
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Residual cells and nutrient availability guide wound healing in bacterial biofilms†
Yusong
Ye
 ab,
Mnar
Ghrayeb
cd,
Sarah
Miercke
e,
Sania
Arif
f,
Susann
Müller
f,
Thorsten
Mascher
e,
Liraz
Chai
*cd and
Vasily
Zaburdaev
*ab
ab,
Mnar
Ghrayeb
cd,
Sarah
Miercke
e,
Sania
Arif
f,
Susann
Müller
f,
Thorsten
Mascher
e,
Liraz
Chai
*cd and
Vasily
Zaburdaev
*ab
aDepartment of Biology, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany. E-mail: vasily.zaburdaev@fau.de
bMax-Planck-Zentrum für Physik und Medizin, Erlangen, Germany
cInstitute of Chemistry, The Hebrew University of Jerusalem, Jerusalem, Israel. E-mail: liraz.chai@mail.huji.ac.il
dThe Center for Nanoscience and Nanotechnology, The Hebrew University of Jerusalem, Jerusalem, Israel
eGeneral Microbiology, TU Dresden, Dresden, Germany
fDepartment of Environmental Microbiology, Helmholtz-Centre for Environmental Research, Leipzig, Germany
First published on 22nd December 2023
Abstract
Biofilms are multicellular heterogeneous bacterial communities characterized by social-like division of labor, and remarkable robustness with respect to external stresses. Increasingly often an analogy between biofilms and arguably more complex eukaryotic tissues is being drawn. One illustrative example of where this analogy can be practically useful is the process of wound healing. While it has been extensively studied in eukaryotic tissues, the mechanism of wound healing in biofilms is virtually unexplored. Combining experiments in Bacillus subtilis bacteria, a model organism for biofilm formation, and a lattice-based theoretical model of biofilm growth, we studied how biofilms recover after macroscopic damage. We suggest that nutrient gradients and the abundance of proliferating cells are key factors augmenting wound closure. Accordingly, in the model, cell quiescence, nutrient fluxes, and biomass represented by cells and self-secreted extracellular matrix are necessary to qualitatively recapitulate the experimental results for damage repair. One of the surprising experimental findings is that residual cells, persisting in a damaged area after removal of a part of the biofilm, prominently affect the healing process. Taken together, our results outline the important roles of nutrient gradients and residual cells on biomass regrowth on macroscopic scales of the whole biofilm. The proposed combined experiment–simulation framework opens the way to further investigate the possible relation between wound healing, cell signaling and cell phenotype alternation in the local microenvironment of the wound.
Introduction
Biofilms are complex aggregates of microbial cells that form in association with almost any kind of a surface and are held together by a self-secreted extracellular matrix (ECM).1–5 Prominent examples from our daily life are dental plaque that we remove when brushing teeth,6 biofilms helping to ferment cheese7 or protecting roots of plants.8,9 Biofilms are highly heterogeneous in their architecture as well as in their cell phenotype distribution, both conveying remarkable degrees of spatio-temporal organization during their growth and development.1,10–13 Heterogeneity is often linked to the physical durability and tolerance of biofilms to environmental stresses, which makes biofilms very difficult to eradicate both chemically and mechanically.14–16 Biofilms are therefore often undesired, for example, in many technological settings when contaminating pipes and devices or in biomedical contexts when posing a huge challenge and threat as they colonize catheters or cause microbial infections that are difficult to treat.17–21 However, biofilms also contribute to positive utilities, including wastewater bio-filtration, remediation of contaminated soil, and microbial fuel cells.22–25Biofilm development
Biofilm growth is a complex, multi-stage process involving coordinated changes in the phenotype of bacterial cells and in their physiological state.12,26 Biofilms are shaped by physical forces and augmented by the cross-talk with the environment as a source of water and nutrients as well as a sink of waste products.27–30 While there is no single universal way of how broadly varying bacterial species form biofilms, it typically involves the following events. During initiation of biofilm formation, swimming planktonic bacteria adhere to a (solid) surface‡, alter their motility mode, and proliferate as the bacterial colony expands.12,31 When a subpopulation of bacteria switches their phenotype to ECM production, it is often referred to as a biofilm forming state.32 Secretion of matrix fundamentally changes the material and biophysical properties of aggregates and contributes to further biofilm spread.33,34 Other cell phenotypes emerge as groups of cells with certain functions in a spatially organized heterogeneous manner, in a process that is often compared to eukaryotic differentiation.35Biofilm biomass increases at the cost of nutrient consumption and differential metabolic activity creates gradients of nutrients and waste products in the biofilm and in its immediate vicinity. In addition to metabolic changes across biofilms, physiological states of cells change from metabolically active and proliferating cells to stationary (not growing), and in some bacteria, such as Bacillus subtilis to dormant (sporulating state) and to cell death (programmed or compulsory).36,37 While it is important to always keep track of the whole intrinsic complexity of biofilm development, here we will focus on the phenotype of ECM production and ability of cells to proliferate, as well as on nutrient availability. The fact that many of the above processes also take place during development of eukaryotic organisms and tissues pushed the appreciation of biofilms as models for differentiation of multi-cellular organisms.35,38 One of the spectacular traits of eukaryotic tissues, recently attributed also to biofilms, is their ability to regenerate and recover from damage.39,40
Wound healing in biofilms
A significant amount of research has been devoted to wound healing and regeneration after damage in eukaryotic organisms. It concerns multiple facets such as sensing of damage,41 mechanical force exertion,42,43 response of cellular signaling and directed cell migration,44–46 secretion and modification of the extracellular matrix,47 and immune response.48 While everyday laboratory practice suggests that biofilms are capable of regeneration after a mechanical damage, systematic studies of this process are very limited.49–51 Considering that biofilm-forming B. subtilis bacterial colonies growing on agar substrates, it has been studied how the colonies recover after cuts of different geometries, different ages of a wild type biofilm and several matrix mutants.49–51In this work, we propose a combination of experiment and whole-biofilm lattice-based simulations to investigate the dynamics of wound healing in bacterial colonies grown on an agar substrate. Here we specifically focus on the large scale dynamics of biomass, effects of cell physiological state and nutrient gradients. We use the experimental observations on B. subtilis biofilm wound cuts to setup the theoretical model and then use the model to identify the key processes that affect the healing and test those in experiments. With the help of model simulations, we pinpoint the critical effects of nutrient gradient and the physiological state of the cells with respect to their ability to proliferate and generate matrix.
A significant and probably unavoidable experimental result in any natural setting, which we also include in our simulations, is the remaining amount of cells in the wound after the cut, which essentially affects the biofilm regrowth dynamics. We corroborate the theoretical predictions by comparing biofilm regeneration following the formation of a cut on an original agar plate to the growth of a biofilm piece that was cut from a whole biofilm and moved to a fresh agar plate. Growth on a fresh plate eliminates any pre-existing gradient in nutrient, waste, and signaling molecules in the environment. Stochastic simulations of our multi-layer, lattice-based model can be adjusted to reproduce growth dynamics in space and time scales directly comparable to experiments while staying numerically efficient. A lattice-based model has the potential for developing towards further complexity, such as accounting for cell-signaling, additional cell phenotypes, and directed water/nutrient transport. At the same time, it provides a basis for rigorous coarse-graining and developing space–time continuum theory of biofilm growth.
The remaining parts of the manuscript are organized as follows. We first introduce the experimental system of B. subtilis biofilms grown on agar substrate and demonstrate the wound closure dynamics. Based on these observations and existing knowledge on biofilm growth, we setup a three-dimensional, coarse-grained, lattice-based model of biofilm growth on the agar substrate. We then recapitulate the wound closure dynamics and test which processes have the central effects on healing. We verify the predictions of the model in the experiments, by moving the wounded biofilms to fresh agar substrates and use spatially resolved fluorescence activated cell sorting (FACS) to quantify cellular dynamics during wound healing. We conclude with the summary of the results and the discussion of the future research directions.
Results
In this work, we consider recovery of B. subtilis biofilms from macroscopic damage, where a significant fraction (25 to 50%) of the biofilm biomass is removed. We are thus interested in the global response at the multicellular scale and how it is affected by the interaction of the biofilm with its environment. Without strong affinity for any particular term we can also formulate this setting as a regeneration process.The macroscopic scale of the damage also allows us to formulate a theoretical model that is coarse-grained and does not require single cell level of resolution. We next describe the experiments on the recovery of the damage, then formulate the theoretical model and see how both can be combined to provide insights into the processes underlying the recovery process.
Wound recovery in B. subtilis
Biofilms grow on agar plates that are supplemented with media, providing nutrients to cells in biofilms. In our laboratory models, biofilm formation starts with a droplet of cell suspension, originating from liquid pre-culture, spotted onto the surface of an agar plate. Cells then grow and divide and they spread into a circular shape until they reach a mature form after 72 hours. We chose to perform cuts in 24 h old biofilms (see the Materials and methods section below) because at this time point most of the biofilm cells make extracellular matrix and there is almost no sporulation yet.13 Indeed, it has been previously observed experimentally that the degree of healing is much more pronounced in biofilms that were cut after 24 h of growth than in older biofilms.49 Specifically, we performed cuts of a quarter and of half the area of the biofilm to better visualize the time evolution of the closing wound (relative to smaller cuts).Fig. 1 shows a 24 h old biofilm prior to performing a quarter of a biofilm cut, the biofilm after performing the cut and removing the cut biofilm piece, and the propagation of the healing process, as the gap that was created by the cut area is filled with biomass (cells and matrix). In addition to observing the gap in the biofilm being recovered with time, a few additional characteristics were noted. First, removal of the cut biofilm piece was not complete. Rather, cells remained in the cut area (arrows in Fig. 1, top row, t = 24 h and bottom row, t = 48 h). Furthermore, the healing process was not uniform in all directions and the healing process was progressing from the periphery of the biofilm inward (opposite to the direction of biofilm expansion) as it was greater and faster in the biofilm edges than in the center.
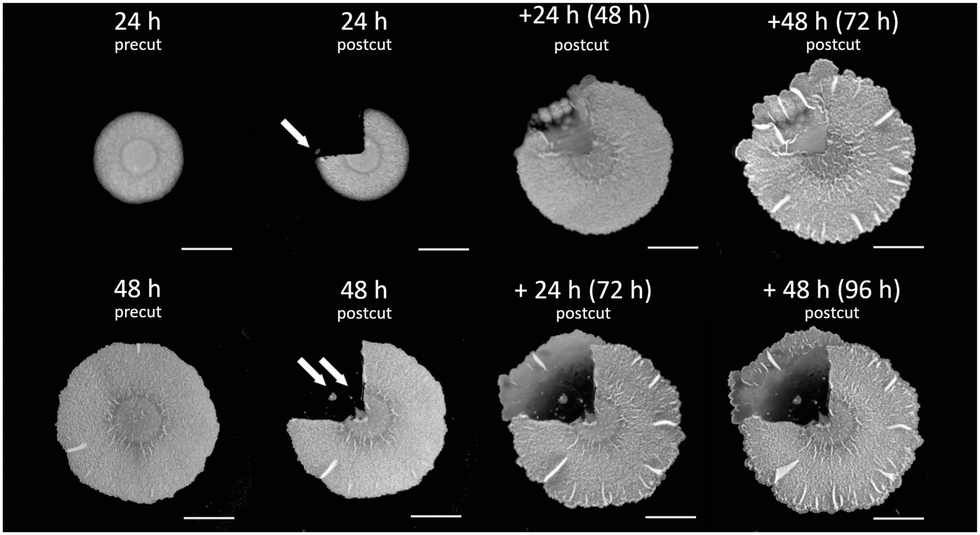 | ||
| Fig. 1 Wound healing in biofilms. B. subtilis biofilm healing following a 1/4 cut. 1/4 of the biofilm mass was cut following 24 h (top row) and 48 h (bottom row) of growth on agar plates. Images show biofilms before (precut) and after (postcut) they were cut together with the healing process along time. Top row: a 24 h old biofilm (pre- and postcut) along with +24 h following the cut (48 h from the original biofilm formation), and +48 h following the cut (72 h from the original biofilm formation). Bottom row: a 48 h biofilm (pre- and postcut) along with +24 h following the cut (72 h from the original biofilm formation), and +48 h following the cut (96 h from the original biofilm formation). Scale bars correspond to 0.5 cm. Representative images are shown, further repeats can be found in Fig. S1 (ESI†). | ||
Finally, we often observed that the biofilm in the wound sector has outgrown the outline of the normal biofilm growth. This might be the accidental result of transporting cells during the removal of the cut biofilm piece but can also be a competitive advantage of cells getting access to more nutrients being further away from the colony center.
A previous study49 has demonstrated that older biofilms heal more slowly and to a smaller degree, and therefore we wondered whether a similar effect is also observed for a different cut geometry where more of the biofilm is being removed. We repeated the experiment of cutting a quarter of the biofilms at later time points, namely at 48 h from the beginning of the biofilm growth.
Interestingly, as we performed the cut in older biofilms (see Fig. 1), the recovered area appeared thinner and it also extended further than the periphery of the original biofilm. Such a healing pattern may result from a combined effect of smaller number of cells remaining in the cut area that still can proliferate, overall depletion of nutrients, as well as migration of cells towards locations that are richer in nutrients, away from the original biofilm where they have been depleted.
To rationalize the above observations and help to determine which mechanisms can potentially contribute to the dynamics of the recovery, we proposed a theoretical model of biofilm growth on an agar substrate which we discuss below.
Lattice-based model of biofilm growth
Following our observations of how B. subtilis biofilms growing on nutrient-containing agar recover from damage, we aimed to find a quantitative framework to describe this process. Our experimental observations and existing knowledge on biofilm growth suggest a certain set of minimal ingredients that the model should be able to cover. We aim to describe macroscopic growth of whole biofilms in a quasi-two-dimensional setting combined with explicit consideration of nutrient transport. Biomass consists of two major components, cells and ECM produced by cells. Different physiological states/phenotypes of the cells are also important to mirror the heterogeneity of the biofilm. The model should be able to describe the dynamics of growth over the scale of several days but does not need to capture single cell level detail.To date there exists a broad range of theoretical approaches for biofilm modeling.52–55 Arguably, for the space and time scale of the problem of wound healing the choice reduces to continuum theory or coarse-grained lattice-based models.34,56–58 However, a need to include cell/matrix components and different physiological states of the cells makes it too complex for the continuum theory yet manageable for lattice-based models. Note that advanced hybrid models coupling lattice and continuum fields, taking into consideration biomass, water fluxes, osmotic effects, and biofilm mechanics were also developed specifically for B. subtilis.59 Here, we aim to address a novel biological question of how biofilms recover from damage. In particular, we are interested in the effects of nutrients and the ability of cells to produce biomass during wound healing. To study these effects closely we chose a non-trivial lattice model with a minimal number of unknown parameters which still is able to capture experimentally observed behaviors. A lattice model is essentially a phenomenological model which absorbs the above mentioned complex transport and growth phenomena in effective diffusion, which, however, in combination with energy functional, can recapitulate the growing phenotypes of quasi-two-dimensional colonies.
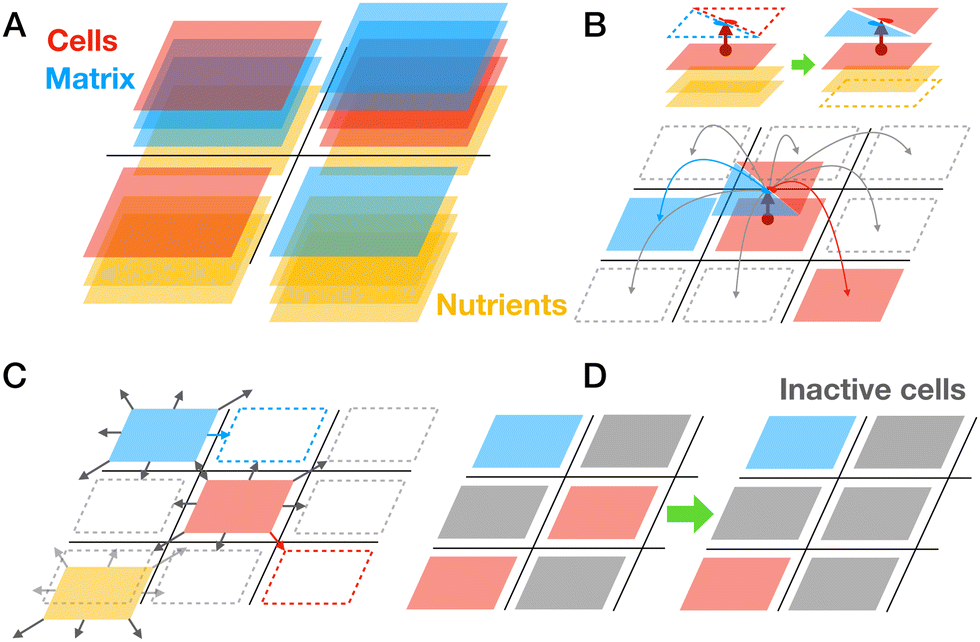 | ||
| Fig. 2 The lattice-based model of biofilm growth and basic schematics of simulations. (A) Two components in biofilm are cells (red), and matrix (blue) distributed on a two-dimensional square lattice in multiple layers. Single lattice site can be occupied by multiple elements of different species. Nutrients (yellow) evolve on their own lattice. (B) A cell can produce another cell or matrix element. Newly generated biomass can be deposited on the same site or on 8 neighboring sites. Production of new biomass requires consumption of nutrients. (C) Biomass and nutrients can diffuse in two dimensions by random hopping to 8 neighboring sites. (D) Cell inactivation happens irreversibly at a constant low rate; however, this rate is increased if the local microenvironment contains more inactive cells (grey). Inactive cells cannot duplicate and produce matrix elements and they also do not move. | ||
Both cell and matrix lattice-species and nutrients can stochastically hop to 8 neighboring lattice sites representing their diffusion (Fig. 2C). The number of biomass layers accumulating in a lattice site is regulated by the energetic cost that aims to mimic the fact of reduced nutrient availability for too thick biofilms (see details of energetic cost discussed below). Finally, we assume that cell species may turn into an inactive state, which biologically can correspond to the stationary phase, persister cells, competent cells, spores, or dead cells. Cell species will turn inactive with a small basal rate; however, we also assume that accumulation of inactive cells will locally increase this rate, mimicking the impact of the local unfavorable microenvironment (Fig. 2D). We also assume that inactive cells cannot migrate or vanish, meaning that they still occupy a specific space in the lattice.
Modeling of biofilm growth
To test the basic functionality of the lattice model we first simulated the normal biofilm growth with several basic perturbations (see Fig. 3A and Movie S1, ESI†). Consistent with multiple previous work the growth of the biofilm under the competition for space and for resources is slower than exponential (Fig. 3B). We see that in simulation time corresponding to 72 h, biofilms attain a near circular shape with a typical diameter of ∼2 cm. Due to introduced repulsion between cell and matrix species we see a formation of locally heterogeneous pattern in the biofilm. By making a virtual cut through a biofilm we can visualize the distribution of total biomass in cell (red) and matrix (blue) fractions (see inset in Fig. 3B). We can also see that in the middle part of the colony there is an accumulation of inactive cells (grey). We can also clearly observe the depletion of the nutrient concentration beneath the biofilm (Fig. 3A, bottom panels). To make the predictions of our model more quantitative we used the experimentally measured colony sizes (diameter) and tuned the parameters of the model to reproduce this growth curve, see Fig. 3C.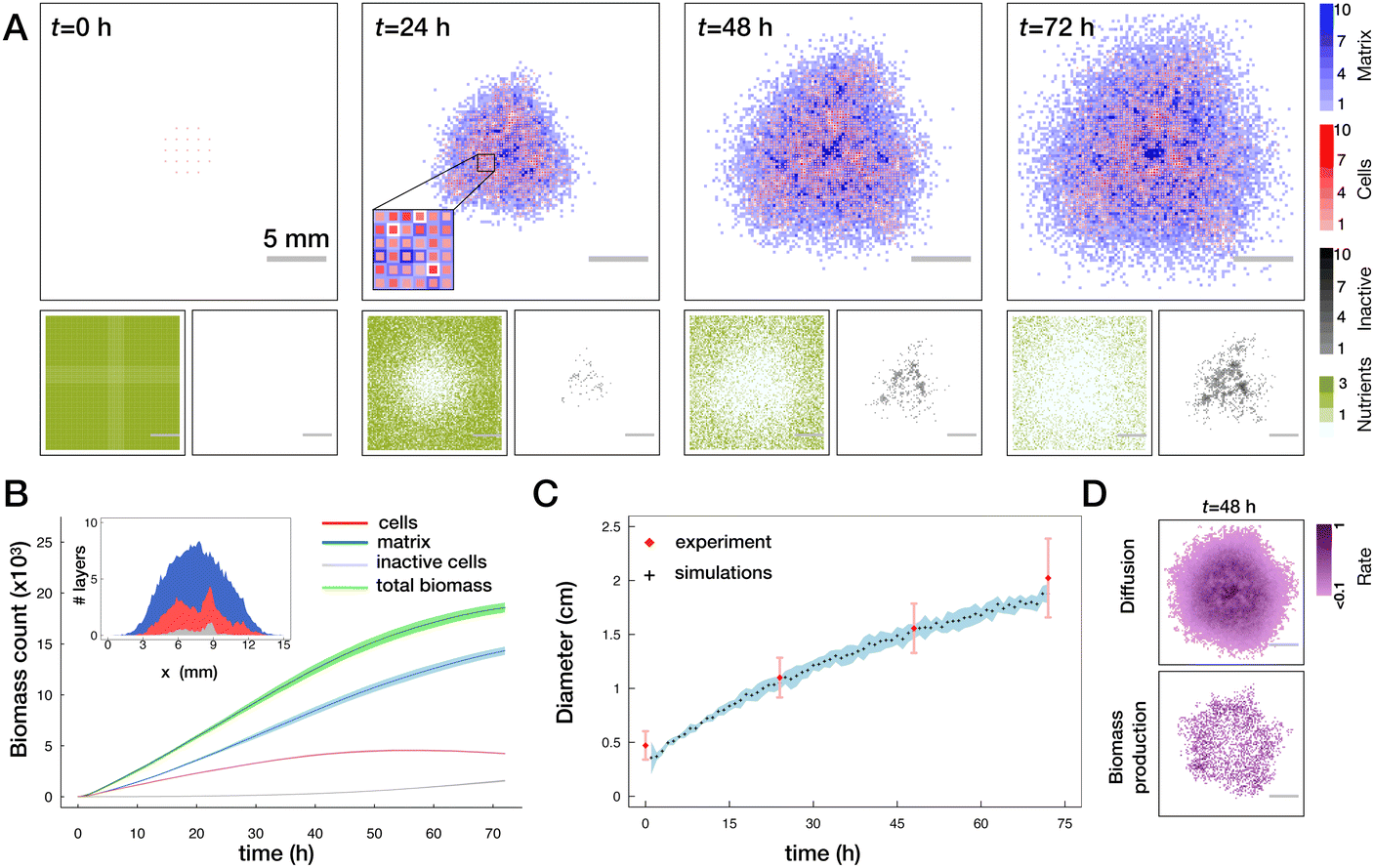 | ||
| Fig. 3 Biofilm growth in the lattice model. (A) Top panels show one realization of the distribution of biomass layers in cells (red) and matrix (blue). In the first panel, uniformly distributed cell layers mimic the initial inoculation droplet. In the second panel, the inset shows the zoom in on an area of a biofilm. Smaller panels in the bottom show the corresponding distribution of nutrients (olive) and inactive cells (grey) (see also Movie S1, ESI†). (B) Evolution of biomass as a function of time quantified by the total number of occupied layers for total biomass (green), active and inactive cells (red and grey respectively) and the matrix (blue). Lines with shades show mean ± SD (n = 5 runs). The inset shows the cut through the center of the biofilm and the respective spatial distribution of the biomass. (C) Comparison of the biofilm diameter in the model (black crosses with a blue shade, mean ± SD (n = 5 runs)) to the experimentally measured colony diameters (red diamonds mean ± SD, see Materials and methods). In simulations, diameter is defined as an average size of horizontal and vertical cross-sections where the biofilm is at least one layer thick. In experiments, diameter is the average of vertical and horizontal colony extension obtained from biofilm images. (D) Frequency of diffusion (top) and of biomass production events (bottom) averaged from 46 to 48 h for n = 5 runs. | ||
Two major drivers of colony expansion are the production of new biomass and its diffusion. For sufficiently old biofilms where the mid part already approaches the height limited by energetic penalty and availability (lack) of nutrients, mostly the cells at the periphery contribute to growth and further expansion of the colony, as shown in Fig. 3D.
Modeling of the wound healing
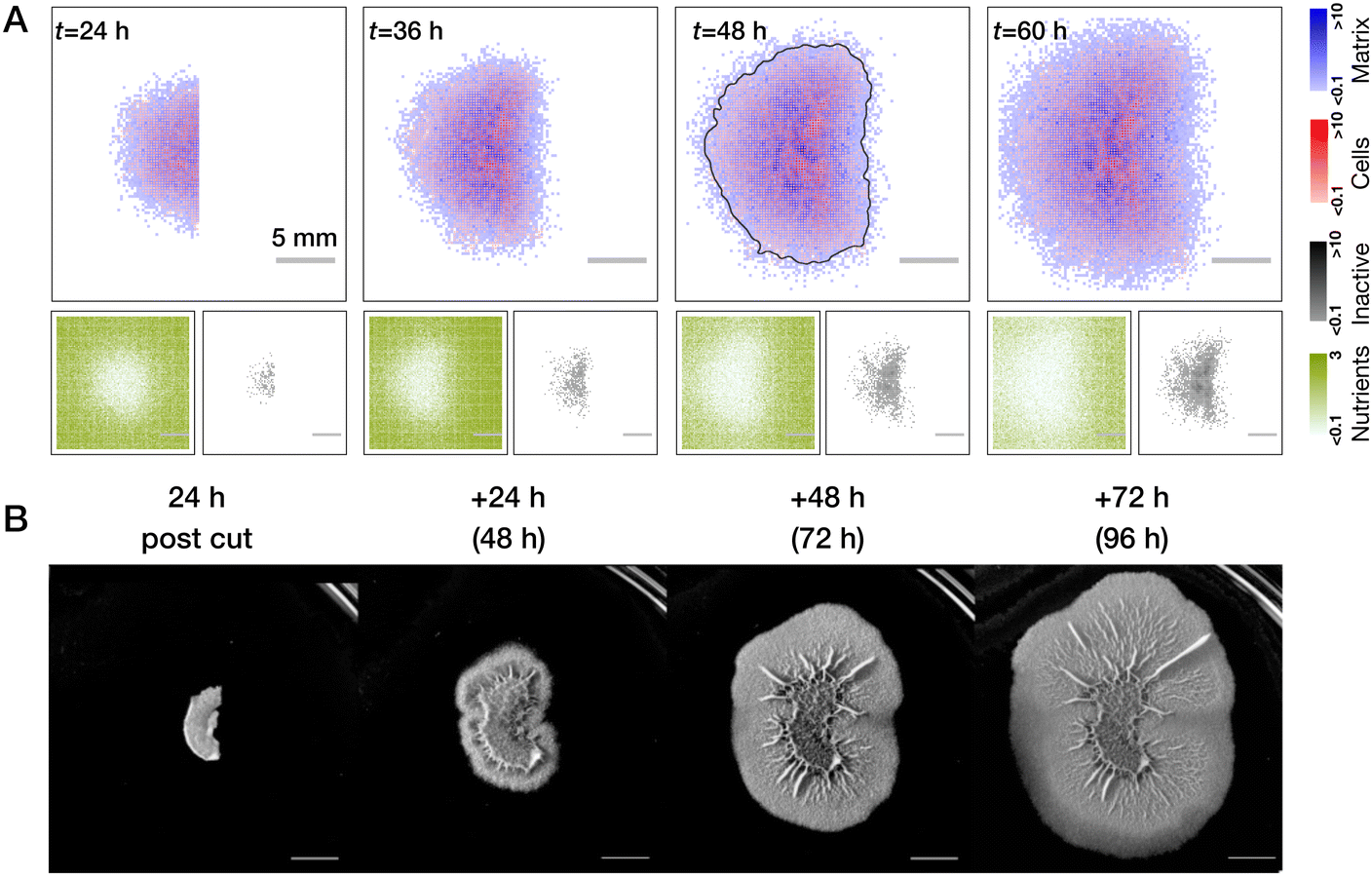
To investigate the wound healing in the model, we first follow the experimental protocol. The colonies are left to grow unperturbed for the time corresponding to t = 24 h in physical time, then a single quadrant of the biofilm is removed (all cell and matrix species), then the regrowth of the biofilm is followed over an extended period of time (see Fig. 4A).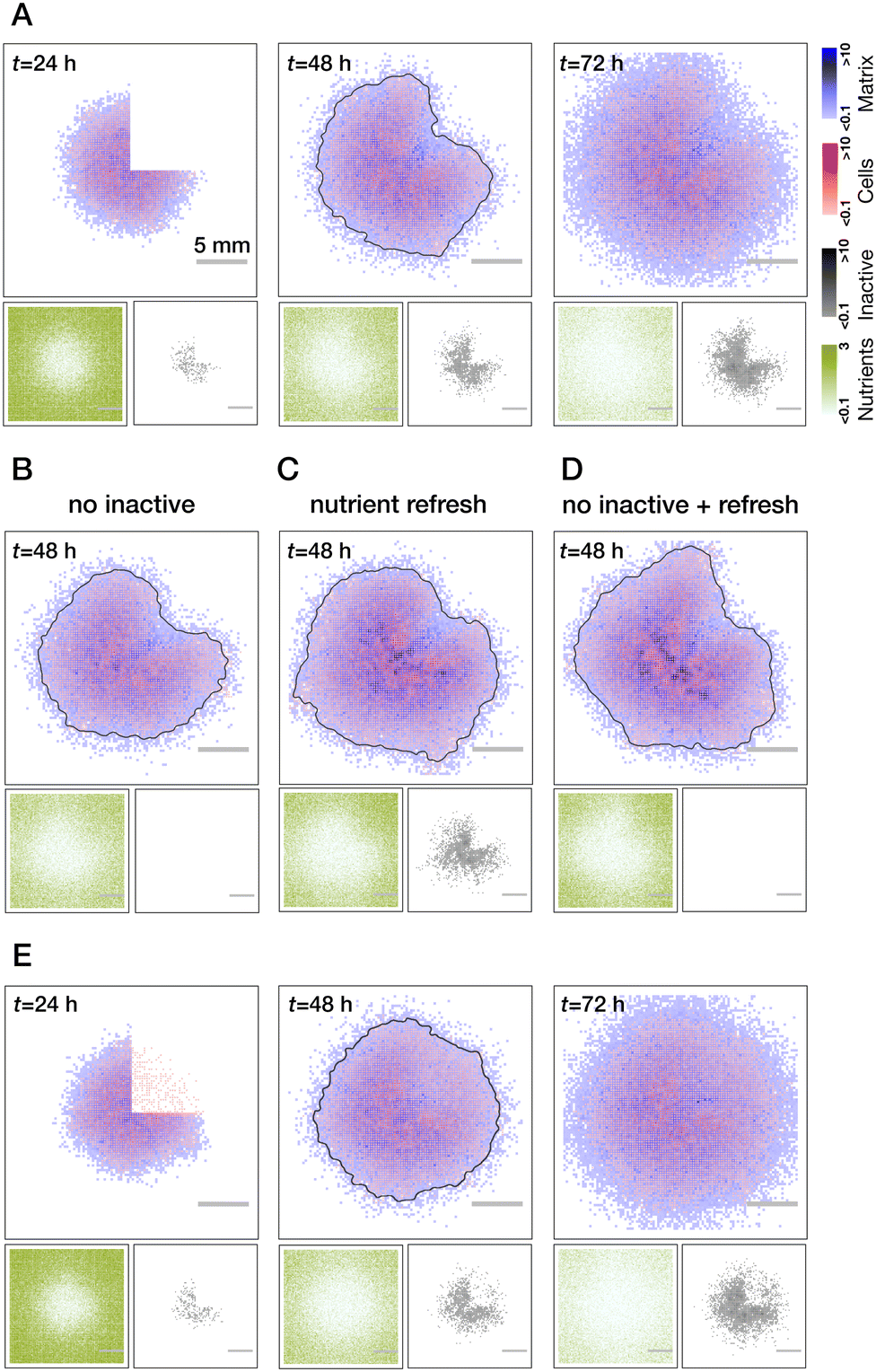 | ||
| Fig. 4 Modeling of wound healing. (A) Biofilm healing from a clean quarter cut (no residual cells in the wound). Wound recovery is inhomogeneous with respect to the radius: faster biomass production at the periphery of the colony due to the gradient of nutrients and inactive cells (see also Movie S2, ESI†). Testing of mechanisms leading to the inhomogeneous wound recovery: no inactive cells (B), refresh of the nutrients right after the cut (C) and both combined (D). While every perturbation alone reduces the degree of inhomogeneity, their combined effect leads to a more uniform wound closure. (E) Effect of the residual cells on wound recovery. Regrowth of biomass proceeds uniformly through the whole area of the wound (see Movie S3, ESI†). Grey lines in all t = 48 h images show a threshold of the overall biomass to better indicate the outline of the colony. All results are averaged over n = 5 realisations. | ||
We first consider the case of clean wounds where no residual cells are left behind. For the parameter settings that were informed by existing literature and reproducing normal growth dynamics, we clearly see an effect of spatially inhomogeneous wound recovery. In particular, there is a faster re-growth of the biomass at the edges of the wound further away from the colony center. Such a behavior was previously experimentally observed for narrow, rectangular cuts along the biofilm radius.49 Based on our observations of the normal growth we can propose (and test) two major mechanisms responsible for such behavior: pre-existing gradient of the nutrients that was created by 24 hours of biofilm growth at the moment of the cut and the accumulation of inactive cells in the center of the colony. In simulations, we can easily control these two effects (see Fig. 4B–D). Indeed, we see a gradual reduction of inhomogeneity if we switch off the inactivation of cells (B) or refresh nutrients right after the cut was made (C). When both are implemented, effects complement each other (D) and regrowth is rather uniform (compare grey outlines of the colonies at t = 48 h). These are the behaviors of the wound healing that would be predicted by the model in the case of an ideal cut.
 | ||
| Fig. 5 Comparison of half-cut biofilms with nutrient gradients and residual cells in theory and experiment. (A) Biofilm recovers from removal of its half and with pronounced effect of residual cells, which is similar to earlier Fig. 4C (see also Movie S4, ESI†). Average over n = 5 realisations is shown. (B) Same setup realized in the experiment. Scale bars in (B) correspond to 0.5 cm. Representative images are shown, further repeats can be found in Fig. S1 (ESI†). | ||
Regrowth of the half-biofilm transferred to the fresh plate (modeled as a refresh of nutrients and no residual cells) proceeds more interestingly, see Fig. 6A and Movie S5 (ESI†). We see that for intermediate times, the outline of the colony takes up a “croissant” shape (see grey outlines of colonies at 48 h in Fig. 5A and 6A) although we are not expecting any nutrient gradients to be present in this case (note that in Fig. 6A we show average over n = 5 simulation runs. In individual realisations, a croissant shape is more pronounced, see Fig. S2, ESI†). Explanation of this shape in simulations, lies in the higher number of inactive cells at the middle of the colony, which, despite fully available refreshed nutrients can only generate a moderate amount of biomass compared to a larger number of active cells at the periphery of the biofilm. However, over time, due to the outgrowth of the colony the inhomogeneity of active/inactive cells is “forgotten” and the colony starts to approach a more symmetric circular shape. Importantly, we also see mirroring of the theoretical predictions in our experiment, where a half cut biofilm is transferred to a fresh agar plate. Here, the croissant shape is even more pronounced than in simulations, see Fig. 6 (see, however, Fig. S2, ESI†).
 | ||
| Fig. 6 Comparison of half-cut biofilms with no nutrient of gradients and no residual cells in theory and experiment. (A) Regrowth of the biofilm half with fully refreshed nutrients and no residual cells. Interestingly, despite full nutrient availability, regrowth at intermediate time is non-uniform, croissant-shaped (see, for example, the grey outline of biomass threshold at t = 48 h). This is due to the effect of inactive cells in the center of the colony. At larger times this effect is screened by new regrown cells and growth becomes homogeneous (see also Movie S5, ESI†). Averages over n = 5 simulations runs are shown. (B) Experiment exhibits very similar dynamics, where croissant shape is even more pronounced. Scale bars in (B) correspond to 0.5 cm. Representative images are shown, further repeats can be found in Fig. S1 (ESI†). | ||
Now that we could rationalize our major qualitative findings with the model, we next wanted to better quantify the difference between the growth dynamics of healing and normal biofilms.
Quantification of healing dynamics
Healing dynamics have been previously quantified using optical imaging, based on segmentation and light transmission through the biofilm mass.50 The presence of the residual cells in the wound does not allow for the image segmentation of the wound outline in our case. Furthermore, optical methods would not be fully suitable because quantifying the thickness of the biofilm by an intensity of transmitted light also relies on the assumption of identical and homogeneous mass distribution which we cannot do a priori, especially when comparing healed to normal biofilms. Instead, we decided to rely on the methods allowing for direct cell counting and biomass quantification.First, we performed optical density measurements of the biofilm samples (1/4 of biofilm) taken from biofilms healed after being wounded and control (unperturbed) areas at different time points, sonicated to break the biomass into individual cells, and then re-suspended in liquid medium at a given dilution. Optical density (turbidity) measurements of these cell suspensions thus serve as a proxy for the total number of cells (see Materials and methods for technical details) and are shown in Fig. 7A. In two independent experiment sets, we recovered the same remarkable trend that the number of cells in the healing wound is initially higher than in the normally growing biofilm, with a crossover happening at around 40 h where the normal growth takes over. We wondered if such a behavior is also recapitulated in our model. And indeed, by counting the number of active cells in healed vs. normal quarter of a biofilm we recover the same trend in our simulations and with a very similar crossover time (see Fig. 7B and overlay of simulations and data in Fig. S3, ESI†).
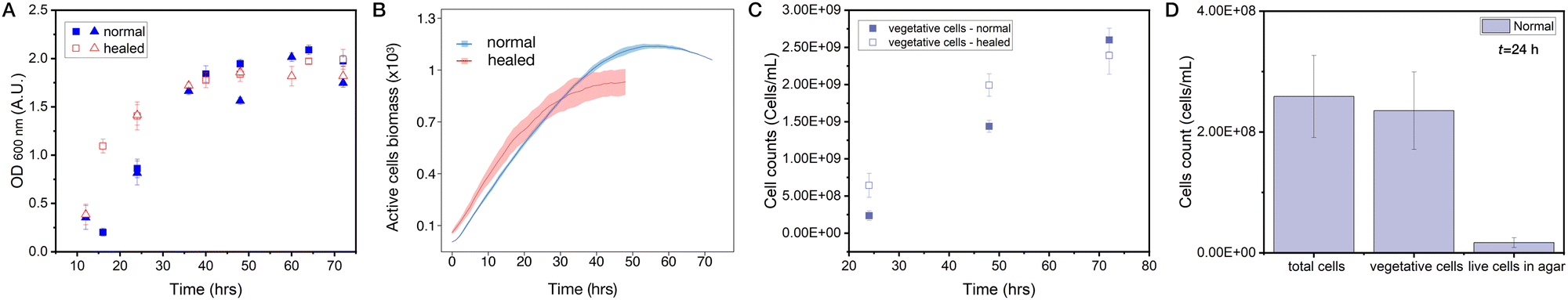 | ||
| Fig. 7 Quantification of wound healing dynamics. (A) Optical density (turbidity) quantification of cell suspensions taken from 1/4 biofilm samples of a normal growing biofilm (solid symbols) and biofilms that healed from damage (open symbols). Two different symbol shapes represent two independent experimental sets (n = 4 in each set). The healing biofilm initially has a higher number of cells and then at around t ∼ 40 h there is a cross over and the number of cells derived from normal biofilms surpasses that derived from healed biofilms. (B) Counts of active cell species in the lattice model in normally growing biofilms (blue) vs. biofilms healed from damage (red). Mean ± SD (n = 5 runs) are shown. For an overlay of simulations and turbidity data, see Fig. S3 (ESI†). (C) FACS cell counting as a function of time of cell suspensions from quarters of normal and healed biofilms also shows the same trend as turbidity (A) and simulations (B) data. (D) Quantification of cell subtypes at t = 24 h using FACS cell sorting to dead cells, vegetative cells and spores, see also Fig. S5 (ESI†). Around 90% of all cells in a normal growing biofilm are vegetative cells (meaning that the rest are spores and dead cells). In the agar after removal of the biofilm during the cut, we can detect live cells (vegetative cells and spores) that mount to approximately 10% of vegetative cells in the biofilm. | ||
Second, we sought the ultimate way to cross-check our optical density measurements (which are still an indirect measure of cell numbers) and deployed recently developed, spatially resolved fluorescence activated cell sorting technique (FACS) to compare cell counts in the healing wounded and normal biofilms, as well as in the agar (to test for residual cells). Here we can specifically focus on counts of live, vegetative cells which contribute to growth and biomass production, see Fig. 7C. These results confirmed our observation that the healing biofilm initially grows faster than the normal biofilm of the same age and that there is a crossover where normal biofilm growth takes over. However, crossover in these data occurs later in time relative to the turbidity measurements, which can be explained by different biofilm growth conditions in the two laboratories where these experiments took place.
We think there is an intuitive explanation of the observed crossover behavior. Residual cells and spores left on/in agar after the removal of a biofilm quarter serve as an initial condition for the wound healing. This number of cells exceeds the number of cells found in the initial droplet from which the biofilm is inoculated. Provided with still enough nutrients, these cells drive rapid biomass regrowth in the wound. At a later stage, however, they face the nutrient deficit (the cells removed during the cut were consuming nutrients for 24 hours) and thus give up the lead to the normal growing biofilms.
Finally, we can also use FACS setup to quantify the amount of different cell types. Here we focus on total cells (including dead cells), their vegetative fraction, and number of live cells (vegetative and spores) in the agar beneath the removed biofilm at t = 24 h, see Fig. 7D. Consistent with our simulations we see that ∼90% of all living cells are vegetative (active in the model). Remarkably, a number of living cells trapped in the agar when biofilm is removed is ∼10% of vegetative cells in the biofilm – also the number we initially used in our simulations. To us it was a surprisingly high number, which we have not seen mentioned in the literature. It would be an exciting direction of future research to further phenotype those remaining cells and their distribution in the agar.
So far we exclusively focused on the cell counts; however, biofilms also contain matrix. Experimentally, it is very difficult to properly identify the ECM fraction, but not in simulations. Interestingly, we observe that in the healing area there is a significant overproduction of the matrix as compared to normal growth (see Fig. S4, ESI†), an effect known to be present in the eukaryotic tissue wound healing process. It will be interesting to also check this model prediction in future experiments on biofilms.
Discussion
In this work, we explored how a biofilm recovers a biomass that was removed by an areal cut. A combination of experiments with a numerical model allowed us to suggest and test the relevance of several key factors that affect the regeneration process.There is a clear effect of the pre-existing gradients. While in the model we only account for nutrient gradients, in experiment, it is a combination of two: nutrients and waste products, including potentially harmful or inhibitory secondary metabolites.65,66 Generally, the effects of waste products on biofilm growth are still poorly understood and would be very important to consider for the problem of wound healing. When the gradients, at least in the agar environment, are removed, we still see transient heterogeneity which we attribute to the ability of cells to proliferate.
Perhaps the most unexpected observation is the dramatic effect of the residual cells which remain in agar and drive the regrowth in the wound and which we quantified to be of the order of 10% of number of cells in the biofilm. There are two potential sources of such cells. First, the removal of the biofilm material from the agar surface is never perfect and some cells embedded in the matrix might just stay in the wound area. However, there is a second possibility, where bacteria invade the agar beneath the biofilm.67,68 In that case, it is impossible to remove these cells by mechanically cutting out the biofilm piece and peeling it off the agar surface. We have suggested that an analogy between the eukaryotic tissue wound healing and comparable processes occurring in biofilms may exist. However, the key questions that explore analogy of wound healing in biofilms and eukaryotic tissues still remain open: (i) whether biofilms can sense damage (ii) whether there is a programmed response (iii) what is the role of different cell phenotypes and matrix (iv) what are the roles of mechanical forces and biofilm material properties and, finally, (v) what is the effect of nutrients, waste and signaling molecule gradients on wound healing. These open questions guide the general direction of research and addressing them will require a consolidated interdisciplinary effort in the future.
Our model could make semi-quantitative predictions about biofilm growth and wound healing focusing on major effects of nutrients and activity of cells. Specifically, we recover the normal biofilm growth, predict croissant shaped colonies on fresh agar, and recover the interesting effect of faster healing with a delayed taking over of normal growth, which is remarkable for that of a simple model. However, a large advantage of the lattice- and agent-based models is their extreme flexibility to include more cell phenotypes and/or signaling chemicals. Interestingly, effects of repair from damage and ageing can be potentially included at a single-cell level.69 Furthermore, mechanics and water transport (and thus nutrient and waste transport) can be also included in the model.59 The most obvious next step is to push the model to more explicitly incorporate the agar-fraction of cells which should be paralleled by the biological inquiry in their identity, distribution, and quantification. Finally, the lattice model can serve as the starting point for developing continuum mechanical theory of tissues combining cells and ECM and thus can be further applied in the context of eukaryotic wound healing.
Materials and methods
Biofilm formation experiments
A liquid culture of Bacillus subtilis (NCIB3610) was grown overnight at 37 °C to optical density (OD) 1.2–1.4. A 2 μL drop from that liquid culture was spotted onto a 1.5% agar plate containing MSgg medium.70 Plates were incubated at 30 °C for 24 h or 48 h as described in the text, and 1/4 or 1/2 pieces of biofilms were cut and removed using a spatula. Plates were then placed back into incubation at 30 °C following the cut and 1/4 or 1/2 piece removal. In addition, for the nutrient elimination experiment (Fig. 6), half cut biofilms were transferred to new MSgg plates and incubated further at 30 °C as indicated in the text. Biofilm images were taken using a P.CAM 360 or Nikon D3300 camera equipped with Nikkor 18–55 mm lens (Nikon, Thailand). The final images were brightness and contrast corrected.Biofilm diameter measurements
The diameter of biofilms was measured using images of normally growing colonies at different time points. We measured biofilms’ maximum extension in x- and y-directions using ImageJ software (https://imagej.net/ij/) and took the mean of those as the report for the diameter. Multiple colonies from different experiments were used for the measurements at t = 24 h (n = 86), t = 48 h (n = 12), t = 72 h (n = 18). To estimate the size of the initial drop we used the diameter of the smooth circular part at colonies’ center in a random subset of t = 24 h images, which correspond to the size of the inoculation region (n = 48). For every time point mean ± SD is reported.Biofilm turbidity measurements
A liquid culture of Bacillus subtilis was grown overnight at 37 °C, 250 RPM, then diluted to optical density (OD) 0.8 using LB Broth. A 2 μL drop of the liquid culture was placed onto a 1.5% MSgg-agar plate.70 Plates were then incubated at 30 °C for different time points. For normal biofilm cell count estimation, a 1/4 biofilm piece was cut and removed from the biofilm, resuspended with 1 mL of LB broth and sonicated using a Sonics VCX 750 probe sonicator (Sonics & Materials, Newtown, CT, USA) with 1 s pulse on, 1 s pulse off for 4 s in total (30% duty cycle, 18J). Optical density (turbidity) of cell suspension was measured at 600 nm. For healed biofilm cell count estimation, biofilms were re-incubated at 30 °C following the removal of a 1/4 biofilm piece at t = 24 h. A 1/4 biofilm piece was cut from the healed area after different times, resuspended with 1 mL of LB broth and sonicated using a Sonics VCX 750 probe sonicator with 1 s pulse on, 1 s pulse off for 4 s in total (30%, duty cycle, 18J). Optical density (turbidity) of the cell suspensions was measured at 600 nm.Fluorescence-activated cell sorting (FACS)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells were recorded per cell gate.
000 cells were recorded per cell gate.
The flow cytometric raw data are available at the FlowRepository website: https://flowrepository.org/id/RvFrFKou3x4p9X9O0FmVwSymsn62Hi2k7gB01rn1PqIQnkB5OHRboQ9XLntDpSyB.
Repository ID number: FR-FCM-Z72B.
| #cells mL−1 = f × C(count) × B × V/[B(YG)V(stain)], |
Initial configuration. Cells and extracellular matrix are required for the biofilm growth. In our simulations, all the initial configurations are made up of 21 lattices sites with one layer of cells uniformly and sparsely distributed in the center of the simulation box in the circle with the diameter of ∼20 lattice sites. This was made to mimic the initial inoculation drop of diameter 4.7 mm put on the agar to start the biofilm colony. It's size (lattice-sites vs. physical length) is determined by matching the parameters of the model to the growth data from experiments, see below.
Allowed moves on the lattice. When a cell produces another cell or a matrix it will consider 8 nearby lattices sites and the current lattice site (total of 9 potential placements) where the newly produced biomass can be deposited. However, only 8 neighboring lattice sites are considered for the diffusive movements of the biomass. Additionally, we assume that inactive cells are not allowed to move.
Hamiltonian of the system. We define the Hamiltonian (energy cost function) as

Three different energy functionals are invoked for cases of new cell production, matrix production and for diffusive moves. Cw, Jw, Nc,mw are three weight parameters for space competition, interaction, and nutrient consumption respectively (different for cells and matrix), while i, j are the lattice indices. Amax is the initial number of nutrient layers, and As shows the lattice's current number of nutrient layers (only for those who produce new biomass). Notice that if As = 0, then it is impossible to produce new biomass.
To summarize, the first part of H describes the competition for space where higher density of biomass (more occupied layers) will lead to a larger Hamiltonian. Here, the weight of any biomass (σi, cells or matrix) is 1. Thus, during biomass production or diffusive moves, it is more likely to jump to the site with smallest number of layers, as it will lead to smaller energy penalty. The second term in H is the classical expression of interaction. Here, we introduce a small repulsion (Jw < 0) between cells and matrix lattice-species.64 This allows us to produce heterogeneous (in terms of cell/matrix distribution) biomass. The third term in H (when relevant) reflects the reduction of the ability to produce new biomass when nutrients get scarce.
And the rates expressions for ki are set to (see also below):

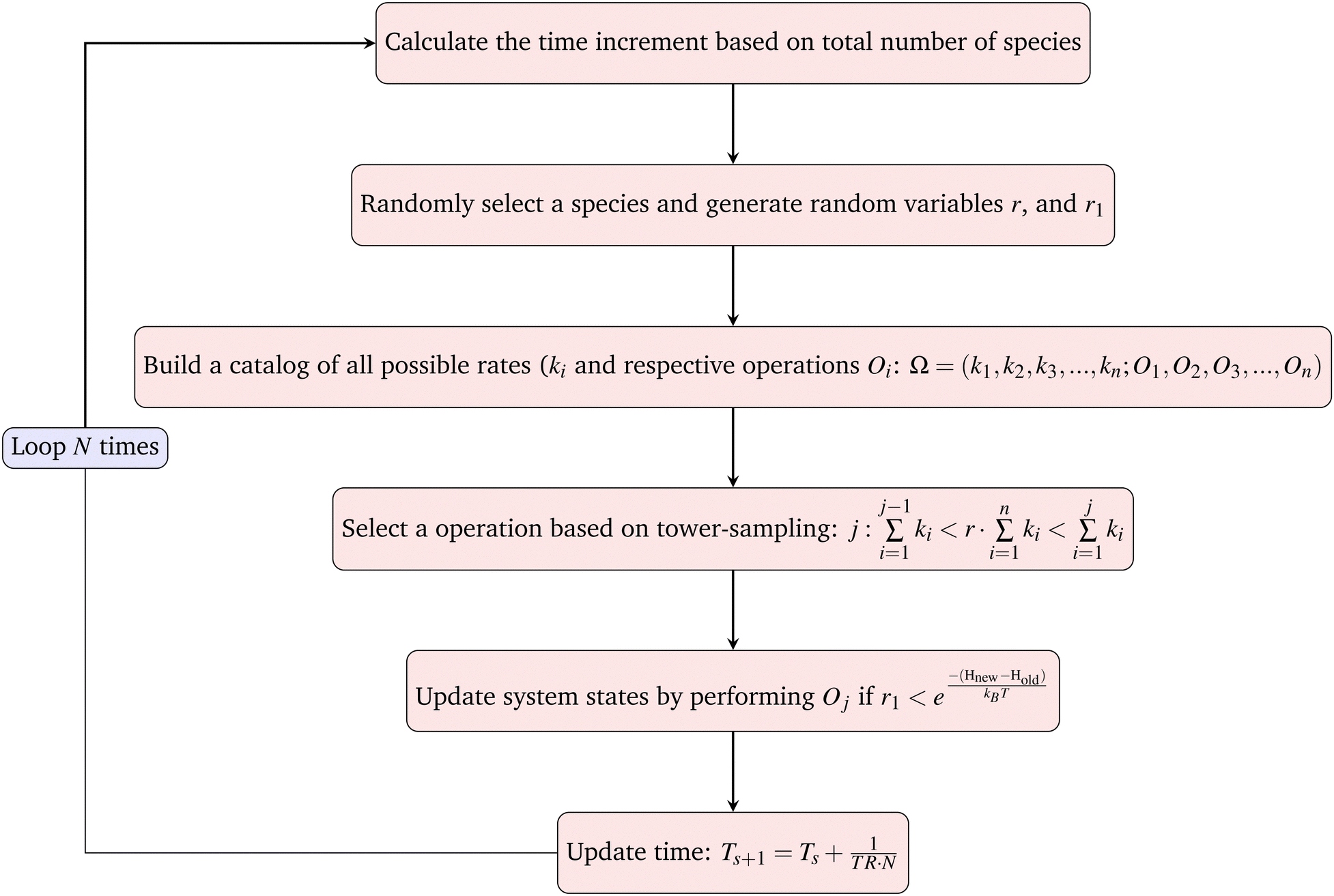
The goal of one Monte-Carlo sweep is to go through all the species in the system to update the system state. We first calculate the total rate of reactions and moves in the system TR (see below), then randomly select a species from the current system and build the catalog of all possible operations. For cells, the possible operations include division, matrix production and diffusion. And for matrix and agar, the only possible operation is diffusion. By using the ‘local update’ rule (see below), expressions for Hamiltonian and rates, one can easily calculate all the possible reaction rates ki and build the catalog. By randomly choosing an operation from the catalog based on tower-sampling we perform the corresponding operation Oj. A full Monte-Carlo sweep should traverse all the species in the system, thus for a single simulation loop the updated ΔT should be the inverse of TR and divided by the total number of all species N. We then have the increment of real physical time and can sum up all the increments and connect it to the physical time scale.
Rates of growth, secretion, and diffusion: our simulations rely on several rate constants: growth rate GR (inverse of cell doubling time), matrix secretion rate SR (rate at which matrix particles are produced), and diffusion rates DRc,m,n (the rate at which cells, matrix, and nutrient particles respectively hop to neighboring lattice sites).
We can now assemble all the rates to the total rate TR:
| TR = GR + SR + DRc + DRm + DRn |
We next define the three relative rate coefficients for cell division (α), secretion (β) and diffusion (γ):
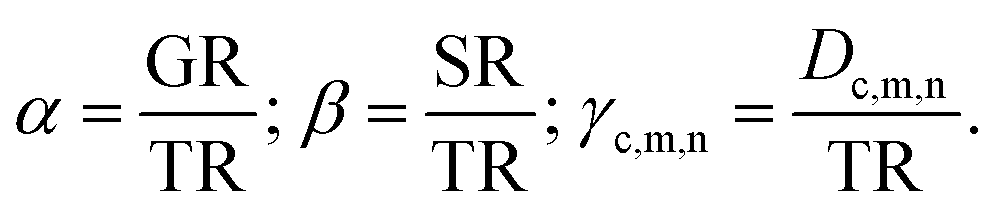
The respective experimental values for the rates will help us to define the values of α, β and γ (see below).
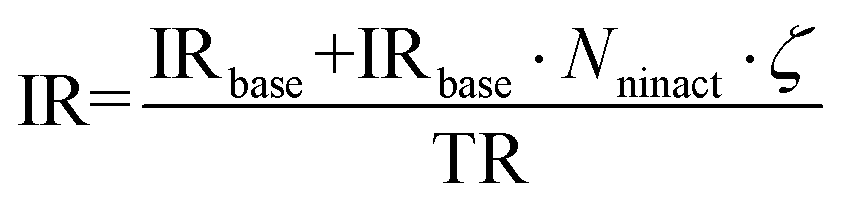
The parameter ζ encodes the positive feedback from inactive cells (i.e. rate of inactivation is higher if there are more inactive cells in the environment) mimicking, for example, a local accumulation of toxic compounds. Inactive cells do not move and do not produce new biomass. The number of surrounding inactive cells Nninact is defined as the total number of inactive cells in 9-sites neighborhood.
Local update. After each iteration, the system's energy is calculated. To improve computation efficiency, we only need to calculate the local updates. The only two lattices and their direct neighborhoods will be accounted for as there is only one particle that can be produced or moved in one Monte Carlo loop. We then only need to calculate the local energy differences to determine whether or not the particle will move.
Mapping of Monte Carlo steps to the physical time: for a particle of type p = c, m, n diffusing on a lattice, the diffusion constant can be written as72
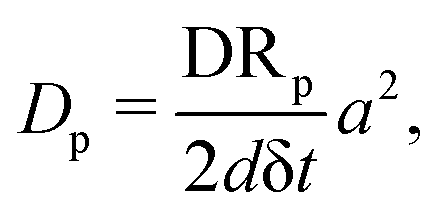
| Description | Value | Symbol |
|---|---|---|
| Lattice size | 100 × 100 | |
| Single lattice dimension | 0.23 mm | a |
| Doubling time of B. subtilis | 7200 s73,74 | |
| Diffusion coefficient of nutrients | 7.096 × 10−5 (mm2 s−1)75 | D A |
| Diffusion coefficient of cells | 5.322 × 10−7 (mm2 s−1)75 | D c |
| Diffusion coefficient of matrix | 3.326 × 10−6 (mm2 s−1)75 | D m |
| Phantom cells fraction | 0.1 or 0 | κ |
| Death rate of cells |
D
b = 6.05 × 10−6 s−176 |
D b |
| Initial layers of agar | 3 | A max |
| Layers of agar in the i’th lattice | — | A s(i) |
| The i’th species in simulation (cells or ECM) | — | σ i |
| Weight parameter of space competition | 0.1kBT | C w |
| Weight parameter of nutrient consumption | 1.5kBT (cells); 1kBT (ECM) | N w |
| Cell and matrix interaction | 0.03kBT | J w |
| Coupling constant for inactivation | 0.954 | ζ |
| Total particles | — | N |
In every Monte Carlo simulation loop, the updated physical time step is
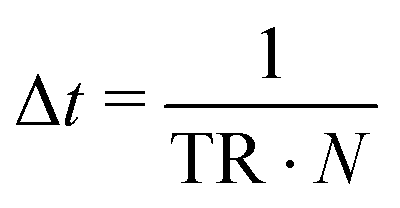
Here, N represents to total number of all particles of all species, including cells, matrix and nutrients. Thus, because the total number of lattice-species in our system is not conserved over time, the rules for physical time updates need to be adjusted as the number of particles changes.
Parameters of the biofilm model. We should stress that we set a goal for the model to achieve semi-quantitative agreement with the experimental data in order to draw biologically meaningful conclusions about the processes that might affect wound healing. The advantage of the coarse-grained lattice-model with kinetic Monte Carlo algorithm for particle moves is the possibility to link stochastic time in the model to real physical time. While the lattice model is one of the most efficient systems to simulate, yet at the level of the whole biofilm, calculations corresponding to several days of biofilm growth get computationally extensive. Thus, in this study, we did not aim to perform extensive scan of the parameter space to get the best fit of the experimental data.
Instead, we started with the parameter values known from the literature and then used our own experimental data on biofilm diameter as a function of time to define the final parameter values to be used in the model. We use the biofilm size (i.e. diameter of the colony measured at a given time) to set the size scale of the lattice, the diffusion constants of nutrients to set the scale for the rates of lattice species’ moves and adjust those to capture the growth dynamics as shown in Fig. 3C. The doubling time and death rate also inform our choice of the corresponding model parameters.
To illustrate how the model reacts to changes in the major parameters we are providing Fig. S6 (ESI†) where we altered biomass production, diffusion of biomass, cell inactivation rate and degree of phase separation of cells and matrix. Fig. S6A and B (ESI†) show the low/high production with GR, SR = 6.94 × 10−5 and GR, SR = 2.08 × 10−4 respectively. Fig. S6C and D (ESI†) show the low/high diffusion of biomass with Dc = 2.66 × 10−7 (mm2 s−1) and 7.98 × 10−7 (mm2 s−1), Dm = 1.66 × 10−6 (mm2 × s−1) and 4.99 × 10−6 (mm2 × s−1) respectively. Fig. S6E and F (ESI†) show the low/high inactivation rate with Db = 3.025 × 10−6 s−1 and 1.815 × 10−5 s−1 respectively. Finally, Fig. S6G and H (ESI†) show the low/high interaction with Jw = 0kBT and 0.015kBT respectively.
Choosing parameter values. We assume the average doubling time of around 2 h73,74 corresponding to the dimensionless growth rate: GR ≃ 1.33 × 10−4. Next, for simplicity we assume that the matrix secretion rate is the same as the growth rate GR = SR.
The diffusion coefficient for a stereotypical molecule in agar is 3 × 10−5 (mm2 s−1).75 While there are no directly measured values for cell or matrix diffusion within the biofilm we assume this process to happen roughly 10−3 to 10−4 times slower than nutrient diffusion, we take diffusion coefficient of cells Dc as 2.25 × 10−7 (mm2 s−1), and diffusion of matrix by an additional factor 6, 25 slower. Thus, the corresponding dimensionless rates of diffusion can be derived as DRc = 4 × 10−5, DRm = 2.5 × 10−4 and DRn = 5.33 × 10−3.
Numerical code availability. The numerical code is available by the following link: https://github.com/MikawaYys/Biofilm_simulation.
Author contributions
LC and VZ conceptualized the project, MG, SMi, and SA performed experiments, MG, SMü, LC analyzed data, YE developed numerical model and performed simulations, YE, VZ, and LC wrote the original draft, TM, SMü, LC, and VZ supervised the work, and all authors contributed to reviewing and editing the final manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Grants 504222949 (to YE, MG, VZ and LC), 504017689 (to SMi and TM) and 503905203 (to SA and SMü) in the framework of the Priority Program SPP2389 “Emergent functions of bacterial multicellularity”. MG acknowledges the support of the Neubauer Family Foundation for the PhD fellowship. We thank Kathrin Stückrath for her assistance with FACS data acquisition and analysis.Notes and references
- D. López, H. Vlamakis and R. Kolter, Cold Spring Harbor Perspect. Biol., 2010, 2, a000398 CrossRef PubMed.
- R. M. Donlan, Emerging Infect. Dis., 2002, 8, 881 CrossRef PubMed.
- H.-C. Flemming, J. Wingender, U. Szewzyk, P. Steinberg, S. A. Rice and S. Kjelleberg, Nat. Rev. Microbiol., 2016, 14, 563–575 CrossRef CAS PubMed.
- H.-C. Flemming, E. D. van Hullebusch, T. R. Neu, P. H. Nielsen, T. Seviour, P. Stoodley, J. Wingender and S. Wuertz, Nat. Rev. Microbiol., 2023, 21, 70–86 CrossRef CAS PubMed.
- S. S. Branda, Å. Vik, L. Friedman and R. Kolter, Trends Microbiol., 2005, 13, 20–26 CrossRef CAS PubMed.
- T. Larsen and N.-E. Fiehn, APMIS, 2017, 125, 376–384 CrossRef CAS PubMed.
- J. E. Button and R. J. Dutton, Curr. Biol., 2012, 22, R587–R589 CrossRef CAS PubMed.
- B. E. Ramey, M. Koutsoudis, S. B. von Bodman and C. Fuqua, Curr. Opin. Microbiol., 2004, 7, 602–609 CrossRef CAS PubMed.
- T. Danhorn and C. Fuqua, Annu. Rev. Microbiol., 2007, 61, 401–422 CrossRef CAS PubMed.
- M. J. Franklin and P. S. Stewart, Nat. Rev. Microbiol., 2008, 6, 199–210 CrossRef PubMed.
- C. M. Toutain, N. C. Caiazza and G. A. O'Toole, Microb. Biofilms, 2004, 43–63 CAS.
- K. Sauer, P. Stoodley, D. M. Goeres, L. Hall-Stoodley, M. Burmølle, P. S. Stewart and T. Bjarnsholt, Nat. Rev. Microbiol., 2022, 20, 608–620 CrossRef CAS PubMed.
- H. Vlamakis, C. Aguilar, R. Losick and R. Kolter, Genes Dev., 2008, 22, 945 CrossRef CAS PubMed.
- M. Simoes, M. O. Pereira and M. J. Vieira, Water Res., 2005, 39, 5142–5152 CrossRef CAS PubMed.
- H.-C. Flemming, J. Wingender, T. Griegbe and C. Mayer, Biofilms: recent advances in their study and control, Harwood Academic Publishers, Amsterdam, 2000, pp. 19–34 Search PubMed.
- H. Wolfmeier, D. Pletzer, S. C. Mansour and R. E. Hancock, ACS Infect. Dis., 2018, 4, 93–106 CrossRef CAS PubMed.
- M. G. Trulear and W. G. Characklis, J. - Water Pollut. Control Fed., 1982, 1288–1301 CAS.
- W. G. Characklis, M. Nevimons and B. Picologlou, Heat Transfer Eng., 1981, 3, 23–37 CrossRef CAS.
- Q. Wang and T. Zhang, Solid State Commun., 2010, 150, 1009–1022 CrossRef CAS.
- J. D. Bryers, Biotechnol. Bioeng., 2008, 100, 1–18 CrossRef CAS PubMed.
- P. S. Stewart and J. W. Costerton, Lancet, 2001, 358, 135–138 CrossRef CAS PubMed.
- T. Eisele and K. Gabby, Miner. Process. Extr. Metall. Rev., 2014, 35, 75–105 CrossRef CAS.
- E. J. Gardel, M. E. Nielsen, P. T. Grisdela Jr and P. R. Girguis, Environ. Sci. Technol., 2012, 46, 5222–5229 CrossRef CAS PubMed.
- B. E. Logan, Microbial fuel cells, John Wiley & Sons, 2008 Search PubMed.
- R. Singh, D. Paul and R. K. Jain, Trends Microbiol., 2006, 14, 389–397 CrossRef CAS PubMed.
- H. Vlamakis, Y. Chai, P. Beauregard, R. Losick and R. Kolter, Nat. Rev. Microbiol., 2013, 11, 157–168 CrossRef CAS PubMed.
- T. R. Garrett, M. Bhakoo and Z. Zhang, Prog. Nat. Sci., 2008, 18, 1049–1056 CrossRef CAS.
- G. C. Wong and G. A. O’Toole, MRS Bull., 2011, 36, 339–342 CrossRef PubMed.
- G. C. Wong, J. D. Antani, P. P. Lele, J. Chen, B. Nan, M. J. Kühn, A. Persat, J.-L. Bru, N. M. Høyland-Kroghsbo and A. Siryaporn, et al. , Phys. Biol., 2021, 18, 051501 CrossRef CAS PubMed.
- P. S. Stewart, J. Bacteriol., 2003, 185, 1485–1491 CrossRef CAS PubMed.
- K. Lemon, A. Earl, H. Vlamakis, C. Aguilar and R. Kolter, Bact. Biofilms, 2008, 1–16 CAS.
- H.-C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS PubMed.
- V. D. Gordon, M. Davis-Fields, K. Kovach and C. A. Rodesney, J. Phys. D: Appl. Phys., 2017, 50, 223002 CrossRef.
- A. Seminara, T. E. Angelini, J. N. Wilking, H. Vlamakis, S. Ebrahim, R. Kolter, D. A. Weitz and M. P. Brenner, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1116–1121 CrossRef CAS PubMed.
- D. Claessen, D. E. Rozen, O. P. Kuipers, L. Søgaard-Andersen and G. P. Van Wezel, Nat. Rev. Microbiol., 2014, 12, 115–124 CrossRef CAS PubMed.
- P. J. Piggot and D. W. Hilbert, Curr. Opin. Microbiol., 2004, 7, 579–586 CrossRef CAS PubMed.
- N. Allocati, M. Masulli, C. Di Ilio and V. De Laurenzi, Cell Death Dis., 2015, 6, e1609 CrossRef CAS PubMed.
- J. A. Shapiro, Annu. Rev. Microbiol., 1998, 52, 81–104 CrossRef CAS PubMed.
- I. George Broughton, J. E. Janis and C. E. Attinger, Plast. Reconstr. Surg., 2006, 117, 12S–34S CrossRef PubMed.
- A. Young and C.-E. McNaught, Surgery, 2011, 29, 475–479 Search PubMed.
- G. Y. Chen and G. Nuñez, Nat. Rev. Immunol., 2010, 10, 826–837 CrossRef CAS PubMed.
- A. Brugués, E. Anon, V. Conte, J. H. Veldhuis, M. Gupta, J. Colombelli, J. J. Muñoz, G. W. Brodland, B. Ladoux and X. Trepat, Nat. Phys., 2014, 10, 683–690 Search PubMed.
- V. Nier, M. Deforet, G. Duclos, H. G. Yevick, O. Cochet-Escartin, P. Marcq and P. Silberzan, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9546–9551 CrossRef CAS PubMed.
- L. G. Rodriguez, X. Wu and J.-L. Guan, Cell Migration: Developmental Methods and Protocols, 2005, pp. 23–29 Search PubMed.
- W. S. Krawczyk, J. Cell Biol., 1971, 49, 247–263 CrossRef CAS PubMed.
- V. Hakim and P. Silberzan, Rep. Prog. Phys., 2017, 80, 076601 CrossRef PubMed.
- G. S. Schultz and A. Wysocki, Wound Repair Regen., 2009, 17, 153–162 CrossRef PubMed.
- A. K. Tsirogianni, N. M. Moutsopoulos and H. M. Moutsopoulos, Injury, 2006, 37, S5–S12 CrossRef PubMed.
- X. Wang, F. Dong, J. Liu, Y. Tan, S. Hu and H. Zhao, Arch. Microbiol., 2021, 203, 5635–5645 CrossRef CAS PubMed.
- X. Wang, Y. Tan, J. Liu, S. Hu and H. Zhao, J. Mech. Med. Biol., 2020, 20, 2050048 CrossRef.
- F. Dong, S. Liu, D. Zhang, J. Zhang and X. Wang, Environ. Microbiol. Rep., 2022, 14, 822–827 CrossRef CAS PubMed.
- R. J. Allen and B. Waclaw, Rep. Prog. Phys., 2018, 82, 016601 CrossRef PubMed.
- M. A. Delavar and J. Wang, Sustainability, 2021, 13, 7968 CrossRef CAS.
- F. L. Hellweger, R. J. Clegg, J. R. Clark, C. M. Plugge and J.-U. Kreft, Nat. Rev. Microbiol., 2016, 14, 461–471 CrossRef CAS PubMed.
- J.-U. Kreft, C. M. Plugge, C. Prats, J. H. Leveau, W. Zhang and F. L. Hellweger, Front. Microbiol., 2017, 8, 2299 CrossRef PubMed.
- M. Mattei, L. Frunzo, B. D’acunto, Y. Pechaud, F. Pirozzi and G. Esposito, J. Math. Biol., 2018, 76, 945–1003 CrossRef CAS PubMed.
- J. Dong and S. Klumpp, Soft Matter, 2018, 14, 1908–1916 RSC.
- E. Alpkvista and I. Klapper, Bull. Math. Biol., 2007, 69, 765–789 CrossRef PubMed.
- D. Espeso, A. Carpio and B. Einarsson, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 91, 022710 CrossRef CAS PubMed.
- M. Andersen, C. Panosetti and K. Reuter, Front. Chem., 2019, 7, 202 CrossRef CAS PubMed.
- C. C. Battaile, Comput. Methods Appl. Mech. Eng., 2008, 197, 3386–3398 CrossRef.
- C. A. Miermans and C. P. Broedersz, Soft Matter, 2020, 16, 544–556 RSC.
- M. H. Flamm, Lattice kinetic Monte Carlo simulations of platelet aggregation and deposition, University of Pennsylvania, 2011 Search PubMed.
- P. Ghosh, J. Mondal, E. Ben-Jacob and H. Levine, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E2166–E2173 CrossRef CAS PubMed.
- M. G. Chevrette, C. S. Thomas, A. Hurley, N. Rosario-Meléndez, K. Sankaran, Y. Tu, A. Hall, S. Magesh and J. Handelsman, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2212930119 CrossRef CAS PubMed.
- E. Sansinenea and A. Ortiz, Biotechnol. Lett., 2011, 33, 1523–1538 CrossRef CAS PubMed.
- M. T. Corcuera, F. Gómez-Aguado, M. L. Gómez-Lus, C. Ramos, M. A. de la Parte, M. J. Alonso and J. Prieto, J. Microbiol. Methods, 2013, 94, 267–273 CrossRef CAS PubMed.
- T. Bhattacharjee and S. S. Datta, Nat. Commun., 2019, 10, 2075 CrossRef PubMed.
- R. J. Wright, R. J. Clegg, T. L. Coker and J.-U. Kreft, Msystems, 2020, 5, 10–1128 CrossRef PubMed.
- S. S. Branda, J. E. González-Pastor, S. Ben-Yehuda, R. Losick and R. Kolter, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11621–11626 CrossRef CAS PubMed.
- Z. Liu, N. Cichocki, T. Hübschmann, C. Süring, I. D. Ofiteru, W. T. Sloan, V. Grimm and S. Müller, Environ. Microbiol., 2019, 21, 164–181 CrossRef CAS PubMed.
- C. Merlet, A. C. Forse, J. M. Griffin, D. Frenkel and C. P. Grey, J. Chem. Phys., 2015, 142, 094701 CrossRef PubMed.
- H. Nguyen, A. Ybarra, H. Basagaoglu and O. Shindell, Sci. Rep., 2021, 11, 1–17 CrossRef PubMed.
- C. Deygout, A. Lesne, F. Campillo and A. Rapaport, Ecol. Modell., 2013, 250, 15–24 CrossRef.
- X. Zhang, X. Wang and Q. Sun, Procedia IUTAM, 2017, 23, 33–41 CrossRef CAS.
- P. Servais, G. Billen and J. V. Rego, Appl. Environ. Microbiol., 1985, 49, 1448–1454 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm01032e |
| ‡ However, biofilms can also form at liquid air interfaces and then referred to as pellicles31 |
| This journal is © The Royal Society of Chemistry 2024 |