
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Critical insights into eutectic molten hydroxide electrolysis for sustainable green hydrogen production
Farooq
Sher
 *a,
Imane
Ziani
abc,
Nawar K.
Al-Shara
d,
Alexander
Chupin
e,
Nađa
Horo
cf,
Bohong
Wang
g,
Saba
Rahman
h,
Bilal
Fareed
ci and
Monica R.
Nemţanu
j
*a,
Imane
Ziani
abc,
Nawar K.
Al-Shara
d,
Alexander
Chupin
e,
Nađa
Horo
cf,
Bohong
Wang
g,
Saba
Rahman
h,
Bilal
Fareed
ci and
Monica R.
Nemţanu
j
aDepartment of Engineering, School of Science and Technology, Nottingham Trent University, Nottingham NG11 8NS, UK. E-mail: Farooq.Sher@ntu.ac.uk; Tel: +44 (0) 115 84 86679
bDepartment of Chemistry, Laboratory of Applied Chemistry and Environment, Faculty of Sciences, Mohammed First University, Oujda 60000, Morocco
cInternational Society of Engineering Science and Technology, Nottingham, UK
dDepartment of Chemical and Environmental Engineering, University of Nottingham, University Park, Nottingham NG7 2RD, UK
ePeoples' Friendship University of Russia (RUDN University), Moscow 117198, Russia
fDepartment of Chemistry, Faculty of Science, University of Sarajevo, Sarajevo 71000, Bosnia and Herzegovina
gNational and Local Joint Engineering Research Center of Harbor Oil and Gas Storage and Transportation Technology, Zhejiang Key Laboratory of Petrochemical Environmental Pollution Control, Zhejiang Ocean University, No. 1 Haida South Road, 316022, Zhoushan, P. R. China
hChemical and Biomedical Engineering, Pennsylvania State University, Pennsylvania PA 16801, USA
iDepartment of Chemical Engineering, Pakistan Institute of Engineering and Applied Sciences, Nilore, 45650, Islamabad, Pakistan
jElectron Accelerators Laboratory, National Institute for Laser, Plasma and Radiation Physics, 409 Atomiştilor Street, Bucharest-Măgurele, Romania
First published on 29th August 2024
Abstract
In addressing global energy demands, the focus on hydrogen gas production from renewable sources intensifies. This research review investigates hydrogen production via steam splitting using eutectic molten hydroxide (NaOH–KOH%) electrolysis, a promising solution for escalating energy needs. A pivotal aspect involves developing a novel reference electrode for eutectic molten hydroxide, enveloping Ni/Ni(OH)2 with an alumina or mullite tube ionic membrane. The mullite-covered electrode proves stable and reusable from 225 to 300 °C, showcasing a novel advancement in electrochemical stability. Compared to silver and platinum quasi-reference electrodes, the designed reference electrode demonstrates superior stability and efficacy in controlling the platinum working electrode, marking a significant innovation. Moreover, an intriguing cyclic voltammetry study examines different working electrodes, including Ni, Pt, Ag, Mo, and stainless steel (SS) in eutectic molten hydroxide at different temperature conditions. The observed reduction potential for hydrogen evolution follows the order: Ni > Pt > Ag > SS > Mo, corroborated by chronoamperometry, underscoring the reliability of the findings. In the pursuit of high-temperature eutectic molten hydroxide electrolysis to split steam into hydrogen fuel, cathodes of nickel, platinum, and stainless steel are deployed alongside stainless steel and graphite anodes. Operating within the temperature range of 225 to 300 °C and applying voltages ranging from 1.5 to 2.5 V, stainless steel as an anode yields impressive current efficiencies at 300 °C: 90.5, 80 and 68.6% for nickel, stainless steel, and platinum cathodes, respectively. This study positions steam splitting via molten hydroxides as a promising alternative for hydrogen production, poised for integration with renewable energy sources, marking a transformative step in sustainable energy practices.
1. Introduction
The rapid industrialization of the last century, driven by the widespread use of fossil fuels, has brought about unprecedented development. However, the consequential rise in greenhouse gas pollutants, including CO2, N2O, and SO2 from burning fossil fuels, has prompted growing environmental concerns.1 In recent years, the quest for alternatives has intensified, with a particular focus on hydrogen gas production. Hydrogen is acknowledged as a highly efficient and environmentally friendly fuel, offering significant promise for the future. Unlike conventional fossil fuels, hydrogen has the potential to drastically reduce air pollution, making it a vital contender for a variety of purposes like transportation, heating, and generating power.2 Despite the challenges encountered in hydrogen fuel production, it emerges as a viable solution to address the issue of greenhouse gas emissions detrimental to the environment. Consequently, hydrogen is positioned to significantly impact the future energy sector, addressing the increasing global demand.3The sources of hydrogen, ranging from non-renewable ones like hydrocarbons to renewables such as hydropower, solar, wind, and biomass, offer diverse options for production.4 Terlouw et al.5 emphasize the possibility of substantial reductions in harmful emissions by generating hydrogen from renewable sources. The operational measure of the hydrogen production setup is outlined as one kilogram of hydrogen at 80 bar pressure, guaranteeing a purity surpassing 99.9%. This metric is derived from an average reference flow of 10 tons of production and storage of hydrogen per day.6 Electrolysis, leveraging water as a pristine and environmentally friendly energy source, stands as the foremost method for hydrogen generation, offering exceptional purity in its output, viable for applications ranging from small-scale to large-scale operations. However, challenges arise as electrolysis may contribute to CO2 emissions if non-renewable energy resources power the necessary electricity generation. Additional advancements are required to enable the commercialization of this clean energy approach for hydrogen gas production as fuel, necessitating materials capable of withstanding challenging operating conditions within electrolysis cells.7 Addressing the imperative to mitigate global warming necessitates an alternative fuel that is affordable, user-friendly, clean, and emits minimal greenhouse gases. Hydrogen emerges as a promising alternative, aligning with the criteria for an ideal fuel due to its potential benefits.8
Hydrogen functions as an energy carrier with high efficiency, emitting minimal to zero emissions during utilization, thus playing a pivotal role in combating global warming. Despite its environmental advantages, challenges persist, particularly in ensuring safety throughout production, storage, and transportation.9 Understanding key metrics such as the hydrogen concentration (measured at 15% during storage conditions at 20 MPa pressure and room temperature), global warming potential (GWP), nitrogen oxide emissions, and acidification potential is crucial for evaluating its environmental impact. The GWP is equivalent to 0.058 kg CO2 eq. per kW per h, with nitrogen oxide emissions measured at 7.04 × 105 NO2 eq. per kW per h and acidification potential at 3.63 × 104 kg SO2 eq. per kW per h. These metrics provide valuable insights into hydrogen's broader implications and guide efforts toward sustainable energy practices.9 In fuel cells, hydrogen reacts with oxygen, yielding energy and producing only water as a by-product. Unlike readily available fuels like coal and hydrocarbons, hydrogen exists as a compound in water, fossil hydrocarbons, and biomass products.10
Addressing economic concerns within the hydrogen fuel industry, efficient and affordable storage and distribution remain pivotal.11 Whether produced in small-scale local plants or large-scale central plants, hydrogen gas can be stored and transported as gaseous hydrogen, liquid hydrogen, or metal hydrides.12 The transportation of hydrogen involves three main routes. First, pipelines similar to those used for natural gas provide a convenient means of delivery, constructed from materials like low-carbon steel, aluminium, or brass.13 Second, large-scale storage tanks, akin to those for natural gas, facilitate storage. Gaseous hydrogen can also be stored in cylinders for small-scale operations, often at high pressures ranging from 20 MPa to 80 MPa for industrial use.13 Liquid hydrogen (LH2), with a low density of 70.8 kg/cm3 and a boiling point of −252.3 °C, requires extremely low temperatures for storage.14 Cryostats are employed for this purpose, although the associated conversion and maintenance costs make this method more suitable for aircraft than road vehicles. Finally, certain metal alloys offer an alternative for storing hydrogen.15 These alloys absorb hydrogen gas reversibly, forming metal hydrides, presenting a unique approach to hydrogen storage.
Embarking on a journey at the intersection of innovation and sustainability, this research review delves into the realm of hydrogen gas production through a lens of unprecedented possibilities. Driven by concerns over environmental impact and the ever-increasing demand for clean energy, the focus shifts towards the electrochemical process of splitting steam for hydrogen production via eutectic molten hydroxide electrolysis. This exploration is not merely a scientific pursuit; it is a quest to redefine our energy landscape. Imagine a novel reference electrode, a stable companion crafted from the fusion of Ni/Ni(OH)2 and an ionic membrane. Picture the efficient working electrodes Ni, Pt, Mo, Ag, and stainless steel each revealing its unique prowess in the intricate process of hydrogen evolution. This review aspires to unravel the mysteries of cyclic voltammetry, chronoamperometry, and two-electrode electrolysis within the dynamic environment of eutectic molten hydroxide, where process variables such as applied voltage, electrode material, and molten salt temperature shape the narrative of clean energy production. From the physical measurement of hydrogen gas using water displacement to the theoretical calculations echoing Dalton's law for gases. Through meticulous exploration and theoretical contemplation, this review sets out to redefine the boundaries of hydrogen gas production, laying the groundwork for a sustainable energy future. This review transcends the ordinary, unlocking the secrets that propel us toward a cleaner, brighter tomorrow.
2. Hydrogen production technologies overview
In response to the pressing demand for sustainable energy solutions, the landscape of hydrogen production undergoes a transformative evolution, embracing diverse methodologies each laden with distinct promises and challenges. Anchored in history, Alkaline Electrolysis has evolved into a globally acknowledged technology,16 employing liquid alkaline electrolytes to yield hydrogen efficiently.17 Solid oxide electrolysis cells (SOECs), functioning at elevated temperatures,18 represent a robust approach, marked by recent advancements in hybrid configurations.19 The emergence of proton exchange membrane electrolysis (PEM) signifies a paradigm shift, leveraging solid polymer electrolytes to elevate performance and efficiency standards.20 Ambitiously harnessing sunlight for water splitting, photo-electrolysis navigates challenges on the path to viability.21 Thermochemical Water Splitting strategies, relying on intricate high and low-temperature chemical reactions, set the course for large-scale, sustainable hydrogen production.22 Within this dynamic landscape, water electrolysis, though not mainstream, emerges as a beacon of simplicity, reliability, and environmental friendliness.As we embark on an in-depth exploration detailed in Table 1, a critical analysis unfolds, shedding light on nuanced operating conditions, efficiency benchmarks, persistent challenges, and noteworthy advancements. Notably, Eutectic Molten Hydroxide Electrolysis stands out, offering compelling advantages in energy efficiency and cost-effectiveness, presenting a viable option for hydrogen production. Beyond these merits, it showcases the ability to convert CO2 into hydrocarbons, presenting a sustainable solution for fuel production. However, this method is not without limitations, involving reduced production rates, specific electrode material demands, increased production expenses, decreased durability, and heightened energy consumption. The ensuing sections delve into a comprehensive study of the novelty and performance of Eutectic Molten Hydroxide in the intricate landscape of hydrogen production methods.
| Electrolysis method | Operating conditions | Efficiency | Challenges | Noteworthy advancements | Reference |
|---|---|---|---|---|---|
| Alkaline electrolysis | Immersing two electrodes in a liquid alkaline electrolyte (e.g., 20–30% KOH), typically below 100 °C | Up to 70% | Diaphragm limitations, restricted current density, hindered high-pressure operation | Low-temperature electrochemical water splitting with high efficiency even at 100 °C, low-cost nickel alloy electrocatalysts, insights from magnetic properties, shaping the design of highly efficient electrocatalysts | 17 and 23 |
| Solid oxide electrolysis cells (SOECS) | Leveraging steam at elevated temperatures (750–1000 °C) | Hybrid-SOEC with a current per unit area of 3.16 A/cm2 at 750 °C and 1.3 V | Temperature-dependent efficiency, cation removal challenges | The hybrid-SOEC with BaZr0.1Ce0.7Y0.1Yb0.1O3−δ electrolyte, no performance decline over 60 hours of uninterrupted operation | 24 and 25 |
| Proton exchange membrane electrolysis (PEM) | Solid polymer electrolyte (e.g., Nafion), effective proton conductivity at around 80 °C | 50–70% | Gas crossover reduction, compact system design | Introduction of solid polymer electrolytes, a significant departure from traditional methods | 26 |
| Photo-electrolysis | Harnessing sunlight to separate water into hydrogen and oxygen, typically at 20 °C to 30 °C | Photo-current density of 4.30 mA/cm2, stability demonstrated over 70 hours in seawater splitting | Challenges in yield and efficiency, economic viability | A durable photo-electrode layout featuring an ultra-stable PEC cell with a BiVO4 photo-anode and a MoO3 barrier layer, ensuring stable and efficient hydrogen production from seawater | 27 |
| Thermochemical water splitting | Converting water into hydrogen and oxygen via high-temperature endothermic (500 °C to 1800 °C) and low-temperature exothermic processes | Potential for sustainable, large-scale hydrogen generation | Energy and exergy efficiency variations, design challenges | Favourable assessment of sulfur-iodine and hybrid sulfur cycles, achieving approximately 77% efficiency | 28 |
| Eutectic molten salt electrolysis | Voltage range: 1.5–2 V | 59.30% (molten chloride), 87.70% (molten hydroxide), 99% (molten carbonate) | Variable product rates, CO2 conversion varies with molten electrolyte type | Longer-chain hydrocarbons are exclusively produced within molten carbonates at 1.5 V. Promising for sustainable hydrocarbon fuel production, providing a foundation for fundamental investigations. Adequate heating values of produced fuels for subsequent applications | 29 |
| Temperature range: 225–475 °C | |||||
| Electrolyte: titanium cathode, graphite anode |
2.1. Advancements in small organic molecule electrolysis
In addition to water electrolysis, small organic molecule electrolysis, such as methanol electrolysis and urea electrolysis, has shown significant advancements. Recent studies have demonstrated remarkable progress in green hydrogen fuel production using these methods. The research on MoP-NC nanosphere-supported Pt nanoparticles showcases significant improvements in methanol electrolysis,30 with the Pt/MoP-NC catalyst achieving a peak current density of 90.7 mA/cm2 for methanol oxidation, 3.2 times higher than commercial Pt/C catalysts. It also exhibits excellent hydrogen evolution reaction (HER) performance with a low overpotential of 30 mV to achieve 10 mA/cm2, reducing energy consumption by about 60%. Similarly, the Pt–CoTe/NC catalyst shows a peak current density of 102.2 mA/cm2 for methanol oxidation and a small overpotential of 28 mV for HER,31 significantly lowering the cell potential required for methanol electrolysis compared to water electrolysis. The integration of Pt and CoSe2 in another study further enhances catalytic activity and durability for methanol and ethanol electro-reforming, with peak current densities reaching 87.61 mA/cm2 for methanol and 48.27 mA/ cm2 for ethanol, demonstrating high stability and efficiency.32Additionally, the NiS2–MoS2 hetero-nanorods catalyst, leveraging high valence state Ni and Mo synergism, exhibits a current density of 103.41 mA/cm2 at 1.54 V for urea-assisted water electrolysis, reducing the cell potential by 224 mV compared to general water electrolysis.33 Another promising catalyst, the 15% Ni/ZrO2 synthesized via the wetness impregnation method, shows high conversion rates of methane (62.9%) and carbon dioxide (64.9%) in the dry methane reforming (DRM) process at 750 °C with a gas hourly space velocity (GHSV) of 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL h−1 gcat−1, indicating its potential for efficient hydrogen production with enhanced stability.34 The NiSe2/MoSe2 heterostructured catalyst demonstrated optimized interfacial electron redistribution and enhanced urea adsorption energies,35 achieving a current density of 10 mA/cm2 at a potential of 1.33 V for urea oxidation, and requiring a cell voltage of only 1.47 V for the urea–water electrolyzer to drive 10 mA/cm2, highlighting significant energy savings.
