLSM micrographs of swollen neutral and charged microgel capsules adsorbed at the glass coverslip. (A)–(D) 2D micrographs captured at different magnifications of the neutral (A) and (C) and charged (B) and (D) microgel capsules. (E) and (F) Maximum intensity projections of the neutral (E) and charged systems obtained from 3.02 μm thick z-stacks. The dashed lines indicate the cross-sections shown in (G) and (H). | ||
Having characterized the structure of both hollow microgel capsules at surfaces both in their dried and swollen state, we further investigate their behaviour in dispersion in their swollen state. The fluorescence imaging of both hollow microgel capsules with a concentration about c ≈ 0.003 wt% is shown in Fig. 5. The neutral microgel capsules were dispersed in Milli-Q water (Video S1, ESI†), whereas the charged microgel capsules were first investigated in buffer at pH = 9 (Video S2, ESI†) at 20 °C. Fluorescence imaging evidences the good dispersion of both systems, that appear well-defined and the presence of the cavity surrounded by the fluorescent microgel shell. When both microgel capsules appear in the majority rather than monodisperse in size and shape, we noted that charged hollow microgel capsules were more polydisperse with several particles presenting an irregular shape (see Fig. 5B and Video S2, ESI†). Strikingly, not only do the charged microgels appear significantly larger, but also much softer as characterized by thermal shape fluctuations that could be evidenced via CLSM measurements performed at 25 °C (Video S3, ESI†).
 | ||
| Fig. 5 Fluorescence imaging of neutral hollow PNIPAM capsules dispersed in pure Milli-Q (A) and charged hollow P(NIPAM-co-IA) capsules in pH = 9 buffer (B) at 20 °C together with a schematic representation of their respective conformation. Scale bars: 5 μm. | ||
We then turn our attention to the responsivity of the microgel capsules starting with the neutral microgel system. To explore the temperature responsiveness outlined in Fig. 6A, we initiated our investigation by systematically conducting DLS on the diluted neutral PNIPAM capsules. When performing such measurements for large particles, one should be aware that the calculated diffusion may be presenting a large q-dependence around minima of the particle form factor. An example of the first-cumulant analysis of the f(q,τ) measured at 20 °C is provided in the ESI† (Fig. S6A). The analysis was therefore performed on the available q-range and the diffusion coefficient defined as the average diffusion over all angles. This was motivated by the large dimensions of the investigated systems. As DLS is influenced by the form factor of the particles large variation of the measured hydrodynamic radius are expected for scattering angles in the vicinity of the form factor minima.61,62 In addition, an additional relaxation mode may be related to shape fluctuations as observed for instance in the case of lipid vesicles (see the ESI† for further details).63 As the low q region is limited, we therefore decided to average over the whole q-range. The swelling curves were measured first with increasing temperature and then by decreasing temperature to check the reversibility of the response. PNIPAM capsules show the typical behaviour expected from conventional PNIPAM microgels with a slightly higher VPTT around 33–34 °C (Fig. 6B). In their swollen state at 20 °C and collapsed state at 41 °C, RH was determined at 837 nm and 531 nm, respectively. In addition, SLS measurements were performed under similar conditions to some further insights into the conformation of the capsules. The measurements were conducted at 20 and 40 °C and as shown in Fig. 6C. The form factor measured at 20 °C is well resolved with many maxima confirming the narrow size and conformation distribution of the capsules.52,64 At 40 °C, the maxima and minima become more pronounced and shift to higher q-values indicating the decrease of the capsule size, thickness and fuzziness. Fits with a fuzzy capsule model appropriately capture the main scattering features at the exception of the more smeared out first minimum in the experiments. The corresponding normalized polymer density profiles are shown in Fig. 6D. The obtained dimensions of the overall capsule radius are in good agreement with the DLS measurements. In addition, the size polydispersity derived from the fits was found to decrease from 11.5 to 6.0%.
