
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Local dynamics and failure of inhomogeneous polymer networks†
Ziyu
Ye‡
a,
Han
Zhang‡
 b and
Robert A.
Riggleman
*b
b and
Robert A.
Riggleman
*b
aDepartment of Chemistry, University of Pennsylvania, Philadelphia, PA 19104, USA
bDepartment of Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, PA 19104, USA. E-mail: rrig@seas.upenn.edu
First published on 22nd May 2024
Abstract
Inhomogeneous crosslinked polymers are powerful platforms for materials design, because they can be synthesized from materials that provide complimentary properties to the resulting gel. For example, a membrane with both glassy and rubbery domains will be mechanically robust while enabling transport. The dynamics, and mechanical and failure properties of rubbery/glassy conetworks are only beginning to be studied, and there is likely to be strong heterogeneities in the dynamics and mechanical response. In this study, we use coarse-grained molecular dynamics simulations to generate microphase separated rubbery/glassy polymer networks with a bicontinuous morphology via in silico crosslinking. We study the effect of phase boundary on the local mobility gradient, and our simulation results reveal an asymmetric shift in the local mobility gradient across the interface that extends deeper into the phase with a lower Tg when the system temperature is between the glass transition temperatures of the two phases. Moreover, by employing a model that allows bond breaking, we examine the microscopic mechanism for failure in these networks as a function of the molecular weight of polymer strands between crosslinks and the number fraction of the glassy domain. Under uniaxial extension, we find that the stress is initially larger in the glassy domain. As the deformation proceeds, the segmental dynamics of the two phases homogenize, and subsequently bond breaking begins.
1 Introduction
Crosslinked polymer networks are important in diverse applications that range from membranes for selective transport to biological scaffolds to the matrix used in 3D-printing.1–4 There are numerous strategies to control the properties of polymer networks; simple strategies include varying the crosslinking density or the polymer strand length, and sophisticated strategies include tuning the architecture of the polymers that comprise the network,5–7 creating interpenetrating double networks,8,9 or creating microphase separation in the network that is locked in place by the crosslinking process.10 Networks with microphase separation between rubbery and glassy polymers are a promising class of materials that are similar to block copolymers in which they can potentially create co-continuous structures with complimentary properties.10,11 Much of the recent progress in tuning the properties of networks has been driven by advances in chemical synthesis, but a molecular picture of the influence of the phase boundary on the local dynamics and mechanical response of these emerging inhomogeneous networks is lacking.Part of the challenge in predicting the properties of inhomogeneous networks with rubbery and glassy domains is that the dynamics will be strongly inhomogeneous. Since the pure polymer glass transition temperatures Tg can vary upwards of 100 K, one can expect many orders of magnitude difference in the segmental dynamics. Recent measurements in block copolymers have shown that Tg can vary by over 60 K and do not follow a local version of the Fox equation,12 and experiments in layered polymer films show perturbations in Tg in a glassy polymer placed near a rubbery polymer over 100s of nm.13 These findings further highlight the need to understand gradients in mobility from a fundamental level, which remains an outstanding challenge.14–19 Moreover, it is known that deformation imparts changes to the segmental dynamics of a glass,20–22 which will further complicate predicting and understanding the mechanical response of inhomogeneous networks with glassy domains.
While molecular modeling has played an important role in our understanding of the structure and properties of inhomogeneous polymeric materials such as block copolymers, modeling approaches for realistic polymer networks, and inhomogeneous polymer networks in particular, have only recently become available.23–32 Part of the challenge lies in efficiently preparing a simulated network with a realistic distribution of network defects, such as loops and dangling ends, that play an essential role in the properties of polymer networks.24,25,33–35 Simulations where the crosslinking process is explicitly simulated have been employed to generate systems with more realistic defect compositions, showing good agreement with properties observed in experiments.29,30,36,37
In this study, we have generated and studied the local dynamics and the mechanical response of inhomogeneous polymer networks containing rubbery and glassy domains. By mimicking an experimental protocol to form co-continuous networks,10 we are able to generate networks with a realistic distribution of defects and domain sizes. In quiescent systems, our simulation results reveal an asymmetric shift in the local mobility gradient across the interface that extends deeper into the phase with a lower Tg when the system temperature is between the glass transition temperatures of the two phases. We also examine the dynamic and mechanical responses until failure of the samples as a function of the number fraction of the high-Tg domain. We find that during material failure, the stress initially concentrates in the glassy domains. As the strain is increased, the segmental dynamics of the two phases become nearly homogeneous near the point where bonds begin breaking. Surprisingly, we find that the fraction of the number of broken bonds in each phase is approximately the same as the overall number fraction, and there is no observed tendency for failure to occur in either the glassy or rubbery domains, at least under the tensile deformations considered here.
2 Methods
Here we implement coarse-grained molecular dynamics simulation in the LAMMPS package38,39 to generate phase separated, end-linked polymer networks. The networks are constructed using a bead-spring model in a simulation box with periodic boundaries imposed in all directions. Our network formation protocol closely follows that of our previous studies,29,30,35 and the protocol approximately mimics an experimental system where linear polymer strands with reactive end groups are reacted with tetra-functional crosslinkers.10 While systems that undergo phase separation during polymerization and/or crosslinking will necessarily be more inhomogeneous, we do not expect such differences to qualitatively affect our results. We begin with a polymer melt of 2n identical linear polymer chains with N = 5, 10 or 20 monomers per chain and n “cross-linkers” that are chemically identical to the polymer monomers. All non-bonded interactions are taken through the 12-6 Lennard-Jones (LJ) cut-and-shifted potential with parameters εij, a unity σ for all species, and a cutoff distance rcut = 2.5σ. Initially harmonic bonds with a unity equilibrium length of σ and force constant kbond = 2000(ε/σ2) are employed, and a time step of δt = 0.002 in LJ units is employed for all simulations.After equilibrating the melt, we perform molecular dynamics simulations at T = 0.7 where we allow reactions to occur between the chain ends and the crosslinkers. During a crosslinking event, covalent bonds are formed any time reactive species come within 1.1σ of each other, and bond breaking is not allowed. Polymer chain ends are only allowed to form one new bond, and the crosslinkers are limited to a maximum of four new bonds. Complete details of the extent of the reaction29 and defect concentrations40 present after network formation can be found in our previous works. After forming the networks, we select at random a fraction fg of our polymer chains and modify the interactions to render them chemically dissimilar and create a difference in their glass transition temperatures, Tg. We retain the interactions of the high-Tg polymer to have εHH = 1.0, while we reduce εLL = 0.3 and change εHL to 0.4. Thus the two species of polymers are now distinguished as high-Tg and low-Tg polymers with pure, bulk state properties THg = 0.46 and TLg = 0.16, respectively (see the ESI† for the determination of Tg). Microphase separation is induced in the network system via changing the cross-interaction energy scale εHL such that the high and low Tg polymers are immiscible. During this step, the system temperature Tsys is maintained above the glass transition temperatures of both high and low Tg polymers. We also modify the bonding interactions to use the breakable quartic potential30,41 so that bonds will break once upon stretching to have an energy of approximately 70εHH. Finally, we allow the inhomogeneous structure with the quartic potential to develop for 2000τ before cooling to the testing temperature.
The local dynamics of the network polymers are studied as a function of four system temperatures at Tsys = [0.50, 0.45, 0.40, 0.30] for the symmetric blend system of fg = 0.50. During the quenching step, the system is quenched from the system formation temperature to the target temperature at a cooling rate of Γ = 5 × 10−5. The mechanical response and dynamics during deformation are analyzed at the temperature of Tsys = 0.30, which is approximately midway between the two glass transition temperatures, THg = 0.46 and TLg = 0.16. At Tsys = 0.30, the microphase separated network system has a distinct glassy and rubbery phase formed by the high and low Tg polymers. Prior to deformation, the systems are aged for an additional 2000τ at Tsys = 0.30.
Uniaxial tensile deformations are then performed to characterize the fracture properties of the network samples where the simulation box is expanded along the deformation axis α at a constant true strain rate (![[small epsi, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a1.gif) ) while maintaining a constant pressure along the other two dimensions. A true strain rate of = 1 × 10−4 is used to deform the simulation box to a final extension ratio λ = [5.0,6.5,8.5] for the N = 5, 10, and 20 networks, respectively, to ensure a complete plane of bonds are broken in the samples during fracture. Three fractions of the glassy domain are considered for each chain length, fg = [0.25,0.5,0.75] by number.
) while maintaining a constant pressure along the other two dimensions. A true strain rate of = 1 × 10−4 is used to deform the simulation box to a final extension ratio λ = [5.0,6.5,8.5] for the N = 5, 10, and 20 networks, respectively, to ensure a complete plane of bonds are broken in the samples during fracture. Three fractions of the glassy domain are considered for each chain length, fg = [0.25,0.5,0.75] by number.
3 Results and discussion
3.1 Morphology and domain characterization
Images of our simulations prior to deformation at each fraction of glassy polymers and each chain length are shown in Fig. 1a. Images are rendered using the OVITO software.42 As expected for an inhomogeneous crosslinked system, we observe finite-sized microdomains that are approximately static with time. For the symmetric system (fg = 0.5), one expects a percolating, cocontinuous network of both phases, while discrete domains are observed for the asymmetric compositions. Next, we quantify the domain spacing d of the fg = 0.5 systems as a function of the linear chains' molecular weight. From the structure factor computed on the high-Tg domain of the network (Fig. 1b), q* is determined from fitting the primary low-q peak to a Lorentzian function | (1) |
 | ||
| Fig. 1 (a) Visualization of microphase separated networks at various chain lengths N and number fractions of the glassy domain fg. Images are rendered using the OVITO software.42 (b) Structure factor of the phase separated domains calculated on the glassy phase for the symmetric blend fg = 0.5 formed with linear chain length N = 5, 10, and 20 (blue, orange, green, respectively) at Tsys = 0.30. Arrows, colored respectively, point to q* for each of the microphase separated networks. The inset shows the domain spacing as a function of N. The scaling relationship between the d-spacing and N is indicated by the black line with a slope of 0.5. | ||
3.2 Temperature dependence of local dynamics
We first explore the local dynamics of the microphase separated structures by examining the polymer segmental dynamics for the two polymer phases with different Tg and at varying system temperatures. We quantify the segmental dynamics by measuring the intermediate scattering function , where δr(t) is the displacement of a monomer over time t, 2π/q corresponds approximately to the length scale over which monomers must move to decorrelate Fs(q,t), and the average is taken over time origins and monomers. We chose q = 7.14σ−1, which approximately corresponds to the first primary peak in the static structure factor of a homogeneous polymer glass. Because some of our system temperatures are below the Tg of one of the domains, Fs(q,t) does not fully decay in all cases, and so we estimate the effective relaxation time by fitting the measured response to a Kohlrausch–Williams–Watts (KWW) stretched exponential and taking the time where the fitted function decays to 0.2 as τα. The unit of τα is given in Lennard-Jones (LJ) time units.
, where δr(t) is the displacement of a monomer over time t, 2π/q corresponds approximately to the length scale over which monomers must move to decorrelate Fs(q,t), and the average is taken over time origins and monomers. We chose q = 7.14σ−1, which approximately corresponds to the first primary peak in the static structure factor of a homogeneous polymer glass. Because some of our system temperatures are below the Tg of one of the domains, Fs(q,t) does not fully decay in all cases, and so we estimate the effective relaxation time by fitting the measured response to a Kohlrausch–Williams–Watts (KWW) stretched exponential and taking the time where the fitted function decays to 0.2 as τα. The unit of τα is given in Lennard-Jones (LJ) time units.
Looking at the system with the smallest domain spacing (N = 5) at the lowest system temperature (Tsys = 0.30), the low-Tg polymers are expected to form a rubbery phase and the high-Tg polymers form a glassy phase as Tsys = 0.30 is well below THg but above TLg. However, at this system temperature, the segmental dynamics behavior of the rubbery phase polymers resembles what one would expect for a glassy material rather than a rubbery one (Fig. 2, left). A typical rubbery material is expected to have a rapidly decaying Fs(q,t) profile, which is consistent with what is seen for the highest temperature Tsys = 0.50 system that is above the Tg of both polymer phases across all domain spacings. At Tsys = 0.30, the low-Tg polymers' Fs(q,t) profiles all exhibit a more glassy-like behavior as seen in the change of the profile's concavity and the emergence of a caging plateau. This is most evident in the N = 5 system but also present in the N = 10 and 20 structures and indicates a reduction in the overall mobility of the rubbery low-Tg phase when in the presence of glassy neighbors. Comparing across the three structures with different domain sizes (N = 5, 10, 20) at the same temperature Tsys = 0.30, the glassy phase's Fs(q,t) have approximately the same profile for all domain spacing values. The glassy phase remains mostly unaltered in its overall mobility by its rubbery neighbors.
 | ||
| Fig. 2 Self part of the intermediate scattering function for the symmetric blend (fg = 0.5) microphase separated systems in the undeformed state at varying system temperatures (Tsys = 0.50, red; Tsys = 0.30, blue). The Fs(q,t) profile is shown for the high-Tg (open circle) and the low-Tg polymers (star) for all molecular weights N = (5, 10, 20). | ||
We then look at τα extracted from the intermediate scattering functions for a quantitative measure of the polymer mobility in the glassy and rubbery phases. Fig. 3 reveals that the mobility varies widely between the high and low Tg phases and across structures with different domain spacing. As a function of system temperature, τα monotonically decreases with increasing temperature for both high and low Tg polymers as expected. For all network structures N = 5, 10, and 20, the high-Tg phase exhibits an approximately Arrhenius scaling τα ∼ eA/T with a fitted slope of A = 2.7. The values of τα at each given temperature are approximately the same for structures with varying domain spacings. This indicates that the presence of a low-Tg neighbor does not influence the overall mobility of the high-Tg phase. For the low-Tg phase, the temperature dependence begins to deviate more from an Arrhenius scaling with an increase in domain spacing or molecular weight of the polymer chain leading to greater deviation. The non-Arrhenius temperature dependence is described using the Williams–Landel–Ferry (WLF) equation given by
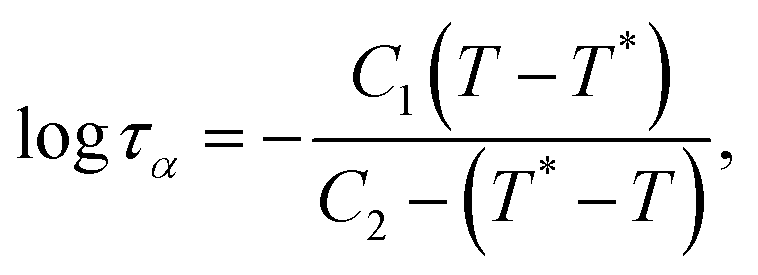 | (2) |
 | ||
| Fig. 3 The α-relaxation time τα as a function of the inverse system temperature for the high and low Tg polymers (open circles, stars, respectively) in the microphase separated symmetric blend (fg = 0.5) structures. The high-Tg phase data are fitted to the Arrhenius function (solid) and the low-Tg phase data are fitted to the WLF equation (dashed). The three domain spacing sizes are indicated by the corresponding molecular weight of the chains N = 5, 10, and 20 in blue, orange and green, respectively. | ||
3.3 Influence of interface on mixed Tg phases
Given the dependence of mobility change on the domain spacing of the structures, we explore the local dynamics in greater detail by examining the segmental dynamics as a function of the distance away from the high-low Tg interface. In the symmetric blend (fg = 0.5), the domain spacing on both sides of the interface is the same, and we expect similar domain geometry for both high and low Tg phases due to the random connectivity of the network structure prior to microphase separation. Using the domain spacing d for each structure sample formed from N = 5, 10, and 20 polymer strands, the maximum distance away from the interface is taken as zmax = 0.5d. We categorize the polymer monomers by their pure state Tg and group them by their distance z to the nearest monomer with the other Tg, where z < 0.5d. At the highest system temperature Tsys = 0.50, which is slightly above THg = 0.46 for the high-Tg polymers, and Fs(q,t) at each position from the interface z exhibits rapid decay in both phases, indicative of rubbery behavior (Fig. 4, top row). For polymers in the high-Tg phase closer to the interface, the local mobility is enhanced. In a similar manner, polymers in the low-Tg phase near the interface exhibit a reduced local mobility. | ||
| Fig. 4 Local mobility, represented by the self-part of the intermediate scattering function, Fs(q,t), is shown for the symmetric blend (fg = 0.5) microphase separated systems in the undeformed state for all molecular weights N = 5, 10, and 20. Fs(q,t) is plotted as a function of the distance z away from the interface between high (positive z direction) and low Tg (negative z direction) domains. The color scheme indicates the distance |z| away from the interface. The top row corresponds to the high system temperature (Tsys = 0.50) and the bottom row corresponds to the low system temperature (Tsys = 0.30). The Fs(q,t) profile is shown for the high-Tg (open circle) and the low-Tg polymers (star). | ||
At the lowest system temperature Tsys = 0.30 shown in the bottom row of Fig. 4, an asymmetric shift of the local mobility profile is observed on either side of the interface. While polymers in the high and low Tg phases experience increased or decreased local mobility, respectively, as they approach the interface, the magnitude of the shift in Fs(q,t) is unequal for the two phases. In the low-Tg phase where polymers are expected to be in the rubbery state, as the strands approach the interface, the shift in Fs(q,t) becomes increasing glassy-like with strong stretching of the segmental relaxation. This shift is the strongest in the N = 5 structure, which has the smallest domain spacing and thus more interference from multiple nearby interfaces.
The relaxation time of the segmental process τα extracted from each layer away from the interface as a function of inverse system temperature reveals that the high-Tg polymers follows the same temperature dependence at all distances z (Fig. 5). There is a small spread in τα across the layers at different distances from the interface for all temperatures studied in the high-Tg phase. Fittings to the Arrhenius scaling give activation energies of 2.7 ± 0.1, 2.7 ± 0.1 and 2.6 ± 0.1 when averaged over all layers in z for the N = 5, 10, and 20 samples. For the low-Tg phase polymers, there is significant variation in the spread of τα among the layers from the interface. In the layer closest to the high-low Tg interface, z = −0.5σ, the temperature dependence of τα approaches that of the high-Tg layers. A linear fit of the z = −0.5σ layer in the low-Tg phase gives slopes of 2.7, 2.4 and 2.1 for the N = 5, 10, and 20 structures, respectively. In the N = 5 structure, the z = −0.5σ layer approximates the slope observed for the high-Tg phase in the same structure. The mobility in the domains farther from the interface in the low-Tg phase exhibits a weaker temperature dependence and high mobility levels continue at all temperatures considered here. Comparing the N = 10 plot in Fig. 5 with Figure S5 (ESI†), which plots the extracted τα(T) values as a function of temperature for the single component high-Tg network, the τα(T) values from the single component high-Tg network are consistently higher than both the high-Tg phase and low-Tg phases in the mixed Tg network system. The values of the orange circle points in Fig. 5, which represent those high-Tg polymers farthest from the glassy–rubbery interface, are closest to the τα(T) values of the single component high-Tg polymers. For the high-Tg phase whose Tg is around 0.42, the τα(Tg) for the bulk system is approximately 105.2 as depicted in Fig. S5 (ESI†).
 | ||
| Fig. 5 The α-relaxation time (τα) as a function of the distance z away from the high-low Tg interface. For N = 5, 10, and 20, τα is plotted against the inverse system temperature for the high and low Tg polymers (open circles, stars, respectively) in the microphase separated symmetric blend (fg = 0.5) structures. Data points are colored according to the distance |z| away from the interface with more green being the closest and more orange the farthest to the interface as in Fig. 4 above. | ||
Focusing on two system temperatures, we examine how the local mobility gradient is affected by the high-low Tg interface as Tsys decreases through the glass transition temperature of the high-Tg phase, THg = 0.46. At the highest system temperature Tsys = 0.50, as the mobility gradient crosses the interface from the middle of the high-Tg phase to the low-Tg phase, a gradual enhancement of local mobility is observed for both N = 5 and 20 structures (Fig. 6). When both high and low Tg phases are in the rubbery state, the mobility gradient is approximately symmetric about the interface. In this relatively high-temperature state, the domain spacing does not impact the mobility gradient of the structures as seen in the similar τα values extracted from the same z-layer in both the N = 5 and 20 structures.
 | ||
| Fig. 6 Local mobility gradient of polymers at an average distance z away from the high-low Tg interface in the microphase separated symmetric blend (fg = 0.5) structures. Two structure samples with varying domain spacing (N = 5,20; closed and open symbols) are shown for two system temperatures at Tsys = 0.30 and 0.50 (blue and red, respectively). The high and low Tg polymer phases are indicated by the circle and star, respectively, with the dashed line indicating the position of the interface. | ||
As the system is quenched below the glass transition temperature of the high-Tg phase, the high-Tg phase becomes a glassy material accompanied by a dramatic increase in the segmental relaxation time while the low-Tg phase remains rubbery. This reduction in local mobility is seen in the shift of the high-Tg phase τα from the order of 101 to above 104 as the system is quenched from Tsys = 0.50 to 0.30. At a large enough distance from the glassy–rubbery interface in the N = 20 system, the low-Tg phase local mobility approaches that of the bulk rubbery material. The τα of the layer at z = −3.5σ remains largely unaffected by the appearance of the glassy neighbor as the system is quenched to Tsys = 0.30 when compared with the same layer at Tsys = 0.50. However, as the domain spacing shrinks, the presence of glassy neighbors significantly impacts the local mobility gradient in the rubbery phase. In the sample with the smallest domain spacing (N = 5), τα extracted from all layers in the rubbery phase is increased from the corresponding layers' τα in the N = 20 structure. This further reduction in local mobility due to the shrinking domain spacing is most evident in the z = −2.5σ layer, which is the layer farthest away from the glassy–rubbery interface for the N = 5 sample. In contrast to the large shift in the mobility gradient of the rubbery domain, the glassy domain's local mobility remains unaffected by the variation in domain spacing; the τα values in the corresponding layers of the N = 5 and N = 20 structures are nearly identical. Overall, the local mobility gradient is asymmetric about the glassy–rubbery interface with the rubbery phase experiencing a much greater change in local mobility as a function of distance away from the interface at Tsys = 0.30. Within approximately 2σ of the glassy–rubbery interface, the local mobility of the rubbery phase approaches that of a glassy material as the estimated τα approaches or exceeds 103.
3.4 Mechanical response
Having characterized the effect of the glassy–rubbery interface on the local mobility of the microphase separated structure, we now explore how the mechanical properties of the networks depend on both the fraction of glassy domains and the length of the polymer strands. The stress/strain response for each system under tensile deformation at Tsys = 0.30 is shown in Fig. 7a. For each system, there is an initial elastic response where the stress increases rapidly with a modulus comparable to a glassy polymer. After the glassy domains yield, there is a prolonged strain period of plastic flow where the stress gradually increases before a pronounced strain hardening regime begins. As fg increases, the stress during the plastic flow increases, and the strain where strain hardening begins occurs at smaller strains (see the ESI†). Interestingly, for a given fg the plateau stress increases as the chain length decreases. As we show below, this is due to the coupling of the dynamics between the phases, where the mobility in the rubbery domain is slower in the systems with shorter polymer strands. | ||
| Fig. 7 System stress and bond scission during fracture for microphase separated networks formed at number fractions of the glassy domain of fg = 0.25, 0.50, 0.75 and chain lengths of N = 5, 10, and 20 (blue, orange, and green, respectively). (a) The total system stress response of the samples during uniaxial tensile deformation and fracture. (b) Fraction of newly broken glassy and rubbery (solid and dashed lines, respectively) chains as a function of system strain during fracture. The curves begin in each case where the chains first begin to break. | ||
Surprisingly, the fraction of chains that are broken in both the rubbery and glassy domains is simply proportional to the overall composition, and chain scission under tensile deformation does not occur preferentially in either phase in our simulations. Fig. 7b shows a plot of the number of chains that have undergone scission in each phase normalized by the total number of broken chains at the end of the simulation. In all cases and for all strains, the ratio of the fraction of broken chains in the rubbery and glassy phase is simply determined by the overall composition, and the values are tabulated in Table 1. This trend holds across all N and fg, and the strain where chains begin to fail increases with N and is largely independent of fg. This observation aligns with our recent findings, which suggests that the topological and geometric factors predominantly determine the failure locations in end-linked polymer networks.35
| f g | N | f b,glassy | f b,rubbery |
|---|---|---|---|
| 0.25 | 5 | 0.26 | 0.74 |
| 10 | 0.25 | 0.75 | |
| 20 | 0.21 | 0.79 | |
| 0.5 | 5 | 0.49 | 0.51 |
| 10 | 0.51 | 0.49 | |
| 20 | 0.49 | 0.51 | |
| 0.75 | 5 | 0.75 | 0.25 |
| 10 | 0.76 | 0.24 | |
| 20 | 0.75 | 0.25 | |
To directly compare the difference in mechanical response of the two phases, we isolate the stress contribution from each domain. The stress contributions of the rubbery/glassy domains are calculated by aggregating stress on all rubbery/glassy monomers in the deformed direction and normalized by the volume fractions of the respective polymer type (details are provided in the ESI†). As shown in Fig. 8, we see that the stress response of the two phases is initially concentrated in the glassy phase (see the ESI† for a zoomed in version showing the small-strain response). On approach to failure, the stress becomes more homogeneous regardless of composition or strand length. During the initial deformation process, strain ε < 0.2, the glassy phase exhibits a sharp upturn in its normalized stress while the rubbery phase contribution to the stress gradually increases for all molecular weights and blend ratios. At larger strains where strain hardening sets in, we find that the rate of stress increase is larger in the rubbery domain, and the stresses in the two domains become essentially equal. We also note that chain scission begins well before the stress maximum, where the material begins to fail.
 | ||
| Fig. 8 Stress contribution by each of the glassy and rubbery phases shown by the solid and dashed lines, respectively. The stress contribution by each of the glassy and rubbery phases is normalized by the volume fraction of the respective polymer type to correct for the prefactor of 1/V that arises in the virial contribution to the stress. Colored arrows mark the strains at which bond scission first occurs for each of the chain lengths. | ||
3.5 Dynamic properties during deformation
Having characterized the stress distribution in the microphase separated structure, we now explore how deformation affects the segmental dynamics of the polymers in both the glassy and rubbery phases. The effective relaxation times (τeff) are estimated in the quiescent state and are indicated as horizontal lines in Fig. 9. For the glassy domain, our estimate of the τeff is insensitive to the matrix chain length, while the mobility in the rubbery domain increases as the chain length increases. | ||
| Fig. 9 Segmental relaxation time of the polymer monomers (τeff) as a function of strain for the microphase separated glassy (open circles) and rubbery domains (stars) during uniaxial deformation for the symmetric blend fg = 0.50 at Tsys = 0.30 for N = 5, 10, and 20 (blue, orange, and green, respectively). Horizontal black bars indicate τeff values of the undeformed glassy (solid) and rubbery (dashed) domains, and colored arrows indicate the strains at which bond scission first occurs for each of the chain lengths. The inset to the right panel is the intensity of the first peak of the radial distribution function between the two domains for N = 5, 10, and 20 in blue, orange, and green, respectively. | ||
Upon deformation, in all systems the mobility homogenizes up to failure, though the homogenization process depends on the strand length in the networks. In the glassy domains the mobility is rapidly enhanced by a factor of 10, consistent with previous simulations showing rapid changes in mobility in glasses during the initial elastic response,44 and the mobility continues to speed up as the deformation proceeds and is only weakly sensitive to the chain length. In contrast, the mobility changes in the rubbery domain depend on the chain length. For the networks with the shortest strands, the rubbery phase exhibits a significantly enhanced mobility upon deformation, which slightly accelerates further as strain increases. For the N = 10 systems, the enhancement compared to the undeformed sample is reduced, and the mobility in the rubbery domain is essentially independent of strain. In contrast to the N = 5 and 10 systems, the rubbery domains in the networks with the longest strands (N = 20) slows upon deformation. This surprising result is apparently due to the deformation leading to an increased mixing of the two domains. To support this explanation, we have calculated the radial distribution function between the rubbery and glassy monomers gAB(r), and we take the value of the first peak of gAB(r) as a measure of the extent of mixing of the domains (Fig. 9, inset). The peak is significantly lower in the N = 20 system prior to deformation and increases as the deformation proceeds, indicating an increasing amount of contact between the rubbery and glassy domains, which slows the rubbery monomers.
4 Conclusions
In this work, we have quantified the local mobility of microphase separated inhomogeneous polymer networks containing mixed high and low Tg phases as a function of distance to the interface. We have also studied the failure of these networks as a function of the network strand length and composition under tensile deformation.The morphology of the microphase separated structures visualized in 3D confirms the bicontinuous nature of randomly end-linked networks presumed by Tew and coworkers in their experimental work.10 Quantifying the domain spacing d of the networks post-phase separation gives the predicted scaling of d ∼ N0.5 by de Gennes.43 In the quiescent structures, the domain spacing of the structures has negligible influence on the local mobility of the high-Tg domain at all system temperatures. The temperature dependence of the segmental relaxation time also follows the same Arrhenius behavior for the high-Tg phase in all structures. In contrast, the low-Tg phase exhibits a wide spread in the temperature dependence of the segmental relaxation time with the overall mobility experiencing a significant reduction as the system temperature is decreased.
For a more detailed look at the local mobility gradient, we have quantified the segmental relaxation time as a function of distance z to the high-low Tg interface. Looking at the highest and lowest system temperatures, the degree of confinement (as measured by the typical domain size) has little to no effect on the local segmental mobility of the high-Tg domain away from the interface. In the low-Tg phase, however, the change in the length scales of confinement strongly impacts the local mobility away from the interface, especially in the lowest temperature case where there are distinct glassy and rubbery phases. The gradient in segmental dynamics across the interface shows that systems with smaller microphase separated domains exhibit a more spatially homogeneous mobility profile. We observe that the perturbation of the local dynamics by the glassy–rubbery interface extends further into the rubbery domain. This is in contrast to observations from experimental studies that probed the dynamic profiles in layered polymer materials13,45 and in diblock copolymer systems12 with dissimilar polymer–polymer interfaces. In the bilayered polymer material with a flat, planar interface studied in the experiments, the glassy phase experiences a more drastic change in local mobility away from the glassy–rubbery interface.13,45 However, recent molecular simulation attempts measuring the local mobility in stacked coarse-grained polymer films have revealed diverse behaviors: fairly symmetric dynamic gradients across the interface46 and more pronounced asymmetry into the high-Tg phase.47 In our system, the bicontinuous morphology of the randomly end-linked networks may locally confine the rubbery phase within a rigid glassy matrix, especially when combined with the small length scales of the domain spacing that are on the order of 10 nm. This structural morphology effectively surrounds a single phase with an interface in all directions. The shift of the mobility gradient towards the rubbery phase in the simulated structures may be due to the drastic change in both the length scale of the microphase separated domains and the geometry of the interface. We speculate that the increase in Tg of the rubbery phase is similar to the significant increase in Tg observed in polymers extremely confined in nanoparticle packings.48
During material failure, we find that the stress initially concentrates in the glassy domains. As the deformation proceeds, bond scission begins when the segmental dynamics in both domains are approximately commensurate with each other. In the glassy domain, the dynamics significantly increase upon deformation, consistent with prior experiments21,22,49 and simulations50–52 on homogeneous materials. In contrast, due to the influence of the glassy domain on the dynamics of the rubbery domain, the changes in the mobility in the rubbery domain depend on the strand length. For the shortest strands (N = 5), the glassy domain slows the mobility in the rubbery domain, and as a result there is an acceleration in this domain upon deformation. In contrast, the network formed with longer strands (N = 20) has domains sufficiently large for the center to exhibit a high mobility. Upon deformation in this system, the dynamics in the rubbery domain actually slow down due to increased mixing of the two domains. Presumably many of the predictions of the simulations here could be tested experimentally using fluorescence dye measurements,12,53–55 and the domains could be isolated by designing dyes that are either integrated into particular locations of the polymer strands or which segregate to one of the two domains. Overall, our work on using molecular dynamics to simulate randomly end-linked polymer networks with irregular phase boundaries highlights how structural morphology influences the local dynamics and the mechanical response of model inhomogeneous materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the Office of Naval Research via ONR-N00014-17-1-2056. Computational resources were made available through XSEDE Award TG-DMR150034. This work was supported in part by high-performance computer time and resources from the DoD High Performance Computing Modernization Program. Valuable discussions with Gregory Tew, Alfred Crosby, Christopher Barney, Ipek Sacligil, and Samuel Layding are gratefully acknowledged.References
- M. Rubinstein and R. H. Colby, Polymer Physics, OUP Oxford, Oxford, 2003 Search PubMed.
- L. Ionov, Mater. Today, 2014, 17, 494–503 CrossRef.
- Y. Gu, J. Zhao and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 5022–5049 CrossRef CAS PubMed.
- T. Zhou, H. Yuk, F. Hu, J. Wu, F. Tian, H. Roh, Z. Shen, G. Gu, J. Xu, B. Lu and X. Zhao, Nat. Mater., 2023, 22, 895–902 CrossRef CAS PubMed.
- A. N. Keith, M. Vatankhah-Varnosfaderani, C. Clair, F. Fahimipour, E. Dashtimoghadam, A. Lallam, M. Sztucki, D. A. Ivanov, H. Liang, A. V. Dobrynin and S. S. Sheiko, ACS Cent. Sci., 2020, 6, 413–419 CrossRef CAS PubMed.
- A. V. Dobrynin, A. Stroujkova, M. Vatankhah-Varnosfaderani and S. S. Sheiko, ACS Macro Lett., 2023, 12, 1510–1516 CrossRef CAS PubMed.
- A. V. Dobrynin, Y. Tian, M. Jacobs, E. A. Nikitina, D. A. Ivanov, M. Maw, F. Vashahi and S. S. Sheiko, Nat. Mater., 2023, 22, 1394–1400 CrossRef CAS PubMed.
- J. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- M. A. Haque, T. Kurokawa and J. P. Gong, Polymer, 2012, 53, 1805–1822 CrossRef CAS.
- C. N. Walker, K. C. Bryson, R. C. Hayward and G. N. Tew, ACS Nano, 2014, 8, 12376–12385 CrossRef CAS PubMed.
- G. Zhang, J. Steck, J. Kim, C. H. Ahn and Z. Suo, Sci. Adv., 2023, 9, eadh7742 CrossRef CAS PubMed.
- D. Christie, R. A. Register and R. D. Priestley, ACS Cent. Sci., 2018, 4, 504–511 CrossRef CAS PubMed.
- R. R. Baglay and C. B. Roth, J. Chem. Phys., 2015, 143, 111101 CrossRef PubMed.
- R. J. Lang, W. L. Merling and D. S. Simmons, ACS Macro Lett., 2014, 3, 758–762 CrossRef CAS PubMed.
- K. S. Schweizer and D. S. Simmons, J. Chem. Phys., 2019, 151, 240901 CrossRef PubMed.
- R. J. S. Ivancic and R. A. Riggleman, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 25407–25413 CrossRef CAS PubMed.
- A. Ghanekarade, A. D. Phan, K. S. Schweizer and D. S. Simmons, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2104398118 CrossRef CAS PubMed.
- A. Ghanekarade, A. D. Phan, K. S. Schweizer and D. S. Simmons, Nat. Phys., 2023, 19, 800–806 Search PubMed.
- Y. J. Gagnon, J. C. Burton and C. B. Roth, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2312533120 CrossRef CAS PubMed.
- K. Chen and K. S. Schweizer, J. Chem. Phys., 2007, 126, 014904 CrossRef PubMed.
- H.-N. Lee, K. Paeng, S. F. Swallen and M. D. Ediger, Science, 2009, 323, 231–234 CrossRef CAS PubMed.
- H.-N. Lee, R. A. Riggleman, J. J. de Pablo and M. D. Ediger, Macromolecules, 2009, 42, 4328–4336 CrossRef CAS.
- C. Nowak and F. A. Escobedo, Macromolecules, 2016, 49, 6711–6721 CrossRef CAS.
- M. Zhong, R. Wang, K. Kawamoto, B. D. Olsen and J. A. Johnson, Science, 2016, 353, 1264–1268 CrossRef CAS PubMed.
- R. Wang, A. Alexander-Katz, J. A. Johnson and B. D. Olsen, Phys. Rev. Lett., 2016, 116, 188302 CrossRef PubMed.
- E. Wang and F. A. Escobedo, Macromolecules, 2016, 49, 2375–2386 CrossRef CAS.
- A. A. Gusev, Macromolecules, 2019, 52, 3244–3251 CrossRef CAS.
- M. Lang and T. Müller, Macromolecules, 2020, 53, 498–512 CrossRef CAS.
- Z. Ye and R. A. Riggleman, Macromolecules, 2020, 53, 7825–7834 CrossRef CAS.
- C. W. Barney, Z. Ye, I. Sacligil, K. R. McLeod, H. Zhang, G. N. Tew, R. A. Riggleman and A. J. Crosby, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2112389119 CrossRef CAS PubMed.
- A. Arora, T.-S. Lin and B. D. Olsen, Macromolecules, 2022, 55, 4–14 CrossRef CAS.
- V. Sorichetti, A. Ninarello, J. Ruiz-Franco, V. Hugouvieux, E. Zaccarelli, C. Micheletti, W. Kob and L. Rovigatti, J. Chem. Phys., 2023, 158, 074905 CrossRef CAS PubMed.
- F. A. Escobedo and J. J. de Pablo, J. Chem. Phys., 1996, 104, 4788–4801 CrossRef CAS.
- F. A. Escobedo and J. J. de Pablo, Phys. Rep., 1999, 318, 85–112 CrossRef CAS.
- H. Zhang and R. A. Riggleman, Phys. Rev. Mater., 2024, 8, 035604 CrossRef CAS.
- G. S. Grest, K. Kremer and E. R. Duering, Phys. A, 1993, 194, 330–337 CrossRef CAS.
- E. R. Duering, K. Kremer and G. S. Grest, J. Chem. Phys., 1994, 101, 8169–8192 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in't Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- H. Zhang and R. A. Riggleman, Mol. Syst. Des. Eng., 2023, 8, 115–122 RSC.
- T. Ge, G. S. Grest and M. O. Robbins, Macromolecules, 2014, 47, 6982–6989 CrossRef CAS.
- A. Stukowski, Modell. Simul. Mater. Sci. Eng., 2010, 18, 015012 CrossRef.
- P. G. de Gennes, J. Phys., Lett., 1979, 40, 69–72 CrossRef CAS.
- R. A. Riggleman, K. S. Schweizer and J. J. de Pablo, Macromolecules, 2008, 41, 4969–4977 CrossRef CAS.
- R. R. Baglay and C. B. Roth, J. Chem. Phys., 2017, 146, 203307 CrossRef PubMed.
- D. D. Hsu, W. Xia, J. Song and S. Keten, MRS Commun., 2017, 7, 832–839 CrossRef CAS.
- A. Ghanekarade and D. S. Simmons, Macromolecules, 2023, 56, 379–392 CrossRef CAS.
- H. Wang, J. L. Hor, Y. Zhang, T. Liu, D. Lee and Z. Fakhraai, ACS Nano, 2018, 12, 5580–5587 CrossRef CAS PubMed.
- L. S. Loo, R. E. Cohen and K. K. Gleason, Science, 2000, 288, 116–119 CrossRef CAS PubMed.
- G. N. Toepperwein, K. S. Schweizer, R. A. Riggleman and J. J. de Pablo, Macromolecules, 2012, 45, 8467–8481 CrossRef CAS.
- R. A. Riggleman, H.-N. Lee, M. D. Ediger and J. J. de Pablo, Phys. Rev. Lett., 2007, 99, 215501 CrossRef PubMed.
- M. Warren and J. Rottler, Phys. Rev. Lett., 2010, 104, 205501 CrossRef PubMed.
- H.-N. Lee, K. Paeng, S. F. Swallen and M. D. Ediger, J. Chem. Phys., 2008, 128, 134902 CrossRef PubMed.
- D. Christie, R. A. Register and R. D. Priestley, Phys. Rev. Lett., 2018, 121, 247801 CrossRef PubMed.
- Y. J. Gagnon and C. B. Roth, ACS Macro Lett., 2020, 9, 1625–1631 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00087k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |