
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Harnessing solid-state technology for next-generation iron–air batteries
Bingqian
Sun
 ab,
Hao
Wang
a and
Cheng
Peng
*a
ab,
Hao
Wang
a and
Cheng
Peng
*a
aKey Laboratory of Interfacial Physics and Technology, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201800, China. E-mail: pengcheng@sinap.ac.cn
bDepartment of Chemistry, School of Science, Shanghai University, Shanghai 200444, China
First published on 31st October 2024
Abstract
Iron–air batteries are increasingly recognized as a significant technological advancement for renewable energy due to their substantial potential for large-scale energy storage. This review summarizes the current status of iron–air battery technology, with a particular emphasis on the trend toward solid-state configurations. We categorize and analyze various types of iron–air batteries and their respective characteristics, followed by an exploration of how solid-state technology has facilitated technological advancements and theoretical innovations across these battery types. Furthermore, we engage in a comprehensive discussion on the challenges currently confronting solid-state iron–air batteries, along with proposed innovative solutions to address these obstacles. The objective of this review is to shed light on the development prospects of solid-state iron–air batteries and to suggest potential avenues for enhancing their overall performance.
1. Introduction
Renewable energy sources are crucial in facilitating the global energy transition and mitigating the impacts of climate change.1–10 These energy sources, including solar, wind, and hydropower, possess clean and low-carbon attributes that efficiently diminish dependence on fossil fuels and decrease greenhouse gas emissions, thereby safeguarding the environment and fostering sustainable development.11–17 However, the utilization of renewable energy sources encounters challenges such as intermittency and instability, necessitating the development of large-scale energy storage technologies to balance supply and demand and enhance the flexibility and stability of energy systems.18–22 Emerging as a promising energy storage technology, metal–air batteries offer higher theoretical energy density compared to traditional lithium-ion batteries, enabling longer storage durations, particularly suitable for large-scale grid storage and long-duration applications.23–28 Moreover, metal–air batteries exhibit high safety standards, being less prone to combustion or explosions, generating minimal waste during discharge, and reducing environmental impact.29–32 Therefore, metal–air batteries have been considered as an optimal solution for future large-scale energy storage technologies to address the intermittency issues associated with renewable energy sources.33–35Iron–air batteries (IABs), a longstanding presence in battery technology, exhibit considerable promise and future growth opportunities in the field of long-duration energy storage owing to their distinctive advantages.36 Iron–air batteries leverage the earth-abundant metal iron as the negative electrode material, offering both cost-effectiveness and environmental friendliness attributed to the widespread availability, accessibility, and recyclability.37 Furthermore, the iron negative electrode facilitates multi-electron transfer, allowing each iron atom to release multiple electrons during battery discharge, significantly boosting energy output.38 Moreover, the design of iron–air batteries allows them to produce only oxygen as the sole byproduct during discharge, which gives them an advantage in environmental friendliness and helps reduce carbon emissions and other environmental pollution during energy storage.39 These features position iron–air batteries as a promising alternative for future renewable energy storage solutions.
The history of iron–air batteries can be traced back to the mid-20th century, with the inception of the first iron–air battery by the National Aeronautics and Space Administration (NASA) in 1968 (Fig. 1).40 This type of iron–air battery was designed to operate at room temperature using an alkaline solution as the electrolyte, intended for traction and vehicle applications. Until 2010, in addition to the further development of room temperature aqueous iron–air batteries,41–46 Matsuda et al. also proposed room-temperature all-solid iron–air batteries.47 Both aqueous iron–air batteries and room-temperature all-solid iron–air batteries utilize an alkaline electrolyte, with hydroxide ions participating in the reactions of the positive and negative electrodes. In 2011, Huang et al. proposed a solid-oxide iron–air redox battery (SOIARB) working at high temperatures,48 expanding the operating temperature range of iron–air batteries, and then Inoishi et al. also carried out research on solid-oxide iron–air redox batteries.49 In 2013, Licht et al. built a molten-salt iron–air battery (MAB) that operates at high temperatures.50 In 2017, Trocino et al. proposed a high temperature ceramic iron–air battery.51 These high-temperature batteries are based on O2− chemistry. From the overview of their development history, room temperature iron–air batteries and high temperature iron–air batteries are gradually developing in the direction of solid state batteries, and they tend to have a minimalist sandwich structure. To date, MABs have evolved to incorporate dual-phase electrolyte structures and semi-solid configurations, while SOIARBs have effectively demonstrated their capability for long-duration energy storage (LDES).52–55
 | ||
| Fig. 1 The development of iron–air batteries. | ||
To date, various technical pathways, including aqueous, solid-oxide redox, and molten-salt iron–air batteries, have been systematically reviewed independently.56–59 Nevertheless, from the perspective of the development of the entire field of iron–air batteries, there is a lack of essential analysis and discussion regarding their solid-state characteristics. Considering the substantial influence of the solid-state characteristics of iron–air batteries on their development, design optimization, and performance enhancement, this paper seeks to rectify the current gaps in the analysis and discussion on these solid-state characteristics.
In this review, we provide a comprehensive overview of the development of various iron–air batteries since 2010, including an outline of their reaction mechanisms and a discussion of the key factors that impact the performance of these batteries. Through a comparative analysis of the performance of different iron–air batteries, we extensively discuss the advantages of the solid-state characteristics. Furthermore, we conduct an analysis of the challenges and strategies for the advancement of iron–air batteries in response to the solid-state characteristics, considering the electrolyte, electrode material, and the three-phase interface. Finally, we provide an outlook on the applicability of iron–air batteries with solid-state characteristics for large-scale energy storage.
2. Room-temperature iron–air batteries
2.1 Aqueous iron–air batteries
Room-temperature iron–air batteries are composed of an iron anode, an air cathode, and an aqueous electrolyte. The electrolyte of this battery is typically an alkaline solution, with excellent ionic conductivity and low corrosiveness towards the iron anode.56 Throughout the charge–discharge cycle, the iron anode undergoes an oxidation–reduction process from Fe to Fe(OH)2, while the air cathode undergoes an oxidation–reduction reaction between O2 and OH− (Fig. 2a).43,56,63–70 In an alkaline electrolyte environment, the theoretical open circuit voltage (OCV) is 1.28 V, which is lower than that of lithium–air and zinc–air batteries.71 It should be noted that the use of alkaline electrolyte is prone to trigger carbonate deposition on the air cathode side, which can deteriorate the overall battery performance.72 Using neutral or acidic electrolytes can prevent the deposition of carbonate on the air electrode side. However, the presence of a high concentration of hydrogen ions in neutral and acidic electrolytes results in hydrogen evolution and corrosion of the iron electrode. Manthiram et al. proposed a hybrid design for an iron–air battery that incorporates an alkaline electrolyte on the iron electrode side and an acidic electrolyte on the air electrode side, with both separated by a solid electrolyte membrane (such as a Li+ or Na+ solid electrolyte membrane).60 In this configuration, alkali metal cations serve as ionic mediators to sustain the redox reactions of the battery, resulting in an elevated operating voltage of the battery to 2.11 V. This approach harnesses the benefits of both electrolytes to optimize battery performance (Fig. 2b).60 | ||
| Fig. 2 (a) Charging and discharging process of iron–air batteries using alkaline electrolytes.56 (b) Schematic of the hybrid iron–air battery design.60 (c) Impact of the Na2S additive on the discharge curves of iron electrodes.61 (d) Potential–capacity profiles of iron electrodes with and without 3,6-dioxa-1,8-octanediol.62 (e) Comparison of the discharge capacity of the iron electrode with bismuth sulfide, standard iron electrode and commercial iron electrodes.43 | ||
The hydrogen evolution potential of the iron anode is −0.83 V (vs. the standard hydrogen electrode, SHE), slightly exceeding the reduction potential of Fe(OH)2 at −0.88 V, potentially initiating the hydrogen evolution reaction during the charging process.40,73,74 This not only diminishes the charging efficiency but also lowers the discharge voltage. Moreover, the formation of Fe(OH)2 during discharge can lead to the passivation of the iron anode, impeding the redox reaction kinetics and subsequently impairing the charge–discharge performance of the battery.75 Incorporating sulfides into alkaline aqueous electrolytes is a viable approach to improving the electrochemical performance of iron electrodes and inhibiting the hydrogen evolution reaction.63,76,77 The addition of Na2S or K2S has been found to inhibit the hydrogen evolution reaction, leading to improved charging efficiency.42,45,61,78,79 Tian et al. developed an aqueous iron–air battery that incorporates Na2S as an additive in the electrolyte.61 The introduction of Na2S effectively suppressed the hydrogen evolution reaction, thereby enhancing the overall performance of the battery. Specifically, the discharge capacity increased significantly to approximately 1.43 μA h cm−2, substantially exceeding the performance of the system without Na2S (Fig. 2c).61 This addition also promotes the formation of a conductive layer on the iron electrode, which enhances the utilization of active materials and contributes to the cycling stability of the battery. Moreover, organic sulfur compounds, including thiols, disulfides, thioethers, and aromatic thiols, contribute to the inhibition of the hydrogen evolution reaction by restricting the interaction of water molecules with the electrode surface.80,81 Narayanan et al. investigated the performance of an aqueous iron–air battery that incorporates 3,6-dioxa-1,8-octanediol as an additive in the electrolyte.62 The results indicate that the inclusion of 3,6-dioxa-1,8-octanediol significantly enhanced the discharge capacity of the battery, achieving a value of 0.2 A h g−1. This capacity is markedly superior to that of the battery configuration without the use of organic sulfide additives (Fig. 2d).62 Nevertheless, it is essential to consider the potential concern regarding the utilization of organic sulfur additives in alkaline aqueous electrolytes, as it may lead to catalyst poisoning in the air electrode. Incorporating metallic sulfides into the iron electrode presents an alternative strategy to avoid the potential side effects on the air electrode. The presence of Bi2S3 promotes the formation of a conductive FeS layer on the iron electrode surface, thereby improving the utilization of the iron electrode and reducing the insulating effect caused by passivation.82,83 Manohar et al. prepared an iron electrode with the addition of bismuth sulfide (Bi2S3).43 Compared to standard iron electrodes, the Bi2S3-enhanced iron electrode demonstrated superior performance, exhibiting a higher discharge rate and enhanced discharge capacity (Fig. 2e).43 Additionally, other metallic sulfides such as CuS, ZnS, and MoS2 can serve as additives in the iron electrode, contributing to the formation of a conductive layer on the iron electrode to enhance the performance of the iron electrode.43,84–86 In addition, a method such as in situ sulfide modification can reduce the outer layer of Fe2O3 to amorphous FeS, leading to a significant reduction in the ohmic overpotential and ion diffusion overpotential during Fe2O3 reduction.87,88
2.2 Room-temperature all-solid iron–air batteries
Metal–air batteries typically use alkaline aqueous solutions as electrolytes, i.e., sodium hydroxide and potassium hydroxide solutions. However, these solutions can compromise the safety and lifespan of batteries due to electrolyte leakage and freeze-induced volume change. To address these issues, alternative alkaline solid electrolytes, including layered double hydroxides and absorbent polymers, have been investigated to assemble all-solid metal–air batteries.47,89–93 In 2014, Matsuda et al. reported on a series of room-temperature all-solid iron–air batteries that utilized KOH-based solid composite materials as hydroxide ion conductors.47 The matrix materials used were ZrO2 and Mg–Al layered double hydroxides. During the discharge process, iron is oxidized to form Fe(OH)2 in the anode, followed by further oxidation to form Fe3O4.47,90 During the charging process, these reactions are reversed, and the metallic iron is regenerated. The preparation process of the iron–air battery utilizing KOH–ZrO2 composites as the solid electrolyte is illustrated in Fig. 3a.89 The resulting battery achieved an initial discharge capacity of 70 mA h per g-Fe, demonstrating superior capacity retention compared to room-temperature iron–air batteries employing aqueous electrolytes (Fig. 3b and c).89 Detailed parameters and performance metrics for both room-temperature aqueous iron–air batteries and room-temperature solid iron–air batteries are presented in Table 1. | ||
| Fig. 3 (a) Assembly steps of a room-temperature all-solid iron–air battery.89 (b) Charge–discharge curves of a room-temperature all-solid iron–air battery.89 (c) Comparison of the capacity retention between an aqueous iron–air battery and room-temperature all-solid iron–air battery.89 | ||
| Electrolyte materials | Anode materials | Cathode materials | Specific capacity (mA h g−1) | Charge efficiency (%) | Ref. |
|---|---|---|---|---|---|
| KOH–K2S | Fe2O3–C | Pt | 810 | — | 42 |
| KOH | Fe–Bi2S3 | NiO | 300 | 96 | 43 |
| KOH | Fe–Bi2O3–Fe2S3 | NiO | 200 | 92 | 46 |
| KOH | Fe–Bi2S3 | Pd–C | 814 | — | 71 |
| NaOH∥Na–Na3Zr2Si2PO12∥H3PO4/NaH2PO4 | Fe2O3/carbon nanofiber | IrO2–Ti//Pt–C | 1200 | — | 60 |
| KOH–ZrO2 | Fe3O4–C | Pt–C | 70 | — | 89 |
| KOH-1-ethyl-3-methylimidazolium L-(+)-lactate | Fe2O3 | Pt | 416 | — | 92 |
| KOH | Fe–Bi2S3 | Pt | 200 | 96.7 | 69 |
| KOH | Fe–ZnS | NiO | 240 | 95 | 84 |
| KOH | Fe2O3-multi-walled carbon nanotube | NiO | 350 | 82 | 82 |
| KOH–Na2S | Fe3O4-reduced graphene oxide | Pt–C | 420 | — | 94 |
| KOH-1-octanethiol | Fe2O3–Bi2S3 | NiO | 171 | — | 80 |
Despite the advantages offered by all-solid iron–air batteries, including enhanced safety, elimination of the risk of electrolyte leakage, and the potential for improved cycling stability and mechanical strength, there are challenges that need to be addressed. For instance, solid electrolytes exhibit lower ionic conductivity compared to liquid electrolytes, leading to increased interface resistance between solid electrolytes and electrode materials. Furthermore, the presence of alkaline compounds like KOH in the electrolyte composition of room-temperature all-solid iron–air batteries could result in the accumulation of carbonate deposits on the air electrode side. Moreover, the high water absorption characteristics of KOH necessitate the regulation of the relative humidity within the operational conditions. Future research should prioritize enhancing the ionic conductivity, stability, and environmental resilience of solid electrolytes, optimizing the interface between electrodes and electrolytes, and improving the electrochemical reaction kinetics of iron electrodes.
3. High-temperature iron–air batteries
Room-temperature iron–air batteries are constrained by aqueous electrolytes, leading to undesirable side reactions such as the hydrogen evolution and surface passivation on the iron anode side and carbonate deposition on the air electrode side.72,95,96 These side reactions significantly reduce the battery performance. In contrast, high-temperature iron–air batteries employ molten salts or solid oxides as electrolytes, effectively circumventing these issues. These high-temperature electrolytes exhibit excellent ionic conductivity and possess a wide electrochemical stability window, demonstrating high resistance to water and carbon dioxide in the air. Unlike the hydroxide ion conduction observed in aqueous electrolytes, these high-temperature electrolytes facilitate oxygen ion conduction, thereby exhibiting different reaction mechanisms. To date, three types of high-temperature iron–air batteries have been developed, including MABs,50 SOIARBs,48 and ceramic IABs.51 Their structure, reaction mechanism, and performance are comprehensively introduced, and the challenges of battery design and key materials encountered by each type of battery are discussed.3.1 Solid-oxide iron–air redox batteries
The solid-oxide iron–air redox battery is a device that utilizes fuel cell technology in rechargeable iron–air batteries.48,97–114 As shown in Fig. 4a,52 it comprises two primary elements: the solid-oxide electrochemical cell (SOEC) unit and the energy storage unit (ESU). The SOEC unit is composed of a solid electrolyte, a hydrogen anode, and an air cathode, facilitating the reversible hydrogen–oxygen reaction. The ESU unit consists of a mixture of Fe/FeOx powder, which entails the redox reaction of iron with steam. In contrast to conventional batteries, the charging and discharging mechanism of the SOIARB relies on the utilization of the H2–H2O redox pair as an oxygen shuttle system. This system circulates in a closed loop between the SOEC and ESU to facilitate energy storage and release. During the discharge process, the iron within the ESU is oxidized by steam, resulting in the production of hydrogen and iron oxide. Subsequently, the generated hydrogen enters the SOEC unit, where it undergoes electrochemical oxidation at the anode. This oxidation process generates electrical energy and steam. During the charging process, the SOEC unit facilitates the electrolysis of steam to generate hydrogen. Subsequently, the produced hydrogen facilitates the reduction of iron oxide within the ESU unit, leading to the restoration of iron and the formation of steam. | ||
| Fig. 4 (a) Schematic of the working principle of the SOIARB.52 (b) Charge and discharge characteristic of the SOIARB at 800 °C.48 (c) Performance of an intermediate-temperature SOIARB at 650 °C.115 (d) Schematic illustration of an intermediate-temperature SOIARB assembly using YSZ as the electrolyte, NiFe-91 as the anode, and BLC oxide as the cathode.116 (e) TEM images of fresh α-Fe2O3 nanoparticles derived from MOFs.117 (f) Schematic illustration of the reduction process in H2 over a proton-conducting BZC4YYb support and Ir catalyst.118 (g) TEM and X-ray images of Fe2O3 powder with 5 wt% Ce0.6Mn0.3Fe0.1O2.119 (h) Results of 200 automated galvanostatic charge–discharge cycles of a two-cell redox battery with tape-cast storage components based on Fe2O3 and 30 vol% 8YSZ.120 (i) Arrhenius plots showing the reaction rate constant for the oxidation of reduced Fe2O3 with and without the addition of Cr2O3 and PrBaMn2O5.121 | ||
The charging and discharging processes of the SOIARB are carried out by the SOEC unit, while energy storage and release take place within the ESU unit. The performance of the SOIARB predominantly relies on the redox reaction between Fe and FeOx within the ESU. In 2011, Xu et al. reported the first SOIARB based on yttria-stabilized zirconia (YSZ).48 This battery exhibited stable charge–discharge cycling over 20 cycles at 800 °C, delivering a discharge capacity of 348 W h per kg-Fe and an energy efficiency of 91.5% (Fig. 4b).48 Zhao et al. utilized the solid-oxide electrolyte La0.8Sr0.2Ga0.83Mg0.17O3−δ (LSGM) for the construction of an intermediate-temperature SOIARB.115 When operated at 650 °C and a current density of 50 mA cm−2, the battery exhibited stable charge–discharge cycling for 100 cycles. It achieved an average specific energy of 760 W h per kg-Fe and a round-trip efficiency of 55.6% (Fig. 4c).115 Sakai et al. employed YSZ as the solid-oxide electrolyte, a NiFe-91 bimetallic electrode as the anode, and Ba0.6La0.4CoO3−δ (BLC) oxide as the cathode for assembling an intermediate-temperature SOIARB (Fig. 4d).116 This battery exhibited the ability to endure around 100 hours (10 charge–discharge cycles) at 600 °C. It exhibited a capacity of approximately 700 mA h per g-Fe and an energy density of 600 mW h per g-Fe.
Temperature plays a crucial role in influencing the resistance at solid–solid interfaces within SOIARBs. High temperatures, while reducing interfacial resistance, also have the potential to expedite chemical reactions and diffusion processes, which can adversely affect the durability of the battery system over time. Therefore, the strategic choice of operating at intermediate temperatures emerges as a promising solution for SOIARBs. This temperature regime strikes a delicate equilibrium between ensuring high battery capacity through sufficient ionic and electronic conductivity and mitigating rapid material degradation caused by excessive thermal reactions. By adopting this approach, SOIARBs can achieve not only stable and efficient performance but also long-term reliability, highlighting the critical importance of temperature management in optimizing the functionality and longevity of these advanced energy storage systems.
However, the redox reaction of Fe/FeOx involves a gas–solid–solid three-phase interface process, and lowering the temperature would decelerate the kinetics. Enhancing the activity of the three-phase interface is a potential way to improve the kinetics of the redox reaction at low temperatures. Zhang et al. synthesized nano-iron materials derived from metal–organic frameworks (MOFs) with high-density iron atomic crystal planes (Fig. 4e).117 Such a unique structure significantly enhanced the reduction activity of Fe/FeOx, enabling the SOIARB to exhibit good rechargeability at 500 °C. The battery exhibited a discharge specific energy of 226.5 W h per kg-Fe (C/4) and 214.8 W h per kg-Fe (C/3). Incorporating catalytically active transition metals, such as iridium (Ir) and palladium (Pd), into the ESU unit can substantially enhance the reduction kinetics of Fe/FeOx.52,122 When operating at 500 °C, the incorporation of Pd into the SOIARB resulted in a discharge specific energy of 960.3 W h per kg-Fe with 80% Fe utilization.122 The addition of Ir significantly reduced the energy barrier of H2 overflow and Fe–O bond breaking and promoted the reduction kinetics of FeOx. The SOIARB showed multiple 12.5 h long cycles at 550 °C.52 When metal Ir and the proton conductor BaZr0.4Ce0.4Y0.1Yb0.1O3 are used together, the former enhances the reduction reaction kinetics, while the latter improves the diffusion of protons at the three-phase interface (Fig. 4f).118 Under the conditions of 500 °C, 50% iron utilization and 0.2C, the discharge energy density reaches 520 W h per kg-Fe, the round-trip efficiency is 61.8%, and the charge–discharge cycle time is 500 hours.118 Moreover, Kim et al. incorporated Ce0.6Mn0.3Fe0.1O2 powder in the ESU unit to construct a SOIARB operating at 400 °C (Fig. 4g).119 This implementation notably enhanced the oxidation rate of iron powder, resulting in a discharge potential of around 1 V and a discharge capacity of 600 mA h per g-Fe at 400 °C.
It is important to highlight that Fe/FeOx nanoparticles within the ESU unit are prone to thermal coarsening and sintering, leading to diminished reaction rates and a decrease in capacity. In order to tackle this issue, Berger et al. integrated YSZ powder into the redox materials (Fig. 4h).120 The ESU unit, consisting of 8YSZ and Fe2O3, exhibited outstanding performance during cyclic charge–discharge testing, completing more than 200 cycles with a current density of 150 mA cm−2 and a cycle duration of 70 minutes. Kim et al. incorporated 3 wt% Cr2O3 into the ESU unit, which effectively suppressed the growth of Fe particles by forming Fe–Cr oxides. This process ultimately improved the cyclic charge–discharge stability of the battery. Furthermore, the incorporation of Cr2O3 can synergistically enhance the cyclic charge–discharge stability and discharge capacity of the battery when combined with other catalysts. When Fe2O3 powder is mixed with 3 wt% Cr2O3 and 3 wt% PrBaMn2O5, the battery exhibited significant improvements in discharge capacity (exceeding 770 mA h per g-Fe) and cycle stability over 50 cycles at 350 °C (Fig. 4i).121 The detailed parameters and battery performance of SOIARBs are shown in Table 2.
| Temp. (°C) | Electrolyte materials | Redox materials | Anode materials | Cathode materials | Energy density (W h kg−1) | Energy efficiency (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 800 | YSZ | Fe2O3–ZrO2 | Ni–YSZ | La0.6Sr0.4Co0.2Fe0.8O3−δ–Ce0.8Sm0.2O2−δ | 348 | 91.5 | 48 |
| 600 | La0.9Sr0.1Ga0.8Mg0.2O3 | Fe2O3 | NiO–Fe2O3 | Sm0.5Sr0.5CoO3 | 752 | — | 49 |
| 550 | La0.8Sr0.2Ga0.83Mg0.17O3−δ | CeO2–Fe2O3–ZrO2 | Ce0.6La0.4O2−δ–Ni–Ce0.8Gd0.2O2−δ–Ni | La0.6Sr0.4Co0.2Fe0.8O3−δ | 1026 | 52.2 | 123 |
| 650 | La0.8Sr0.2Ga0.83Mg0.17O3−δ | Fe2O3–ZrO2 | Ni–CeO2 | Sm0.5Sr0.5CoO3–Sm0.2Ce0.8O1.9 | 760 | 55.5 | 115 |
| 550 | La0.8Sr0.2Ga0.83Mg0.17O3−δ | Fe2O3–C–ZrO2 | Ni–CeO2 | Sm0.5Sr0.5CoO3–Sm0.2Ce0.8O1.9 | 1188 | 76.3 | 124 |
| 550 | La0.8Sr0.2Ga0.83Mg0.17O3−δ | Fe–C–ZrO2 | Ni–CeO2 | Sm0.5Sr0.5CoO3–Sm0.2Ce0.8O1.9 | 1056 | 59.8 | 124 |
| 800 | 8YSZ | Fe2O3-30%YSZ | Ni-8YSZ | La0.6Sr0.4Co0.2Fe0.8O3−δ | 900 | — | 120 |
| 400 | La0.9Sr0.1Ga0.8Mg0.2O3 | Ce0.6Mn0.3Fe0.1O2–Fe | NiO–Fe2O3 | Ba0.6La0.4CoO3 | 420 | 85 | 125 |
| 400 | La0.9Sr0.1Ga0.8Mg0.2O3 | Ce0.6Mn0.3Fe0.1O2–Fe | NiO–Fe2O3 | Ba0.6La0.4CoO3 | 600 | — | 119 |
| 600 | 8YSZ | Fe3O4 | NiO–Fe2O3 | Ba0.6La0.4CoO3 | 600 | — | 116 |
| 500 | (ZrO2)0.89(Sc2O3)0.1(CeO2)0.01 | Pd– Fe2O3–ZrO2 | Ni–(ZrO2)0.89(Sc2O3)0.1(CeO2)0.01 | La0.8Sr0.2Ga0.83Mg0.17O3–(Bi0.75Y0.25)0.93Ce0.07O1.5 | 960.3 | 65.8 | 122 |
| 500 | (ZrO2)0.89(Sc2O3)0.1(CeO2)0.01 | Pd– Fe2O3–ZrO2 | Ni–(ZrO2)0.89(Sc2O3)0.1(CeO2)0.01 | La0.8Sr0.2Ga0.83Mg0.17O3–(Bi0.75Y0.25)0.93Ce0.07O1.5 | 606.4 | 62.9 | 122 |
| 350 | La0.9Sr0.1Ga0.8Mg0.2O3 | Fe2O3–Cr2O3–PrBaMn2O5 | NiO–Fe2O3 | Ba0.6La0.4CoO3 | 550 | 50 | 121 |
| 500 | ZrO2-based | MOF-Fe2O3 | Ni-based | — | 226.5 | 65.4 | 117 |
| 500 | ZrO2-based | MOF-Fe2O3 | Ni-based | — | 214.8 | 56.4 | 117 |
| 500 | ZrO2-based | MOF-Fe2O3 | Ni-based | — | 548.8 | 58.1 | 117 |
| 550 | (Sc2O3)0.1(CeO2)0.01(ZrO2)0.89 | Fe2O3–BaZr0.4Ce0.4Y0.1Yb0.1O3–IrO2 | NiO–(Sc2O3)0.1(CeO2)0.01(ZrO2)0.89 | La0.8Sr0.2MnO3–(Bi0.75Y0.25)0.93Ce0.07O1.5 | 601.9 | 82.9 | 118 |
| 500 | (Sc2O3)0.1(CeO2)0.01(ZrO2)0.89 | Fe2O3–BaZr0.4Ce0.4Y0.1Yb0.1O3–IrO2 | NiO–(Sc2O3)0.1(CeO2)0.01(ZrO2)0.89 | La0.8Sr0.2MnO3–(Bi0.75Y0.25)0.93Ce0.07O1.5 | 520 | 61.8 | 118 |
| 550 | (Sc2O3)0.1(CeO2)0.01(ZrO2)0.89 | Fe2O3–IrO2 | NiO–(Sc2O3)0.1(CeO2)0.01(ZrO2)0.89 | La0.8Sr0.2MnO3–(Bi0.75Y0.25)0.93Ce0.07O1.5 | 625 | 87 | 52 |
| 500 | La0.9Sr0.1Ga0.8Mg0.2O3 | Fe2O3 | NiO–Fe2O3–Ce0.6Mn0.3Fe0.1O2 | Ba0.6La0.4CoO3 | 450 | 82 | 126 |
Multiphysics field simulation is essential for the comprehensive investigation of SOIARBs.127–134 These simulations are instrumental in attaining a profound comprehension of the complex phenomena transpiring within the battery, thereby providing insights that are invaluable to the processes of design and optimization. Guo et al. presented a two-dimensional multiphysics model based on tubular SOIARBs, integrating mass and momentum transfer to effectively simulate the discharge behavior under conditions of elevated current density.127 The results exhibit a robust correlation with empirical data. Despite the substantial contributions of this pioneering effort to the field, the model does not encompass all relevant physical processes within the battery. Constructing upon this foundational research, Jin et al. refined a more nuanced two-dimensional axisymmetric multiphysics model. This model not only incorporates current density, mass transfer, and thermal equilibrium but also places a significant emphasis on the redox kinetics within ESUs.128,130,132
A significant achievement of multiphysics field simulation is the elucidation that the reduction reaction of Fe3O4 during the charging process serves as the rate-controlling step for the entire battery.135 This finding was derived from a comprehensive analysis of a typical SOIARB configuration including a reversible solid oxide fuel cell (RSOFC) and a metal–metal oxide redox cycle unit (RCU), as shown in Fig. 5a, and a finite element method (FEM) model that details the actual dimensions and configuration of the RSOFC and RCU (Fig. 5b).131 This FEM model was validated by an experimental V–I curve of the RSOFC (Fig. 5c) and an experimental voltage vs. Depth of Discharge (DoD) curve of the RCU (Fig. 5d).131 Furthermore, the model developed by Jin et al. accounts for the impact of temperature on battery performance, thereby enhancing the simulation's precision through the integration of thermodynamic and chemical kinetic coupling effects. In addition, Jin et al. have conducted an exploration into strategies aimed at augmenting battery performance. This was achieved by optimizing the geometric structure and operating parameters, including enhancements in charging and discharging efficiencies through fine-tuning of the battery's thickness, electrode porosity, and electrolyte formulation. The development of these strategies is based on an exhaustive analysis of data derived from multiphysics field simulations. This integrated simulation approach, which considers multiple physical fields in tandem, enables a more comprehensive understanding of the performance variations of batteries under diverse operating conditions. Consequently, it offers substantial theoretical support for material selection, structural design, and the engineering optimization of battery systems.
 | ||
| Fig. 5 (a) Schematic of the SOIARB configuration.131 (b) The finite element method model of the SOIARB (unit: mm).131 (c) Validation of the V–I curve at 550 °C.131 (d) Validation of the E vs. Fe-utilization curve.131 | ||
Nowadays, Siemens has developed energy storage systems based on SOIARB technology, demonstrating its potential for practical applications.136 The key structural components of a typical SOIARB energy storage system include battery stacking, a thermal management system, and a gas supply and management system (Fig. 6a).136 The collaborative functioning of these components ensures that the battery system operates stably at high current densities while maintaining efficient thermal cycling and gas exchange. For instance, the SOIARB stack developed has demonstrated a power output capability exceeding 1 kW in laboratory tests and has achieved multiple kilowatt-level demonstrations (Fig. 6b).136 Furthermore, it has exhibited remarkably low degradation rates of only 1–2% per 1000 charge and discharge cycles in a small-scale stack measuring 10 cm × 10 cm (Fig. 6c).136
 | ||
| Fig. 6 (a) Short stack arrangement of the SOIARB energy storage system.136 (b) Laboratory tests of the 1 kW SOIARB stack.136 (c) Storage capacity degradation rates of the SOIARB system.136 | ||
Undoubtedly, the development of SOIARBs has not only propelled their commercial viability and scalability, but also provided valuable insights and benchmarks for the development of other high-temperature iron–air battery technologies. Advancements in material selection, battery configuration, system integration, and thermal management have set a precedent for the progression of subsequent technologies. With ongoing research dedicated to enhancing SOIARB performance and minimizing costs, these batteries are anticipated to play a significant role in areas such as smart grids, renewable energy storage, and backup power systems, thereby demonstrating promising engineering application prospects.
3.2 Molten-salt iron–air batteries
MABs have garnered significant attention due to their simplified design and cost-effectiveness.50,137–143 The battery is composed of an air cathode, a molten salt electrolyte, and an anode current collector (Fig. 7a).50 The most commonly used electrolyte is molten carbonates, which exhibit effective oxygen ion conductivity and facilitate the dissolution of the active material FeO2−. The air cathode primarily consists of nickel metal, which is able to form a three-phase interface to facilitate oxygen evolution and reduction reactions when immersed in molten electrolyte.145–147 During the charging process, FeO2− is reduced to metallic iron on the surface of the anode current collector, releasing oxygen ions (O2−) that subsequently participate in the oxygen evolution reaction at the air cathode. Conversely, during the discharging process, oxygen on the air cathode is reduced to O2−, leading to the oxidation of metallic iron on the anode current collector to produce FeO2−, which then dissolves in the molten carbonate electrolyte. | ||
| Fig. 7 (a) Illustration of the MAB during charge/discharge in molten carbonate.50 (b) Cycle performance for the MAB at 500 °C.144 (c) Coulombic efficiency of the MAB over 450 cycles.144 (d) Schematic of the triple-phase interface and oxygen reduction reaction (ORR) characteristics on the fin type air electrode.144 (e) Schematic of a dual-phase MAB in the discharging mode.53 (f) Scanning electron microscopy (SEM) image of the interface between the molten salt and YSZ in a dual-electrolyte MAB.53 (g) The charge–discharge curves of a quasi-solid-state MAB in the 10th (red), 20th (blue), 30th (green), 40th (violet), 50th (gray), 60th (brown), and 70th (orange) cycles, respectively.54 (h) Long-term cycling performance of the quasi-solid-state MAB at 800 °C for 70 cycles.54 (i) Illustration of the negative electrode in the quasi-solid-state MAB.54 | ||
The precise control of the chemical composition of the molten salt electrolyte can significantly influence the operating temperature of MABs. For instance, Cui et al. initially developed a lithium carbonate-based MAB with an operating temperature of 730 °C.50 Subsequently, by employing a ternary eutectic carbonate electrolyte Li0.87Na0.63K0.50CO3, the operating temperature of the MAB was reduced to 600 °C.138 Further advancements were made by utilizing a quaternary eutectic molten-salt electrolyte (KCl–LiCl–LiOH–NaOH) with a melting point of 283 °C, resulting in a successful reduction of the operating temperature to 500 °C.148 However, it is important to note that this electrolyte system, based on the KCl–LiCl eutectic, has limitations in dissolving oxygen ions and exhibits strong corrosiveness to the air electrode. To address these challenges, the binary eutectic chloride KCl–LiCl was substituted by a ternary eutectic carbonate Li0.87Na0.63K0.50CO3, resulting in the formation of a quinary eutectic molten-salt electrolyte Li0.87Na0.63K0.50CO3–LiOH–NaOH with a melting point of 340 °C (Fig. 7b).144 This pentagonal eutectic molten-salt electrolyte enabled the MAB to exhibit remarkable cycling stability with a ∼100% coulombic efficiency over 450 cycles (Fig. 7c).144
The design of the air electrode is a crucial factor in enhancing the battery performance as it directly impacts the OER and ORR kinetics, which in turn determines the charging efficiency and cycling stability of the battery.145,146 Cui et al. improved the geometric structure of the nickel air electrode (Fig. 7d),144 thereby increasing the active surface area and reducing electrode polarization. This improvement significantly enhanced the coulombic efficiency, rate performance, and cycling stability of the battery. Moreover, they developed dual-functional catalysts, such as PdO, amorphous MnO2/lithiated NiO nanosheets, and NiMnO3–La2O3 composite materials, which further improved the OER and ORR performance of the air electrode, leading to a significant enhancement in the overall performance of the battery.141,147,149
In high-temperature molten salt iron–air batteries, the air cathode is in direct contact with the molten salt electrolyte. However, the highly corrosive nature of high-temperature molten salts when exposed to air severely restricts the selection of materials for the air cathode. Peng et al. developed a dual-phase electrolyte structured MAB that incorporates a solid-oxide electrolyte layer to isolate the air cathode from the high-temperature molten-salt electrolyte (Fig. 7e).53 This design effectively prevents molten-salt corrosion of the air cathode (Fig. 7f).53 Importantly, it enables the utilization of cost-effective and high-performance air cathode materials, such as perovskite-structured La0.8Sr0.2MnO3 (LSM), in MABs. Moreover, the incorporation of a solid electrolyte layer establishes a gas–solid–solid three-phase interface. This interface, as opposed to the gas–liquid–solid three-phase interface formed in conventional MABs, offers advantages in terms of control, optimization, and expansion of the active reaction area.
