An efficient particulate photocatalyst for overall water splitting based on scandium and magnesium co-doped strontium titanate†
Received
1st November 2023
, Accepted 27th November 2023
First published on 29th November 2023
Abstract
Effects of co-doping of aluminum (Al), scandium (Sc), and magnesium (Mg) into SrTiO3 particles by a high temperature (1200 °C) flux treatment in a molten SrCl2 on the structural properties and photocatalytic activities for overall water splitting were investigated. Isotropically-rounded polygonal-shaped particles of almost phase-pure SrTiO3 crystals were obtained from SrTiO3 particles co-doped with Al and Sc (SrTiO3:Al,Sc) and Al and Mg (SrTiO3:Al,Mg), whereas, the cubic-shaped particles having specific nanosized steps on the edge of each particle were obtained by co-doping with Sc and Mg (SrTiO3:Sc,Mg). Apparent quantum yields (AQYs) for overall water splitting at a band edge region (365 nm) were examined using these SrTiO3 samples loaded with Rh/Cr2O3 and CoOOH cocatalyst nanoparticles (for H2 and O2 evolution, respectively), and it reached the highest (66%) when the photocatalyst based on SrTiO3:Sc,Mg was used. The best photocatalytic performance obtained over the photocatalyst is attributed to the achievement of the separation of reaction sites for reduction and oxidation of water, i.e., the former reaction occurred on the Rh/Cr2O3 cocatalyst selectively deposited on the flat {100} facets of the SrTiO3:Sc,Mg particle, whereas the later O2 evolution occurred on the CoOOH cocatalyst that was only deposited on the nanosized step part on the particle.
Introduction
Strontium titanate (SrTiO3) has been investigated as an efficient photocatalytic material for overall water splitting since the pioneering finding in the 1980s on the use of a commercially available SrTiO3 powder loaded with partially oxidized nickel oxide nanoparticles (NiOx-SrTiO3) for inducing the reaction in an alkaline suspension system.1–3 During the same period, simultaneous productions of H2 and O2 from water vapor by using a Pt-loaded SrTiO3 (Pt-SrTiO3) single crystal covered with a deliquescent electrolyte, such as NaOH, was demonstrated.4 Afterward, the induction of water splitting over a Pt-SrTiO3 powder photocatalyst was achieved in a highly concentrated Na2CO3 solution though the effect of the carbonate addition was more intense for a Pt-loaded titanium oxide (Pt-TiO2) photocatalyst than that for Pt-SrTiO3.5 We have demonstrated the stoichiometric production of H2 and O2 from pure water by assembling a Pt-SrTiO3-based photocatalyst composed of Pt-SrTiO3 (core)–silica (shell) particle partially modified with a fluoroalkylsilylation agent at a gas–water interface.6 An attempt to provide a visible-light response to the SrTiO3-based photocatalyst was also successful by the co-doping of rhodium (Rh3+) and antimony (Sb5+) ions followed by the loading of an IrO2 catalyst. The photocatalyst thus obtained induced overall water splitting under visible light giving 0.1% of the apparent quantum yield (AQY) at 420 nm.7 The Rh-doped SrTiO3 powders have been studied intensively as the photocatalyst for water reduction into H2 in a Z-scheme system, which consisted of a heterojunction of two different semiconductors for catalyzing water oxidation and reduction combined with an electron mediator.8–12
Recently, a significant enhancement of the photocatalytic activity for the overall splitting of pure water over the SrTiO3-based system was reported by applying a high-temperature flux treatment in an SrCl2 molten salt containing Al2O3 powders followed by the loading of a Rh (core)–Cr2O3 (shell) nanoparticle (Rh/Cr2O3) cocatalyst for H2 evolution reaction (HER) together with a Co oxyhydroxide (CoOOH) cocatalyst for O2 evolution reaction (OER).13–17 The flux treatment is known to improve crystallinity, leading to the reduction of the detrimental defect(s) to induce the reaction. Intentionally added Al2O3 powders during the flux treatment resulted in the suppression of the crystalline growth as well as selective exposures of {100} and {110} crystal facets. The Rh/Cr2O3 HER cocatalyst was photodeposited on the {100} facets and subsequently, the mediated electron transfer to water produced H2. Whereas, the CoOOH OER cocatalyst was oxidatively photodeposited on the {110} facets on which O2 evolution efficiently occurred.14,17 The added Al2O3 was also considered to act as a p-type dopant to reduce the concentration of the majority carrier (surplus electrons), i.e., doped Al ions (Al3+) are selectively replaced with the B site Ti ions (Ti4+) in the perovskite lattice of SrTiO3 owing to the relative similarity of ionic radii between them compared to those between Al3+ and A site Sr ions (Sr2+).17,18
The best AQY reported for the above-mentioned Al-doped SrTiO3 (SrTiO3:Al)-based photocatalyst, i.e., flux-mediated SrTiO3:Al loaded with Rh/Cr2O3 and CoOOH, for water splitting of pure water reached almost unity.15 However, the subsequent trials in our laboratory using several SrTiO3 samples purchased from different suppliers did not reach the high level of AQY for the flux-mediated SrTiO3:Al-based photocatalysts even under various conditions, such as duration and/or temperature of the flux treatment and amount of Al2O3 addition during the flux treatment. Moreover, the use of other p-type dopants such as Mg2+ and Sc3+ reported previously13,19 could not attain the photocatalysts showing their benchmark AQYs. These results and facts motivated us to seek a way to achieve a high AQY. Among the variety of approaches for this purpose, we chose simultaneous additions of two different divalent or trivalent oxides during the flux treatment. As a result, we found specific effects of co-doing of Mg and Sc in an Al-free SrTiO3 particle for the enhancement of AQYs under monochromatic-light irradiation at a band-edge region (365 nm). In this study, results on the photocatalytic properties of the Mg- and Sc-doped photocatalyst are discussed in relation to its morphological and electric structures.17,18
Experimental
Flux process to prepare doped-SrTiO3 powders
For the preparation of Al-doped SrTiO3 (SrTiO3:Al), 1.835 g (10 mmol) of SrTiO3 powder (FUJIFILM Wako) was mixed with 20.39 mg (0.2 mmol) of Al2O3 (Sigma-Aldrich) in an agate mortar, followed by blending with 15.85 g (100 mmol) of SrCl2 (Kanto Chemical). The mixture was heated in an alumina crucible at 1200 °C for 30 h at the heating ramp of ca. 600 °C h−1. The lumped sample, thus obtained, was washed repeatedly with distilled water to separate the residual SrCl2 flux until the disappearance of Cl− ions in the supernatant, which was confirmed by adding the AgNO3 solution. For doping of Sc and Mg, 13.91 mg (0.1 mmol) of Sc2O3 (Sigma-Aldrich) and 2.02 mg (0.05 mmol) MgO (FUJIFILM Wako) were used as sources of these dopants, respectively. It should be noted that a magnesia crucible was used for preparations of Mg-doped and Sc, Mg-co-doped SrTiO3 (SrTiO3:Mg and SrTiO3:Sc,Mg) to avoid contamination of Al from the used crucible.