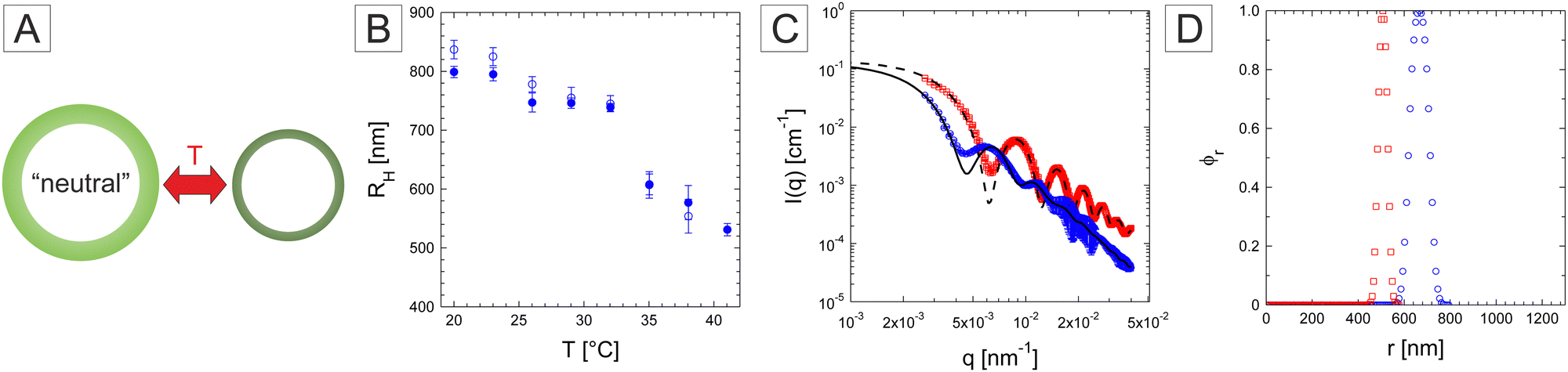 | ||
| Fig. 6 (A) Schematic swelling behaviour of the neutral PNIPAM capsules, (B) swelling behaviour of the neutral PNIPAM microgel capsules probed by DLS. The hydrodynamic radius was measured with increasing (full symbols) and decreasing temperature (hollow symbols). (C) Form factor of the capsules measured at 20 °C (blue circles) and 40 °C (red squares) fitted with a fuzzy core–shell model (full and dashed lines). (D) Relative radial density profile obtained from fits at 20 °C (blue circles) and 40 °C (red squares). | ||
The responsivity of the ionic P(NIPAM-co-IA) capsules to ionic strength, pH and temperature schematically displayed in Fig. 7A was further investigated. To test the weak polyelectrolyte properties of the P(NIPAM-co-IA) capsules, confocal spinning disk fluorescence microscopy (SDFM) measurements were conducted in aqueous buffer solution. The dimensions of the capsules were determined at pH = 3 (I = 10 mM) (Fig. 7A) and pH = 9 (I = 10 mM) (Fig. 7B) from the intensity profiles captured in the equatorial plane. The evaluation of 25 capsules at pH = 3 gives a mean radius of 0.69 ± 0.02 μm and the measurement of 25 capsules at pH = 9 gives a mean radius of 0.94 ± 0.02 μm. Electrophoretic mobility measurements were performed in a buffer with a fixed ionic strength concentration of 10 mM at 20 °C. The ζ-potential is a theoretical quantity and defined for hard particles. However, it loses its physical meaning for soft particles such as microgels and electrophoretic mobility is alternatively used as a quantity that provides information about the surface charge.65,66 The mean electrophoretic mobility was determined at pH = 3 at (0.11 ± 0.03) × 108 m2 V−1 s−1 and at pH = 9 at (−1.11 ± 0.02) × 108 m2 V−1 s−1. The measurements clearly show the ionization of the capsules, which leads to a negative charge at pH = 9. Similar results can be found in the literature for small P(NIPAM-co-IA) hollow microgels.50
 | ||
| Fig. 7 Confocal spinning disk fluorescence microscopy (SDFM) images (top) and associated intensity profiles (below) of P(NIPAM-co-IA) capsules in aqueous solution at pH = 3, I = 10 mM (A) and pH = 9, I = 10 mM (B). | ||
Suggesting a two-stage deprotonation with increasing pH-values corresponding to the step-by-step deprotonation of the two acid groups (–COOCH3) in the IA moiety. It was found that the P(NIPAM-co-IA) microgels are mainly protonated at pH = 3 and fully deprotonated at pH = 9.50 The observed change in size when the pH value increases from 3 to 9 can be attributed to the charges in the microgels. At a low pH value (pH = 3), two acidic groups (–COOCH3) are fully neutralized through protonation, facilitating the formation of hydrogen bonds with the amide (–CONH) groups of PNIPAM rather than relying on electrostatic interactions.67 At pH = 9, the presence of deprotonated acidic groups does not favour the formation of similar hydrogen bonds. Counterions are located in the surrounding of fixed charges but are still mobile for entropic reasons and create an osmotic pressure between the network and solution. Thus, the network swells. In addition, electrostatic repulsion works between the fixed charges, which tend towards the periphery of the capsules and pull the network outwards until expansion is compensated by elastic forces.50
The thermoresponsivity of the P(NIPAM-co-IA) microgels was first tested at fixed ionic strength (I = 10 mM) via DDM (see Fig. S4 and S5 and the ESI† for further details). At pH = 9, the P(NIPAM-co-IA) capsules are swollen with a constant RH ≈ 950 nm (see Fig. S5B, ESI†) up to 40 °C. However, the P(NIPAM-co-IA) capsules at pH = 3 become responsive to temperature in their protonated state and their size (RH) decreases from about 750 nm at 20 °C to 500 nm at 35 °C (see Fig. S5A, ESI†) before the capsule dispersion become unstable at higher temperatures.50 In their collapsed state the protonated capsules are only electrostatically stabilized by residual initiator fragments that are screened by the high ionic strength of the solution leading to the coagulation of the capsules. Note that the ionic strength also seems to play a role at pH = 9, leading to moderate deswelling from 10 to 100 mM buffer by screening the electrostatic repulsion between the deprotonated network moieties.
To minimize this effect, we performed DLS measurements under highly dilute conditions with a 10 mM buffer without adding any extra NaCl salt. The P(NIPAM-co-IA) capsules at pH = 9 did not show any temperature dependence similarly to the DDM results, with however a larger radius determined around 1.3 μm (Fig. 8C). Examples of the fitted f(q,τ) for both pH conditions measured at 20 °C are provided in Fig. S6 (ESI†). The large error bars are here a consequence of the large variation observed in the determination of D at the different angles, which could be related to the form factor of the capsules and to the larger polydispersity and fluctuations of the capsules in size and shape. We note that this value is significantly larger than the DDM measurements performed with the addition of NaCl. Measurements at pH = 3 (Fig. 8D) confirmed the response in temperature with a large size reduction from 941 nm to 296 nm above the VPTT also detected around 33–34 °C pointing to the complete collapse of the capsules. Interestingly, unlike the other systems, the response is not reversible and the measured radius by decreasing temperature became significantly smaller than the initial recorded values below the VPTT.
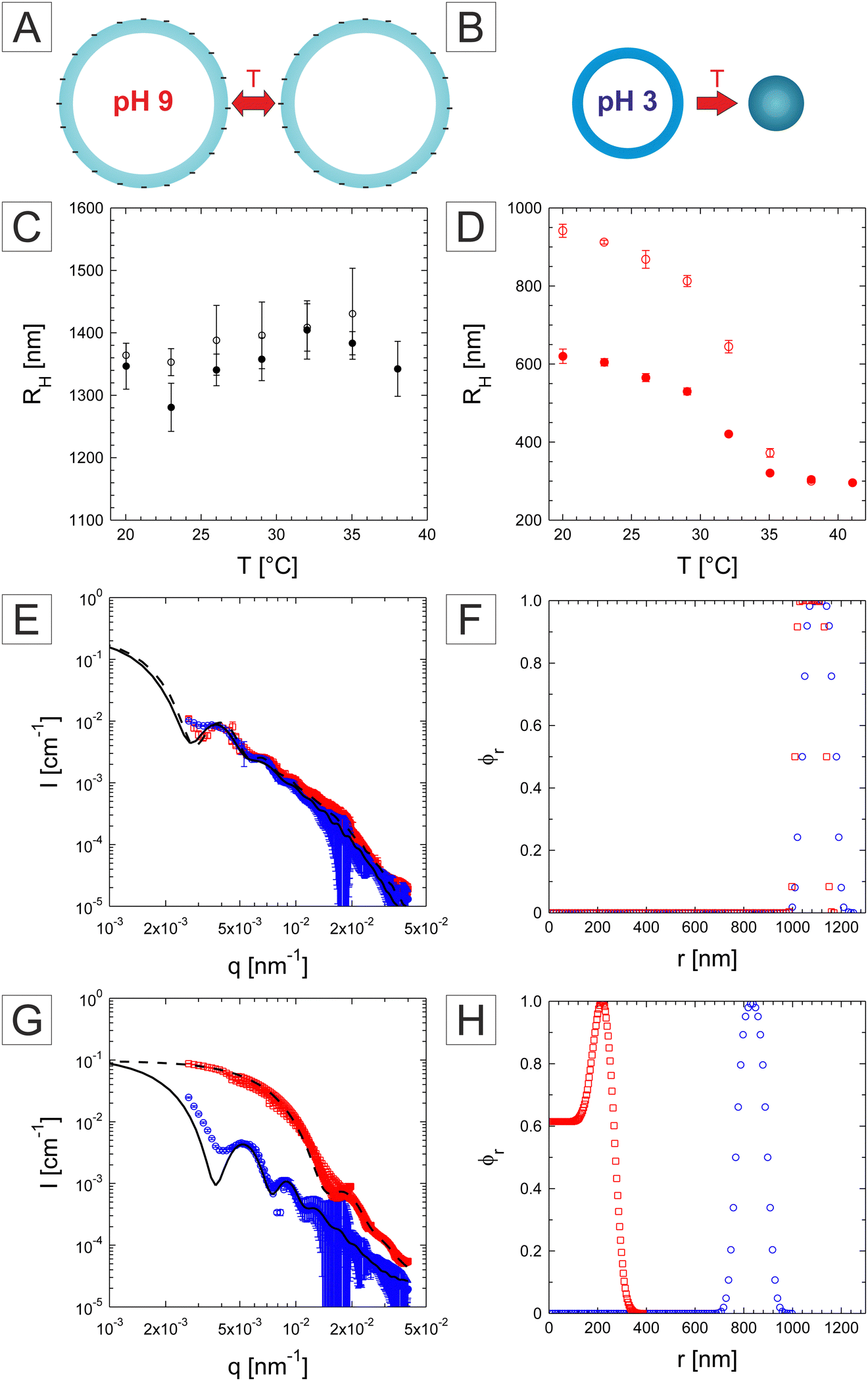 | ||
| Fig. 8 Schematic of the swelling behaviour of P(NIPAM-co-IA) capsules at pH = 9 (A) and pH = 3 (B) with temperature. Swelling behaviour of the P(NIPAM-co-IA) capsules at pH = 9 (C) and pH = 3 (D) measured by DLS. Hollow symbols correspond to measurements of the hydrodynamic radius first performed with increasing temperature, whereas full symbols correspond to the subsequent measurements with decreasing temperature. (E)–(H) Static light scattering curves and relative radial density profile obtained from the fuzzy-core–shell fit of the P(NIPAM-co-IA) capsules at pH = 9 (E), (F) and pH = 3 (G), (H) at 20 °C (blue circles) and 40 °C (red squares). | ||
The SLS analysis was applied to the P(NIPAM-co-IA) capsules at pH = 9, and the measurements at the two temperatures are almost similar confirming that the capsules do not respond significantly to temperature in this temperature range (see Fig. 8E). The data could be fitted with a similar model leading to an overall size of about 1.21 and 1.16 μm and shell thickness of circa 211 and 167 nm at 20 and 40 °C, respectively (Fig. 8F). The corresponding polydispersity was derived at 20 °C and 13.9%. Note that the large dimensions of the capsules do not allow us to probe the forward scattering affecting the robustness of the fits. Finally, the SLS measurements performed at pH = 3 support the size reduction and the formation of more defined capsules characterized by better defined minima and maxima at 20 °C (Fig. 8G). The forward scattering could not be fully reproduced by the fit, however, the other features are well described by the analysis. The overall size was derived at 958 nm, which is significantly larger than the DLS results, with a shell thickness of 251 nm (Fig. 8H). The scattering intensity profile changes significantly at 40 °C following the capsule collapse. The data could not be fitted with a simple capsule model anymore pointing to the filling up of the cavity. Similar observations were reported for smaller charged hollow microgels.50
In our previous study, it was observed that neutral microgel capsules buckle under sufficient osmotic pressure set by the addition of high molecular weight Dextran (150000 g mol−1).52 This transition is characterized in most of the observations by the development of a single instability leading to a shape transformation from spherical to bowl-shaped. To investigate the influence of charged moieties on the buckling of microgel capsules, dispersions of both neutral and charged capsules were prepared with 10 wt% Dextran, resulting in an osmotic pressure of approximately 16.5 kPa.52 Experiments on neutral capsules were conducted in pure double distilled water, while charged microgel capsules were dispersed in 1 mM, pH = 9 and 1 mM, pH = 3 buffer solutions. Fluorescence microscopy experiments were performed at 22 °C for the statistical evaluation of the proportion of buckled microcapsules, fB. Characteristic micrographs are presented in Fig. 9. The analysis was based on the evaluation of more than 100 individual capsules diffusing in solution. Under these conditions, nearly all neutral microcapsules buckle (fB = 89%) (see Fig. 9A), which is consistent with our previous observations.52 Similarly, under acidic conditions (Fig. 9B), charged microcapsules predominantly fully buckle (fB = 93%). Note that in both experiments, we did observe the presence of many aggregates that we ascribe to the onset of depletion interactions.68,69 Interestingly, as depicted in Fig. 9C, under basic conditions at pH = 9, the majority of the capsules remain spherical (fB = 18%) and fully dispersed. The additional contribution of electrostatic interactions and the increased osmotic pressure within the microgel network seem to be at the origin of the reduced buckling under these conditions. In addition, pH jump experiments were performed under high osmotic pressure using the same Dextran concentration to check the reversibility of the buckling process with pH (see Fig. S7 and the Experimental section for further details, ESI†). In this case, instead of a buffer solution, the pH of a charged microgel capsule dispersion was adjusted by the addition of sodium hydroxide (NaOH) and hydrochloric acid (HCl). After the addition of 1 mM NaOH, the microgel capsules were mostly unbuckled with fB = 14% and well dispersed similar to our former observations at pH = 9 (Fig. 9C). Adding 2 mM HCl to the former dispersion, i.e., setting the pH to circa pH = 3, the capsules were mostly buckled (fB = 91%) and aggregated similar to Fig. 9B. Finally, the consecutive addition of 2 mM NaOH, again to the same dispersion, bringing it back to basic conditions was found to result in the unbuckling of the capsule (fB = 25%) and their redispersion from the aggregates. Such proof of concept experiments clearly establish the versatility of the response of such systems to different stimuli and the possibility for further tuning the conformation, mechanical properties, and eventually the permeability of capsules. A more thorough investigation of the buckling transition of the charged microgel capsules and their kinetics will be undertaken in the future.
 | ||
| Fig. 9 Microgel capsules under osmotic stress set by the addition of 10 wt% Dextran (πDextran ≈ 16.5 KPa). Fluorescent micrographs of neutral microgels (A), charged microgels capsules at pH = 3 (B) and charged microgel capsules at pH = 9 (C). The insets show the higher magnification of the capsules recorded under the CLSM imaging mode. The proportion of buckled capsules, fB, was determined from statistical analysis of the fluorescence micrographs. | ||
3 Conclusion
In this study, well-defined and monodisperse hollow soft microgel capsules were synthesized consisting of a micrometric size range showing temperature and pH responsiveness. The neutral PNIPAM capsules exhibited thermoresponsive behaviour, whereas, the charged P(NIPAM-co-IA) capsules were thermoresponsive at low pH (pH = 3) and no temperature responsivity was observed at high pH (pH = 9). The size of the neutral capsules is smaller than the charged capsules and the hollow architecture with extreme softness makes them strongly flatted on glass surfaces. In addition, the hollow responsive microgels were widely characterized using different methods, ranging from direct imaging using confocal, fluorescence, and atomic force microscopy to scattering techniques involving DLS and SLS. The direct imaging confirmed their size range as well as the unique structural behaviour due to softness. The dynamic scattering measurement ensured the temperature-dependent swelling behaviour of PNIPAM and P(NIPAM-co-IA) capsules at pH = 3 and the capsule form factor was measured by static light scattering experiments. Moreover, the large swelling of the charged microgel capsules due to very soft behaviour is reflected by thermal fluctuation and high deformability. Both PNIPAM and P(NIPAM-co-IA) microgel capsules were found to buckle under a high osmotic pressure of 16.5 kPa. However, while P(NIPAM-co-IA) microgel capsules buckle at pH = 3, they remain mostly unbuckled at pH = 10. These initial experiments demonstrate the significance of additional charged moieties in the buckling behaviour. A more systematic investigation of the buckling behaviour and permeability of the P(NIPAM-co-IA) microgel capsules, depending on their degree of protonation and ionic strength, will be undertaken in the future. Such a study will further contribute to establishing their application as cargo for nanomaterials transport and delivery. Besides, due to their extreme softness and responsivity toward pH, ionic strength and temperature, they should present some fascinating properties and serve as unique soft model systems to study their assembly at fluid interfaces and their phase behaviour, glass transition and jamming in dispersions at high volume fractions. Hereby, the single particle deformation could be resolved in situ in real space with respect to their large dimensions and hollow structure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the SFB 985 “Functional Microgels and Microgel Systems” of Deutsche Forschungsgemeinschaft is greatly acknowledged. The authors thank the RWTH Start-Up-Grant initiative financed by the Deutsche Forschungsgemeinschaft for the kind support within the project. We would like to thank Rebecca Hengsbach and Prof. Dr Simon from the Institute of Inorganic Chemistry, RWTH Aachen for the SEM measurements and Frédéric Dux for the development of the DDM software.Notes and references
- F. A. Plamper and W. Richtering, Acc. Chem. Res., 2017, 50, 131–140 CrossRef CAS PubMed.
- R. Pelton, Adv. Colloid Interface Sci., 2000, 85, 1–33 CrossRef CAS PubMed.
- M. Karg, A. Pich, T. Hellweg, T. Hoare, L. A. Lyon, J. Crassous, D. Suzuki, R. A. Gumerov, S. Schneider and I. I. Potemkin, et al. , Langmuir, 2019, 35, 6231–6255 CrossRef CAS PubMed.
- B. R. Saunders and B. Vincent, Adv. Colloid Interface Sci., 1999, 80, 1–25 CrossRef CAS.
- A. Scotti, M. F. Schulte, C. G. Lopez, J. J. Crassous, S. Bochenek and W. Richtering, Chem. Rev., 2022, 122, 11675–11700 CrossRef CAS PubMed.
- H. Senff and W. Richtering, J. Chem. Phys., 1999, 111, 1705–1711 CrossRef CAS.
- M. Brugnoni, A. C. Nickel, L. C. Kröger, A. Scotti, A. Pich, K. Leonhard and W. Richtering, Polym. Chem., 2019, 10, 2397–2405 RSC.
- T. Hoare and R. Pelton, Macromolecules, 2004, 37, 2544–2550 CrossRef CAS.
- M. Karg, I. Pastoriza-Santos, B. Rodriguez-Gonzalez, R. von Klitzing, S. Wellert and T. Hellweg, Langmuir, 2008, 24, 6300–6306 CrossRef CAS PubMed.
- S. Lally, R. Bird, T. J. Freemont and B. R. Saunders, Colloid Polym. Sci., 2009, 287, 335–343 CrossRef CAS.
- A. P. Gelissen, A. Scotti, S. K. Turnhoff, C. Janssen, A. Radulescu, A. Pich, A. A. Rudov, I. I. Potemkin and W. Richtering, Soft Matter, 2018, 14, 4287–4299 RSC.
- J. Pinheiro, L. Moura, R. Fokkink and J. Farinha, Langmuir, 2012, 28, 5802–5809 CrossRef CAS PubMed.
- E. Daly and B. R. Saunders, Langmuir, 2000, 16, 5546–5552 CrossRef CAS.
- D. Kleinschmidt, K. Nothdurft, M. V. Anakhov, A. A. Meyer, M. Mork, R. A. Gumerov, I. I. Potemkin, W. Richtering and A. Pich, Mater. Adv., 2020, 1, 2983–2993 RSC.
- M. Barth, M. Wiese, W. Ogieglo, D. Go, A. J. Kuehne and M. Wessling, J. Membr. Sci., 2018, 555, 473–482 CrossRef CAS.
- L. V. Sigolaeva, S. Y. Gladyr, A. P. Gelissen, O. Mergel, D. V. Pergushov, I. N. Kurochkin, F. A. Plamper and W. Richtering, Biomacromolecules, 2014, 15, 3735–3745 CrossRef CAS PubMed.
- D. Buenger, F. Topuz and J. Groll, Prog. Polym. Sci., 2012, 37, 1678–1719 CrossRef CAS.
- D. Keskin, O. Mergel, H. C. Van der Mei, H. J. Busscher and P. van Rijn, Biomacromolecules, 2018, 20, 243–253 CrossRef PubMed.
- M. Dirksen, C. Dargel, L. Meier, T. Brändel and T. Hellweg, Colloid Polym. Sci., 2020, 298, 505–518 CrossRef CAS.
- R. Dave, G. Randhawa, D. Kim, M. Simpson and T. Hoare, Mol. Pharmaceutics, 2022, 19, 1704–1721 CrossRef CAS PubMed.
- N. M. Smeets and T. Hoare, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3027–3043 CrossRef CAS.
- D. Klinger and K. Landfester, Polymer, 2012, 53, 5209–5231 CrossRef CAS.
- Y. Hertle and T. Hellweg, J. Mater. Chem. B, 2013, 1, 5874–5885 RSC.
- J. Oberdisse and T. Hellweg, Colloid Polym. Sci., 2020, 298, 921–935 CrossRef CAS.
- S. Nayak, D. J. Gan, M. J. Serpe and L. Lyon, Small, 2005, 1, 416–421 CrossRef CAS PubMed.
- H. Gao, W. Yang, K. Min, L. Zha, C. Wang and S. Fu, Polymer, 2005, 46, 1087–1093 CrossRef CAS.
- J. Dubbert, T. Honold, J. S. Pedersen, A. Radulescu, M. Drechsler, M. Karg and W. Richtering, Macromolecules, 2014, 47, 8700–8708 CrossRef CAS.
- A. Scotti, M. Brugnoni, A. A. Rudov, J. E. Houston, I. I. Potemkin and W. Richtering, J. Chem. Phys., 2018, 148, 174903 CrossRef CAS PubMed.
- K. C. Bentz and D. A. Savin, Polym. Chem., 2018, 9, 2059–2081 RSC.
- Y. Su, O. F. Ojo, I. K. M. Tsengam, J. He, G. L. McPherson, V. T. John and J. A. Valla, Langmuir, 2018, 34, 14608–14616 CrossRef CAS PubMed.
- L. Zha, Y. Zhang, W. Yang and S. Fu, Adv. Mater., 2002, 14, 1090–1092 CrossRef CAS.
- M. Windbergs, Y. Zhao, J. Heyman and D. A. Weitz, J. Am. Chem. Soc., 2013, 135, 7933–7937 CrossRef CAS PubMed.
- S. K. Wypysek, S. P. Centeno, T. Gronemann, D. Wöll and W. Richtering, Macromol. Biosci., 2023, 2200456 CrossRef CAS PubMed.
- M. Deloney, K. Smart, B. A. Christiansen and A. Panitch, J. Controlled Release, 2020, 323, 47–58 CrossRef CAS PubMed.
- A. J. Schmid, J. Dubbert, A. A. Rudov, J. S. Pedersen, P. Lindner, M. Karg, I. I. Potemkin and W. Richtering, Sci. Rep., 2016, 6, 22736 CrossRef CAS PubMed.
- H. Masoud and A. Alexeev, ACS Nano, 2012, 6, 212–219 CrossRef CAS PubMed.
- A. Moncho-Jorda, A. Germán-Bellod, S. Angioletti-Uberti, I. Adroher-Bentez and J. Dzubiella, ACS Nano, 2019, 13, 1603–1616 CAS.
- S. Seiffert, J. Thiele, A. R. Abate and D. A. Weitz, J. Am. Chem. Soc., 2010, 132, 6606–6609 CrossRef CAS PubMed.
- W. Xu, A. A. Rudov, R. Schroeder, I. V. Portnov, W. Richtering, I. I. Potemkin and A. Pich, Biomacromolecules, 2019, 20, 1578–1591 CrossRef CAS PubMed.
- Z. Li, M.-H. Kwok and T. Ngai, Macromol. Rapid Commun., 2012, 33, 419–425 CrossRef CAS PubMed.
- M. Horecha, V. Senkovskyy, M. Stamm and A. Kiriy, Macromolecules, 2009, 42, 5811–5817 CrossRef CAS.
- Q. Sun and Y. Deng, J. Am. Chem. Soc., 2005, 127, 8274–8275 CrossRef CAS PubMed.
- Z. Cao, U. Ziener and K. Landfester, Macromolecules, 2010, 43, 6353–6360 CrossRef CAS.
- F. Lu, Y. Luo, B. Li, Q. Zhao and F. J. Schork, Macromolecules, 2010, 43, 568–571 CrossRef CAS.
- C.-J. Huang and F.-C. Chang, Macromolecules, 2009, 42, 5155–5166 CrossRef CAS.
- J. Vialetto, F. Camerin, F. Grillo, S. N. Ramakrishna, L. Rovigatti, E. Zaccarelli and L. Isa, ACS Nano, 2021, 15, 13105–13117 CrossRef CAS PubMed.
- V. Lapeyre, N. Renaudie, J.-F. Dechezelles, H. Saadaoui, S. Ravaine and V. Ravaine, Langmuir, 2009, 25, 4659–4667 CrossRef CAS PubMed.
- J. Dubbert, K. Nothdurft, M. Karg and W. Richtering, Macromol. Rapid Commun., 2015, 36, 159–164 CrossRef CAS PubMed.
- M. Brugnoni, F. Fink, A. Scotti and W. Richtering, Colloid Polym. Sci., 2020, 298, 1179–1185 CrossRef CAS.
- S. K. Wypysek, A. Scotti, M. O. Alziyadi, I. I. Potemkin, A. R. Denton and W. Richtering, Macromol. Rapid Commun., 2020, 41, 1900422 CrossRef CAS PubMed.
- M. P. Neubauer, M. Poehlmann and A. Fery, Adv. Colloid Interface Sci., 2014, 207, 65–80 CrossRef CAS PubMed.
- F. Hagemans, F. Camerin, N. Hazra, J. Lammertz, F. Dux, G. Del Monte, O.-V. Laukkanen, J. J. Crassous, E. Zaccarelli and W. Richtering, ACS Nano, 2023, 17, 7257–7271 CrossRef CAS PubMed.
- C. Gao, E. Donath, S. Moya, V. Dudnik and H. Möhwald, Eur. Phys. J. E: Soft Matter Biol. Phys., 2001, 5, 21–27 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- C. Bonnet-Gonnet, L. Belloni and B. Cabane, Langmuir, 1994, 10, 4012–4021 CrossRef CAS.
- W. Burchard and W. Richtering, Progr. Colloid Polym. Sci., 1989, 80, 151–163 CAS.
- O. L. J. Virtanen, PhD thesis, RWTH Aachen University, 2016, pp. 1–186.
- A. Van Helden and A. Vrij, J. Colloid Interface Sci., 1980, 78, 312–329 CrossRef CAS.
- I. Berndt, J. S. Pedersen and W. Richtering, J. Am. Chem. Soc., 2005, 127, 9372–9373 CrossRef CAS PubMed.
- W. Burchard, Light scattering from polymers, 1983, pp. 1–124 Search PubMed.
- W. Van Megen and P. Pusey, Phys. Rev. A: At., Mol., Opt. Phys., 1991, 43, 5429 CrossRef CAS PubMed.
- G. Bryant, S. Martin, A. Budi and W. van Megen, Langmuir, 2003, 19, 616–621 CrossRef CAS.
- P. Brocca, L. Cantù, M. Corti, E. Del Favero and S. Motta, Langmuir, 2004, 20, 2141–2148 CrossRef CAS PubMed.
- J. Park, E. Lee, N.-M. Hwang, M. Kang, S. C. Kim, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim and J.-H. Park, et al. , Angew. Chem., 2005, 117, 2932–2937 CrossRef.
- H. Ohshima, Colloid Polym. Sci., 2007, 285, 1411–1421 CrossRef CAS.
- H. Ohshima, Electrophoresis, 1995, 16, 1360–1363 CrossRef CAS PubMed.
- V. Nigro, R. Angelini, B. Rosi, M. Bertoldo, E. Buratti, S. Casciardi, S. Sennato and B. Ruzicka, J. Colloid Interface Sci., 2019, 545, 210–219 CrossRef CAS PubMed.
- P. Jenkins and M. Snowden, Adv. Colloid Interface Sci., 1996, 68, 57–96 CrossRef CAS.
- H. N. W. Lekkerkerker, R. Tuinier and M. Vis, Depletion Interaction, Springer International Publishing, Cham, 2024, pp. 67–120 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00111g |
| ‡ https://github.com/ovirtanen/fitit |
| § https://github.com/duxfrederic/ddmsoft |
| This journal is © The Royal Society of Chemistry 2024 |