Based on the dual-phase electrolyte architecture, Zhang et al. developed a quasi-solid-state MAB by mixing a molten-salt electrolyte with solid-oxide powder.54 The adsorption of solid-oxide powder thermodynamically inhibits the fluidity and volatility of the molten salt electrolyte, thereby significantly enhancing the cycling stability of the battery (Fig. 7g and h).54 Additionally, the molten lithium carbonate (Li2CO3) was substituted with the K2CO3–Na2CO3 eutectic,54 leading to a significant reduction in material expenses. On the iron anode side, a reversible solid-state electrochemical redox reaction takes place as a result of the limited solubility of FeO2− in the molten eutectic of K2CO3–Na2CO3. This reaction entails a three-phase interface mechanism involving solid–solid–solid interactions, which fundamentally differs from the dissolution–deposition mechanism typically observed in lithium carbonate-based melts (Fig. 7i).54 The composition of the molten salt electrolyte and the battery performance of MABs are provided in Table 3.
| Temp. (°C) | Electrolyte materials | Electrolyte melting point (°C) | Coulombic efficiency (%) | Voltage efficiency (%) | Ref. |
|---|---|---|---|---|---|
| 730 | Li2CO3–Fe2O3–Li2O | 723 | 75 | 84 | 50 |
| 600 | Li0.87Na0.63K0.50CO3–Fe2O3–Li2O | 393 | 92 | — | 138 |
| 500 | KCl–LiCl–LiOH–NaOH–Fe2O3 | 283 | 99.10 | — | 148 |
| 500 | Li0.87Na0.63K0.50CO3–LiOH–NaOH–Fe2O3 | 340 | 95.10 | 59.9 | 144 |
| 500 | KCl–Li0.87Na0.63K0.50CO3–LiOH–NaOH–Fe2O3 | 322 | 88.60 | 76.20 | 145 |
| 500 | KCl–Li2SO4–LiOH–NaOH–Fe2O3 | 321 | 79.90 | 73 | 145 |
3.3 High-temperature solid-state ceramic iron–air batteries
In 2017, Trocino et al. proposed solid-state ceramic iron–air batteries operating at high temperatures (500–800 °C).51 This battery employs Gd0.1Ce0.9O2(CGO) or LSGM as the solid-oxide electrolyte layer, with the anode layer consisting of an Fe/Fe2O3-CGO composite material and the cathode layer composed of La0.6Sr0.4Fe0.8Co0.2O3, forming an extremely simplified sandwich structure (Fig. 8a).150 During the charging process, iron oxide is reduced in the anode layer, resulting in the production of iron. Concurrently, the O2− ions migrate across the solid-oxide electrolyte layer towards the cathode layer, where they are released as oxygen. During the discharging process, the cathode layer undergoes an oxygen reduction reaction to generate O2− ions. Subsequently, these O2− ions traverse the solid-oxide electrolyte layer and oxidize the iron in the anode layer. Specifically, the LSGM-based ceramic IAB exhibited a stable cycling performance over 100 cycles at 40% iron utilization, with an average discharge capacity of 508 mA h per g-Fe (Fig. 8b) and an average energy efficiency of approximately 53.7% (Fig. 8c).150 | ||
| Fig. 8 (a) Configuration and SEM images of the interfaces in the ceramic iron–air battery.150 (b) Specific capacity and specific energy of the ceramic IAB at 650 °C.150 (c) Faradaic efficiency and voltage efficiency of the ceramic IAB versus the number of cycles at 650 °C.150 | ||
The absence of high-temperature fluids, such as H2/steam and molten salts, in ceramic IABs contributes to a more compact configuration, facilitating increased energy and power densities. Furthermore, the all-solid-state design mitigates concerns associated with sealing and corrosion, thereby simplifying the battery manufacturing procedure. However, the ceramic IAB features solid-state electrochemical redox reactions at the anode, characterized by low reactivity and sluggish charge transfer kinetics. Currently, ceramic IABs underperform in critical performance metrics such as iron utilization, discharge capacity, and cyclic charge–discharge stability compared to MABs and SOIARBs. Despite these challenges, ceramic IABs are regarded as a promising energy storage technology due to their all-solid-state nature and potential for high safety. Table 4 presents the detailed battery performance of ceramic IABs under various operating conditions.
3.4 Comparison between room-temperature and high-temperature iron–air batteries
Compared with the normal temperature battery, the reaction mechanism of the battery has changed fundamentally, from a normal temperature hydroxide ion chemical reaction to a high temperature oxygen ion chemical reaction. The utilization of high temperatures has been shown to improve the kinetics of electrochemical reactions by increasing operating temperatures. This enhancement results in a notable improvement in the charge–discharge rates of the battery, the reversibility of reactions, and the overall efficiency. Consequently, it strengthens both the charging effectiveness and cycling stability of the battery. Moreover, the elevated operating temperatures expand the range of materials suitable for both electrodes and electrolytes, offering greater flexibility in the structural configuration of iron–air batteries and significantly improving their overall performance.Moreover, high temperatures offer significant advantages for the thermal regulation and safety of iron–air batteries. The heat produced by the battery during charging and discharging cycles is harnessed to sustain the required high-temperature conditions, reducing the need for external heat inputs and improving the energy self-reliance of the system. The intrinsic self-heating characteristic not only reduces energy dissipation but also contributes to operational effectiveness. Furthermore, elevated temperature conditions establish comparatively isolated operational settings for batteries, thereby reducing the possible adverse effects of external environmental variations on battery performance. Moreover, increased temperatures have the ability to mitigate harmful side reactions taking place in room-temperature iron–air batteries such as the hydrogen evolution reaction, consequently decreasing safety risks. Concerning safety protocols, the design of high-temperature iron–air batteries typically incorporates mechanisms to avert thermal runaway. Operating batteries at elevated temperatures enables prompt implementation of cooling measures upon detecting abnormal conditions, such as excessively high temperatures, effectively regulating and forestalling thermal runaway occurrences. This proactive design element enhances the safety integrity of the battery system.
4. Solid-state advantages
The solid-state approach improves battery safety and stability by replacing liquid electrolytes with solid alternatives.11 This substitution helps reduce the risks of corrosion and leakage while also simplifying battery design and manufacturing processes. The solid-state approach primarily involves two fundamental elements: solid-state electrolytes and solid-state electrode reactions (Fig. 9a). Solid-state electrolytes facilitate the advancement of iron–air batteries towards all-solid-state configurations. In contrast to conventional liquid electrolytes, solid electrolytes like YSZ demonstrate significant chemical stability and non-volatility at high temperatures. This characteristic enhances battery safety, structural durability, and thermal stability. Liquid electrolytes, including aqueous solutions and high-temperature molten salts, present challenges related to battery failure and performance decline because of their volatility and fluidic nature.151 In dual-phase structured molten-salt iron–air batteries, the incorporation of solid electrolytes presents a feasible approach to addressing the issue of molten salt corrosion on air electrode materials.53 In quasi-solid-state molten-salt iron–air batteries, the combination of solid electrolytes with molten salt electrolytes leads to a notable decrease in volatility and fluidity.54 This, in turn, improves battery stability by mitigating the risks typically associated with liquid electrolytes. Moreover, the adoption of solid electrolytes simplifies the design of battery structures, consequently improving both energy density and power density. | ||
| Fig. 9 (a) Illustration of the advantages conferred by solid electrolyte and solid-state electrode reactions in iron–air batteries. (b) Solid-state battery challenges and potential solutions. | ||
Solid-state electrode reactions involve alterations in electrode materials and reaction mechanisms in pursuit of improving the overall battery performance. In iron–air batteries, the electrochemical behavior of the iron electrode is characterized by a complex redox process. Solidification is attained by establishing a stable three-phase interface that includes gas, electrolyte, and iron electrode materials. Efficient electron and ion transport at the three-phase interface, in conjunction with chemical reactions involving active substances, is crucial for optimizing electrode reaction kinetics. In aqueous iron–air batteries and semi-solid molten salt batteries, a three-phase interface reaction involving liquid–solid–solid phases takes place on the ferroelectric electrode side. This reaction leads to the electrochemical reduction of iron oxides to metallic iron. In molten–air batteries, the reaction occurring on the iron electrode side follows a typical dissolution–deposition process. This process involves the dissolution of metallic iron in the molten salt after oxidation. In SOIARBs, the three-phase interface reaction involving gas, solid, and solid phases takes place on the anode side. The participation of gas accelerates the chemical reaction between iron and iron oxides. In all-solid-state iron–air batteries operating at both room temperature and high temperature, a three-phase interface reaction involving solid–solid–solid interfaces takes place on the ferroelectric electrode side. The stability, ease of design and control, and resistance to electrolyte volatilization and corrosion provided by the solid–solid–solid three-phase interface reaction present diverse opportunities for the categories and configurations of iron–air batteries. Moreover, solid-state electrode reactions expand the range of electrode materials that can be utilized, especially those that are susceptible to instability in liquid electrolytes. Solid-state electrode reactions represent an essential technological pathway for enhancing design versatility and maximizing the overall performance in iron–air batteries.
5. Solid-state challenges and potential solutions
The application of solid-state integration technology has been promoted by the research of solid-state electrolyte materials. Although solid electrolytes help improve the safety of batteries, the ionic conductivity of electrolytes and their interface compatibility with electrode materials seriously affect the charging and discharging performance and long-term stability of batteries. We will comprehensively analyze the challenges brought about by solid-state batteries from the perspective of key materials of iron–air batteries, including electrolytes, iron electrodes, and their interfaces (Fig. 9b).5.1 Electrolyte
The electrolyte materials utilized in room-temperature iron–air batteries typically consist of alkaline aqueous solution electrolytes and solid electrolytes based on potassium hydroxide or layered double hydroxides (LDHs). Alkaline aqueous electrolytes are preferred due to their high ionic conductivity and low corrosive nature. Nevertheless, obstacles like carbonate deposition and hydrogen evolution corrosion impede the overall performance and safety of batteries. Solid electrolytes based on potassium hydroxide or LDHs have been developed to improve battery safety and enhance cycling stability.47,89–91 However, these materials encounter challenges such as low ionic conductivity, high interface resistance, intricate preparation procedures, and increased costs. Despite their advantageous characteristics, solid electrolytes face challenges related to carbonate deposition and long-term cycling stability. Future research should prioritize improving the ionic conductivity, stability, and environmental durability of solid electrolytes. Additionally, optimizing the interface between electrodes and electrolytes is crucial for comprehensively boosting the performance of iron–air batteries.Polymer electrolyte membranes have emerged as a promising electrolyte material for room-temperature iron–air batteries. These membranes exhibit selective ion conduction and have been investigated in alternative battery systems, such as zinc–air batteries.152–156 Polymer electrolyte membranes are distinguished by their remarkable mechanical processing capabilities, enabling them to be co-processed with electrode materials to form membrane electrode assemblies (MEAs). This process simplifies the battery structure. Moreover, these membranes exhibit outstanding physical and chemical stability, allowing them to function effectively over a broad temperature range from room temperature to 300 °C. This characteristic extends the operating temperature range of iron–air batteries. The utilization of polymer electrolyte membranes could result in elevated material expenses and require meticulous control of environmental humidity, given their high humidity prerequisites during operation. Hence, it is imperative for battery system design to take into account humidity control in order to mitigate performance deterioration.
High-temperature iron–air batteries employ molten salts and solid oxide electrolytes to facilitate the conduction of oxygen ions, showcasing excellent ionic conductivity under high-temperature conditions.48–50,157 Nevertheless, the corrosive nature of molten salt electrolytes towards electrode materials presents a notable challenge, particularly in air environments where corrosion is more pronounced.158–160 This accelerates the deterioration of electrode materials, thereby affecting battery performance and lifespan significantly. The corrosiveness of the molten salt electrolyte limits the selection of electrode materials suitable for battery applications, as only a few highly corrosion-resistant materials remain stable under such conditions. Moreover, the molten-salt corrosion on current collectors and sealing materials would reduce the mechanical strength of battery components, thereby resulting in battery failure. While employing a dual-phase electrolyte structure can avoid direct exposure of the molten salt electrolyte to the air cathode, the corrosive nature of the molten salt affecting the solid-oxide electrolyte can still result in the deterioration of the performance of the solid-oxide electrolyte.53
Solid electrolytes such as YSZ and LSGM have been effectively utilized in high-temperature iron–air batteries (including MABs, SOIARBs, and ceramic IABs) due to their exceptional oxygen ion conductivity. However, these electrolytes encounter various challenges in practical implementation. The ionic conductivity of solid-oxide electrolytes generally lags behind that of liquid electrolytes, which limits the current density and overall performance of iron–air batteries. YSZ electrolytes are prevalent in solid-oxide fuel cells because of their exceptional high-temperature stability and high ionic conductivity (Fig. 10).161 At a temperature of 1000 °C, the ionic conductivity of YSZ electrolytes can reach 0.1 S cm−1; however, it diminishes as the temperature decreases. In accordance with Ohm's law, the resistance of ions is directly correlated with the thickness of the electrolyte layer. Therefore, decreasing the thickness of the layer effectively reduces the resistance. Studies have shown that decreasing the thickness of the YSZ electrolyte layer to 300 nm leads to a notable improvement in ionic conductivity, even at reduced temperatures such as 350 °C.162
 | ||
| Fig. 10 Ionic conductivities of solid-oxide electrolytes.161 | ||
Moreover, the introduction of scandium (Sc) into zirconia can result in the production of solid-oxide electrolyte materials characterized by enhanced conductivity. The addition of scandium increases the concentration of oxygen vacancies in YSZ electrolytes, thereby enhancing the ionic conductivity. Sc-stabilized zirconia (ScSZ) demonstrates not only improved ionic conductivity but also provides superior mechanical strength and thermal stability in comparison with YSZ.163,164
LSGM is recognized as a superior solid-oxide electrolyte material, distinguished by its exceptional chemical and thermal stability, robust mechanical properties, remarkably high ionic conductivity in the range of 500–800 °C, and a wide electrochemical stability window that facilitates steady operation under challenging oxidative and reductive conditions.165–167 These attributes not only ensure the long-term stability and reliability of the battery system under demanding operational circumstances but also position LSGM as a promising alternative for solid-oxide electrolytes in high-temperature iron–air batteries.
As another solid-oxide electrolyte candidate, Bi2O3 exhibits a conductivity 1–2 orders of magnitude higher than that of YSZ at similar temperatures. This enhanced conductivity is mainly due to its abundant oxygen vacancies and the increased rate of anion migration in Bi2O3.168 At 730 °C, a phase transition of Bi2O3 occurs, transforming from monoclinic α-phase to cubic δ-phase crystals. However, the fluctuation in volume due to the phase transition could limit the application of Bi2O3. The introduction of rare earth ions such as Dy3+ and W6+ serves to stabilize the structure of the electrolyte, ensuring structural integrity at lower temperatures.169 Cerium dioxide (CeO2) also emerges as a promising candidate for solid-oxide electrolytes.170 Similar to Bi2O3, a significant phase transition of CeO2 occurs at 700 °C. In addition to its ionic conductivity, CeO2 exhibits significant electronic conductivity, which could lead to battery self-discharge during electrochemical reduction. To improve the performance of CeO2-based solid-oxide electrolyte materials, structural stabilization is attained by introducing low-valent cations such as Sm3+ or Gd3+.171,172 The ionic conductivity of Sm-doped cerium (SDC) and Gd-doped cerium (GDC) can achieve a value of 0.1 S cm−1 at 800 °C. Moreover, the incorporation of secondary-phase materials such as carbonates or semiconductors into CeO2-based electrolytes to form heterogeneous structured composite materials markedly improves the ionic conductivity of the materials, reaching 0.1 S cm−1 at 600 °C. These composite materials are designed with a core–shell structure, utilizing the characteristics of the interface mechanism to effectively control the grain growth of cerium-based electrolytes, resulting in an optimal microstructure and enhanced overall ionic conductivity.
It is noteworthy that Bi2O3 and CeO2 demonstrate electron conductivity under electrochemical reduction conditions, presenting a challenge to their direct application as solid-oxide electrolytes in high-temperature iron–air batteries. To address this constraint, a method involving the deposition of a protective YSZ layer on the Bi2O3 and CeO2 solid-oxide electrolytes can be adopted. The YSZ layer functions to prevent direct contact between Bi2O3/CeO2 and electrode materials, thereby avoiding the electron conductivity of Bi2O3 and CeO2. The deposition of the YSZ layer can be achieved through various techniques, such as chemical vapor deposition (CVD), atomic layer deposition (ALD), or sputtering deposition. These methods facilitate precise manipulation of the thickness and uniformity of the YSZ layer, ensuring optimal preservation of the performance of the electrolyte layer.
5.2 Electrode materials
In iron–air batteries, the air electrode is essential for enabling the reversible oxygen reduction reaction (ORR) and oxygen evolution reaction (OER).146 Room temperature iron–air batteries typically utilize bifunctional metal catalysts, such as precious metals and transition metal alloys, at the air electrode. High-temperature iron–air batteries often employ metal oxide catalysts such as perovskite-structured metal oxides (such as LSM and LSCF) and valuable metals such as Ag and Pt at the air electrode. With the rapid progress in room-temperature metal–air battery technology, extensive research efforts have been focused on investigating bifunctional air electrode catalyst materials. The progress in high-temperature technologies, such as solid-oxide fuel cells (SOFCs), has prompted investigations into oxide-based catalysts for high-temperature air electrodes. The literature extensively discusses air electrode materials, while there has been limited discussion on iron electrode materials so far. Considering the significance of iron electrode materials in iron–air batteries, this chapter provides a comprehensive review and discussion on the challenges and possible solutions associated with iron electrode materials.The performance of iron–air batteries is predominantly determined by the activity and stability of the iron electrode materials. The activity of the iron electrodes significantly influences the electrochemical reaction kinetics, while the stability of the electrodes is crucial for the cycling stability and reliability of the battery. The interface between the electrolyte and the iron electrode determines the electrochemical activity of the iron electrode, with the charge transfer process at this interface being particularly significant. During cyclic charge and discharge, the stability of the iron electrodes is challenged by structural evolution that can potentially lead to a decline in battery performance. This section will specifically concentrate on the stability of iron electrodes during charge–discharge cycles. Strategies for enhancing the activity of electrode reactions through interface regulation will be discussed in detail in the following chapters. By tackling the challenges associated with the stability and activity of iron electrode materials, it is possible to enhance the overall performance of iron–air batteries.
The stability of iron electrodes is crucial for the performance of iron–air batteries. In aqueous iron–air batteries, significant volume changes occur during charge–discharge cycling, leading to increased mechanical strain on the iron electrode. In SOIARBs, the Fe/FeOx particles within the ESU are susceptible to hot coarsening and sintering during high-temperature redox cycles. However, the presence of a physical gap filled with a H2–H2O mixture between the Ni-based anode and the Fe-bed prevents the solid–solid contact. This design can avoid the impact of volume changes on battery stability that would be problematic in an all-solid-state configuration. In contrast, in ceramic IABs, the hot coarsening and sintering of the iron electrode can lead to significant volume and morphological changes, which can reduce the effective contact with the collector and the solid electrolyte, thereby affecting the stability and reliability of the battery.120 Additionally, in high-temperature iron–air batteries, the iron electrode material may undergo chemical composition changes due to thermal diffusion after prolonged exposure to high-temperature conditions (Fig. 11a and b).173 These changes in the chemical composition can decrease the electrochemical activity of the electrode, impacting the energy conversion efficiency and cycling stability of the battery.
 | ||
| Fig. 11 SEM images of the cross-section of an iron particle before (a) and after oxidation (b).173 Comparison of SEM images of Fe2O3 (c) and 1% Cu-doped Fe2O3 (d) before and after redox reactions.174 Comparison of SEM images of Fe2O3 (e) and La-doped Fe2O3 (f) before and after redox reactions.175 | ||
Strategies such as composite material design and doping are effective in mitigating structural changes in iron electrode materials during cyclic charge and discharge processes. These strategies not only enhance the stability of the iron electrode but also increase the energy density, cycling stability, and long-term reliability of the battery. The composite material design strategy involves incorporating oxide additives into the iron electrode to suppress coarsening and sintering during high-temperature electrochemical redox reactions. In SOIARBs, integrating YSZ powder as an inert support material with the ESU effectively mitigates the sintering of Fe/FeOx particles.120 This approach not only strengthens the structural stability of the ESU but also improves the cycling stability of the battery. Moreover, the addition of Cr2O3 to form Fe–Cr oxides can inhibit the thermal coarsening of Fe/FeOx particles, further enhancing the stability of the ESU during cyclic charge and discharge processes.121 By optimizing the microstructure of the ESU, these strategies offer a novel approach to enhancing the overall performance of iron–air batteries and prolonging their operational lifespan. Additionally, for ceramic IABs, the design of composite materials should consider the interfacial compatibility between the electrode, current collector, and solid electrolyte. Optimizing these interfaces can improve the stability of the electrode and enhance its electrochemical activity.
Doping strategies have been demonstrated to effectively mitigate volume changes within iron electrodes during high-temperature redox processes. Notably, the doping of copper (Cu) (Fig. 11c and d) and lanthanum (La) (Fig. 11e and f) at low concentrations also serves to suppress structural and morphological changes in iron oxides during high-temperature redox reactions, which in turn improves the cycling stability of the iron oxides.173–176 However, it is crucial to note that high concentrations of dopants may induce dopant segregation during cyclic high-temperature redox processes, which can adversely affect electrode performance. For example, iron oxide doped with titanium (Ti) may undergo phase separation at elevated temperatures, potentially exerting a detrimental impact on the redox reactions of the iron oxide.177 Therefore, the careful regulation of dopant concentrations is crucial for optimizing the structural integrity and electrochemical reactivity of iron electrodes.
5.3 Electrode/electrolyte interfaces
In iron–air batteries, the three phase interface (TPI) serves as the reactive site for electrochemical redox reactions. The TPI refers to the interface where the electrode material, electrolyte, and reactive gases (i.e., oxygen and hydrogen) are in contact. This interface facilitates concurrent charge and mass transfer, which is crucial for the efficiency and cycling stability of the battery. The solid-state electrode reactions in iron–air batteries include the reactions between iron and ferrous hydroxide with magnetite in aqueous iron–air batteries, the anodic reactions involving iron and potassium ferrate in quasi-solid-state molten–air batteries, and the interactions between metallic iron and iron oxide in both SOIARB and ceramic IAB configurations.48,50,51,54 It is evident that these solid-state reaction mechanisms are complex, and the kinetic processes are sluggish, which significantly limits the battery performance. Therefore, optimizing the design of the TPI and exploring methods to enhance the stability and kinetics of the electrode reactions are of significant importance for improving the performance of these batteries.A common challenge across both room-temperature and high-temperature solid-state iron–air batteries is the sluggish charge diffusion at the TPI within the iron electrode. This issue significantly hampers the battery performance in terms of their charge–discharge rates and power density. In aqueous iron–air batteries, the iron electrode undergoes a phase transition from Fe to Fe(OH)2 and then to Fe3O4 during the charge–discharge processes. However, the electrode reactions are characterized by slow kinetics due to the limited charge diffusion within these phases. In high-temperature iron–air batteries, despite the enhanced reaction kinetics at elevated temperatures, the inherent diffusion limitation of solid-state materials remains.
Another important issue in high-temperature iron–air batteries is the potential for thermal diffusion at the interface between the electrode and the solid electrolyte, which can result in structural and compositional changes of the TPI. Such changes can compromise the integrity of the TPI and may also trigger parasitic reactions at the interface. In high-temperature molten–air batteries, the direct contact between the iron electrode and the molten salt electrolyte can lead to the corrosion and subsequent dissolution of the iron electrode, which may enhance the electronic conductivity of the electrolyte. This increase in electronic conductivity has the potential to induce self-discharge or internal short-circuiting within the battery. In ceramic IABs, high operating temperatures can induce side reactions at the interface between the solid electrolyte and the electrode. These reactions can lead to the formation of a passivation layer, which diminishes the transport of electrons and ions, increases the resistance associated with electrode reactions, and subsequently reduces the overall performance of the battery. Additionally, the disparity in thermal expansion coefficients between the electrolyte and electrode materials at high temperatures could lead to interfacial cracking, which can jeopardize the integrity of TPI in the iron electrode.
Strategically optimizing the TPI to enhance both ionic and electronic conductivities has the potential to significantly increase the rate of charge diffusion within the iron electrode. The incorporation of CeO2 into the iron electrode layer in ceramic IABs has been found to be highly effective in enhancing the diffusion of O2− ions.150 CeO2, renowned for its effectiveness as an O2− ion conductor, establishes accelerated pathways for the transportation of O2− ions at high temperatures. This enhancement not only accelerates the kinetics of the electrode reactions but also increases the Fe utilization within the iron electrode.
The redox reactions at iron electrodes in iron–air batteries involve the solid-state redox reactions between iron and oxides. These reactions are characterized by limited ionic and electronic conductivities. Nanotechnology provides a sophisticated approach to addressing this concern through the optimization of the size, morphology, and structural composition of iron electrode materials. This optimization results in a notable reduction in the charge transfer distance within the iron oxide phase.64,70 Moreover, nanostructured iron electrodes, characterized by their increased surface area, extend the TPI length within the iron electrode. This extension improves the charge diffusion and enhances the reaction kinetics of the iron electrode.
Doping strategies can substantially improve the ionic and electronic conductivity of iron oxides, thereby enhancing charge diffusion at the TPI within the iron electrode. Ti-doping can introduce oxygen vacancies within the iron oxide, leading to a notable enhancement in its electronic conductivity and potentially an improvement in its ionic conductivity (Fig. 12a and b).177 Additionally, the deliberate addition of dopants can introduce catalytically active heteroatoms, thereby enhancing the reactivity of the electrode material at the TPI. Furthermore, the incorporation of low concentrations of La and Cu (Fig. 12c) has been shown to optimize the electronic structure and surface activity of the iron electrode material, leading to a pronounced enhancement in the redox capabilities of iron oxides.174,175
 | ||
Fig. 12 Vacancy formation energy on the surface and on the subsurface as a function of temperature at a pressure of 1 atm for (a) FeTiO3 and (b) α-Fe2O3 phases. Yellow ball represents the surface vacancy  and green ball represents the subsurface vacancy and green ball represents the subsurface vacancy 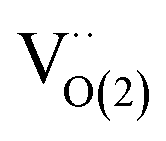 .177 (c) Variation in oxygen vacancy formation energy on the Cu–Fe2O3 surface.174 (d) High-resolution transmission electron microscopy (HRTEM) image of fresh Fe2O3/ZrO2–IrO2.52 (e) HRTEM image of Fe3O4/ZrO2–Ir after 100 h of testing.52 (f) Schematic diagram of the Fe3O4/ZrO2–Ir reduction process.52 (g) Bright field transmission electron microscopy image of Pd-impregnated Fe2O3–ZrO2. The PdO nanoparticles are marked with arrows, ∼6 nm in size. The incorporated Pd oxide nanoparticles are attached to the surface of Fe2O3–ZrO2.122 (h) Temperature-programmed reduction (TPR) profiles of baseline and Pd-impregnated Fe2O3–ZrO2 samples.122 .177 (c) Variation in oxygen vacancy formation energy on the Cu–Fe2O3 surface.174 (d) High-resolution transmission electron microscopy (HRTEM) image of fresh Fe2O3/ZrO2–IrO2.52 (e) HRTEM image of Fe3O4/ZrO2–Ir after 100 h of testing.52 (f) Schematic diagram of the Fe3O4/ZrO2–Ir reduction process.52 (g) Bright field transmission electron microscopy image of Pd-impregnated Fe2O3–ZrO2. The PdO nanoparticles are marked with arrows, ∼6 nm in size. The incorporated Pd oxide nanoparticles are attached to the surface of Fe2O3–ZrO2.122 (h) Temperature-programmed reduction (TPR) profiles of baseline and Pd-impregnated Fe2O3–ZrO2 samples.122 | ||
The incorporation of catalysts into the TPI represents a strategic approach to significantly enhancing the reaction kinetics within the iron electrode. In SOIARBs, the utilization of noble metal catalysts, such as iridium (Ir) (Fig. 12d–f) or palladium (Pd) (Fig. 12g and h), in conjunction with iron oxide, is instrumental in creating a highly active TPI.52,118,122 This synergy not only boosts the reduction activity of iron oxides but also facilitates efficient recharging at reduced temperatures. Consequently, this leads to an enhancement in the charge–discharge efficiency and a notable improvement in the cycling stability.
6. Conclusion and perspectives
Iron–air batteries have attracted considerable interest due to their potential for large-scale energy storage. Despite the theoretical advantages in energy density and environmental compatibility, persistent challenges include suboptimal charge–discharge efficiency and limited cycle life. To overcome these challenges, there has been a pronounced trend towards the development of solid-state battery designs. This review provides a comprehensive discussion on the development of iron–air battery technology, emphasizing the important impact of solid-state integration at room temperature and high temperature on improving energy storage performance.At present, the design range of all-solid-state iron–air batteries includes high-temperature and room-temperature applications. Room-temperature methods, including polymers or an alkaline substance that combines solid oxides, greatly improve the safety and stability of the battery. High-temperature approaches, which involve the use of molten salts or solid oxide electrolytes, have significantly enhanced the efficiency, stability, and operating temperature range of batteries. The integration of solid-state electrolytes, such as YSZ and LSGM, has improved safety and reliability, minimized maintenance expenses, and alleviated safety hazards, laying a strong foundation for long-term operation.
As an emerging technology, all-solid-state iron–air batteries confront several significant challenges that are crucial for the research community to address. The low ionic conductivity of solid electrolytes poses a primary obstacle, directly impacting the performance of these batteries by hindering efficient ion transport. Additionally, the high-temperature durability of electrolytes is a critical concern and the susceptibility of electrolytes to degradation at elevated temperatures can compromise the battery's integrity and safety. The activity of the iron electrode plays a pivotal role in the electrochemical reaction kinetics, with the battery's performance fundamentally limited by the electrode's reactivity. Furthermore, the stability of the iron electrode is paramount, as it determines the feasibility of applying iron–air batteries to long-term energy storage applications. Moreover, the sluggish charge diffusion at the TPI impedes the reaction kinetics, affecting the battery's overall efficiency and power output. Tackling these challenges is essential for the advancement of all-solid-state iron–air batteries and their integration into future energy storage solutions.
The advancement of solid-state technology has greatly expanded the opportunities for iron–air batteries. Future research should prioritize the development of high-performance electrolyte materials to enhance ionic conductivity, thermal stability, and chemical durability. This can be achieved through the exploration of novel compositions, the implementation of doping strategies, and the utilization of nanoengineering techniques. Simultaneously, optimizing the stability of iron electrodes is crucial for sustaining long-term battery performance, which might include developing corrosion-resistant electrode materials, applying protective coatings to prevent degradation, and designing composite structures to accommodate volumetric changes during operation. Additionally, nanotechnology presents a promising avenue for enhancing the TPI active sites, which are critical for efficient electrochemical reactions. This enhancement can be achieved by manipulating nanostructures to expedite charge transfer, creating hierarchical porous architectures to expand the electrochemically active surface area, and integrating catalysts to reduce overpotentials and accelerate reaction kinetics.
Moreover, the development trend of solid-state iron–air batteries is expected to have a significant impact on the metal–air battery field. These trends introduce new strategies aimed at improving the efficiency, stability, and safety of batteries by integrating solid electrolytes. This strategic shift expands the range of materials that can be used for electrode and electrolyte design, increasing flexibility and reducing risks associated with corrosion and leakage, thereby improving battery stability and adaptability to a variety of environments. The development of these trends is expected to significantly broaden the potential applications of metal–air batteries in large-scale energy storage, in line with the growing demand for efficient and reliable energy storage solutions driven by the increasing integration of renewable energy sources.
Looking forward, a critical aspect of solid-state iron–air batteries will be the implementation of cost-effective manufacturing strategies. These strategies are essential not only for reducing overall production costs but also for upholding the high standards of performance and safety inherent to these batteries. A key strategy involves the use of earth-abundant materials for electrode and electrolyte fabrication, leveraging their availability and low cost to significantly reduce material expenses. Additionally, exploring scalable and low-cost printing techniques for electrode deposition, such as screen printing, inkjet printing, and roll-to-roll processing, offers a promising approach to enhancing cost efficiency. These methods provide the benefits of expedited manufacturing and minimized waste, which are crucial for cost reduction in large-scale battery production.
Furthermore, the streamlining of cell assembly processes is crucial for driving down costs and enhancing the efficiency of battery manufacturing. By implementing streamlined procedures, adopting monolithic cell designs, and introducing automation in production lines, manufacturers can significantly boost the scalability and consistency of the manufacturing process. These refinements not only lower the cost of production but also ensure that solid-state iron–air batteries can be produced in larger quantities while maintaining uniform quality. The collective impact of these cost-reducing strategies is likely to be substantial, thereby enhancing the economic feasibility of solid-state iron–air batteries and facilitating their broader adoption.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
B. S. wrote the manuscript. H. W. contributed to the discussion on the manuscript. C. P. supervised and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDA0400000.References
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- S. Kozarcanin, H. Liu and G. B. Andresen, Joule, 2019, 3, 992–1005 CrossRef.
- C. A. Hunter, M. M. Penev, E. P. Reznicek, J. Eichman, N. Rustagi and S. F. Baldwin, Joule, 2021, 5, 2077–2101 CrossRef.
- H. Yang, M. Jing, L. Wang, H. Xu, X. Yan and X. He, Nanomicro Lett., 2024, 16, 127 Search PubMed.
- C. Zhang, Y. Yang, X. Liu, M. Mao, K. Li, Q. Li, G. Zhang and C. Wang, Innovation, 2023, 4, 100518 Search PubMed.
- C. Sun, R. Hui and J. Roller, J. Solid State Electrochem., 2009, 14, 1125–1144 CrossRef.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- C. Sun, Adv. Mater. Lett., 2018, 9, 336–344 CrossRef CAS.
- K. Tuo, C. Sun and S. Liu, Electrochem. Energy Rev., 2023, 6, 17 CrossRef CAS.
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696–2767 RSC.
- S. S. Shinde, N. K. Wagh, S. H. Kim and J. H. Lee, Adv. Sci., 2023, 10, e2304235 CrossRef PubMed.
- M. A. Alemu, D. T. Ebissa, M. Z. Getie, A. K. Worku, H. M. Wassie and M. S. Alem, Energy Rep., 2024, 12, 517–528 CrossRef.
- A. K. Koech, G. Mwandila and F. Mulolani, Heliyon, 2024, 10, e34806 CrossRef CAS PubMed.
- Y. Zhang, S. Jing, H. Shen, S. Li, Y. Huang, Y. Shen, S. Liu, Z. Zhang and F. Liu, ChemElectroChem, 2024, 11, e202300495 CrossRef.
- W. Zhang, J. Wei, F. Yin and C. Sun, Mater. Chem. Front., 2023, 7, 1943–1991 RSC.
- K. Gao, C. Sun and Z. Wang, Mater. Chem. Front., 2024, 8, 3082–3105 RSC.
- Y. Luo, Z. Rao, X. Yang, C. Wang, X. Sun and X. Li, Energy Environ. Sci., 2024, 7543–7565, 10.1039/d4ee02358g.
- F. Öksüzoğlu, Ş. Ateş, O. M. Özkendir, G. Çelik, Y. R. Eker and H. Baveghar, Ceram. Int., 2024, 50, 31435–31441 CrossRef.
- L. Xia, H. Liu and Y. Pei, Nanoscale, 2024, 16, 15481–15501 RSC.
- X. Zhou, Y. Zhou, L. Yu, L. Qi, K.-S. Oh, P. Hu, S.-Y. Lee and C. Chen, Chem. Soc. Rev., 2024, 53, 5291–5337 RSC.
- S. Zou, Y. Yang, J. Wang, X. Zhou, X. Wan, M. Zhu and J. Liu, Energy Environ. Sci., 2024, 17, 4426–4460 RSC.
- W. Sun, F. Wang, B. Zhang, M. Zhang, V. Küpers, X. Ji, C. Theile, P. Bieker, K. Xu, C. Wang and M. Winter, Science, 2021, 371, 46–51 CrossRef CAS PubMed.
- J. Fu, Z. P. Cano, M. G. Park, A. Yu, M. Fowler and Z. Chen, Adv. Mater., 2016, 29, 1604685 CrossRef PubMed.
- M. Gao, Y. Song, X. Zou, M. Salla, H. Zhang, Z. Wang, S. Huang and Q. Wang, Adv. Energy Mater., 2023, 13, 2301868 CrossRef CAS.
- G. M. Wu, S. J. Lin and C. C. Yang, J. Membr. Sci., 2006, 280, 802–808 CrossRef CAS.
- X. Zhao, Y. Gong, X. Li, N. Xu and K. Huang, J. Mater. Chem. A, 2013, 1, 14858 RSC.
- A. Inoishi, T. Sakai, Y.-W. Ju, S. Ida and T. Ishihara, J. Mater. Chem. A, 2013, 1, 15212 RSC.
- L. Otaegui, L. M. Rodriguez-Martinez, L. Wang, A. Laresgoiti, H. Tsukamoto, M. H. Han, C. L. Tsai, I. Laresgoiti, C. M. López and T. Rojo, J. Power Sources, 2014, 247, 749–755 CrossRef CAS.
- J. S. Lee, S. Tai Kim, R. Cao, N. S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2010, 1, 34–50 CrossRef.
- Y. Li, M. Gong, Y. Liang, J. Feng, J.-E. Kim, H. Wang, G. Hong, B. Zhang and H. Dai, Nat. Commun., 2013, 4, 1805 CrossRef PubMed.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- Y. Li and J. Lu, ACS Energy Lett., 2017, 2, 1370–1377 CrossRef CAS.
- Y. Sun, X. Liu, Y. Jiang, J. Li, J. Ding, W. Hu and C. Zhong, J. Mater. Chem. A, 2019, 7, 18183–18208 RSC.
- H.-F. Wang and Q. Xu, Matter, 2019, 1, 565–595 CrossRef CAS.
- A. I. Ikeuba, P. C. Iwuji, I.-I. E. Nabuk, O. E. Obono, D. Charlie, A. A. Etim, B. I. Nwabueze and J. Amajama, J. Solid State Electrochem., 2024, 28, 2999–3025 CrossRef CAS.
- J. Jiang and J. Liu, Interdiscip. Mater., 2022, 1, 116–139 Search PubMed.
- R. Chen, R. Luo, Y. Huang, F. Wu and L. Li, Adv. Sci., 2016, 3, 1600051 CrossRef PubMed.
- Y. Xu, X. Wu, S. K. Sandstrom, J. J. Hong, H. Jiang, X. Chen and X. Ji, Adv. Mater., 2021, 33, 2003733 CrossRef PubMed.
- H. A. F. Rodríguez, R. D. McKerracher, C. P. de León and F. C. Walsh, J. Electrochem. Soc., 2019, 166, A107–A117 CrossRef.
- S. R. Narayanan, G. K. S. Prakash, A. Manohar, B. Yang, S. Malkhandi and A. Kindler, Solid State Ionics, 2012, 216, 105–109 CrossRef CAS.
- H. Kitamura, L. Zhao, B. T. Hang, S. Okada and J.-i. Yamaki, J. Electrochem. Soc., 2012, 159, A720–A724 CrossRef CAS.
- A. K. Manohar, S. Malkhandi, B. Yang, C. Yang, G. K. Surya Prakash and S. R. Narayanan, J. Electrochem. Soc., 2012, 159, A1209–A1214 CrossRef CAS.
- A. K. Manohar, C. Yang, S. Malkhandi, B. Yang, G. K. Surya Prakash and S. R. Narayanan, J. Electrochem. Soc., 2012, 159, A2148–A2155 CrossRef CAS.
- B. T. Hang, D. H. Thang and E. Kobayashi, J. Electroanal. Chem., 2013, 704, 145–152 CrossRef CAS.
- A. K. Manohar, C. Yang, S. Malkhandi, G. K. S. Prakash and S. R. Narayanan, J. Electrochem. Soc., 2013, 160, A2078–A2084 CrossRef CAS.
- A. Matsuda, H. Sakamoto, T. Kishimoto, K. Hayashi, T. Kugimiya and H. Muto, Solid State Ionics, 2014, 262, 188–191 CrossRef CAS.
- N. Xu, X. Li, X. Zhao, J. B. Goodenough and K. Huang, Energy Environ. Sci., 2011, 4, 4942 RSC.
- A. Inoishi, S. Ida, S. Uratani, T. Okano and T. Ishihara, Phys. Chem. Chem. Phys., 2012, 14, 12818 RSC.
- S. Licht, B. Cui, J. Stuart, B. Wang and J. Lau, Energy Environ. Sci., 2013, 6, 3646 RSC.
- S. Trocino, S. C. Zignani, M. Lo Faro, V. Antonucci and A. S. Aricò, Energy Technol., 2017, 5, 670–680 CrossRef CAS.
- Q. Tang, Y. Zhang, N. Xu, X. Lei and K. Huang, Energy Environ. Sci., 2022, 15, 4659–4671 RSC.
- C. Peng, C. Guan, J. Lin, S. Zhang, H. Bao, Y. Wang, G. Xiao, G. Z. Chen and J. Q. Wang, ChemSusChem, 2018, 11, 1880–1886 CrossRef CAS PubMed.
- S. Zhang, Y. Yang, L. Cheng, J. Sun, X. Wang, P. Nan, C. Xie, H. Yu, Y. Xia, B. Ge, J. Lin, L. Zhang, C. Guan, G. Xiao, C. Peng, G. Z. Chen and J.-Q. Wang, Energy Storage Mater., 2021, 35, 142–147 CrossRef.
- C. Morey, Q. Tang and K. Huang, ECS Trans., 2023, 111, 1771–1783 CrossRef CAS.
- R. D. McKerracher, C. Ponce de Leon, R. G. A. Wills, A. A. Shah and F. C. Walsh, ChemPlusChem, 2014, 80, 323–335 CrossRef.
- C. Zhang and K. Huang, Adv. Energy Mater., 2020, 11, 2002896 CrossRef.
- H. Wang and C. Peng, Sustainable Energy Fuels, 2023, 7, 330–354 RSC.
- H. Wang, B. Q. Sun and C. Peng, J. Electrochem. Soc., 2024, 171, 052501 CrossRef.
- X. Yu and A. Manthiram, ACS Energy Lett., 2017, 2, 1050–1055 CrossRef CAS.
- B. Tian, J. Światowska, V. Maurice, S. Zanna, A. Seyeux and P. Marcus, Electrochim. Acta, 2018, 259, 196–203 CrossRef CAS.
- B. Yang, S. Malkhandi, A. K. Manohar, G. K. Surya Prakash and S. R. Narayanan, Energy Environ. Sci., 2014, 7, 2753 RSC.
- A. K. Manohar, C. Yang and S. R. Narayanan, J. Electrochem. Soc., 2015, 162, A1864–A1872 CrossRef CAS.
- R. D. McKerracher, C. Alegre, V. Baglio, A. S. Aricò, C. Ponce de León, F. Mornaghini, M. Rodlert and F. C. Walsh, Electrochim. Acta, 2015, 174, 508–515 CrossRef CAS.
- S. Velraj and J. H. Zhu, J. Electroanal. Chem., 2015, 736, 76–82 CrossRef CAS.
- H. Weinrich, J. Come, H. Tempel, H. Kungl, R.-A. Eichel and N. Balke, Nano Energy, 2017, 41, 706–716 CrossRef CAS.
- D. Mitra and S. R. Narayanan, Top. Catal., 2018, 61, 591–600 CrossRef CAS.
- H. T. Bui and T. M. Vu, J. Electron. Mater., 2019, 48, 7123–7130 CrossRef CAS.
- H. Weinrich, J. Pleie, B. Schmid, H. Tempel, H. Kungl and R. A. Eichel, Batteries Supercaps, 2022, 5, e202100415 CrossRef CAS.
- C. Fang, X. Tang, J. Wang and Q. Yi, Front. Energy, 2023, 42–53, DOI:10.1007/s11708-023-0913-5.
- H. A. Figueredo-Rodríguez, R. D. McKerracher, M. Insausti, A. G. Luis, C. P. de León, C. Alegre, V. Baglio, A. S. Aricò and F. C. Walsh, J. Electrochem. Soc., 2017, 164, A1148–A1157 CrossRef.
- H.-K. Lim, H.-D. Lim, K.-Y. Park, D.-H. Seo, H. Gwon, J. Hong, W. A. Goddard, H. Kim and K. Kang, J. Am. Chem. Soc., 2013, 135, 9733–9742 CrossRef CAS PubMed.
- C. Yang, A. K. Manohar and S. R. Narayanan, J. Electrochem. Soc., 2017, 164, A418–A429 CrossRef CAS.
- H. Weinrich, M. Gehring, H. Tempel, H. Kungl and R.-A. Eichel, J. Appl. Electrochem., 2018, 48, 451–462 CrossRef CAS.
- N. I. Villanueva-Martínez, C. Alegre, J. Rubín, R. McKerracher, C. P. de León, H. A. F. Rodríguez and M. J. Lázaro, Electrochim. Acta, 2023, 465, 142964 CrossRef.
- B. T. Hang and D. H. Thang, J. Electroanal. Chem., 2016, 762, 59–65 CrossRef CAS.
- K. Hayashi, Y. Wada, Y. Maeda, T. Suzuki, H. Sakamoto, W. K. Tan, G. Kawamura, H. Muto and A. Matsuda, J. Electrochem. Soc., 2017, 164, A2049–A2055 CrossRef CAS.
- T. A. Trinh and T. H. Bui, J. Mater. Eng. Perform., 2020, 29, 1245–1252 CrossRef CAS.
- B. T. Hang, T. Van Dang and N. Van Quy, J. Electron. Mater., 2022, 51, 2168–2177 CrossRef CAS.
- R. D. McKerracher, H. A. Figueredo-Rodriguez, C. Alegre, A. S. Aricò, V. Baglio and C. Ponce de León, Electrochim. Acta, 2019, 318, 625–634 CrossRef CAS.
- R. D. McKerracher, H. A. Figueredo-Rodriguez, K. Dimogiannis, C. Alegre, N. I. Villanueva-Martinez, M. J. Lázaro, V. Baglio, A. S. Aricò and C. Ponce de León, J. Solid State Electrochem., 2020, 25, 225–230 CrossRef.
- P. S. Arunkumar, T. Maiyalagan, S. Kheawhom, S. Mao and Z. Jiang, Mater. Sci. Energy Technol., 2021, 4, 236–241 CAS.
- H. Weinrich, M. Gehring, H. Tempel, H. Kungl and R.-A. Eichel, Electrochim. Acta, 2019, 314, 61–71 CrossRef CAS.
- D. Mitra, A. S. Rajan, A. Irshad and S. R. Narayanan, J. Electrochem. Soc., 2021, 168, 030518 CrossRef CAS.
- E. O. Aremu and K.-S. Ryu, Electrochim. Acta, 2019, 313, 468–477 CrossRef CAS.
- W. K. Tan, K. Asami, K. Maegawa, G. Kawamura, H. Muto and A. Matsuda, Energy Storage, 2020, 2, e196 CrossRef CAS.
- Q. Wang and Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 10334–10342 CrossRef CAS PubMed.
- N. Villanueva, C. Alegre, J. Rubin, H. A. Figueredo-Rodríguez, R. D. McKerracher, C. P. de León and M. J. Lázaro, ACS Appl. Energy Mater., 2022, 5, 13439–13451 CrossRef CAS.
- W. K. Tan, K. Asami, Y. Maeda, K. Hayashi, G. Kawamura, H. Muto and A. Matsuda, Appl. Surf. Sci., 2019, 486, 257–264 CrossRef CAS.
- T. Tsuneishi, T. Esaki, H. Sakamoto, K. Hayashi, G. Kawamura, H. Muto and A. Matsuda, Key Eng. Mater., 2014, 616, 114–119 CAS.
- T. Tsuneishi, H. Sakamoto, K. Hayashi, G. Kawamura, H. Muto and A. Matsuda, J. Asian Ceram. Soc., 2018, 2, 165–168 CrossRef.
- M. A. Deyab and Q. Mohsen, Renewable Sustainable Energy Rev., 2021, 139, 110729 CrossRef CAS.
- K. Hayashi, Y. Maeda, T. Suzuki, H. Sakamoto, T. Kugimiya, W. K. Tan, G. Kawamura, H. Muto and A. Matsuda, ECS Trans., 2017, 75, 111–116 CrossRef CAS.
- W. K. Tan, K. Asami, K. Maegawa, R. Kumar, G. Kawamura, H. Muto and A. Matsuda, Mater. Today Commun., 2020, 25, 101540 CrossRef CAS.
- S. Xu, S. Lau and L. A. Archer, Inorg. Chem. Front., 2015, 2, 1070–1079 RSC.
- L. Li and A. Manthiram, Adv. Energy Mater., 2015, 6, 1502054 CrossRef.
- H. Ohmori, S. Uratani and H. Iwai, J. Power Sources, 2012, 208, 383–390 CrossRef CAS.
- X. Zhao, N. Xu, X. Li, Y. Gong and K. Huang, RSC Adv., 2012, 2, 10163 RSC.
- A. Inoishi, Y. W. Ju, S. Ida and T. Ishihara, J. Power Sources, 2013, 229, 12–15 CrossRef CAS.
- H. Ohmori and H. Iwai, ECS Trans., 2013, 57, 233–242 CrossRef.
- A. Inoishi, T. Sakai, Y.-W. Ju, S. Ida and T. Ishihara, Int. J. Hydrogen Energy, 2014, 39, 21352–21357 CrossRef CAS.
- X. Zhao, X. Li, Y. Gong, N. Xu and K. Huang, RSC Adv., 2014, 4, 22621–22624 RSC.
- F. Kosaka, H. Hatano, Y. Oshima and J. Otomo, Chem. Eng. Sci., 2015, 123, 380–387 CrossRef CAS.
- H. Ohmori and H. Iwai, J. Power Sources, 2015, 286, 264–275 CrossRef CAS.
- C. M. Berger, A. Mahmoud, R. P. Hermann, W. Braun, E. Yazhenskikh, Y. J. Sohn, N. H. Menzler, O. Guillon, M. Bram and T. Troczynski, J. Am. Ceram. Soc., 2016, 99, 4083–4092 CrossRef CAS.
- Q. Fang, C. M. Berger, N. H. Menzler, M. Bram and L. Blum, J. Power Sources, 2016, 336, 91–98 CrossRef CAS.
- F. Kosaka, S. Isogai, H. Hatano, Y. Oshima and J. Otomo, J. Chem. Eng. Jpn., 2016, 49, 243–250 CrossRef CAS.
- H. Ohmori, H. Iwai, K. Itakura, M. Saito and H. Yoshida, J. Power Sources, 2016, 309, 160–168 CrossRef CAS.
- C. Zhang, C. Ji, W. Wang, D. Schmidt, X. Jin, J. P. Lemmon and K. Huang, Energy Environ. Sci., 2016, 9, 3746–3753 RSC.
- W. Braun, V. Erfurt, F. Thaler, N. H. Menzler, R. Spatschek and L. Singheiser, ECS Trans., 2017, 75, 59–73 CrossRef CAS.
- Q. Tang, Y. Ma and K. Huang, ACS Appl. Energy Mater., 2021, 4, 7091–7100 CrossRef CAS.
- T. Ishihara, H. Kim, Y. Inoishi and J. Matsuda, J. Am. Ceram. Soc., 2022, 105, 6718–6731 CrossRef CAS.
- Q. Tang and K. Huang, Chem. Eng. J., 2022, 434, 134771 CrossRef CAS.
- Y. Wen, Y. Zhang and K. Huang, ECS Trans., 2023, 111, 621–633 CrossRef CAS.
- X. Zhao, Y. Gong, X. Li, N. Xu and K. Huang, J. Electrochem. Soc., 2013, 160, A1716–A1719 CrossRef CAS.
- T. Sakai, A. Inoishi, M. Ogushi, S. Ida and T. Ishihara, J. Energy Storage, 2016, 7, 115–120 CrossRef.
- C. Zhang and K. Huang, Chem. Commun., 2017, 53, 10564–10567 RSC.
- Q. Tang, C. Morey, Y. Zhang, N. Xu, S. Sun and K. Huang, Adv. Sci., 2022, 9, 2203768 CrossRef CAS PubMed.
- H. Kim, A. Inoishi, S. Ida and T. Ishihara, J. Mater. Chem. A, 2016, 4, 5482–5488 RSC.
- C. M. Berger, O. Tokariev, P. Orzessek, A. Hospach, Q. Fang, M. Bram, W. J. Quadakkers, N. H. Menzler and H. P. Buchkremer, J. Energy Storage, 2015, 1, 54–64 CrossRef.
- H. Kim, S. Ida, Y.-W. Ju, J. Matsuda, G. Kim and T. Ishihara, J. Mater. Chem. A, 2017, 5, 364–371 RSC.
- C. Zhang and K. Huang, ACS Energy Lett., 2016, 1, 1206–1211 CrossRef CAS.
- X. Zhao, Y. Gong, X. Li, N. Xu and K. Huang, J. Electrochem. Soc., 2013, 160, A1241–A1247 CrossRef CAS.
- X. Zhao, X. Li, Y. Gong and K. Huang, Chem. Commun., 2014, 50, 623–625 RSC.
- A. Inoishi, J. Hyodo, H. Kim, T. Sakai, S. Ida and T. Ishihara, J. Mater. Chem. A, 2015, 3, 8260–8264 RSC.
- A. Inoishi, T. Sakai, Y. W. Ju, S. Ida and T. Ishihara, J. Power Sources, 2014, 262, 310–315 CrossRef CAS.
- M. Guo, X. Zhao, R. E. White and K. Huang, J. Electrochem. Soc., 2013, 160, A2085–A2092 CrossRef CAS.
- X. Jin, A. M. Uddin, X. Zhao, R. White and K. Huang, J. Electrochem. Soc., 2015, 162, A1476–A1484 CrossRef CAS.
- X. Jin, X. Zhao, R. E. White and K. Huang, J. Electrochem. Soc., 2015, 162, F821–F833 CrossRef CAS.
- X. Jin, X. Zhao, J. Tong, F. Yasmeen, R. E. White and K. Huang, J. Energy Storage, 2015, 3, 1–9 CrossRef.
- X. Jin, X. Zhao, C. Zhang, R. E. White and K. Huang, Electrochim. Acta, 2015, 178, 190–198 CrossRef CAS.
- X. Jin, X. Zhao and K. Huang, J. Power Sources, 2015, 280, 195–204 CrossRef CAS.
- X. Jin and K. Huang, J. Electrochem. Soc., 2020, 167, 124501 CrossRef CAS.
- X. Jin and K. Huang, Sci. Bull., 2016, 61, 1345–1354 CrossRef CAS.
- C. Morey, Q. Tang, S. Sun and K. Huang, J. Electrochem. Soc., 2023, 170, 104504 CrossRef CAS.
- W. Drenckhahn, H. Greiner, M. Kühne, H. Landes, A. Leonide, K. Litzinger, C. Lu, C. Schuh, J. Shull and T. Soller, ECS Trans., 2013, 50, 125–135 CrossRef.
- S. Liu, W. Han, B. Cui, X. Liu, H. Sun, J. Zhang, M. Lefler and S. Licht, J. Electrochem. Soc., 2018, 165, A149–A154 CrossRef CAS.
- B. Cui and S. Licht, J. Mater. Chem. A, 2014, 2, 10577–10580 RSC.
- H. Liu, X. Zhang, S. He, D. He, Y. Shang and H. Yu, Mater. Today, 2022, 60, 128–157 CrossRef CAS.
- Y. Yu, Y. Zhang, H. Wang and G. Z. Chen, Electrochemistry, 2024, 92, 043003 CrossRef CAS.
- S. Liu, S. Gao, B. Cui, X. Liu, Z. Yu, Y. Wang and Z. Zhang, J. Power Sources, 2020, 473, 228572 CrossRef CAS.
- Y. G. Zhu, G. Leverick, A. Accogli, K. Gordiz, Y. Zhang and Y. Shao-Horn, Energy Environ. Sci., 2022, 15, 4636–4646 RSC.
- S. Liu, W. Han, B. Cui, X. Liu, F. Zhao, J. Stuart and S. Licht, J. Power Sources, 2017, 342, 435–441 CrossRef CAS.
- B. Cui, W. Xiang, S. Liu, H. Xin, X. Liu and S. Licht, Sustainable Energy Fuels, 2017, 1, 474–481 RSC.
- B. Cui, J. Zhang, S. Liu, H. Xin, X. Liu, M. Lefler and S. Licht, J. Electrochem. Soc., 2018, 165, A235–A243 CrossRef CAS.
- B. Cui, H. Xin, S. Liu, X. Liu, Y. Hao, Q. Guo and S. Licht, J. Electrochem. Soc., 2016, 164, A88–A92 CrossRef.
- B. Cui, Y. Shao, W. Xiang, S. Liu, X. Liu, W. Han, J. Zhang and S. Licht, J. Phys. Chem. C, 2018, 122, 8109–8115 CrossRef CAS.
- S. Liu, X. Li, B. Cui, X. Liu, Y. Hao, Q. Guo, P. Xu and S. Licht, J. Mater. Chem. A, 2015, 3, 21039–21043 RSC.
- S. Liu, S. Gao, B. Cui, W. Xiang, X. Liu, W. Han and J. Zhang, Energy Technol., 2019, 7, 1800932 CrossRef.
- S. Trocino, M. Lo Faro, S. C. Zignani, V. Antonucci and A. S. Aricò, Appl. Energy, 2019, 233–234, 386–394 CrossRef CAS.
- S. N. Jagadeesan, G. D. Barbosa, F. Guo, L. Zhang, A. M. M. Abeykoon, G. Kwon, D. Olds, C. H. Turner and X. Teng, Chem. Mater., 2023, 35, 6517–6526 CrossRef CAS.
- C. M. Phela, R. Sigwadi and P. F. Msomi, Polym. Adv. Technol., 2023, 34, 2044–2056 CrossRef CAS.
- J. Fu, D. U. Lee, F. M. Hassan, L. Yang, Z. Bai, M. G. Park and Z. Chen, Adv. Mater., 2015, 27, 5617–5622 CrossRef CAS PubMed.
- M. Wu, G. Zhang, L. Du, D. Yang, H. Yang and S. Sun, Small Methods, 2021, 5, 2000868 CrossRef CAS PubMed.
- P. Zhang, Z. Chen, N. Shang, K. Wang, Y. Zuo, M. Wei, H. Wang, D. Zhong and P. Pei, Mater. Chem. Front., 2023, 7, 3994–4018 RSC.
- Y. Wei, Y. Shi, Y. Chen, C. Xiao and S. Ding, J. Mater. Chem. A, 2021, 9, 4415–4453 RSC.
- J. Xu, Q. Cao, L. Wang, B. Ouyang, T. Wei, J. Hao, J. Chen, L. He, L. Liu and K. Huang, Energy Adv., 2024, 3, 263–272 RSC.
- V. S. Saji, ChemElectroChem, 2023, e202300136, DOI:10.1002/celc.202300136.
- R. Kong, X. Su, S. Li, Y. Fan, J. He, H. Jiao and J. Song, J. Electrochem. Soc., 2024, 171, 032504 CrossRef CAS.
- T. Ghaznavi, S. Y. Persaud and R. C. Newman, J. Electrochem. Soc., 2022, 169, 061502 CrossRef CAS.
- S. M. Haile, Acta Mater., 2003, 51, 5981–6000 CrossRef CAS.
- S. Ryu, I. W. Choi, Y. J. Kim, S. Lee, W. Jeong, W. Yu, G. Y. Cho and S. W. Cha, ACS Appl. Mater. Interfaces, 2023, 15, 42659–42666 CrossRef CAS PubMed.
- L. Mathur, S.-Y. Jeon, Y. Namgung, M. P. G. Hanantyo, J. Park, M. S. Islam, S. Sengodan and S.-J. Song, Solid State Ionics, 2024, 408, 116507 CrossRef CAS.
- A. Kežionis, T. Šalkus, M. Dudek, D. Madej, M. Mosiałek, B. D. Napruszewska, W. Łasocha, M. B. Hanif and M. Motola, J. Power Sources, 2024, 591, 233846 CrossRef.
- X. Chen, C. Wu, S. Hao, B. Liu, T. Lu, P. Dong, Y. Zhang, J. Xiao, X. Zeng and S. Zhai, ACS Appl. Energy Mater., 2023, 6, 11043–11050 CrossRef CAS.
- G. Chasta, Himanshu and M. S. Dhaka, Int. J. Energy Res., 2022, 46, 14627–14658 CrossRef CAS.
- S. He, Y. Zou, K. Chen and S. P. Jiang, Interdiscip. Mater., 2023, 2, 111–136 Search PubMed.
- P. Shuk, H. D. Wiemhofer, U. Guth, W. Gopel and M. Greenblatt, Solid State Ionics, 1996, 89, 179–196 CrossRef CAS.
- N. X. Jiang, E. D. Wachsman and S. H. Jung, Solid State Ionics, 2002, 150, 347–353 CrossRef CAS.
- L. Zhang, J. Meng, F. Yao, W. Zhang, X. Liu, J. Meng and H. Zhang, Inorg. Chem., 2018, 57, 12690–12696 CrossRef CAS PubMed.
- J.-G. Li, T. Ikegami and T. Mori, Acta Mater., 2004, 52, 2221–2228 CrossRef CAS.
- S. Kuharuangrong, J. Power Sources, 2007, 171, 506–510 CrossRef CAS.
- L. Qin, A. Majumder, J. A. Fan, D. Kopechek and L.-S. Fan, J. Mater. Chem. A, 2014, 2, 17511–17520 RSC.
- L. Qin, M. Q. Guo, Y. Liu, Z. Cheng, J. A. Fan and L. S. Fan, Appl. Catal., B, 2018, 235, 143–149 CrossRef CAS.
- L. Qin, Z. Cheng, M. Guo, M. Xu, J. A. Fan and L.-S. Fan, ACS Energy Lett., 2016, 2, 70–74 CrossRef.
- L. Zeng, Z. Cheng, J. A. Fan, L.-S. Fan and J. Gong, Nat. Rev. Chem, 2018, 2, 349–364 CrossRef CAS.
- L. Qin, Z. Cheng, J. A. Fan, D. Kopechek, D. Xu, N. Deshpande and L.-S. Fan, J. Mater. Chem. A, 2015, 3, 11302–11312 RSC.
| This journal is © The Royal Society of Chemistry 2024 |