Characterizations
Crystallographic analyses were performed by powder XRD using a Rigaku Mini Flex X-ray diffractometer (Cu Kα, Ni filter). Morphological properties were observed using a scanning electron microscope, JSM-IT800, JEOL. Atomic compositions of some selected samples were determined by inductively coupled plasma (ICP) analysis on a Shimadzu ICPS-8100 ICP emission spectrometer. Photoabsorption properties were examined by measuring DR spectra using a Shimadzu UV-2600i UV-visible spectrophotometer equipped with an integrating sphere. The ionization potential of SrTiO3:Sc,Mg was analysed using a Bunkoukeiki BIP-KV201 photoelectron yield spectrometer. For the measurement, SrTiO3:Sc,Mg powder was spread on carbon tape on a metal base plate to prevent charge-up. The photoelectron yield (Y) can be expressed using the following equation:where IP is the difference between the valence band maximum (VBM) and the vacuum levels, and A is the proportionality constant.20 Therefore, the VBM level of the SrTiO3:Sc,Mg sample with reference to the vacuum level was deduced from the extrapolation of linear slopes of the plot to its baseline.21
Transmittance depth profile analyses
Transmittance depth profiles of a SrTiO3 bulk crystal at various photon energies were obtained using the following equation:where I, I0, α, and x denote the intensity of the monochromatic light after passing through the SrTiO3 crystal, the intensity of the incident monochromatic light, and photoabsorption coefficient extracted from the experimental data reported by Zollner et al.,22 and the distance from the surface of the SrTiO3 crystal, respectively.
Evaluation of the photocatalytic activity
The doped SrTiO3 powder (100 mg) was dispersed in 100 ml of distilled water in a glass vessel with a quartz window at the top part. Into the suspension, aqueous solutions of RhCl3·3H2O (FUJIFILM Wako), K2CrO4 (Kanto Chemical), and Co(NO3)2·6H2O (Kanto Chemical) were sequentially added under irradiation using a CERMAX PE300BF Xe lamp (300 W) through the quartz window. Sequential reductive deposition of Rh(III) and Cr(VI) species resulted in the formation of Rh/Cr2O3 HER cocatalysts,23 whereas oxidative deposition of Co(II) led to the formation of the CoOOH cocatalysts.16 Amounts of these solutions were adjusted to deposit 0.1 wt% as Rh, 0.05 wt% as Cr, and 0.05 wt% as Co. The durations for the deposition of Rh(III), Cr(VI), and Co(II) species were fixed at 10 min, 5 min, and 5 min, respectively. The vessel containing the thus-obtained photocatalyst suspension was connected to an air-free closed gas circulation and evaluation system to perform a photocatalytic reaction. Prior to the reaction, the system was evaluated to remove air and then filled with Ar at the pressure of ca. 20 kPa. The suspension was then illuminated with light using the above-mentioned Xe lamp through the quartz window. Evolved H2 and O2 accumulated in the reaction system were analysed using a GL Sciences GC3210 gas chromatography equipped with a TCD and an MS-5A column (Ar carrier). For the measurement of AQY, monochromatic light irradiation was performed through a bandpass filter at required wavelengths. As a light source, a Hamamatsu Photonics LC8 high-pressure mercury (Hg) lamp or a CERMAX PE300BF Xe lamp (300 W) was used. The AQY was determined from photocurrent using the following equation:| | | AQY (%) = {(NH2 × 2) / Np} × 100 | (3) |
where NH2 and Np denote the number of H2 molecules produced and the number of photons that reached the surface of the reaction solution, respectively. The Np value was calculated from the light intensity of the used monochromatic light by using an Ophir PD300 photodiode detector.
Time-resolved absorption measurements
Time-resolved absorption measurements were carried out using laboratory-built spectrometers.24,25 Briefly, the band gap of the SrTiO3 powder was excited using the 355 nm laser pulses from a Continuum Surelite I Nd:YAG laser (6 ns in duration, repetition rate of 10–0.2 Hz). In the visible region (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–10000 cm−1) and the near-infrared region (NIR; 10000–6000 cm−1), light from a halogen lamp (50 W) was focused on the SrTiO3 powder, and the diffusely reflected light was monochromatized by the spectrometer. In the mid-IR region (6000–1000 cm−1), light from a MoSi coil was focused onto the SrTiO3 powder and the transmitted light was monochromated by the spectrometer. The output visible and near-infrared monochromatic light was detected by Si and InGaAs photodiode detectors, respectively, whereas the output IR light was detected by a Kolmar MCT detector equipped with a Stanford Research Systems SR560 AC coupled amplifier. The time profiles of the absorption changes were recorded with a digital oscilloscope. The temporal resolution of this spectrometer in the visible region was 4 ms, limited by the stray light from the pumping pulse and/or strong short-duration emission from the sample, and that of the NIR and IR regions was 1–2 ms, limited by the AC coupled amplifier.
000–10000 cm−1) and the near-infrared region (NIR; 10000–6000 cm−1), light from a halogen lamp (50 W) was focused on the SrTiO3 powder, and the diffusely reflected light was monochromatized by the spectrometer. In the mid-IR region (6000–1000 cm−1), light from a MoSi coil was focused onto the SrTiO3 powder and the transmitted light was monochromated by the spectrometer. The output visible and near-infrared monochromatic light was detected by Si and InGaAs photodiode detectors, respectively, whereas the output IR light was detected by a Kolmar MCT detector equipped with a Stanford Research Systems SR560 AC coupled amplifier. The time profiles of the absorption changes were recorded with a digital oscilloscope. The temporal resolution of this spectrometer in the visible region was 4 ms, limited by the stray light from the pumping pulse and/or strong short-duration emission from the sample, and that of the NIR and IR regions was 1–2 ms, limited by the AC coupled amplifier.
Results and discussion
A commercial SrTiO3 powder obtained from FUJIFILM Wako (FW SrTiO3) with a particle size of ca. 100–200 nm (Fig. S1†) was used as a source material. During the flux treatment at 1200 °C in SrCl2, 2 mol% of Al2O3, 1 mol% of Sc2O3, and 0.5 mol% of MgO were added for doping Al, Mg, and Sc, respectively. Based on the reported ionic radii,26 all the dopants used in this study should be replaced preferentially with the B site Ti ion (Ti4+) but not with the A site Sr ion (Sr2+). Thus, they were p-type dopants to reduce the concentration of the majority carrier of the electrons in SrTiO3.13,19 Powder XRD patterns of the obtained samples doped with Al (SrTiO3:Al), Sc (SrTiO3:Sc), Mg (SrTiO3:Mg) as well as those co-doped with Al and Sc (SrTiO3:Al,Sc), Al and Mg (SrTiO3:Al,Mg), Sc and Mg (SrTiO3:Sc,Mg) indicated almost the same profiles corresponded to the cubic perovskite SrTiO3 (ICSD 56092)27 except for weak reflections assigned to ScCO3 derived from the used ScCl2 flux (Fig. S2†). On the other hand, SEM observations of these samples exhibited appreciable differences in their morphologies, as shown in Fig. 1. The SrTiO3:Al sample was composed of isotropic rounded-polygonal-shaped particles with relatively uniform particle size distribution (Fig. 1a). The average diameter of the particles (Davg) and its standard deviation (SD) of the SrTiO3:Al sample determined by measuring more than 100 particles were 150 nm and 41 nm, respectively (Fig. S3a†). As reported previously,17 the incompletely cubic morphology of the sample indicated exposures of various non-equivalent crystal facets in addition to the most stable {100} facets. As estimated from the orientation of the equivalent {100} facets forming the cubic shape, both {110} and {111} facets were also likely to be exposed for these samples. As shown in Fig. 1b and S3b,† the Sc-doped sample (SrTiO3:Sc) had almost the same morphology and size distribution as those of SrTiO3:Al, suggesting that the added Sc2O3 during the flux treatment exhibited similar functions for size and shape controls of the resulting particle to those of Al2O3. In contrast, the SrTiO3:Mg sample consisted of cubic-shaped crystalline grains with relatively large particle sizes and size distribution (Fig. 1c and S3c†) probably due to the lack of Al or Sc components. As expected from the prediction, such an appreciable grain growth observed on SrTiO3:Mg was suppressed by co-doping of Al or Sc with Mg, i.e., SrTiO3:Al,Sc and SrTiO3:Al,Mg samples thus-obtained were composed of relatively uniform-sized particles of 110 nm in diameter with SDs comparable to SrTiO3:Al and SrTiO3:Sc samples (Fig. 1d, e, S3d and S3e†). In addition, a distinct difference observed on SrTiO3:Al,Mg and SrTiO3:Sc,Mg samples compared to those of Mg-free samples is that the shape of each particle is close to a cube, indicating that preferential exposures of the equivalent {100} facets occurred in the presence of MgO during the flux treatment. In addition to the uniformly sized cubic morphology, a specific morphology was also observed for the SrTiO3:Sc,Mg sample, i.e., the sample showed the presence of nanosized steps on the edges of each particle (Fig. 1f and S3f†). Since orientations of these nanosized steps were orthogonal to the flat {100} facets, these steps should be composed of {100} facets. As for the results on the above-mentioned morphological changes, the Sc2O3 additive should act as an agent for controlling particle sizes of the obtained SrTiO3:Mg,Sc sample, whereas the co-presence of the MgO additive was likely to act as an agent for selective exposure of the {100} facet, leading to inducing the formation of the unique nanosized step structure.
 |
| | Fig. 1 SEM images of (a) SrTiO3:Al, (b) SrTiO3:Sc, (c) SrTiO3:Mg, (d) SrTiO3:Al,Sc, (e) SrTiO3:Al,Mg, and (f) SrTiO3:Sc,Mg. | |
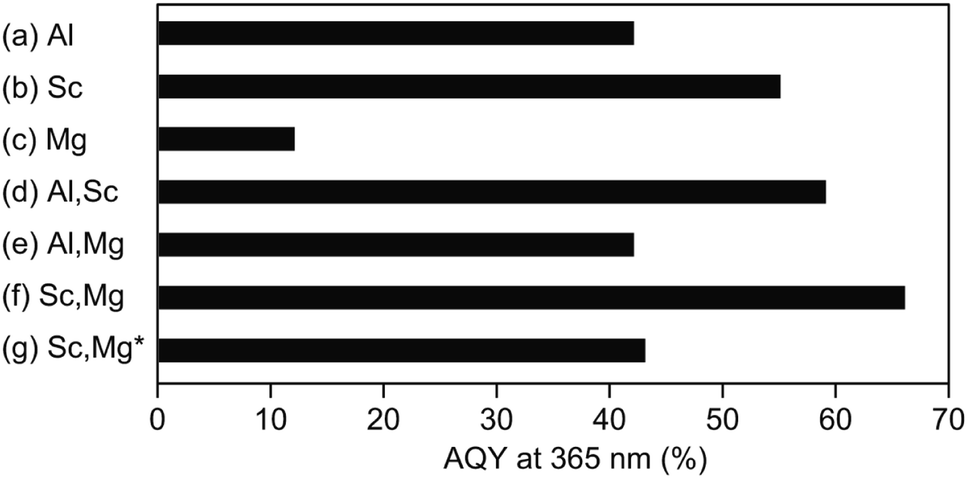
Fig. 2 shows AQYs for the overall water splitting over photocatalysts based on the above-discussed doped-SrTiO3 particles with surface-loaded Rh/Cr2O3 HER and CoOOH OER cocatalysts under the irradiation of monochromatic light at 365 nm. The SrTiO3:Al-based photocatalyst showed AQY of 42% (Fig. 2a), the value of which was less than half of the best photocatalyst reported previously,17 probably due to the difference in the source SrTiO3 powder used. As expected from the previous study,19 the use of Sc instead of Al (i.e., a photocatalyst based on SrTiO3:Sc) resulted in an enhancement of AQY (55%), irrespective of the morphological similarity of this samples to SrTiO3:Al (Fig. 2b). Appreciable drop in the AQY value (17%) compared to that of the SrTiO3:Al-based photocatalyst was obtained over the SrTiO3:Mg-based photocatalyst (Fig. 2c). In accordance with the above morphological features, the low AQY of the photocatalyst was attributed to the lack of the exposure of the {110} crystal facets, which acted as the O2 evolution site after deposition of the CoOOH OER cocatalyst as well as the overgrowth of the particle, leading to insufficient diffusion of the photoexcited carriers generated at the inner part of the particle. Similar to the above-mentioned SrTiO3:Sc-based photocatalyst, enhancement of AQY of 59% was achieved over the photocatalyst based on SrTiO3:Al,Sc (Fig. 2d) To obtain the factor(s) of observed AQY increments over photocatalysts based on SrTiO3:Sc and SrTiO3:Al,Sc, the contents of Al and Sc elements in the SrTiO3:Al,Sc sample were examined by the ICP measurement. For comparison, the same analysis was also performed on the raw SrTiO3 sample (FW SrTiO3). These results are summarized in Table 1. Since the amounts of both Al and Sc in the source FW SrTiO3 were under detection limits, these components in the SrTiO3:Al,Sc sample were intentionally included for the flux treatment. Despite the lower charged content of Sc than that of Al included in the flux, the content of Sc in the SrTiO3:Al,Sc sample thus obtained was found to be much higher than that of Al, indicating the effectiveness of the addition of Sc as the p-type dopant for the reduction of the surplus electrons in the crystalline bulk of SrTiO3 in the present sample when compared to that of the Al dopant. Regarding the co-doping of Mg with Al, there was no gain of the enhancement of AQY: 42% of AQY obtained over the SrTiO3:Al,Mg-based photocatalyst and it was comparable to that for the SrTiO3:Al-based photocatalyst (Fig. 2e). Although the added Mg was assumed to be a p-type dopant to increase AQY, insufficient exposure of the {110} facets, which should act as the reaction site for water oxidation (see Fig. 1e), cancelled out the positive effect of the Mg-doping. On the other hand, enhanced AQY to 66% was achieved over the photocatalyst based on SrTiO3:Sc,Mg (Fig. 2f). Previous reports on SrTiO3-based photocatalytic systems for overall water splitting with high AQYs used SrTiO3 crystals doped with Al as the base materials without exception.13–17 However, As confirmed by the ICP analyses (Table 1), the SrTiO3:Sc,Mg sample did not contain Al; thereby this is the first demonstration to achieve a high AQY for photocatalytic overall water splitting over an Al-free SrTiO3-based photocatalyst. It should be noted that the excess inclusion of Mg in the sample compared to its charged content was thought to be derived from the used MgO crucible. Concentrations of Sc and Mg dopants estimated from the ICP results are given in Table 1 are 2.9 × 1020 cm3 and 1.4 × 1020 cm3, respectively. Since a non-doped SrTiO3 powder should be a conventional insulator with its carrier (i.e., electron) density of below 1014 cm3, amounts of these dopants are sufficient to cancel out the excess electrons when the complete replacement of the B site Ti ions (Ti4+) with both Sc3+ and Mg2+ is assumed. In this case, moreover, one half oxygen vacancy (VO) per atom of Sc and one VO per atom of Mg were generated to valence electroneutrality, ca. 4.0 × 1020 cm3 of VOs are formed by the incorporation of these dopants. Although such VOs are predicted to be electron-trapping sites, they would not promote carrier recombination (see below).
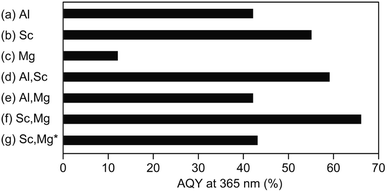 |
| | Fig. 2 AQYs of the photocatalysts based on the flux treated SrTiO3 powders after loading with Rh/Cr2O3 (for HER) and CoOOH (for OER) cocatalysts. Base SrTiO3 samples used are (a) SrTiO3:Al (labelled Al), (b) SrTiO3:Sc (labelled Sc), (c) SrTiO3:Mg (labelled Mg), (d) SrTiO3:Al,Sc (labelled Al,Sc), (e) SrTiO3:Al,Mg (labelled Al,Mg), and (f) SrTiO3:Sc,Mg (labelled Sc,Mg), respectively. The photocatalyst (g) composed of the Rh/Cr2O3-loaded SrTiO3:Sc,Mg sample without the CoOOH OER cocatalyst (labelled Sc,Mg*). | |
Table 1 Al, Sc, and Mg contents in SrTiO3 powders
| Sample |
Chargeda/mol% |
Crucibleb |
Measuredc/mol% |
| Al |
Sc |
Mg |
Al |
Sc |
Mg |
|
Charged contents for applying the flux treatment.
Materials of crucible used for the flux treatment.
Contents calculated from the results obtained by ICP-AES analyses.
Source SrTiO3 powder purchased from FUJIFILM Wako Pure Chemicals.
Not detected.
Not measured.
|
| FW SrTiO3d |
— |
— |
— |
— |
NDe |
NDe |
NMf |
| SrTiO3:Al,Sc |
4.0 |
2.0 |
0 |
Al2O3 (4N) |
0.23 |
1.66 |
NMf |
| SrTiO3:Sc,Mg |
0 |
2.0 |
0.5 |
MgO (4N) |
NDe |
1.84 |
0.91 |
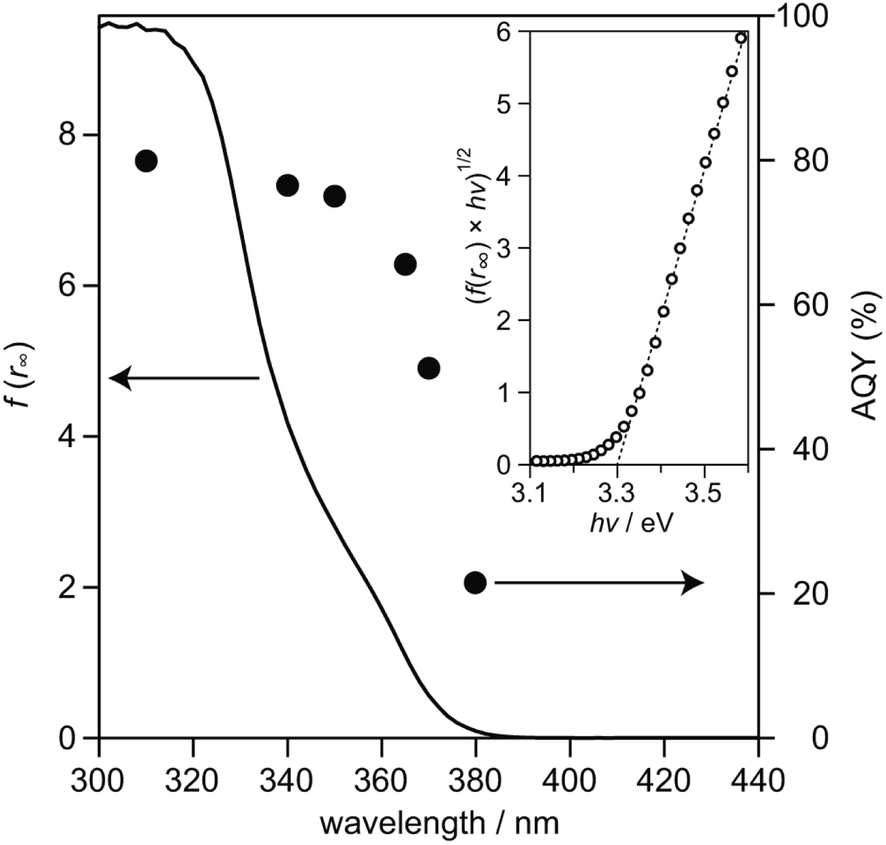
To examine relationship between the photoabsorption characteristic and photocatalytic activity of SrTiO3:Sc,Mg-based system, diffuse reflection (DR) spectrum of the SrTiO3:Sc,Mg powder was compared to the dependence of photoirradiation wavelengths of monochromatic light ranging from 310 nm to 380 nm on AQYs for overall water splitting over the SrTiO3:Sc,Mg-based photocatalyst. As shown in Fig. 3, the DR spectrum of SrTiO3:Sc,Mg showed a photoabsorption onset at ca. 380 nm; a gradual increase in photoabsorption to the shorter wavelength region was observed. For analyzing the details of the interband transition characteristics and optical bandgap (Eg) of the SrTiO3:Mg,Sc sample, a Tauc plot expressed as the following equation was applied to the DR spectrum.
| | | (f(r∞) × hν)n = k × (hν − Eg) | (4) |
where
f(
r∞) is the Kubelka–Munk (KM) function (
i.e., the vertical axis of the DR spectra) which is proportional to the absorption coefficient,
hν is the incident photon energy,
k is a proportionality constant, and
n varies from 0.5 to 2.0 depending on the nature of the optical transition. A straight line for the (
f(
r∞) ×
hν)
1/2vs. hν plot (
i.e.,
n = 1/2) was obtained at a band-edge region of the present sample, indicating indirectly allowed transition characteristics coinciding with those of SrTiO
3 (inset of
Fig. 3).
28,29 From an intersect of the linear portion of the (
f(
r∞) ×
hν)
1/2vs. hν curve with the photon energy axis, the
Eg value of the present sample was determined to be 3.30 eV, the value of which corresponded to be the reported range from 3.2 eV to 3.4 eV.
22,28–31 In addition, photoelectron yield spectroscopy (PYS) was applied to determine the valence band maximum (VBM) energy of the SrTiO
3:Sc,Mg (Fig. S4
†). As a result, the VBM energy of the sample was estimated to be −6.9 eV (
vs. vacuum), the value of which is in good agreement with that obtained for the non-doped SrTiO
3 sample reported in the literature.
32 Based on these results, electron energy structures of SrTiO
3 were confirmed to be unchanged essentially by additions of Sc and Mg dopants. With respect to the wavelength dependence of AQY over the SrTiO
3:Sc,Mg-based photocatalyst, the onset wavelength of
ca. 380 nm coincided with that of the photoabsorption onset. A proportional increase in AQYs along with the DR spectrum of SrTiO
3:Sc,Mg was obtained and the AQY value was saturated to reach 81% at 310 nm. As estimated by the absorption coefficients of the SrTiO
3 bulk crystal at various wavelengths,
22 the penetration length of photons with a wavelength close to the band edge region was found to be much larger than that in a deep UV region (Fig. S5
†). Therefore, lowering the AQY in a region close to the band edge of SrTiO
3:Sc,Mg was due to probabilistic occurrences of carrier recombination generated at an inner part of the SrTiO
3:Sc,Mg particle. Thus, there is room for further enhancements of AQY values of the present SrTiO
3:Sc,Mg-based photocatalyst by improvements of bulk qualities of the particle.
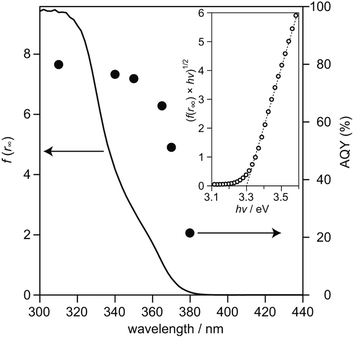 |
| | Fig. 3 DR spectra of SrTiO3:Sc,Mg (solid line) and AQYs during water splitting on SrTiO3:Sc,Mg loaded with Rh/Cr2O3 HER and CoOOH OER cocatalysts (filled symbols). The inset shows a (f(r∞) × hν)1/2vs. hν plot obtained from the DR spectrum. | |
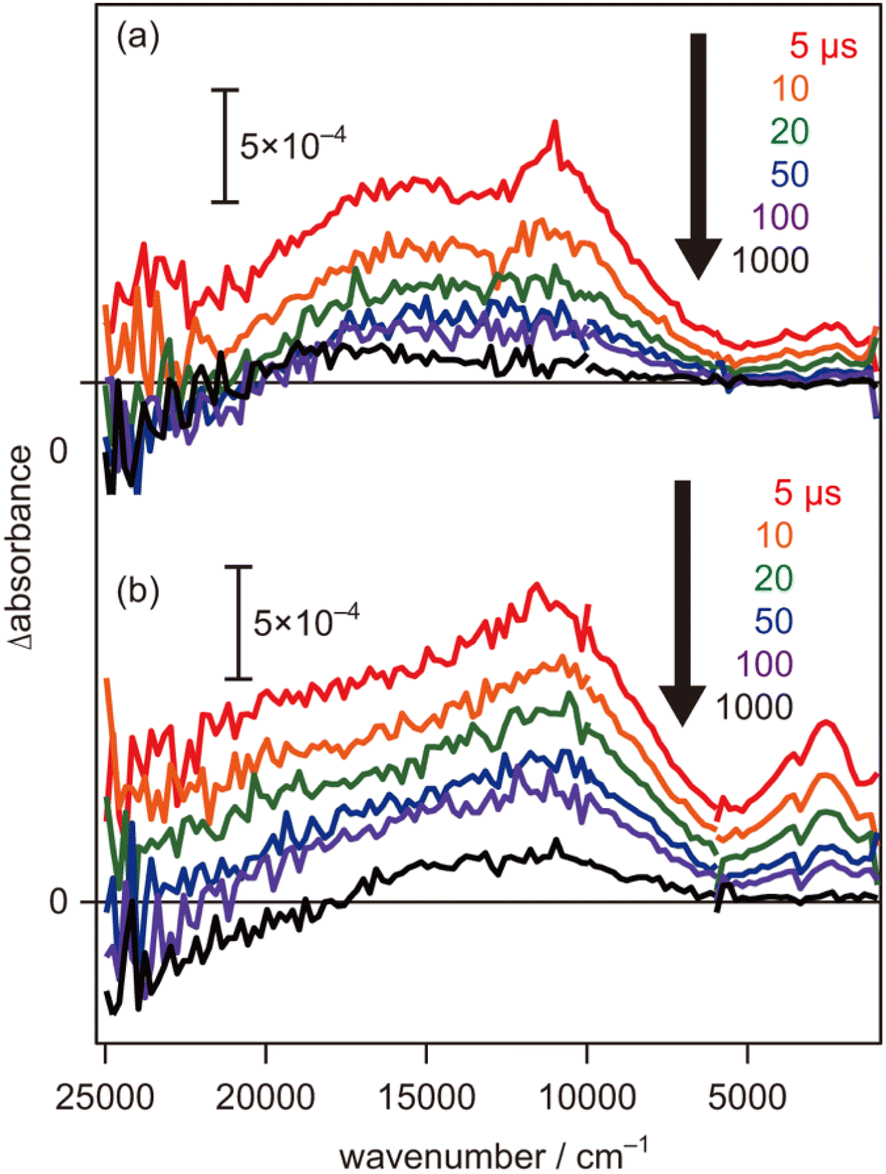
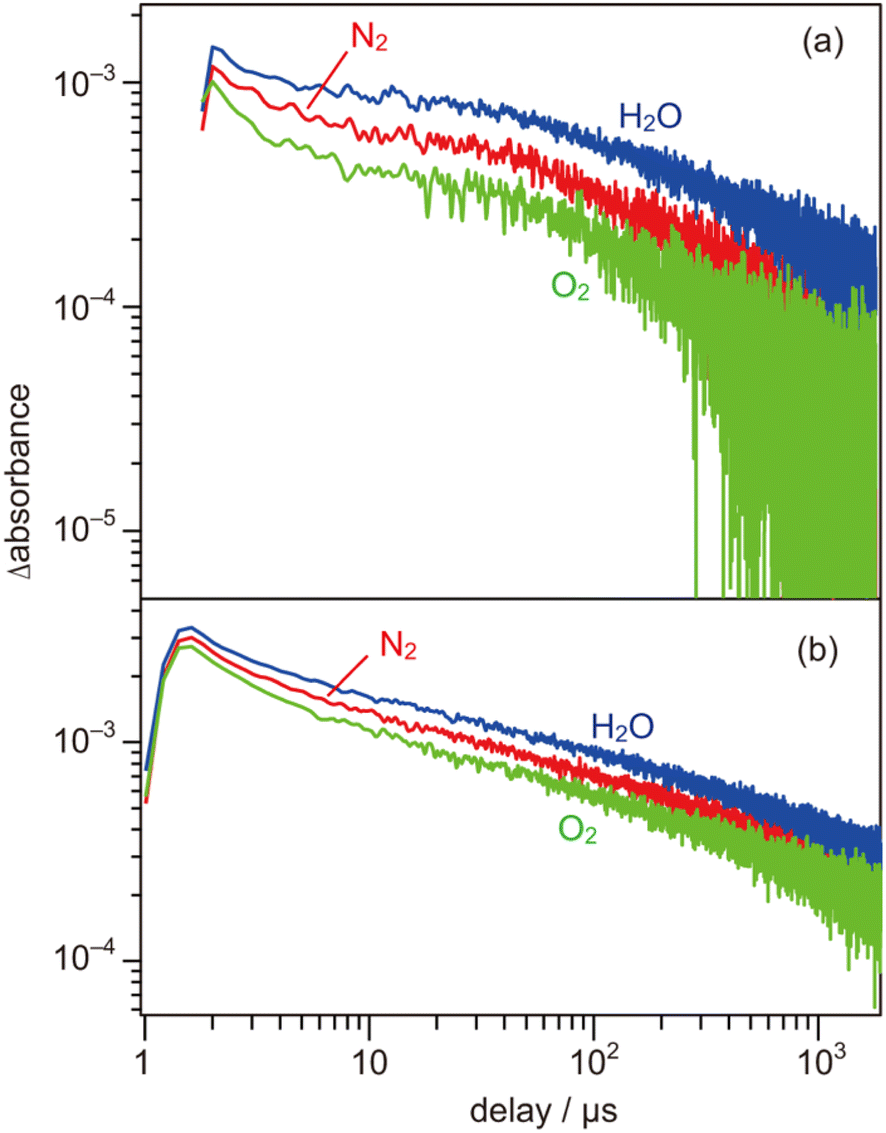
To further evaluate the electron energy characteristics of the SrTiO3:Mg,Sc sample, time-resolved absorption spectroscopy was performed. Fig. 4a shows the transient absorption spectra of the SrTiO3:Mg,Sc powder obtained under an N2 atmosphere (2.7 kPa) after 355 nm laser pulse irradiation. In accordance with previous results for SrTiO3 powders,24,33,34 absorption bands observed at 5000–1000 cm−1 can be assigned to free and/or shallowly trapped electrons, whereas, a broad absorption band at >6000 cm−1 can be identified as mixed absorption bands of trapped electrons (centred at 11000 cm−1) and trapped holes (cantered at 20000 cm−1). As shown in Fig. 4b, the shape of the transient absorption spectrum obtained for the SrTiO3:Al powder was almost identical to those for the SrTiO3:Mg,Sc sample except for the relatively intense absorption bands at 5000–1000 cm−1. Due to the increment of VOs induced by the substitution of the B site Ti ions (Ti4+) with lower valence cations, the dissociation of Ti–O bonds creates dangling bonds of Ti-3d, resulting in the formation of mid-gap states that work as electron traps.35 Low intensities of the absorption bands derived from free and/or shallow trapped electrons observed over the SrTiO3:Mg,Sc sample suggest the ease of occurrence of trapping of photoexcited electrons over the sample compared to that over the SrTiO3:Al sample. If this is the case, the difference in reactivities of the trapped electrons would affect the photocatalytic activity. To obtain insights into the reactivities of the trapped electrons, decay curves of the transient absorption at 11000 cm−1 for the SrTiO3:Mg,Sc sample were compared to those for the SrTiO3:Al sample. Although both samples showed similar decay curves when the measurements were performed under an N2 atmosphere, the introduction of O2 (2.7 kPa) or water vapor (2.7 kPa) induced appreciable differences in their decay curves, as shown in Fig. 5. In the presence of O2, the decay over the SrTiO3:Mg,Sc sample was significantly accelerated compared to that over the SrTiO3:Al sample. For example, the ratio of the intensity of the transient absorption at 11000 cm−1 for the SrTiO3:Ma,Sc sample at 100 μs measured in O2 relative to that measured in N2 was 0.76, whereas that for the SrTiO3:Al sample was larger than 0.8 (Table S1†). These results suggest the presence of reactive sites to induce efficient charge transfer of the trapped electrons to O2 on the surface of the SrTiO3:Mg,Sc sample. The high reactivity of the trapped electrons with reactant molecules was also observed on a Na-doped SrTiO3-based photocatalyst34 as well as a carbon nitride (CN)-based photocatalyst.36 The energy of the trapped electrons at 11000 cm−1 (ca. 1.4 eV) below the conduction band minimum (CBM) is thermodynamically unacceptable to induce direct electron transfer from such a deep trap to O2. However, because the observed band spreads out by stepping out to the lower wavenumber region, it implies that the trap levels of the electrons are not localized but are extended continuously to the CBM energy, such an electronic structure is likely to enable possible electron transfer to O2 in the present system. The other feature of the decay curve of the SrTiO3:Mg,Sc sample was a remarkable deceleration induced by the introduction of water vapors (Fig. 5a). Compared to the SrTiO3:Al sample, in the presence of water vapor (Fig. 5b), as shown in Table S1,† the intensity of the decay for the SrTiO3:Ma,Sc sample at 100 μs measured in water vapor was 1.7 times more than that measured in N2, while such an increment was less significant for the SrTiO3:Al sample (below ×1.4). Since water vapor is thought to increase the lifetime of the trapped electrons when it reacts with holes, these results imply that the SrTiO3:Mg,Sc sample can utilize positive holes more efficiently than the SrTiO3:Al sample.
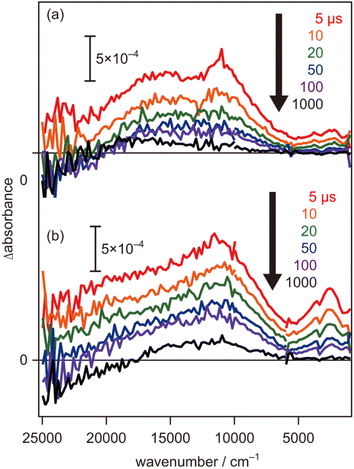 |
| | Fig. 4 Transient absorption spectra of (a) SrTiO3:Sc,Mg and (b) SrTiO3:Al powders irradiated by UV (355 nm) laser pulses in N2 (2.7 kPa). The pump energy was 0.5 mJ per pulse, and the repetition rate was 5 Hz. | |
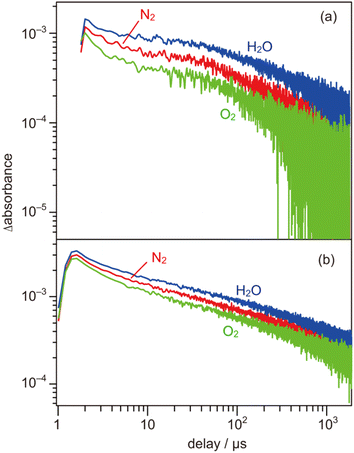 |
| | Fig. 5 Decay curves of the transient absorption of (a) SrTiO3:Sc,Mg and (b) SrTiO3:Al powders irradiated by 355 nm laser pulses (0.5 mJ per pulse at 0.2 Hz). The decay curves were 11000 cm−1, in N2 (2.7 kPa), O2 (2.7 kPa), and water vapor (2.7 kPa). | |
In contrast to the above-mentioned characteristics of the SrTiO3:Mg,Sc sample that are beneficial for inducing overall water splitting, one of the negative features observed in the transient absorption spectra is their relatively low intensities of the mixed absorption bands of the trapped electrons and trapped holes at the initial 5 μs when compared to those obtained for the SrTiO3:Al sample (Fig. 4). As discussed above, these trends suggest that a certain amount of photoexcited carriers would be recombined on a timescale faster than 5 μs, and thereby, further improvements in bulk and/or surface qualities are necessary to obtain AQY reaching 100%.
As mentioned above in morphological analyses, the SrTiO3:Sc,Mg particle has the characteristic nanosized steps. The effectiveness of such a nanosized step structure for the enhancement of photocatalytic activity for the overall water splitting was reported for the system based on a La-doped NaTaO3 perovskite particle (NaTaO3:La) loaded with a NiO HER cocatalyst.37 The result supports the significance of nanostructure to induce efficient overall water splitting over the present SrTiO3:Sc,Mg-based photocatalytic system. However, since the NaTaO3:La particle had a nanosized step on its entire surface, there is the definitive difference in the surface structure of the present SrTiO3:Sc,Mg particle having both flat {100} facets and the nanosized-step form of the {100} facets formed locally on the edge of the particle. Thus, there should be a distinctive mechanism to induce overall water splitting over the SrTiO3:Sc,Mg-based photocatalyst. In order to obtain mechanistic aspects, morphological properties of the SrTiO3:Sc,Mg particle loaded with the Rh/Cr2O3 HER cocatalyst and that loaded with both of the Rh/Cr2O3 HER and the CoOOH OER cocatalysts were compared. Fig. 6 shows the SEM images and corresponding schematic illustrations of these samples. The former sample exhibited deposits of appreciable amounts of nanoparticles on the flat {100} facets, as shown in Fig. 6a and c. Although it is not very clear to observe local morphologies of the nanosized step regions, the loaded Rh/Cr2O3 HER cocatalyst tended to deposit the part of the {100} facets. On the other hand, the later sample obviously indicated the presence of nanoparticulate deposits at the nanosized step region of several particles (Fig. 6b and d), suggesting that the CoOOH OER cocatalyst was loaded preferentially at the nanosized step of the SrTiO3:Sc,Mg particle. These results indicate that efficient photocatalytic overall water splitting over the SrTiO3:Sc,Mg-based photocatalysts are likely to be achieved by the separation of reaction sites for H2 and O2 production. As has been proposed in previous reports,17 the flat {100} facets are the H2 evolution sites because of the occurrence of preferential collection of the photoexcited electrons. Since there are no {110} facets of the SrTiO3:Sc,Mg particle, holes would be attracted to the nanosized steps for inducing O2 evolution. It should be noted that the AQY (at 365 nm) obtained by the Rh/Cr2O3-SrTiO3:Sc,Mg sample, i.e., the photocatalyst without the CoOOH OER cocatalyst, reached 43% (Fig. 2g). The value was comparable to that obtained over the photocatalyst composed of SrTiO3:Al and both Rh/Cr2O3 HER and CoOOH OER cocatalysts, implying possible achievements of efficient charge separation over the equivalent {100} facet having the unique nanosized step structure without using the non-equivalent {110} facet requisite for the Al-doped system. Therefore, this would be the other strategy to design a highly efficient and durable photocatalyst for overall water splitting.
 |
| | Fig. 6 SEM images of (a) SrTiO3:Sc,Mg loaded with Rh/Cr2O3 HER cocatalyst nanoparticles (Rh/Cr2O3-SrTiO3:Sc,Mg) and (b) SrTiO3:Sc,Mg loaded with Rh/Cr2O3 HER and CoOOH OER (Rh/Cr2O3-CoOOH-SrTiO3:Sc,Mg) cocatalyst nanoparticles. Schematic illustrations of these Rh/Cr2O3-SrTiO3:Sc,Mg and Rh/Cr2O3-CoOOH-SrTiO3:Sc,Mg particles are given in panels (c) and (d), respectively. | |
Conclusions
The effectiveness of co-doing of Sc and Mg into the crystalline lattice of the SrTiO3 particle by applying a high temperature (1200 °C) flux treatment in a molten SrCl2 for achieving a high AQY of overall water splitting was demonstrated. The sample co-doped with Sc and Mg (SrTiO3:Sc,Mg) had uniform-sized single crystalline particles with particle sizes of a hundred and several tens of nanometers, similar to that obtained for the conventional Al-doped SrTiO3 particle (SrTiO3:Al). The significant morphological feature of the SrTiO3:Sc,Mg particle is that the sample was cubic having a nanosized step structure on the edge of the particle. The unique morphology realized an efficient separation of the reaction sites for H2 and O2 evolution: the Rh/Cr2O3 HER cocatalyst was reductively photodeposited on the flat {100} facets, and subsequently mediated electron transfer to water for producing H2; the CoOOH OER cocatalyst was oxidatively photodeposited on the nanosized step to induce water oxidation. Since the cubic morphology of the sample was derived from the selective exposure of the equivalent {100} facets, the present nanosized steps with directions orthogonal to the {100} facets were also composed of the {100} facets. Hence, the concept of separating the reduction and oxidation sites in the present system was different from the use of two different facets ({100} and {110} facets) for the SrTiO3:Al-based photocatalytic system reported so far. The use of the stable crystalline facets with controlled nanostructures reported here is, therefore, the other concept for the development of materials for efficient utilization of the photoexcited electrons and holes to induce photocatalytic overall water splitting under visible light.
Author contributions
S. I. and T. M. directed and led the research. R. O. performed structural analyses and photocatalytic experiments. Y. N. and A. Y. performed time-resolved absorption measurements and analyses. All the authors discuss the results. S. I. wrote the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was performed under the Hirao Taro Foundation of KONAN GAKUEN for Academic Research, award no. 202201. This work was also partly supported by JSPS Grants-in-Aid for Scientific Research (KAKENHI), awards no. 21K19033 and 23H02074. We gratefully acknowledge the helpful discussions with Professor Kazunari Domen (Shinshu University and The University of Tokyo) and Professor Tsuyoshi Takaka (Shinshu University).
References
- K. Domen, S. Naito, T. Onishi and K. Tamaru, Chem. Phys. Lett., 1982, 92, 433 CrossRef CAS.
- K. Domen, A. Kudo and T. Onishi, J. Catal., 1986, 102, 92 CrossRef CAS.
- K. Domen, A. Kudo, T. Onishi, N. Kosugi and H. Kuroda, J. Phys. Chem., 1986, 90, 292 CrossRef CAS.
- F. T. Wagner and G. A. Somorjai, J. Am. Chem. Soc., 1980, 102, 5494 CrossRef CAS.
- K. Sayama and H. Arakawa, Chem. Lett., 1992, 21, 253 CrossRef.
- S. Ikeda, K. Hirao, S. Ishino, M. Matsumura and B. Ohtani, Catal. Today, 2006, 117, 343 CrossRef CAS.
- R. Asai, H. Nemoto, Q. Jia, K. Saito, A. Iwase and A. Kudo, Chem. Commun., 2014, 50, 2543 RSC.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054 CrossRef CAS.
- Q. Jia, A. Iwase and A. Kudo, Chem. Sci., 2014, 5, 1513 RSC.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 1 Search PubMed.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, Y. Suzuki, Z. Pan, J. Seo, M. Katayama, T. Minegishi, H. Nishiyama, T. Takata, K. Seki, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2017, 139, 1675 CrossRef CAS PubMed.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2018, 2, 509 CrossRef CAS.
- Y. Ham, T. Hisatomi, Y. Goto, Y. Moriya, Y. Sakata, A. Yamakata, J. Kubota and K. Domen, J. Mater. Chem. A, 2016, 4, 3027 RSC.
- T. H. Chiang, H. Lyu, T. Hisatomi, Y. Goto, T. Takata, M. Katayama, T. Minegishi and K. Domen, ACS Catal., 2018, 8, 2782 CrossRef CAS.
- H. Lyu, T. Hisatomi, Y. Goto, M. Yoshida, T. Higashi, M. Katayama, T. Takata, T. Minegishi, H. Nishiyama, T. Yamada, Y. Sakata, K. Asakura and K. Domen, Chem. Sci., 2019, 10, 3196 RSC.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411 CrossRef CAS.
- T. Takata and K. Domen, J. Phys. Chem. C, 2009, 113, 19386 CrossRef CAS.
- T. Takata, C. Pan, M. Nakabayashi, N. Shibata and K. Domen, J. Am. Chem. Soc., 2015, 137, 9627 CrossRef CAS PubMed.
- G. W. Gobeli and F. G. Allen, Phys. Rev., 1962, 127, 141 CrossRef CAS.
- S. Ikeda, W. Fujita, R. Katsube, Y. Nose, H. Suzuki, R. Abe and K. Yoshino, Electrochim. Acta, 2023, 454, 142384 CrossRef CAS.
- S. Zollner, A. A. Demkov, R. Liu, P. L. Fejes, R. B. Gregory and P. Alluri, J. Vac. Sci. Technol. B, 2000, 18, 2242 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 118, 79703 Search PubMed.
- A. Yamakata, J. J. M. Vequizo and H. Matsunaga, J. Phys. Chem. C, 2015, 119, 24538 CrossRef CAS.
- J. J. M. Vequizo, H. Matsunaga, T. Ishiku, S. Kamimura, T. Ohno and A. Yamakata, ACS Catal., 2017, 7, 2644 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., 1976, 32, 751 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- L. Gracia, J. Andrés, V. M. Longo, J. A. Varela and E. Longo, Chem. Phys. Lett., 2010, 493, 141 CrossRef CAS.
- M. Cardona, Phys. Rev., 1965, 140, A651 CrossRef.
- M. Capizzi and A. Frova, Phys. Rev. Lett., 1970, 25, 1298 CrossRef CAS.
- K. W. Blazey, Phys. Rev. Lett., 1971, 27, 146 CrossRef CAS.
- J.-i. Fujisawa, T. Eda and M. Hanaya, Chem. Phys. Lett., 2017, 685, 23 CrossRef CAS.
- A. Yamakata, H. Yeilin, M. Kawaguchi, T. Hisatomi, J. Kubota, Y. Sakata and K. Domen, J. Photochem. Photobiol., A, 2015, 313, 168 CrossRef CAS.
- K. Kato, J. Jiang, Y. Sakata and A. Yamakata, ChemCatChem, 2019, 11, 6349 CrossRef CAS.
- A. Janotti, J. B. Varley, P. Rinke, N. Umezawa, G. Kresse and C. G. Van de Walle, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085212 CrossRef.
- Z. Teng, Q. Zhang, H. Yang, K. Kato, W. Yang, Y.-R. Lu, S. Liu, C. Wang, A. Yamakata, C. Su, B. Liu and T. Ohno, Nat. Catal., 2021, 4, 374 CrossRef CAS.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Riku
Okamoto
a,
Akira
Kimura
a,
Yuhi
Nakayasu
c,
Akira
Yamakata
*ab,
Riku
Okamoto
a,
Akira
Kimura
a,
Yuhi
Nakayasu
c,
Akira
Yamakata