000 mL h−1 gcat−1, indicating its potential for efficient hydrogen production with enhanced stability.34 The NiSe2/MoSe2 heterostructured catalyst demonstrated optimized interfacial electron redistribution and enhanced urea adsorption energies,35 achieving a current density of 10 mA/cm2 at a potential of 1.33 V for urea oxidation, and requiring a cell voltage of only 1.47 V for the urea–water electrolyzer to drive 10 mA/cm2, highlighting significant energy savings.
In line with these advancements, the conversion of formaldehyde (HCHO) using a direct formaldehyde fuel cell achieved remarkable results, with a selectivity greater than 99% and a faradaic efficiency of 200%. Similarly, methanol (CH3OH) co-electrolysis with water demonstrated a promising reaction potential of approximately 1.3 V vs. RHE, producing hydrogen and formate with a faradaic efficiency exceeding 95%. The co-electrolysis of urea (CO(NH2)2) and water also showed significant potential, resulting in the production of hydrogen and carbon monoxide with a reaction potential of around 1.3 V vs. RHE. Moreover, the use of nickel-based catalysts, such as Ni(OH)2, significantly enhanced the electro-oxidation of methanol, achieving a current density increase of 8.2 times when modified with platinum (Pt).36 These results collectively underscore the potential for efficient and sustainable hydrogen and value-added chemical production using small organic molecules, while maintaining high selectivity and energy efficiency.
2.2. Electrolysis mechanism and catalyst material evolution
The water electrolysis process, fundamental for hydrogen production, involves splitting water molecules into hydrogen and oxygen using an electric current, with the primary reactions being the oxygen evolution reaction (OER) at the anode and the hydrogen evolution reaction (HER) at the cathode. Efficient electrolysis necessitates the use of catalysts to lower the overpotentials of these reactions, with Pt commonly used for HER and IrO2 or RuO2 for OER.37 However, research is actively exploring cost-effective alternatives such as transition metal oxides, phosphides, and sulfides. For instance, Ni-based catalysts, which form surface hydroxides or oxyhydroxides acting as active sites for OER, can dissolve over time, impacting catalytic activity. Fe-doped Ni2P catalysts exhibit enhanced stability and activity due to the formation of a FeNiOOH layer during operation.38 Similarly, advanced catalysts like NiCo@C/MXene/CF electrodes show superior stability and efficiency, achieving a hydrogen production rate of 18.66 mmol/cm2/h at a current density of 1.0 A/cm2, attributed to the mesoporous array of NiCo-decorated carbon nanosheets facilitating rapid gas bubble release during electrolysis. In contrast, the absence of MXene results in irregular microstructures and decreased hydrogen production efficiency of 7.46 mmol/cm2/h at the same current density.38Further, FeCoMoS@NG catalysts, used in zinc–air batteries and water splitting, demonstrate excellent stability and low cell voltages, requiring only 1.58 V to achieve a current density of 10 mA/cm2 and maintaining stability over 100 hours of operation. Rh/RhOx catalysts achieve remarkable turnover frequencies of 2.19 s−1 at 1.53 V and 20.30 s−1 at 1.63 V, reflecting a significant increase in activity compared to standard Rh catalysts and exhibiting exceptional stability over 20 hours at a current density of 50 mA/cm2.39 Additionally, a recent study developed a highly efficient water electrolysis system powered by a cellulose sponge-based hydrovoltaic power generator (CHPG) optimized for long-term stability and high performance. A single CHPG achieved an open-circuit voltage (Voc) of approximately 0.47 V and a short-circuit current (Isc) of approximately 477 μA under relative humidity of 45–50% at 25 °C.40 By connecting six CHPG modules in parallel, the system produced a Voc of 2.09 V and an Isc of 3.11 mA, sufficient to drive water electrolysis for hydrogen production. The HER was facilitated by a cobalt phosphide/nickel foam (PCO/NF) electrode, which showed a significantly lower overpotential and a Tafel slope of 85 mV/dec compared to other electrodes. For OER, the Co5O4/NiFe-LDH/NF electrode demonstrated an overpotential of 1.5 V to achieve 1 mA and a Tafel slope of 74 mV/dec. These optimized electrodes exhibited excellent long-term stability, maintaining performance over 24 hours at a current density of 100 mA/cm2. The integrated system achieved a hydrogen production rate of 81.0 μmol/h, highlighting its potential for sustainable and efficient hydrogen generation.40
Building on recent advancements, several studies have demonstrated significant improvements in hydrogen yield and production efficiency through innovative catalyst designs. For instance, Chai et al.41 reported a 71% increase in hydrogen yield using Fe nanoparticles and a 35.2% increase with NiO nanoparticles in wastewater treatments. Furthermore, they observed a 623% enhancement in hydrogenase activity with 20 mg/L Ec-NiO-NP synthesized from Eichhornia crassipes, significantly boosting hydrogen production. Similarly, Liu et al.42 showed that dual-doped Co3N electrodes achieved high current densities at low overpotentials, retaining 92.3% of their current density after 10 hours of operation. Additionally, Qian et al.43 developed Ni3N–Co3N heterointerfaces on Ni foam, achieving an overpotential of 43 mV for HER at 10 mA/cm2 and an ultralow working potential of −88 mV for HzOR, maintaining 25 mA/cm2 for 40 hours without significant decay. Moreover, Zhu et al.44 demonstrated that Ruc/NiFe-LDH catalysts achieved an industrial-scale current density of 1 A/cm2 at 0.43 V, resulting in an energy saving of 3.94 kW h m3 of H2, while maintaining performance over 120 hours at 5 A/cm2. Zhu et al.45 showcased Ni3N/Co3N nanowires, achieving a 94.6% faradaic efficiency for formate production at 1.35 V, producing 11 mmol/cm2/h of formate and 21.4 mmol/cm2/h of hydrogen, and maintaining stability for over 200 hours at 1 A/cm2. Collectively, these insights underscore the potential of advanced catalysts to significantly enhance hydrogen production efficiency. In situ and ex situ characterizations, along with advanced techniques like Density Functional Theory (DFT), provide a deeper understanding of catalyst evolution and reaction mechanisms, essential for optimizing performance and ensuring long-term stability. Comprehensive testing and analysis are crucial for addressing overpotential, efficiency, and durability in water electrolysis systems.
3. Steam electrolysis using eutectic molten hydroxides
Molten hydroxide electrolysis for water splitting presents numerous advantages. One notable advantage is that a substantial portion of the necessary energy for the electrolysis process is provided in the form of heat, proving to be a more economical option compared to using electricity.46 The exceptional high-temperature conductivity of hydroxide electrolytes improves as temperatures increase. Molten hydroxides demonstrate superior conductivity, even at lower temperatures when compared to other molten salts such as chlorides and carbonates.For instance, at a temperature of 427 °C, the conductivity of NaOH is twice that of NaNO3 at the equivalent temperature. This heightened conductivity facilitates faster reaction kinetics and minimizes energy loss resulting from electrode over-potential. Consequently, it proves advantageous for high-temperature fuel cell systems and water-splitting procedures.47
Consequently, there is a general improvement in system efficiency. Moreover, this technology eliminates the necessity for valuable catalytic metals by employing base metals to generate hydrogen gas, with the molten hydroxide acting as a catalyst instead.48 The increased operational temperature of molten salt reduces the decomposition potential of water, thereby enhancing efficiency even further. By effectively isolating the electrolysis system, sustained energy savings can be consistently attained for prolonged usage.49 Moreover, molten salt, without the need for extra heating, sustains the required temperature through the passage of current during electrolysis, provided that the system is properly insulated.50 The process can also leverage waste heat from other systems. However, the primary drawback limiting its industrial application is the requirement for a working temperature of less than 300 °C.51 According to the Carnot cycle for thermodynamic efficiency, this leads to heat wastage, preventing widespread industrial utilization.
3.1. Thermoelectric analysis of alkaline water electrolysis
In alkaline water electrolysis, a direct current is applied to water, splitting it into hydrogen and oxygen gas molecules (Fig. 1(A)). The process involves two metal electrodes and an electrolyte to facilitate separation, with the electrolyte enhancing ionic conductivity.55 Electrodes must resist corrosion, exhibit efficient electric conductivity, and possess good catalytic properties while maintaining structural integrity. A membrane is used to prevent the synthesis of hydrogen and oxygen gas into water.56 The electrochemical cell comprises electrodes, a membrane, and an electrolyte. The overall water electrolysis reaction is expressed as eqn (1).57 During electrolysis, reduction occurs at the cathode, generating hydrogen, while oxidation occurs at the anode, generating oxygen.58 The process of water electrolysis transforms electrical and thermal energy into chemical energy, which is stored as hydrogen fuel. The energy needs are in line with thermodynamic principles, with the required energy dictated by the enthalpy change (ΔH) of the process (eqn (2)).59 A portion of electric energy is utilized, as indicated by the Gibbs free energy change (ΔG), while the remaining energy is in the form of thermal energy (QE). The minimum cell voltage for electrolysis under specific pressure and temperature conditions can be expressed as eqn (3).59
for electrolysis under specific pressure and temperature conditions can be expressed as eqn (3).59| H2O → H2(g) + ½O2(g) | (1) |
| ΔG = ΔH − QE = ΔH − TΔS | (2) |
 | (3) |
 | (4) |
485 C/mol. In an ideal electrolysis process, the minimum voltage, known as the thermo-neutral voltage (ΔE°), is equivalent to the enthalpy change potential ΔEΔH as highlighted in eqn (4). For an actual electrolysis process, must be greater than ΔEΔH, representing additional energy caused by thermodynamic irreversibility, primarily associated with water vapour in the hydrogen and oxygen streams.60 Thermodynamic properties under standard conditions (1 atm and 25 °C) provide insights into the feasibility of the process, including enthalpy change (ΔH°), Gibbs free energy change (ΔG°), entropy change (ΔS°), and cell voltage  .61 These insights shed light on the energy transformations involved in generating hydrogen and oxygen gases.
.61 These insights shed light on the energy transformations involved in generating hydrogen and oxygen gases.
 | ||
| Fig. 1 (A) Traditional electrocatalytic water splitting in alkaline environments. (B) (a) X-ray powder diffraction (PXRD) patterns of α-Ni(OH)2 obtained from the nonwoven fabric (NF) before (above) and after (below) oxygen evolution reaction (OER). (b) Transmission electron microscopy (TEM), (c) atomic force microscopy (AFM), and (d) high-resolution TEM images of α-Ni(OH)2. (C) (a) TEM and (b) HRTEM images of α-Ni(OH)2 after OER. High-resolution X-ray photoelectron spectroscopy (XPS) spectra for (c) Ni 2p and (d) O 1s of α-Ni(OH)2 before (above) and after (below) OER. The original data is represented by the black curves in panels (c) and (d). (D) Scanning electron microscopy (SEM) images at different magnifications of platinum nanoparticles (Pt NPs) electrodeposited on self-supporting electrodes manufactured at various electrolyte levels (EL): (a and b) 0.05 V, (c and d) −0.2 V, (e and f) −0.35 V, and (g and h) −0.5 V. Edited with permission from ref. 52–54. Copyright ACS ©2018, ©2021 and Elsevier ©2022. | ||
3.2. Alkaline water electrolysis and efficiency analysis
The functioning mechanism of an alkaline water electrolysis cell is characterized by a straightforward configuration consisting of a cathode, anode, electrolyte, and power supply. The application of direct current (DC) maintains electron flow from the negative compartment to the cathode, where hydrogen gas is produced by consuming electrons from hydrogen ions.62 Simultaneously, hydroxide ions migrate towards the anode within the electrolyte, ensuring charge balance.63 Commonly used electrolytes, such as potassium hydroxide, enhance solution conductivity and prevent corrosion loss. Nickel, recognized for its cost-effectiveness and high efficiency, is commonly used as electrode material.64 In their investigation, Cao et al.52 delved into the electrocatalytic properties of α-Ni(OH)2 on a self-supporting electrode for the oxygen evolution reaction (OER), aiming to optimize preparation conditions for heightened electrocatalytic activity and stability. The study employed various measurements and characterizations to assess the catalyst's performance, revealing structural and morphological changes pre- and post-OER. For instance, Fig. 1(B) illustrates PXRD patterns and images of α-Ni(OH)2 from different electrochemical conditions, indicating similar crystalline α-Ni(OH)2 generation.52 Furthermore, Fig. 1(B) also displayed TEM, AFM, and HRTEM images, providing visual evidence of the material's morphology and structure. Additionally, high-resolution XPS spectra indicated the conversion of α-Ni(OH)2 to β-Ni(OH)2 during OER, with a portion converting into NiOOH, the active OER species.52 Similarly, Fig. 1(C) presented TEM and HRTEM images post-OER, along with XPS spectra, clarified the post-reaction composition and phase transition. The observed phase transition, marked by the emergence of NiOOH, substantiated the catalyst's heightened catalytic activity after OER.52In a related study, Zhang et al.65 aimed to creat self-supporting electrodes (SSEs) for use in direct methanol fuel cells (DMFC) and direct alcohol fuel cells (DAFC) by utilizing a simple square-wave potential (SWP) method to electrodeposit platinum nanoparticles (Pt NPs) onto carbon paper.65 The study targeted an enhancement in the utilization rate and catalytic durability of Pt in these fuel cells. Specifically, Fig. 1(D) showcased SEM images of the electrodeposited Pt NPs on SSEs prepared at different lower potentials, revealing morphological changes in Pt NPs with varying potentials (E). For example, At E = 0.05 V, smooth sphere-like Pt NPs were observed. However, as the potential negatively shifted to −0.2 V, the PtNPs surface became rough, presenting a cauliflower-like morphology.65 At E = −0.35 V, observations revealed platinum nanoparticles (Pt NPs) resembling coral formations, with surface protrusions. Finally, at E = −0.5 V, thorn-like Pt NPs were formed.65 These outcomes suggest that managing the potential had a notable effect on the quantity, distribution, and structure of the electrodeposited Pt NPs on carbon paper, potentially influencing the catalytic activity and endurance of the Pt catalysts in fuel cells.
In the process of water electrolysis, hydrogen ions are drawn towards the cathode, while hydroxide ions are directed towards the anode. Meanwhile, gas receivers gather hydrogen and oxygen generated at the cathode and anode, respectively, with a membrane separating them. T notably, the combined potential of the reactions is −1.23 V, indicating the theoretical cell voltage for the procedure. However, various barriers must be overcome, including electrode surface boundary layers, electrode and electrolyte phases, separator, electrical resistance, and activation energies, analyzed in thermodynamic, kinetic, and transport process principles contexts.64 Specifically, resistance in a water electrolysis system includes external electrical circuit resistance (R1), anode overpotential (Ranode), resistance due to oxygen bubbles (Rbubble of O2), electrolyte resistance (Rions), membrane resistance (Rmembrane), resistance due to hydrogen bubbles (Rbubble of H2), cathode overpotential (Rcathode), and electrical resistance at the cathode  . Therefore, the total resistance is expressed in eqn (5).66
. Therefore, the total resistance is expressed in eqn (5).66
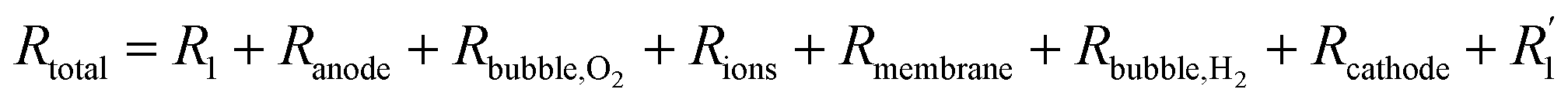 | (5) |
 | (6) |
 | (7) |
4. Thermodynamics assessment and integration of alkaline electrolysis
The electrolyte concepts depicted in Fig. 2(A) play an essential role in shaping the thermodynamics of alkaline water electrolysis cells. These concepts, namely porous diaphragms, anion-exchange membranes (AEM), and materials with narrow electrolyte-filled channels, offer diverse approaches to improve the effectiveness and functionality of alkaline electrolysis cells.69 Porous diaphragms, illustrated in Fig. 2A(a), are crafted from poly(arylene ether sulfone) combined with inorganic hydrophilic particles, creating a highly porous composite structure featuring denser surface layers. These separators aid in establishing ionic conductivity between electrodes while reducing the mixing of product gases.69 The impact on the thermodynamics of alkaline electrolysis cells is notable, particularly in influencing gas crossover and the efficiency of gas discharge. Anion-exchange membranes (AEM), illustrated in Fig. 2A(b), are composed of polymer chemistries with covalently linked cationic properties, typically employing quaternary ammonium-functionalized groups.69 AEMs contribute to improved ion conductivity, reduced gas crossover, and enhanced chemical and mechanical stability, thereby influencing the thermodynamics of alkaline electrolysis cells positively. Fig. 2A(c) introduces the concept of highly alkali-resistant materials with narrow electrolyte-filled channels.69 This involves separators made of such materials, striving to merge superior conductivity with exceptional hydrogen (H2) barrier properties and durability. The potential impact on the thermodynamics of alkaline electrolysis cells includes reduced ohmic losses, suppressed gas crossover, and efficient gas discharge. | ||
| Fig. 2 (A) Illustration demonstrating various electrolyte concepts in distilled water and in combination with aqueous KOH, including (a) porous diaphragms, (b) anion-exchange membranes (AEM), and (c) ion-solvating membranes. (B) Visual representation outlining the sequential steps of high-temperature water electrolysis. (C) Representation of cell potential for hydrogen gas production through the water electrolysis process as a function of temperature. (D) Schematic depicting the electrical double layer near a negatively charged electrode surface. (E) Depiction of the effect of potential change on Gibbs energy: the overall relationship between energy change and reaction state. (F) Enlarged view of the highlighted section in (E). Reproduced with permission from ref. 69–73. Copyright ACS ©2023 and 2019, Elsevier ©2022, 2015, ACS ©2020 and Springer ©2023. | ||
Connecting this insight to the broader context of the section discussing equilibrium potential and thermodynamics, it's evident that these electrolyte concepts play a pivotal role in influencing overpotentials, ion conductivity, and overall efficiency in water electrolysis.69 The interaction among these ideas and the thermodynamic principles deliberated upon provides insights into the complex factors influencing the functionality of alkaline electrolysis cells. The equilibrium potential is derived by deducting the cathode electrode's equilibrium potential from that of the anode electrode, as outlined in eqn (8).74 The equilibrium cell potential correlates with the Gibbs free energy alteration of the entire cell reaction, as delineated in eqn (9).74 In the case of a spontaneous cell reaction, the driving force possesses a negative value of ΔGcell, articulated as eqn (10).74
 | (8) |
 | (9) |
| ΔGcell = ∑ΔGproduct − ∑ΔGreactant = ΔHcell − TΔScell | (10) |
During high-temperature water electrolysis, several reaction steps occur as highlighted in Fig. 2(B). Initially, H2O adheres to the perovskite surface or the three-phase boundary (TPB), leading to the formation of an OH− ion by relinquishing a hydrogen atom. Subsequently, the OH− ion sheds another hydrogen atom, yielding O2− ions, while the liberated hydrogen atoms merge to generate hydrogen gas.78 The generated O2− ions move towards the YSZ phase and traverse the YSZ electrolyte's interior towards the anode side, where they unite to generate oxygen gas. These steps are influenced by factors such as electrode material properties, TPB nature, and microstructure. It's important to highlight that the conversion of water into hydrogen and oxygen is not thermodynamically favourable, necessitating an external supply of electrical energy to initiate the cell reaction.79 The cell potential must exceed the applied potential, and reactions can vary in speed, with some requiring overpotential for the necessary current density.80 The kinetics discussed earlier, reveal an increase in overpotential with rising current density. Additional energy introduces a potential drop influenced by electrolyte properties, electrode shape, and cell design. As expressed in eqn (11), the cell potential consistently ranges between 1.8–2.0 V, with a current density value between 0.001–0.003 A/cm2 for an industrial water electrolysis system.80
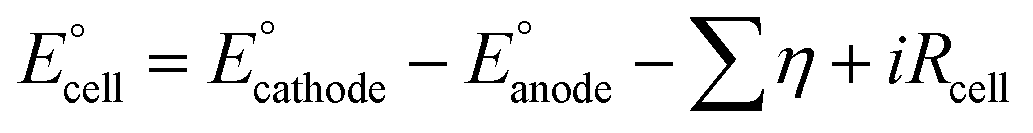 | (11) |
The efficiency of electrolysis can be assessed using different parameters. One crucial indicator is voltage efficiency, which signifies the proportion of the actual voltage employed for water splitting compared to the total voltage applied to the cell. This relationship is articulated in eqn (12).85 Thermal efficiency, as delineated in eqn (13) is grounded in the energy variations of the water electrolysis reaction.85 It covers the complete thermal equilibrium of the process. Another approach to assessing efficiency involves evaluating the rate of hydrogen gas generation compared to the total electrical energy input to the electrolysis system, as depicted in eqn (14).85
 | (12) |
 | (13) |
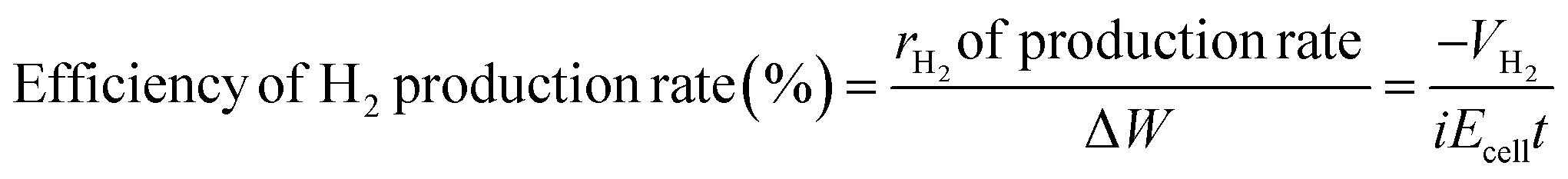 | (14) |
4.1. Electrochemical kinetics of reference electrodes
The electrode reaction rate, determined by current density, relies on the electrode surface nature and pre-treatment. The electrolytic solution composition near the electrodes influences the reaction rate, forming a double layer of ions.88 For instance, a charge layer near the cathode involves hydroxyl and potassium ions during electrolysis. Moreover, the reaction rate is contingent upon the electrode potential, denoted by the reaction overpotential. Investigating electrode kinetics aims to elucidate the macroscopic relationship between current density, the composition of the electrolytic solution near the electrode and surface overpotential.89 In Fig. 2(D), the double layer is depicted, characterized by two mobile layers with accumulated ions and solvent molecules.90 The layer proximal to the electrode, termed the Inner Helmholtz Layer (IHL), exhibits a relatively ordered structure, while the Outer Helmholtz Layer (OHL) is less organized. Ionic countercharges in the vicinity balance the electrical charges on electrode surfaces. The plot in Fig. 2(D) illustrates the potential distribution with respect to the distance from the electrode surface, revealing an interfacial potential variation resulting from the double layer.39 This non-faradic mechanism plays a role in the capacitive characteristics of electrode reactions, which is an important aspect of electrode kinetics. The Butler–Volmer equation illustrates the relationship between current density, surface potential, and the electrolytic solution composition near the electrode surface.91 This equation can be expressed as a one-electron reaction, linking the current to the rate as shown in eqn (15).91| i = icathodic − ianodic = FAK°(C0(0,t)e−αf(E−E°) − CR(0,t)e(1−α)f(E−E°)) | (15) |
The Butler–Volmer equation describes the connection between current density, surface potential, and the composition of the electrolyte near the electrode surface. Expressed as a one-electron reaction, it involves parameters such as A (electrode surface area), K° (typical rate constant), α (transfer coefficient ranging from 0 to 1 for a single-electron reaction), and f (the  ratio). The variables “t” and “0” within the brackets represent the particular time and distance from the electrode, respectively.91
ratio). The variables “t” and “0” within the brackets represent the particular time and distance from the electrode, respectively.91
The Butler–Volmer equation, derived through the transition-state theory, utilizes curvilinear coordinates along reaction pathways (Fig. 2(E)), where potential energy varies as a function of independent coordinates within the system. An increase in potential (ΔE) results in a reduction in the electron's relative energy by F(E − E°) (as shown in Fig. 2(F)), subsequently influencing the Gibbs free energy of hydrogen ions and hydrogen in the reaction as highlighted in Fig. 2(F).92 This behaviour does not impose any restrictions on mass transfer. The Butler–Volmer equation (eqn (16)) can be simplified as follows.92 Where i0 represents the current density of exchange, which is defined as the current related to the reversible process of water cleavage. The overpotential at each electrode is derived from this simplified equation.92 Neglecting one term in the total resistance equation is permissible in the absence of mass transfer influence and at large over-potentials (>118 mV at 25 °C), i.e., when e−αfη > e−(1−α)fη. The relation between i and η(E − E°) is represented by the Tafel eqn (17).92
| i = i0(e−αfη − e−(1−α)fη) | (16) |
| η = a + blogi | (17) |
 and
and  . The linear relationship between overpotential and the logarithm of current density is defined by slope (b), where α represents the exchange current density i0, also recognized as the Tafel slope. In conclusion, the electrolysis rate is directly determined by current or current density, which, in turn, depends on various factors, including overpotential influenced by activation energy (EA).93 The electrode material plays a crucial role in minimizing activation energy, making it a primary focus in research alongside optimizing electrode materials and surface configurations.
. The linear relationship between overpotential and the logarithm of current density is defined by slope (b), where α represents the exchange current density i0, also recognized as the Tafel slope. In conclusion, the electrolysis rate is directly determined by current or current density, which, in turn, depends on various factors, including overpotential influenced by activation energy (EA).93 The electrode material plays a crucial role in minimizing activation energy, making it a primary focus in research alongside optimizing electrode materials and surface configurations.
4.2. Enhancing molten hydroxide electrolysis under diverse conditions
In this section, the intricacies of anodic and cathodic reactions within a molten hydroxide electrolysis cell are investigated, aiming to unravel the nuanced interplay under diverse process conditions. Influencing these orchestrated reactions are critical factors such as salt compositions, temperature, atmosphere, pressure, and electrode materials, each leaving a distinctive imprint. The consequential variable, hydrogen gas production, showcases considerable variability under these conditions, providing a rich terrain for exploration. Choices span from room temperature to high-temperature scenarios, and from aqueous to molten hydroxide salt environments. Meticulously documented in Table 2, the conditions narrate the stories of low-temperature processes in ambient air and high-temperature counterparts immersed in argon, ammonia, or nitrogen atmospheres, capturing developments from the past until the present.| Composition of electrolyte | Temperature (°C) | Atmosphere | Reference electrode | Working electrode | Crucible material | Anode | Cathode | Applied voltage (V) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| NaOH | 550 | Ar | Ni rod | Pt wire, Ni wire, and Ni pallet | Ni | — | — | — | 94 |
| NaOH–KOH | 200 | Ar | Pt wire | Pt, Au, Fe rod | Platinum | — | — | — | 95 |
| NaOH–KOH (50:50) |
347 | N2 | Cu plated Platinum sheet | Pt | Vitreous carbon crucible | — | — | — | 96 |
| LiOH–NaOH, LiOH–KOH, NaOH–KOH | 250–300 | — | Ag/AgCl | Pt, Pd, Ni, Ag, Al | Nickel crucible | — | — | — | 97 |
| KOH | 110–160 | — | Fe | Fe, FeSi, and FeC | PTFE | — | — | — | 98 |
| KOH | 35–400 | Steam | — | — | Ni 400 | Ni, Ni400, Co plated Ni | Ni 400 | 99 | |
| KOH, NaOH, Ba(OH)2, LiOH | 200–700 | Ar | — | — | Alumina | Ni, Pt and lithiated Ni | Ni | 1.1–2.3 | 100 |
| NaOH | 25 | Air | SCE | Cu rod | — | — | — | — | 101 |
| NaOH (8.0 M) | 90 | Hg/HgO/1 M NaOH | Co–Mo alloy | — | — | — | — | 102 | |
| KOH (10 wt%) | 20, 40, 60 | Air | — | — | Vinyl chloride | Ni–Cr–Fe alloy | Ni–Cr–Fe alloy | 6 | 103 |
| NaOH–NaHS (50:50) |
80 | Air | Hg/HgO (1 M NaOH) | — | Glass | Graphite, Ni, Ni–Cr alloy | Ni, graphite | — | 104 |
| NaOH | 320–400 | Argon | —— | — | — | Ni | Ni | 105 | |
| Aqueous, KOH–NaOH, LiOH–NaOH | 110–140, 350, 300–400 | Argon | — | — | — | Activated Ni | Activated Ni | 1.55,1.3, 1.45 | 106 |
| KOH (1.0 M) | 25 | Air | Ag/AgCl | C-felt supported Ni, Co, Ni–Co | — | — | — | — | 107 |
| NaOH (0.1–5.0 M) | 25 | Ag/AgCl | Ni–Mo–Fe coated stainless steel | — | — | — | — | 108 | |
| NaOH–KOH (57:43) |
200–220 | Ar or NH3 | Ag wire | Pt plate | Ni-400 (commercial Monel® alloy) | — | — | — | 109 |
| NaOH | 530 | Ar | —— | — | Ni-covered by alumina | Ni | FeO | 1.7 | 110 |
| KOH (50 wt%) | 80, 150, 208, 264 | — | (DHE) | Ni | —— | — | — | 111 | |
| NaOH–KOH (51:49) |
200–450 | NH3 | — | Ni tube | Ni-400 (commercial Monel® alloy) | —— | — | — | 112 |
| NaOH–KOH (50:50) |
200 | N2 and steam | — | — | — | Planar Ni | Mesh Ni monel | 1.2 | 113 |
| NaOH | 550 | O2 + air | Ag wire | Ni alloy | Alumina | — | — | — | 114 |
| NaOH–KOH (54:46) |
400 | O2 + air | Ag wire | Ni alloy | Alumina | —— | — | — | 114 |
| NaOH (8.0 M) | 70 | Air | Hg/HgO | Ni | Plastic | — | — | — | 115 |
In 1976, Miles et al.116 demonstrated a temperature-dependent influence on electrode kinetics, influencing the oxygen evolution reaction to a greater extent compared to the hydrogen evolution. Subsequently, Divisek et al.117 expanded this inquiry to molten NaOH, achieving an efficiency of approximately 38–39% in water electrolysis at elevated temperatures. Furthermore, in a further study, Divisek et al.118 broadened their scope to the electrolytic splitting of water in aqueous KOH and molten hydroxides (NaOH, LiOH–NaOH) at different temperature ranges. This research achieved a notable current efficiency of 90% in a NaOH melt at 350 °C, revealing a unique side reaction of peroxide production, which was notably diminished within a LiOH–NaOH melt. Moreover, Anani et al.119 demonstrated the potential for hydrogen production by electrolyzing sulfur hydrogen at 80 °C in an equimolar aqueous solution of NaOH. A novel approach was taken by Abouatallah et al.,120 investigating the addition of soluble V2O5 to an 8 M KOH aqueous solution at 70 °C to reactivate a nickel cathode during hydrogen evolution. While the V2O5 additive proved effective, the electro-catalytic activity of vanadium-modified nickel did not surpass that of a fresh nickel electrode.
Additionally, Miles et al.121 explored various eutectic molten hydroxides (NaOH–KOH, LiOH–KOH, LiOH–NaOH) with different working electrodes. Proposing Ag/AgCl as a reference electrode due to its swifter reaction kinetics, the study emphasized the applicability of molten hydroxides in thermal battery applications, underscoring the need to minimize H2O and O2 for the effective use of lithium or sodium anodes. Nagai et al.122 investigated the effect of bubble formation between electrodes on water electrolysis efficiency. Using a 10 wt% KOH aqueous solution and Ni–Cr–Mo alloy electrodes, they found that adjusting the space between electrodes influenced the void fraction, impacting electrolysis efficiency. Similarly, Zabinski et al.123 investigated augmenting hydrogen evolution by incorporating carbon into the Co–Mo alloy cathode. They conducted experiments in an 8 M NaOH solution at a temperature of 90 °C. Despite increased hydrogen evolution activity, preventing molybdenum dissolution proved elusive. Híveš et al.124 pioneered the electrochemical production of ferrate(VI) in eutectic molten hydroxide NaOH–KOH at 200 °C. Ferrate(VI) formation was detected at the inert electrode in contact with ferrate(III) ions and an iron electrode.
Simultaneously, Jayalakshmi et al.125 explored the electrochemical catalytic activity of a stainless steel substrate coated with a composite film of Ni–Mo–Fe in alkali solutions, showcasing improved hydrogen evolution activity. The potential for ferrate(VI) production was further explored by Híveš et al.,126 using a NaOH–KOH eutectic hydroxide at temperatures between 170–200 °C with a stationary iron electrode. The formation of ferrate(VI) at the anodic oxidation of the iron electrode was confirmed, achieving a current efficiency of Fe(VI) formation of up to 72%. In a different domain, Ganley et al.127 pioneered a direct ammonia fuel cell using eutectic molten hydroxides (NaOH–KOH) at temperatures from 200 to 450 °C, achieving a maximum power density of 40 mW/cm2 at 450 °C. On a separate note, Cox and Fray128 effectively converted iron(III) oxide to iron in molten sodium hydroxide at 530 °C, accomplishing a current efficiency of around 90%. In addition, Ganley129 extended the investigation, examining the impacts of elevated temperature and pressure on alkaline electrolysis, employing concentrated KOH at 400 °C across different pressure conditions. The research utilized nickel Monel as a cathode and evaluated different anode materials. It determined that the most optimal cell performance was achieved when employing a cobalt-plated nickel anode at 400 °C and under a steam partial pressure of 8.7 MPa.
Continuing the journey to enhance hydrogen evolution, Döner et al.130 employed a 1.0 M KOH water solution at ambient temperature. They supported cathodes made from various materials (Ni, Co, and Ni–Co alloy) with C-felt coating, resulting in significant increases in current densities, particularly with the C-felt/Ni–Co combination recording the highest current density. Extending the versatility of molten hydroxide, Guo et al.131 demonstrated a direct carbon fuel cell with enhanced performance up to 400 °C by employing the eutectic mixture as an electrolyte. Moreover, Hrnčiariková et al.132 contributed to this area by examining how the composition of the anode affects the electrochemical production of ferrate(VI) using molten KOH. They analyzed three distinct anode materials in molten KOH: pure iron (Fe), white cast iron (FeC), and silicon-rich steel (FeSi) electrodes. The research unveiled the concurrent existence of ferrate(VI) formation alongside significant oxygen evolution. Recent advancements include Yang et al.'s platinum electrode, achieving a maximum power of approximately 16 mW/cm2.133 Licht et al.100,134 in their studies demonstrated ammonia production feasibility while observing a decrease in hydrogen gas evolution efficiency at high temperatures. The hydrogen gas evolution efficiency of the mixed hydroxide electrolyte (NaOH–KOH; 50–50 mol%) dropped from 96% to 13.4% with increasing temperature from 200 to 600 °C, as illustrated in Fig. 3(A). Ultimately, the evolving narrative illustrates the interconnected nature of these studies, providing insights into the intricate world of molten hydroxide electrochemistry.
 | ||
| Fig. 3 (A) The efficiency of hydrogen production as a function of temperature and the selected hydroxide electrolyte under atmospheric pressure was assessed at 1 A between two flat nickel electrodes. (B) A cyclic voltammogram of a nickel wire electrode in the NaOH melt at 550 °C was conducted at a scan rate of 100 mV/s. The reference electrode was a nickel wire, with the potential limited between −0.4 V and 1.3 V. (C) A cyclic voltammogram of a platinum wire electrode in the NaOH melt at 550 °C was conducted at a scan rate of 100 mV/s. The reference electrode was a nickel wire, with the potential limited between −0.4 V and 1.3 V. (D) Cyclic voltammograms of a platinum electrode in NaOH–KOH at 200 °C were performed at a scan rate of 20 mV/s, using argon gas or ammonia as the atmosphere, and with Ag reference electrode. (E) (a) Cyclic voltammograms of a Pt plate electrode under Ar or NH3 at 200 °C (sweep rate: 20 mV/s); (b) an enlarged view of the profiles between −0.75 and −0.35 V from (a). Edited with permission from ref. 100, 109 and 135. Copyright IOPscience ©2016, 2015 and Elsevier ©2014. | ||
5. Electrochemical stability of eutectic molten hydroxide electrodes
5.1. Voltammetric investigation of nickel electrode
Many reactions in molten salt electrolytes have been elucidated through cyclic voltammetry, the recognized as a powerful tool for comprehending electrochemical processes at electrode interface. Cyclic voltammetry plays a pivotal role in shedding light on the fundamental aspects of reactions and contributing to the comprehensive exploration of electrolytic systems.136 In an insightful investigation led by Wang et al.,137 the behaviour of an in-use nickel electrode immersed in molten NaOH–KOH at 280 °C was examined in depth. The outcomes, revealed through cyclic voltammetry, showcased a visually striking result a distinctive black film materialized on the nickel electrode's surface. This captivating phenomenon was attributed to the oxidation process, converting NiO to NiO2, as succinctly expressed in eqn (18).| NiO + 2OH− → NiO2 + H2O + 2e− | (18) |
Litch et al.138 explore alternative molten electrolytes for water splitting to generate hydrogen fuel. Their study demonstrates that the electrolysis potential needed for water splitting at 400 °C in molten NaOH is remarkably low, measuring below 1.4 V (Fig. 3(B)). Under these circumstances, elevated current densities, potentially reaching A/cm2, are readily attainable. Anticipations suggest that utilizing reticulated or textured electrodes with enhanced microscopic or nanoscopic surface areas may further decrease electrolysis overpotentials. Anodes composed of Ni, Pt, monel, and Ir, paired with Ni cathodes, demonstrate notable stability, maintaining effective current densities of 1 A/cm2 under conditions where both anode and cathode limitations are present.138 Notably, at low current density, the electrolysis potential notably drops to less than 1 V in 700 °C LiOH, even yielding some hydrogen gas. In a parallel study, Ge et al.57 delved into the oxidation of nickel-to-nickel oxide in a one-step reaction within molten NaOH, albeit at a higher temperature of 550 °C. This transformative process was succeeded by a secondary reaction involving hydrogen evolution. Fig. 3(C) vividly depicts the cyclic voltammogram of a nickel wire working electrode in molten NaOH at 550 °C, revealing clear redox peaks. The oxidation peak O3 indicates the oxidation of the Ni wire in NaOH, leading to the creation of a thin oxide film on the Ni surface. Concurrently, peak C3 captures the reduction of the oxide C3. Noteworthy is the intensified current observed at the cathode in C2 and at the anode in O2, corresponding to the evolution of hydrogen and oxygen gas, respectively, as articulated in eqn (19) and (20).
| 2H2O + 2e− → H2(g) + 2OH− | (19) |
| 2OH− → ½O2(g) + H2O + 2e− | (20) |
Transitioning to another facet of research, Sayed et al.139 conducted an extensive investigation on an independent nickel-layered double hydroxide characterized by a hierarchical nanosheet architecture. This unique structure, synthesized efficiently on nickel foam via hydrothermal treatment and later transformed into NiO at 500 °C, demonstrated impressive activity in methanol oxidation.139 The synthesized electrodes displayed an onset potential of 0.35 V in a 1 M KOH solution, with the nanosheet structure enhancing charge and mass transfers, resulting in superior overall activity. Notably, these electrodes demonstrated prolonged stability during extended oxidation activity, maintaining a discharge current at 0.5 V for more than 1 hour without any decline in performance represents a noteworthy improvement compared to bare nickel foam.139 These findings emphasize the effectiveness of nickel-layered double hydroxide and nickel oxide electrodes as anodes in alkaline direct methanol fuel cells, suggesting substantial promise for energy conversion systems.
Additionally, Al-Shara et al.140 made a valuable contribution to the field by developing a novel reference electrode, Ni/Ni(OH)2, specifically designed for electrolysis in eutectic molten hydroxides. This electrode was manufactured by employing a eutectic molten hydroxide (NaOH–KOH; 49–51 mol%) at a temperature of 300 °C.140 Cyclic voltammetry tests were conducted to evaluate its performance, revealing exceptional stability and reusability. In a comparative analysis with platinum and silver quasi-reference electrodes, the Ni/Ni(OH)2 electrode emerged as a promising candidate, demonstrating suitability, stability, reproducibility, and reusability as a reference electrode in a molten hydroxide electrolyte. In summary, these crucial findings underscore the potential of the Ni/Ni(OH)2 electrode as a stable and efficient reference electrode for electrolysis in eutectic molten hydroxides.140
5.2. Electrochemical capability platinum electrode in molten hydroxide
In the captivating domain of electrochemical exploration, the platinum electrode assumes the spotlight. A meticulous cyclic voltammetry analysis at 550 °C, immersed in molten NaOH, unveils the nuanced interplay of redox peaks, symbolic of the reduction of a delicate oxide film enveloping the platinum wire's surface.135Fig. 3(D) presents the cyclic voltammograms from Ge et al.'s study135 employing platinum as the working electrode. Each peak narrates a unique story: the cathodic current peak C1 signifies the poetic reduction of the oxide film; the captivating surge in cathodic current at C2 (−0.4 V) unfolds a ballet of hydrogen gas evolution; the anodic current peak O1 depicts the stoic oxidation of the oxide film, and the enchanting O2 serves as a crescendo harmonizing with the birth of oxygen gas. The saga continues beyond platinum, venturing into the realm of noble nickel. Its cyclic voltammetry narrative in molten NaOH reveals a tapestry of redox peaks akin to its platinum counterpart. The cathodic sonnet at C3 serenades the reduction of a wispy oxide film caressing the nickel surface. Meanwhile, the anodic peak O3 resonates with the bold oxidation of the nickel wire in the molten embrace. However, a twist in potential scan limits between 0.3 V and 1.3 V dims the presence of O3, creating an interlude where the generated nickel oxide films seek refuge from reduction in the first cycle.135 Both platinum and nickel trace redox peaks in molten NaOH, a testament to the oxide tales etched on their surfaces. Yet, nuances emerge, with platinum showcasing distinctive choreography of reduction and oxidation peaks, while nickel pirouettes with reduction peaks for the oxide film and an ode to oxidation for the nickel wire. Specific potential ranges embellish each peak with uniqueness.Enter Ge et al.,135 pioneers in this symphony. Cyclic voltammetry, the maestro's baton, gracefully wielded on a platinum electrode basking in molten NaOH at 550 °C. A material chosen for its chemical steadfastness, platinum graced the stage with cyclic voltammograms. The cathodic reverie, C1, whispers the reduction of a platinum-clad oxide film; O1, anodic in nature, resounds oxidation. C2's crescendo echoes the hydrogen evolution reaction, while O2, a sonnet, harmonizes with the oxygen evolution reaction. The saga extends as Ge et al.135 venture into the realms of platinum wire, a virtuoso in three molten hydroxides NaOH–KOH at 280 °C, LiOH–NaOH, and LiOH–KOH at 270 °C. In NaOH–KOH's embrace, a cathodic crescendo at −0.32 V, a ballet of superoxide ions (O−2) reduction, unravelled. Cyclic voltammetry gracefully revealed the nuances of a platinum (Pt) electrode in a molten hydroxide electrolyte, a narrative skillfully crafted by Yang et al..109 Reduction and oxidation peaks pirouetted elegantly at distinctive potentials, with reduction currents exhibiting their dance below −0.55 V. The stage was then seized by a commanding oxidation peak between −0.55 V and 0.1 V, rising dramatically above 0.17 V, where the evolution of oxygen took center stage.109
In the presence of ammonia (NH3), an ethereal onset potential of approximately −0.67 V marked the beginning of anodic currents, with a crescendo leading to a maximum of around −0.2 V. However, the drama unfolded swiftly above 0.15 V as oxygen evolution claimed the spotlight. The forward scan from −0.2 to 0.15 V witnessed a diminishing oxidation current, a subtle interplay involving Pt oxidation and the reduction of the active surface for ammonia oxidation. In a seamless transition, Yang et al.109 continued their electrochemical tale, this time exploring the ammonia-driven drama on a platinum stage immersed in molten NaOH–KOH at 200 °C. Fig. 3(E) gracefully presented cyclic voltammograms, a visual symphony offering choices of argon and ammonia, while Ag stood as the stoic ref. 109. The dashed line served as a blank canvas, encapsulating platinum's narrative in the hydroxide embrace, while the solid line painted the enchanting transformation of ammonia to N2, all harmonized by the eutectic molten hydroxides. The platinum electrode, a versatile protagonist, showcased its prowess in every experiment, contributing a lyrical stanza to the grand saga of electrochemistry.
5.3. Voltammetry of diverse metals in molten hydroxides
Embarking on a comprehensive exploration of electrode materials in molten hydroxides, the utilization of Cyclic Voltammetry (CV), a robust electrochemical method, revealed the complexities of reduction and oxidation processes of molecular species, offering insights into chemical reactions and catalysis initiated by electron transfer.141 In Choi et al.'s investigation,142 the study explores the durability of Ag/Ag+ reference electrodes in MgCl2–KCl–NaCl molten chloride salts, providing crucial insights into their stability. Using open circuit potential (OCP) and cyclic voltammetry (CV) measurements, the research establishes the working electrode potential, ensuring the precision of cyclic voltammetry experiments. The evaluation of reference electrode materials, specifically comparing mullite and MgO in molten chloride salts, reveals in Fig. 4(A) that the pair of reference electrode tubes appear identical before and after the test, while the Mg rod undergoes dissolution/reaction, resulting in a rough surface and a tapered tip. This observation supports the assumption that the Mg rod dissolves and buffers the redox potential of the salt. Furthermore, the mullite-based reference electrode exhibits a drift rate of −1.34 mV per day, emphasizing the importance of appropriate material selection to minimize potential drift.142 Although the focus is on molten chloride salts, these findings hold valuable implications for optimizing cyclic voltammetry experiments on Ag metals in molten hydroxides. | ||
| Fig. 4 (A) Depictions of (a) mullite tube reference electrode (RE), (b) MgO tube RE, and (c) Mg working electrode (WE) following a 31 days testing period. (B) Cyclical voltammograms recorded for a Pt plate electrode under Ar or NH3 at 200 °C with a sweep rate of 20 mV/s. (C) Illustration of the synthesis process of Nb-doped Nb-NiFe-LDH via molten salt. (D) Electrochemical evaluations conducted in 1.0 M KOH, comprising (a) linear sweep voltammetry (LSV) curves for Nb-NiFe-LDH, NiFe-LDH, Nb-Ni-LDH, Ni-LDH, NF, and RuO2 on NF, accompanied by (b) corresponding Tafel plots; (c) comparative assessment of overpotential at 10 mA/cm2 and Tafel slopes compared to other catalysts; (d) calculation of the double-layer capacitance (Cdl) value from cyclic voltammetry (CV) curves at 1.30 V vs. reversible hydrogen electrode (RHE); (e) polarization curves before and after 5000 CV scans; and (f) chronopotentiometric testing at 100 mA/cm2. (E) Comparison of cyclic voltammograms for Ni, Pt, Ag, Mo, and stainless steel working electrodes in molten hydroxide at 300 °C with a scan rate of 100 mV/s, utilizing a 0.5 mm Ni/Ni(OH)2 reference electrode and a 5 mm stainless steel counter electrode with an immersion depth of 14 mm, under Ar gas at a flow rate of 40 cm3/min. Edited with permission from ref. 143–145. Copyright IOPscience ©2023, Elsevier ©2013, ©2022, and ©2020. | ||
In a separate study by Choi et al.,146 the exploration of CV in molten hydroxides, particularly with Mg metals, takes center stage. The cyclic voltammetry (CV) technique allows for the qualitative and quantitative determination of various species in the electrolyte, surpassing open circuit potentiometry. The introduction of hydroxide ions and dissolved magnesium metal to the MgCl2–KCl–NaCl salt mixture induces effects on redox potential and electrochemical behaviour. While presenting a solubility measurement of approximately 0.0205 wt% at 500 °C (Fig. 4(B)), the study acknowledges a high associated error, necessitating further understanding in future studies.143 This study significantly enhances the understanding of the electrochemical behaviour of Mg metals in molten hydroxides. Turning attention to Zhou et al.'s147 investigations, they delve into the synthesis process of Nb-doped Nb-NiFe-LDH via molten salt (Fig. 4(C)) and examine the effect of high-valence Nb doping in NiFe hydroxides on improving the oxygen evolution reaction (OER) in water splitting. The findings depicted in Fig. 4(D) underscore the remarkable electrochemical efficacy of Nb-NiFe-LDH, as demonstrated by a notable decrease in overpotential at 50 mA/cm2 from 280 mV (for NiFe-LDH) to 242 mV with Nb doping, signifying considerable advantages for the oxygen evolution reaction. This observation finds further reinforcement in the overpotential values recorded for Ni-LDH (428 mV) and Nb-Ni-LDH (342 mV). Tafel slopes reveal that Nb-NiFe-LDH (31.3 mV/dec) exhibits enhanced electron transport compared to NiFe-LDH (47.5 mV/dec), NbNi-LDH (42.8 mV/dec), and Ni-LDH (38.8 mV/dec), thus accelerating OER kinetics. Electrochemical Impedance Spectroscopy (EIS) at 1.37 V vs. RHE illustrates the commendable electrode conductivity and electron transfer rate of Nb-NiFe-LDH, as indicated by the smallest fitted semicircle diameter.147
Calculations of double-layer capacitance (Cdl) from cyclic voltammetry curves highlight Nb-NiFe-LDH's larger Cdl value (5.2 mF/cm2) compared to NiFe-LDH (4.5 mF/cm2), Nb-Ni-LDH (3.0 mF/cm2), and Ni-LDH (1.8 mF/cm2), indicating a higher exposure of effective active sites through Nb integration.147 These comprehensive findings significantly advance our understanding of electrochemical processes, providing valuable insights for optimizing cyclic voltammetry experiments in both chloride and hydroxide systems. Comparing various working electrodes (Ni, Pt, Ag, Mo, and St. st) in eutectic molten hydroxide at 300 °C using a potential scan rate of 100 mV/s, each electrode displayed a unique reduction potential value.145 Platinum had the most positive reduction potential value, followed by nickel and then stainless steel (Fig. 4(E)). Silver and molybdenum electrodes demonstrated lower reduction potential values. The obtained reduction potential values are consistent with previous literature findings, highlighting the uniqueness of each working electrode. The values, ranging from approximately −0.47 V for platinum (Pt) to −0.56 V for molybdenum (Mo), underscore the diverse electrochemical characteristics of these materials.145 In conclusion, this collective exploration sheds light on crucial aspects of electrode stability, material selection, and electrochemical behaviour in molten hydroxides, providing valuable insights for optimizing future cyclic voltammetry experiments.
6. Emerging techniques for hydrogen evolution from eutectic hydroxide molten salts
Molten salt electrolysis is recognized as a “clean technology” for extracting metals from waste materials, concurrently generating pure O2 gas. Its adaptability, particularly in noble metal recovery, capitalizes on a broad operating temperature range and an expansive electrochemical window.148 Despite its potential, widespread implementation for metal recovery faces hurdles related to cost-effectiveness. The elucidation of metal recovery methods reveals a reliance on electrodeposition, which deposits metal in pores as small as 10 nm, enabling the characterization of microscopic pore-throat systems. Additionally, electrodeposition allows for the examination of features that cannot be directly observed due to its large observation area.149 Additionally, electrorefining supplements purification, offering advantages like a wider operating temperature range, effective digestion of refractory borides and carbides, and favourable coating formation. This process enhances the purity of crude metals from approximately 99 to over 99.9%.150The versatility of molten salt electrorefining is evident in the recovery of metals such as tungsten, cobalt, manganese, zirconium, lead, uranium, silicon, and ruthenium.151 Tailored techniques like selective electrodeposition and electrorefining for specific metals such as Te, Cu, U, and actinides enhance efficiency and reduce radioactive sludge formation, contributing to overall process sustainability. Additionally, Fig. 5(A) illustrates a schematic diagram delineating the recycling process based on regenerative redox targeting for used LiFePO4 material, facilitating comprehension of the steps and compartments involved in recycling LiFePO4 material using the regenerative redox targeting method.151 Shen et al.156 explore the diverse applications of hydrogen generated in molten salts, showcasing its potential for large-scale production of advanced carbon nanostructures. The molten salt-assisted electrolysis strategy emerges as an innovative pathway for reducing CO2 and H2O into syngas and hydrocarbon fuels. Operating in molten salts as a reaction solvent, this process benefits from high thermal stability and outstanding CO2 dissolving capacity, further amplified by the integration of renewable solar energy.156 This green and efficient electrolysis process converts CO2 into valuable carbon nanostructures, leveraging rapid ion migration and diffusion, high thermal stability, and superior CO2 dissolving capacity offered by molten salts. Moreover, the method shows promise for large-scale production of advanced carbon nanostructures, capitalizing on the hydrogen generated in molten salts. Notably, the glassy carbon retains its initial characteristics post-electrolysis.156 At 650 °C, thick carbon sheets assemble from quasi-spheres, while at 750 °C, fiber-like nanostructures of hollow multi-walled CNTs measuring 20 nm in diameter and having a wall thickness ranging from 5 to 10 nm appear.156
 | ||
| Fig. 5 (A) Illustrated depiction outlining the regenerative redox targeting-based recycling process for spent LiFePO4 material. (B) Schematic representation of hydrogen production through molten salt and its subsequent application in the direct reduction of suspended Fe2O3 powders within the melt. (C) Diagrammatic representation of electrochemical CO2 fixation in molten salts utilizing a liquid Zn cathode. (a) The alumina tube sealed at one end, (b) configuration of the liquid Zn cathode, and (c) formation of Zn@C spheres with a core–shell structure within a carbon flake. (D) Overview emphasizing key aspects of the direct sustainability of metallic materials. (E) Illustration showcasing various scales concerning kinetic and thermodynamic simulations and experiments, focusing on the solid-state direct reduction of iron oxides using hydrogen. Edited with permission from ref. 152–155. Copyright ACS ©2020, Elsevier ©2023, ©2019 and ACS ©2023. | ||
Moreover, carbon materials originating from chitosan via a single-step carbonization process using LiCl–ZnCl2 molten salt exhibit a substantial specific surface area of 2025 m2/g and a significant nitrogen content of 5.1 wt%. These materials demonstrate noteworthy CO2 uptake under various temperatures and pressures.157 The conversion of PET into nitrogen-doped porous carbon using melamine and ZnCl2/NaCl eutectic salts at 550 °C yields a considerable specific surface area of 1173 m2/g and abundant nitrogen dopants. Additionally, porous biochar derived from wood waste with eutectic molten salts shows enhanced CO2 sorption, with the most efficient biochar displaying the highest CO2 uptake and selectivity.157 Furthermore, N-doped microporous carbons synthesized from melamine and sucrose using molten LiCl/KCl salts show significant CO2 uptake under specific conditions. Fig. 5(B) elucidates the specific steps and conditions integral to the borate-enhanced molten salt electrolysis process, directing the mass production of sophisticated carbon nanostructures of high worth using hydrogen produced within molten salts.152 Additionally, Fig. 5(C) illustrates a schematic representation of the process where hydrogen produced in molten salt is employed for the direct reduction of suspended Fe2O3 powders in the melt.155 This sustainable method involves the electrochemical decomposition of water in molten salt, generating hydrogen gas that reduces Fe2O3 particles to metallic iron.
Furthermore, the remarkable faradaic efficiency of approximately 94.5% in the one-pot generation of syngas through CO2 electrolysis at a low temperature of 450 °C is achieved by utilizing renewable solar energy for electricity generation and concentrated solar light for eutectic melting.152 The research also highlights achievements in the electrolysis of CO2 to produce a range of hydrocarbon fuels utilizing eutectic molten salts. Raabe et al.153 further explore the potential of electrochemical hydrogen generation in high-temperature molten salts as a versatile method for producing metals and alloys. The electrolysis procedure entails conducting an electrical current through a molten salt electrolyte containing metal ions, resulting in the deposition of metals and alloys on the cathode. During the electrochemical fabrication of graphene in molten LiCl, the procedure produced interconnected graphene nanosheets/nanoscrolls. The reduced graphene exhibited a specific surface area of 565 m2/g and a specific capacitance of 255 F/g. Even after 5000 charge/discharge cycles in 6 M KOH, it retained 95% of its capacitance.158 Additionally, the electrochemical etching process using molten salt to produce carbide-derived carbon (CDC) demonstrated encouraging outcomes. For example, employing a molten salt (CaCl2) electrochemical etching method on silicon carbide led to the creation of porous nanospheres, exhibiting a specific capacitance of 176 F/g at 1 A/g in a 6 M KOH electrolyte.159 Notably, this electrochemical method excels in producing exceptionally pure metals, utilizing sustainable electrical energy sources like wind or hydropower directly.160 This makes it an attractive alternative to conventional reduction methods reliant on fossil reductants. Moreover, integrating renewable electrical energy in electrolytic cells contributes to higher overall efficiency.
Challenges accompany this technology as the high operating temperatures required for electrolysis can induce aggressive interactions with electrodes and insulation materials. The development of carbon-free electrode materials becomes crucial to minimize CO2 emissions.161 Additionally, the variability in sustainable electrical power supply, particularly from sources such as solar or wind energy, underscores the need to design electrolysis cells capable of adapting to variable power supply and interruptions. Fraction of Renewable Energy System values exhibit variation across different scenarios and months, with a fluctuation range of 30–35%. The fluctuations mainly stem from seasonal shifts in renewable resources and changes in demand.162Fig. 5(D) provides a comprehensive summary of the crucial aspects of direct sustainability in metallic materials production, including various ores, such as low-grade and high-grade, used for synthesizing metals from mineral raw materials, as well as the utilization of scrap and waste as alternative feedstock sources.153 Additionally, Fig. 5(E) illustrates the intricate multi-scale nature of simulations and experiments related to kinetics and thermodynamics involved in the hydrogen-driven solid-state reduction of iron oxides, providing a visual representation of the process's complexity.153 In conclusion, electrochemical hydrogen generation in high-temperature molten salts offers a promising approach to the sustainable production of metals and alloys. This method utilizes renewable electrical energy sources and has different conversion efficiencies depending on the energy source used. For example, solar PV + electrolysis has a conversion efficiency of 10.5%, while wind + electrolysis, hydroelectric + electrolysis, and tidal + electrolysis has a conversion efficiency of 70%.163 These technologies are at different stages of development, as indicated by their technology maturity level (TML) ratings. Ongoing research and development efforts are essential to address challenges related.163
7. Challenges and prospects of molten hydroxide electrolysis
Molten hydroxide electrolysis, which operates at high temperatures, presents both significant challenges and promising prospects. One primary challenge is the need for materials that can withstand the highly corrosive environment created by molten hydroxides, often exceeding 200 °C.164 For example, 316 L stainless steel exhibits a corrosion rate of up to 18 mm per year in aerated 50 wt% NaOH at 90 °C, highlighting the difficulty in maintaining long-term material stability.165 Recent studies have shown that catalysts such as Ag-nanowires/NiFeCrAl foam and certain cobalt oxides can remain stable for several hundred hours at temperatures up to 200 °C, achieving impressive current densities up to 3.75 A/cm2 at 1.75 V.166 Another significant challenge is the efficiency losses due to heat dissipation, which impacts the effective conversion of energy into hydrogen. The Faraday efficiency for mixed NaOH and KOH at 500 °C is only 28%, although using LiOH at higher pressure can improve this efficiency to 88%. Additionally, the high operating temperatures can lead to increased operational costs. For example, operating at 450 °C can increase energy costs by approximately 20–30% compared to lower temperature processes.Scaling up from laboratory to industrial-scale operations also poses difficulties, particularly in maintaining efficiency and managing costs. Laboratory setups achieving high efficiencies often do not translate directly to industrial applications, with efficiency drops of around 5–10%.167 Furthermore, the safe handling of molten salts at high temperatures is critical to prevent accidents, as any spillage or leak can cause severe burns and equipment damage. Managing these materials in large-scale operations introduces additional safety and regulatory concerns. Despite these challenges, the prospects for molten hydroxide electrolysis are compelling. The high operating temperatures significantly enhance reaction kinetics, reducing overvoltages and improving overall efficiency. Studies have reported a reduction in cell voltage by 3.4 to 4 mV/K between 100 °C and 200 °C, substantially higher than the thermodynamic reduction of 0.8 mV/K for liquid water in the same range. Additionally, the conductivity of the electrolyte, particularly KOH solutions, increases with temperature, further boosting efficiency. Ongoing research into more efficient and durable catalysts is critical for advancing molten hydroxide electrolysis. For instance, NiFeOxHy nanosheets have demonstrated impressive performance characteristics, with a low overpotential of 216 mV in freshwater electrolytes and 232 mV in saline solutions.168 These nanosheets show exceptional stability, maintaining functionality for 1000 hours at a current density of 0.1 A/cm2 in freshwater electrolytes and for 550 hours at 1 A/cm2 in saline solutions. The Tafel slope of these nanosheets is as low as 51 mV/dec, indicating efficient kinetics and a rapid increase in current density with increasing overpotential. Their robust nanosheet structure provides abundant electroactive sites, reduces charge transfer resistance (Rct of 1.54 Ω), and enhances mechanical stability, allowing for efficient mass and charge transfer.168
The development of new materials that can better withstand harsh conditions could reduce maintenance costs and improve system longevity. Ni-based alloys, for example, show promise due to their durability in molten hydroxide environments. Integrating advanced materials and nanostructures in these catalysts could significantly enhance the lifespan and efficiency of molten hydroxide electrolysis systems, reducing the need for frequent catalyst replacement and lowering overall operational costs. Furthermore, integrating renewable energy sources, such as solar or wind power, with molten hydroxide electrolysis could significantly lower operational costs, potentially reducing the overall cost of hydrogen production by 15–20%.169 Process optimization techniques, such as advanced thermal management and automated control systems, can improve efficiency by 10–12%, making operations more cost-effective. For instance, an ET-PEMEC system using concentrated solar power and thermal energy storage showed improved overall efficiency and operational flexibility. As technology advances and production scales up, the cost of hydrogen production through molten hydroxide electrolysis could become competitive with traditional methods. With technological advancements and increased production scales, the cost of hydrogen production could potentially lower to below $2 per kilogram of hydrogen, making it a viable option for large-scale hydrogen production.169 For example, a PtM system utilizing a LSGM-based tubular SOEC stack achieved a CO2-to-CH4 conversion ratio of 98.7% and a PtG efficiency of 94.5%, demonstrating the potential for economic feasibility and large-scale application.170
8. Conclusions
In conclusion, this review has provided a comprehensive exploration of hydrogen gas production from steam through eutectic molten hydroxide electrolysis. The investigations incorporated cyclic voltammetry and amperometry techniques to predict electrolysis reactions, establishing correlations between process efficiency and various experimental parameters. The direct electrolysis of steam through eutectic molten hydroxide was examined using different analysis techniques, including hydrogen gas tube detection and hydrogen gas sensors, with quantification performed through the water displacement technique. A significant breakthrough is the study of ionic membranes for the nickel reference electrode. The mullite membrane exhibits superior stability compared to alumina within the eutectic molten hydroxide, extending the reference electrode's lifespan to 9 days. Mullite-covered electrodes effectively control the potential of platinum working electrodes, yielding high cathodic products (hydrogen gas). Cyclic voltammetry results for various working electrodes unveil a distinct order in hydrogen evolution reaction reduction potential: Pt > Ni > SS > Ag > Mo. The nickel reference electrode crafted shows efficiency in molten hydroxide environments, a novel achievement. Direct electrolysis with nickel, platinum, and stainless-steel cathodes shows increased current with higher temperatures and voltages. At 300 °C, current efficiencies are 90.5, 68.6 and 80% for nickel, platinum, and stainless-steel cathodes, respectively. Utilizing a graphite anode doesn't enhance hydrogen production; instead, efficiency decreases. Nickel emerges as the most efficient cathode in eutectic molten hydroxide. The study also discusses various aspects of hydrogen evolution from molten salts, emphasizing its applications in renewable development. Looking forward, future work is essential for process optimization. Integrating the electrochemical cell with renewable sources, exploring diverse molten hydroxide mixtures, investigating electro-catalytic cathodic metals, studying cathode–anode distance impact, and exploring additives to reduce bubble formation are key. These recommendations pave the way for enhancing efficiency and sustainability in hydrogen gas production through eutectic molten hydroxide electrolysis.Nomenclature
| HER | Hydrogen evolution reaction |
| OER | Oxygen evolution reaction |
| PTFE | Polytetrafluoroethylene |
| ΔE | Potential difference |
| ΔG | Gibbs free energy |
| ΔH | Enthalpy change |
| ΔS | Entropy change |
| A 0 | Frequency factor |
| C 0 | Concentration of the reactant |
| C R | Concentration of the product |
| E | Potential |
| E A | Activation energy |

| Equilibrium potential |
| E pa | Anodic peak potential |
| K eq | Reversible constant |
| K 0 | Standard rate constant |
| Q E | Thermal energy |
| R | Gas constant |

| External electric resistance at cathode |
| R anode, Rcathode | Resistance of the electrodes |
| R bubble,H2 | Resistance of hydrogen bubbles |
| R bubble,O2 | Resistance of oxygen bubbles |
| R ions | Resistance of electrolyte |
| R membrane | Resistance of membrane |
| H | Overpotential of the reaction |
| K | Electrical conductivity |
Data availability
Data will be made available on request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research is supported by “Pioneer” and “Leading Goose” R&D Program of Zhejiang (2024C04049), China. The authors are grateful for the financial support from the International Society of Engineering Science and Technology (ISEST) UK. The authors are also thankful for financial support from the Ministry of Research, Innovation and Digitalization (MCID) under the Romanian National Core Program LAPLAS VII-Contract No. 30N/2023.References
- R. York and S. E. Bell, Energy Res. Social Sci., 2019, 51, 40–43 CrossRef.
- S. Mathur, G. Gosnell, B. K. Sovacool, D. D. F. Del Rio, S. Griffiths, M. Bazilian and J. Kim, Energy Res. Social Sci., 2022, 90, 102638 CrossRef.
- S. Öberg, M. Odenberger and F. Johnsson, Int. J. Hydrogen Energy, 2022, 47, 624–644 CrossRef.
- M. Neuwirth, T. Fleiter, P. Manz and R. Hofmann, Energy Convers. Manage., 2022, 252, 115052 CrossRef CAS.
- T. Terlouw, C. Bauer, R. McKenna and M. Mazzotti, Energy Environ. Sci., 2022, 15, 3583–3602 RSC.
- F. Qureshi, M. Yusuf, M. Arham Khan, H. Ibrahim, B. C. Ekeoma, H. Kamyab, M. M. Rahman, A. K. Nadda and S. Chelliapan, Fuel, 2023, 340, 127574 CrossRef CAS.
- F. Sher, I. Ziani, M. Smith, G. Chugreeva, S. Z. Hashimzada, L. D. T. Prola, J. Sulejmanović and E. K. Sher, Coord. Chem. Rev., 2024, 500, 215499 CrossRef CAS.
- H. A. Ibrahim, M. K. Ayomoh, R. C. Bansal, M. N. Gitau, V. S. S. Yadavalli and R. Naidoo, Energy Strategy Rev., 2023, 47, 101085 CrossRef.
- A. M. Abdalla, S. Hossain, O. B. Nisfindy, A. T. Azad, M. Dawood and A. K. Azad, Energy Convers. Manage., 2018, 165, 602–627 CrossRef CAS.
- N. Sazali, Int. J. Hydrogen Energy, 2020, 45, 18753–18771 CrossRef CAS.
- F. Sher, I. Ziani, M. Hameed, S. Ali and J. Sulejmanović, Curr. Opin. Green Sustainable Chem., 2024, 47, 100925 CrossRef.
- A. G. Olabi, A. A. Abdelghafar, A. Baroutaji, E. T. Sayed, A. H. Alami, H. Rezk and M. A. Abdelkareem, Int. J. Hydrogen Energy, 2021, 46, 23498–23528 CrossRef CAS.
- H. Nazir, N. Muthuswamy, C. Louis, S. Jose, J. Prakash, M. E. Buan, C. Flox, S. Chavan, X. Shi and P. Kauranen, Int. J. Hydrogen Energy, 2020, 45, 20693–20708 CrossRef CAS.
- S. Niaz, T. Manzoor and A. H. Pandith, Renewable Sustainable Energy Rev., 2015, 50, 457–469 CrossRef CAS.
- V. A. Yartys, C. J. Webb and F. Cuevas, J. Alloys Compd., 2023, 170133 CrossRef CAS.
- F. M. Sapountzi, J. M. Gracia, H. O. A. Fredriksson and J. W. H. Niemantsverdriet, Prog. Energy Combust. Sci., 2017, 58, 1–35 CrossRef.
- M. D. Rashid, M. K. Al Mesfer, H. Naseem and M. Danish, Int. J. Eng. Adv. Technol., 2015, 4(3), 2249–8958 Search PubMed.
- J. Kim, A. Jun, O. Gwon, S. Yoo, M. Liu, J. Shin, T.-H. Lim and G. Kim, Nano Energy, 2018, 44, 121–126 CrossRef CAS.
- C. H. Wendel, P. Kazempoor and R. J. Braun, J. Power Sources, 2016, 301, 93–104 CrossRef CAS.
- R.-T. Gao, X. Guo, S. Liu, X. Zhang, X. Liu, Y. Su and L. Wang, Appl. Catal., B, 2022, 304, 120883 CrossRef CAS.
- F. Safari and I. Dincer, Energy Convers. Manage., 2020, 205, 112182 CrossRef CAS.
- C. Xiang, K. M. Papadantonakis and N. S. Lewis, Mater. Horiz., 2016, 3, 169–173 RSC.
- F. M. Sapountzi, J. M. Gracia, H. O. A. Fredriksson and J. W. H. Niemantsverdriet, Prog. Energy Combust. Sci., 2017, 58, 1–35 CrossRef.
- J. Kim, A. Jun, O. Gwon, S. Yoo, M. Liu, J. Shin, T.-H. Lim and G. Kim, Nano Energy, 2018, 44, 121–126 CrossRef CAS.
- C. H. Wendel, P. Kazempoor and R. J. Braun, J. Power Sources, 2016, 301, 93–104 CrossRef CAS.
- T. Schuler, C. C. Weber, J. A. Wrubel, L. Gubler, B. Pivovar, F. N. Büchi and G. Bender, Adv. Energy Mater., 2024, 2302786 CrossRef CAS.
- R.-T. Gao, X. Guo, S. Liu, X. Zhang, X. Liu, Y. Su and L. Wang, Appl. Catal., B, 2022, 304, 120883 CrossRef CAS.
- F. Safari and I. Dincer, Energy Convers. Manage., 2020, 205, 112182 CrossRef CAS.
- O. Al-Juboori, F. Sher, U. Khalid, M. B. K. Niazi and G. Z. Chen, ACS Sustain. Chem. Eng., 2020, 8, 12877–12890 CrossRef CAS.
- M. Li, F. Yang, J. Chang, A. Schechter and L. Feng, Acta Phys.-Chim. Sin., 2023, 39, 2301005 Search PubMed.
- Y. Zhou, Y. Kuang, G. Hu, X. Wang and L. Feng, Mater. Today Phys., 2022, 27, 100831 CrossRef CAS.
- Y. Zhou, Q. Wang, X. Tian, J. Chang and L. Feng, J. Energy Chem., 2022, 75, 46–54 CrossRef CAS.
- S. Wang, L. Zhao, J. Li, X. Tian, X. Wu and L. Feng, J. Energy Chem., 2022, 66, 483–492 CrossRef CAS.
- S. Azeem, M. Safdar, R. Aslam, B. Wang, I. Ziani, S. Ansar and F. Sher, Process Saf. Environ. Prot., 2024, 187, 962–973 CrossRef CAS.
- C. Yin, F. Yang, S. Wang and L. Feng, Chin. J. Catal., 2023, 51, 225–236 CrossRef CAS.
- Y. Zhu, Y. Chen, Y. Feng, X. Meng, J. Xia and G. Zhang, Adv. Mater., 2024, 2401694 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach and A. Ludwig, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- J.-T. Ren, L. Chen, H.-Y. Wang, W.-W. Tian and Z.-Y. Yuan, Energy Environ. Sci., 2024, 17, 49–113 RSC.
- K. Veeramani, G. Janani, J. Kim, S. Surendran, J. Lim, S. C. Jesudass, S. Mahadik, T.-H. Kim, J. K. Kim and U. Sim, Renewable Sustainable Energy Rev., 2023, 177, 113227 CrossRef CAS.
- S.-H. Lee, Y. Kwon, S. Kim, J. Yun, E. Kim, G. Jang, Y. Song, B. S. Kim, C.-S. Oh and Y.-H. Choa, Chem. Eng. J., 2024, 153411 CrossRef CAS.
- Y. Chai, Z. Lyu, H. Du, P. Li, S. Ding, Y. Jiang, H. Wang, Q. Min, D. Du and Y. Lin, SusMat, 2022, 2, 392–410 CrossRef CAS.
- Y. Liu, J. Zhang, Y. Li, Q. Qian, Z. Li, Y. Zhu and G. Zhang, Nat. Commun., 2020, 11, 1853 CrossRef CAS PubMed.
- Q. Qian, J. Zhang, J. Li, Y. Li, X. Jin, Y. Zhu, Y. Liu, Z. Li, A. El-Harairy and C. Xiao, Angew. Chem., 2021, 133, 6049–6058 CrossRef.
- Y. Zhu, Y. Chen, Y. Feng, X. Meng, J. Xia and G. Zhang, Adv. Mater., 2024, 2401694 CrossRef CAS.
- Y. Zhu, Q. Qian, Y. Chen, X. He, X. Shi, W. Wang, Z. Li, Y. Feng, G. Zhang and F. Cheng, Adv. Funct. Mater., 2023, 33, 2300547 CrossRef CAS.
- J. Chen, S. Sun, G. Tu and F. Xiao, Miner. Eng., 2023, 204, 108425 CrossRef CAS.
- W. Li, H. Tian, L. Ma, Y. Wang, X. Liu and X. Gao, Mater. Adv., 2022, 3, 5598–5644 RSC.
- N. K. Al-Shara, F. Sher, A. Yaqoob and G. Z. Chen, Int. J. Hydrogen Energy, 2019, 44, 27224–27236 CrossRef CAS.
- S. Niaz, T. Manzoor and A. H. Pandith, Renewable Sustainable Energy Rev., 2015, 50, 457–469 CrossRef CAS.
- J. Knöppel, M. Möckl, D. Escalera-López, K. Stojanovski, M. Bierling, T. Böhm, S. Thiele, M. Rzepka and S. Cherevko, Nat. Commun., 2021, 12, 1–9 CrossRef.
- O. Al-Juboori, F. Sher, S. Rahman, T. Rasheed and G. Z. Chen, ACS Sustain. Chem. Eng., 2020, 8, 19178–19188 CrossRef CAS.
- L. M. Cao, Q. C. Cao, J. Zhang, X. Y. Zhu, R. Z. Sun, Z. Y. Du and C. T. He, Inorg. Chem., 2021, 60, 3365–3374 CrossRef CAS.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS.
- W. Zhang, X. Wang, M. Tan, H. Liu, Q. Ma, Q. Xu, B. G. Pollet and H. Su, Int. J. Hydrogen Energy, 2023, 48, 2617–2627 CrossRef CAS.
- W. Li, H. Tian, L. Ma, Y. Wang, X. Liu and X. Gao, Mater. Adv., 2022, 3, 5598–5644 RSC.
- S. Licht, S. Liu, B. Cui, J. Lau, L. Hu, J. Stuart, B. Wang, O. El-Ghazawi and F.-F. Li, J. Electrochem. Soc., 2016, 163, F1162 CrossRef CAS.
- Ge, J., Zhang, F., Jiao, H. and Jiao, S, Journal of The Electrochemical Society, 2015, 162(9), E185 CrossRef.
- B. K. Chakrabarti, E. Kalamaras, A. K. Singh, A. Bertei, J. Rubio-Garcia, V. Yufit, K. M. Tenny, B. Wu, F. Tariq and Y. S. Hajimolana, Sustainable Energy Fuels, 2020, 4, 5433–5468 RSC.
- Y. Feng, J. Han, M. Xu, X. Liang, T. Jiang, H. Li and Z. L. Wang, Adv. Energy Mater., 2022, 12, 2103143 CrossRef CAS.
- C. Lamy and P. Millet, J. Power Sources, 2020, 447, 227350 CrossRef CAS.
- S. Jiao, X. Fu, S. Wang and Y. Zhao, Energy Environ. Sci., 2021, 14, 1722–1770 RSC.
- K. Hu, J. Fang, X. Ai, D. Huang, Z. Zhong, X. Yang and L. Wang, Appl. Energy, 2022, 312, 118788 CrossRef CAS.
- M. David, C. Ocampo-Martínez and R. Sánchez-Peña, J. Energy Storage, 2019, 23, 392–403 CrossRef.
- T. F. Yi, T. T. Wei, J. Mei, W. Zhang, Y. Zhu, Y. G. Liu, S. Luo, H. Liu, Y. Lu and Z. Guo, Adv. Sustainable Syst., 2020, 4, 1900137 CrossRef CAS.
- W. Zhang, X. Wang, M. Tan, H. Liu, Q. Ma, Q. Xu, B. G. Pollet and H. Su, Int. J. Hydrogen Energy, 2023, 48, 2617–2627 CrossRef CAS.
- L. Shi, R. Rossi, M. Son, D. M. Hall, M. A. Hickner, C. A. Gorski and B. E. Logan, Energy Environ. Sci., 2020, 13, 3138–3148 RSC.
- Y. Ding, P. Cai and Z. Wen, Chem. Soc. Rev., 2021, 50, 1495–1511 RSC.
- M. N. Islam, A. B. Mansoor Basha, V. O. Kollath, A. P. Soleymani, J. Jankovic and K. Karan, Nat. Commun., 2022, 13, 6157 CrossRef CAS.
- D. Aili, M. R. Kraglund, S. C. Rajappan, D. Serhiichuk, Y. Xia, V. Deimede, J. Kallitsis, C. Bae, P. Jannasch, D. Henkensmeier and J. O. Jensen, ACS Energy Lett., 2023, 8, 1900–1910 CrossRef CAS.
- A. Dukić, Int. J. Hydrogen Energy, 2015, 40, 7465–7474 CrossRef.
- P. Cavaliere, in Water Electrolysis for Hydrogen Production, ed. P. Cavaliere, Springer International Publishing, Cham, 2023, pp. 105–157 Search PubMed.
- D. J. Deka, S. Gunduz, J. Kim, T. Fitzgerald, Y. Shi, A. C. Co and U. S. Ozkan, Ind. Eng. Chem. Res., 2019, 58, 22497–22505 CrossRef CAS.
- R. Funari, A. Matsumoto, J. R. de Bruyn and A. Q. Shen, Anal. Chem., 2020, 92, 8244–8253 CrossRef CAS.
- A. Shrivastava, V. Q. Do and K. C. Smith, ACS Appl. Mater. Interfaces, 2022, 14, 30672–30682 CrossRef CAS PubMed.
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernäcker, T. Weißgärber, L. Röntzsch and J. Meier-Haack, Sustainable Energy Fuels, 2020, 4, 2114–2133 RSC.
- Y. Li, X. Gao, X. Feng, X. Han, J. Du, L. Lu and M. Ouyang, J. Energy Chem., 2022, 67, 34–45 CrossRef CAS.
- P. Cerqueira, M. A. Soria and L. M. Madeira, Energy Convers. Manage., 2021, 238, 114146 CrossRef CAS.
- D. J. Deka, S. Gunduz, J. Kim, T. Fitzgerald, Y. Shi, A. C. Co and U. S. Ozkan, Ind. Eng. Chem. Res., 2019, 58, 22497–22505 CrossRef CAS.
- K. Brinkert and P. Mandin, npj Microgravity, 2022, 8, 52 CrossRef PubMed.
- W. Guo, H. Kim, S. Hong, S. Y. Kim and S. H. Ahn, Dalton Trans., 2023, 52, 14039–14046 RSC.
- C. Niether, S. Faure, A. Bordet, J. Deseure, M. Chatenet, J. Carrey, B. Chaudret and A. Rouet, Nat. Energy, 2018, 3, 476–483 CrossRef CAS.
- A. Đukić, Int. J. Hydrogen Energy, 2015, 40, 7465–7474 CrossRef.
- A. J. Shih, M. C. O. Monteiro, F. Dattila, D. Pavesi, M. Philips, A. H. M. da Silva, R. E. Vos, K. Ojha, S. Park and O. van der Heijden, Nat. Rev. Methods Primers, 2022, 2, 84 CrossRef CAS.
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitski, H. N. Alshareef and H. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS.
- W. Yang, R. R. Prabhakar, J. Tan, S. D. Tilley and J. Moon, Chem. Soc. Rev., 2019, 48, 4979–5015 RSC.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem, 2017, 1, 3 CrossRef CAS.
- C. Santoro, P. Bollella, B. Erable, P. Atanassov and D. Pant, Nat. Catal., 2022, 5, 473–484 CrossRef CAS.
- S. Nagappan, S. Yang, A. Adhikari, R. Patel and S. Kundu, Sustainable Energy Fuels, 2023, 7, 3741–3775 RSC.
- A. Nairan, C. Liang, S.-W. Chiang, Y. Wu, P. Zou, U. Khan, W. Liu, F. Kang, S. Guo and J. Wu, Energy Environ. Sci., 2021, 14, 1594–1601 RSC.
- R. Funari, A. Matsumoto, J. R. de Bruyn and A. Q. Shen, Anal. Chem., 2020, 92, 8244–8253 CrossRef CAS PubMed.
- R. Morasch, H. A. Gasteiger and B. Suthar, J. Electrochem. Soc., 2023, 170, 80522 CrossRef.
- P. Cavaliere, in Water Electrolysis for Hydrogen Production, ed. P. Cavaliere, Springer International Publishing, Cham, 2023, pp. 105–157 Search PubMed.
- Y. Bicer and I. Dincer, ACS Sustain. Chem. Eng., 2017, 5, 8035–8043 CrossRef CAS.
- J. Ge, F. Zhang, H. Jiao and S. Jiao, J. Electrochem. Soc., 2015, 162, E185–E189 CrossRef CAS.
- J. Híveš, M. Benová, K. Bouzek and V. K. Sharma, Electrochem. Commun., 2006, 8, 1737–1740 CrossRef.
- G. Schiavon, S. Zecchin and G. G. Bombi, J. Electroanal. Chem. Interfacial Electrochem., 1972, 38, 473–475 CrossRef CAS.
- M. H. Miles, J. Appl. Electrochem., 2003, 33, 1011–1016 CrossRef CAS.
- L. Hrnčiariková, K. Kerekeš, J. Híveš and M. Gál, Int. J. Electrochem. Sci., 2013, 8, 7768–7778 CrossRef.
- J. C. Ganley, Int. J. Hydrogen Energy, 2009, 34, 3604–3611 CrossRef CAS.
- S. Licht, S. Liu, B. Cui, J. Lau, L. Hu, J. Stuart, B. Wang, O. El-Ghazawi and F.-F. Li, J. Electrochem. Soc., 2016, 163, F1162 CrossRef CAS.
- I. Zaafarany and H. Boller, Curr. World Environ., 2009, 4, 277–284 CrossRef CAS.
- P. R. Zabinski, H. Nemoto, S. Meguro, K. Asami and K. Hashimoto, J. Electrochem. Soc., 2003, 150, C717 CrossRef CAS.
- N. Nagai, M. Takeuchi, T. Kimura and T. Oka, Int. J. Hydrogen Energy, 2003, 28, 35–41 CrossRef CAS.
- A. A. Anani, Z. Mao, R. E. White, S. Srinivasan and A. J. Appleby, J. Electrochem. Soc., 1990, 137, 2703 CrossRef CAS.
- J. Divisek, J. Mergel and H. F. Niessen, Int. J. Hydrogen Energy, 1980, 5, 151–164 CrossRef CAS.
- J. Divisek, J. Mergel and H. Schmitz, Int. J. Hydrogen Energy, 1982, 7, 695–701 CrossRef CAS.
- A. Döner, İ. Karcı and G. Kardaş, Int. J. Hydrogen Energy, 2012, 37, 9470–9476 CrossRef.
- M. Jayalakshmi, W.-Y. Kim, K.-D. Jung and O.-S. Joo, Int. J. Electrochem. Sci., 2008, 3, 908–917 CrossRef CAS.
- J. Yang, H. Muroyama, T. Matsui and K. Eguchi, J. Power Sources, 2014, 245, 277–282 CrossRef CAS.
- A. Cox and D. J. Fray, J. Appl. Electrochem., 2008, 38, 1401–1407 CrossRef CAS.
- M. H. Miles, G. Kissel, P. W. T. Lu and S. Srinivasan, J. Electrochem. Soc., 1976, 123, 332 CrossRef CAS.
- J. C. Ganley, J. Power Sources, 2008, 178, 44–47 CrossRef CAS.
- S. Licht, B. Cui, B. Wang, F.-F. Li, J. Lau and S. Liu, Science, 2014, 345, 637–640 CrossRef CAS PubMed.
- L. Guo, J. M. Calo, E. DiCocco and E. J. Bain, Energy Fuels, 2013, 27, 1712–1719 CrossRef CAS.
- R. M. Abouatallah, D. W. Kirk and J. W. Graydon, Electrochem. Solid-State Lett., 2002, 5, E9 CrossRef CAS.
- M. H. Miles, G. Kissel, P. W. T. Lu and S. Srinivasan, J. Electrochem. Soc., 1976, 123, 332 CrossRef CAS.
- J. Divisek, J. Mergel and H. F. Niessen, Int. J. Hydrogen Energy, 1980, 5, 151–164 CrossRef CAS.
- J. Divisek, J. Mergel and H. Schmitz, Int. J. Hydrogen Energy, 1982, 7, 695–701 CrossRef CAS.
- A. A. Anani, Z. Mao, R. E. White, S. Srinivasan and A. J. Appleby, J. Electrochem. Soc., 1990, 137, 2703 CrossRef CAS.
- R. M. Abouatallah, D. W. Kirk and J. W. Graydon, Electrochem. Solid-State Lett., 2002, 5, E9 CrossRef CAS.
- M. H. Miles, J. Appl. Electrochem., 2003, 33, 1011–1016 CrossRef CAS.
- N. Nagai, M. Takeuchi, T. Kimura and T. Oka, Int. J. Hydrogen Energy, 2003, 28, 35–41 CrossRef CAS.
- P. R. Zabinski, H. Nemoto, S. Meguro, K. Asami and K. Hashimoto, J. Electrochem. Soc., 2003, 150, C717 CrossRef CAS.
- J. Híveš, M. Benová, K. Bouzek and V. K. Sharma, Electrochem. Commun., 2006, 8, 1737–1740 CrossRef.
- M. Jayalakshmi, W.-Y. Kim, K.-D. Jung and O.-S. Joo, Int. J. Electrochem. Sci., 2008, 3, 908–917 CrossRef CAS.
- J. Híveš, M. Benová, K. Bouzek, J. Sitek and V. K. Sharma, Electrochim. Acta, 2008, 54, 203–208 CrossRef.
- J. C. Ganley, J. Power Sources, 2008, 178, 44–47 CrossRef CAS.
- A. Cox and D. J. Fray, J. Appl. Electrochem., 2008, 38, 1401–1407 CrossRef CAS.
- J. C. Ganley, Int. J. Hydrogen Energy, 2009, 34, 3604–3611 CrossRef CAS.
- A. Döner, İ. Karcı and G. Kardaş, Int. J. Hydrogen Energy, 2012, 37, 9470–9476 CrossRef.
- L. Guo, J. M. Calo, E. DiCocco and E. J. Bain, Energy Fuels, 2013, 27, 1712–1719 CrossRef CAS.
- L. Hrnčiariková, K. Kerekeš, J. Híveš and M. Gál, Int. J. Electrochem. Sci., 2013, 8, 7768–7778 CrossRef.
- J. Yang, H. Muroyama, T. Matsui and K. Eguchi, J. Power Sources, 2014, 245, 277–282 CrossRef CAS.
- S. Licht, B. Cui, B. Wang, F.-F. Li, J. Lau and S. Liu, Science, 2014, 345, 637–640 CrossRef CAS PubMed.
- J. Ge, F. Zhang, H. Jiao and S. Jiao, J. Electrochem. Soc., 2015, 162, E185–E189 CrossRef CAS.
- Q. Zhu, Y. Zeng and Y. Zheng, Ind. Chem. Mater., 2023, 1, 595–617 RSC.
- H. Wang, N. J. Siambun, L. Yu and G. Z. Chen, J. Electrochem. Soc., 2012, 159, H740 CrossRef CAS.
- S. Licht, S. Liu, B. Cui, J. Lau, L. Hu, J. Stuart, B. Wang, O. El-Ghazawi and F.-F. Li, J. Electrochem. Soc., 2016, 163, F1162–F1168 CrossRef CAS.
- E. T. Sayed, M. A. Abdelkareem, T. Wilberforce, K.-J. Chae and A. G. Olabi, Int. J. Hydrogen Energy, 2024, 52, 241–250 CrossRef CAS.
- N. K. Al-Shara, F. Sher, A. Yaqoob and G. Z. Chen, Int. J. Hydrogen Energy, 2019, 44, 27224–27236 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- S. Choi, J. Steppan and M. F. Simpson, J. Electrochem. Soc., 2023, 170, 057505 CrossRef CAS.
- S. Choi, N. E. Orabona, O. R. Dale, P. Okabe, C. Inman and M. F. Simpson, Sol. Energy Mater. Sol. Cells, 2019, 202, 110087 CrossRef CAS.
- Y. Zhou, F. Wang, S. Dou, Z. Shi, B. Dong, W. Yu, H. Zhao, F. Wang, J. Yu and Y. Chai, Chem. Eng. J., 2022, 427, 131643 CrossRef CAS.
- F. Sher, N. K. Al-shara, S. Z. Iqbal, Z. Jahan and G. Z. Chen, Int. J. Hydrogen Energy, 2020, 45, 28260–28271 CrossRef CAS.
- S. Choi, N. E. Orabona, O. R. Dale, P. Okabe, C. Inman and M. F. Simpson, Sol. Energy Mater. Sol. Cells, 2019, 202, 110087 CrossRef CAS.
- Y. Zhou, F. Wang, S. Dou, Z. Shi, B. Dong, W. Yu, H. Zhao, F. Wang, J. Yu and Y. Chai, Chem. Eng. J., 2022, 427, 131643 CrossRef CAS.
- R. Sharifian, R. M. Wagterveld, I. A. Digdaya, C. Xiang and D. A. Vermaas, Energy Environ. Sci., 2021, 14, 781–814 RSC.
- L. Cui, K. Yliniemi, J. Vapaavuori and M. Lundström, Chem. Eng. J., 2023, 465, 142737 CrossRef CAS.
- Q. Xia, Q. Song and Z. Xu, Waste Manage., 2023, 158, 146–152 CrossRef CAS.
- W. Jin and Y. Zhang, ACS Sustain. Chem. Eng., 2020, 8(12), 4693–4707 CrossRef CAS.
- Y. Shen, Fuel, 2023, 339, 127473 CrossRef CAS.
- D. Raabe, Chem. Rev., 2023, 123, 2436–2608 CrossRef CAS.
- E.-Y. Choi and S. M. Jeong, Prog. Nat. Sci.: Mater. Int., 2015, 25, 572–582 CrossRef CAS.
- K. Xie and A. R. Kamali, Int. J. Hydrogen Energy, 2019, 44, 24353–24359 CrossRef CAS.
- Y. Shen, Fuel, 2023, 339, 127473 CrossRef CAS.
- S. K. Tiwari, M. Bystrzejewski, A. De Adhikari, A. Huczko and N. Wang, Prog. Energy Combust. Sci., 2022, 92, 101023 CrossRef.
- A. M. Abdelkader, J. Mater. Chem. A, 2015, 3, 8519–8525 RSC.
- Z. Pang, G. Li, X. Xiong, L. Ji, Q. Xu, X. Zou and X. Lu, J. Energy Chem., 2021, 61, 622–640 CrossRef CAS.
- G. H. Tariq, G. Asghar, M. S. Shifa, M. Anis-Ur-Rehman, S. Ullah, Z. A. Shah, I. Ziani, A. M. Tawfeek and F. Sher, Phys. Chem. Chem. Phys., 2023, 25, 31726–31740 RSC.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y. M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B. M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C. J. Yang and K. Caldeira, Science, 2018, 360(6396), eaas9793 CrossRef PubMed.
- Z. Medghalchi and O. Taylan, Energy Convers. Manage., 2023, 294, 117594 CrossRef CAS.
- S. Aslam, S. Rani, K. Lal, M. Fatima, T. Hardwick, B. Shirinfar and N. Ahmed, Green Chem., 2023, 25, 9543–9573 RSC.
- L. He, G. Yu, Y. Cheng, N. Wang and W. Hu, Mater. Chem. Front., 2023, 7, 5661–5692 RSC.
- R. Davalos Monteiro, J. van de Wetering, B. Krawczyk and D. L. Engelberg, Met. Mater. Int., 2020, 26, 630–640 CrossRef CAS.
- F. P. Lohmann-Richters, S. Renz, W. Lehnert, M. Müller and M. Carmo, J. Electrochem. Soc., 2021, 168, 114501 CrossRef CAS.
- P. Kondaiah and R. Pitchumani, Renewable Energy, 2023, 205, 956–991 CrossRef CAS.
- J. Deng, Z. Wang, H. Yang, R. Jian, Y. Zhang, P. Xia, W. Liu, O. Fontaine, Y. Zhu and L. Li, Chem. Eng. J., 2024, 482, 148887 CrossRef CAS.
- Y. Shen and X. Yuan, Green Chem., 2023, 25, 2087–2108 RSC.
- W. Zhang, M. Liu, X. Gu, Y. Shi, Z. Deng and N. Cai, Chem. Rev., 2023, 123, 7119–7192 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |