
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe importance of antibody orientation for enhancing sensitivity and selectivity in lateral flow immunoassays
Zhao-Yu
Lu
a and
Yang-Hsiang
Chan
 *abc
*abc
aDepartment of Applied Chemistry, National Yang Ming Chiao Tung University, Hsinchu, 30050 Taiwan. E-mail: yhchan@nycu.edu.tw
bCenter for Emergent Functional Matter Science, National Yang Ming Chiao Tung University, Hsinchu, 30050 Taiwan
cDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung, 80708 Taiwan
First published on 23rd August 2024
Abstract
In the field of point-of-care diagnostics, lateral flow assays (LFAs) stand out as highly promising due to their compact size, ease of use, and rapid analysis times. These attributes make LFAs invaluable, especially in urgent situations or resource-limited regions. However, their Achilles' heel has always been their limited sensitivity and selectivity. To address these issues, various innovative approaches, including sample enrichment, assay optimization, and signal amplification, have been developed and are extensively discussed in the literature. Despite these advancements, the importance of antibody orientation is often neglected, even though improper orientation can significantly impair detection performance. This review article first explores well-established traditional methodologies, such as minor physical adjustments and non-specific chemical bond formations. It then shifts focus to the oriented immobilization of antibodies on probe surfaces. This approach aims to enhance sensitivity and selectivity fundamentally by leveraging protein affinities or complementary amino acid sequences. The review summarizes the impact of antibody orientation on the analytical performance of LFAs in terms of sensitivity, specificity, speed, reliability, cost-effectiveness, and stability. Additionally, we introduce recent modifications to assay membrane materials and discuss the current limitations and future prospects of LFAs.
1. Introduction
The exact origins of the first lateral flow assay (LFA)-like device remains a topic of debate; however, it is widely acknowledged that the foundational framework for LFAs was established in the 1950s by pioneers such as Plotz and Singer, who devised the latex agglutination assay, and Berson and Yalow, who pioneered paper-based immunoassays.1,2 From the latter half of the 20th century to the first decade of the 21st century, a multitude of immunoassays emerged thanks to advancements in technology. Among the commonly used immunoassays are enzyme immunoassay (EIA), enzyme-linked immunosorbent assay (ELISA), fluoroimmunoassay (FIA), and radioimmunoassay (RIA), among others.3,4 These assays target a diverse range of substances, including drugs, small molecules, proteins, nucleic acids, microbes, and viruses. LFAs, with their versatility, find applications in various settings, including laboratories, medical centers, and everyday life.5 Undoubtedly, the evolution of immunoassay technology has played a significant role in human history. Reverse transcriptase PCR (RT-PCR) stands out as the primary diagnostic tool in the field of bioassay.6,7 Its ability to repeatedly reverse transcribe mRNA fragments over extended periods results in remarkable amplification of DNA signals, thereby enhancing sensitivity and selectivity compared to many other immunoserological methods.8 While RT-PCR is commonly employed in non-urgent scenarios such as academic research and paternity testing due to its longer turnaround time, its indispensability becomes evident in urgent situations. The rapid global impact of SARS-CoV-2 serves as a poignant example of RT-PCR's pivotal role in addressing sudden and pressing healthcare needs.9During the COVID-19 pandemic, pre-existing problems were magnified, exacerbating chaos due to the uneven distribution of clinical resources and shortages of medical personnel. This led to the extension of epidemic areas and a soaring death rate.10–12 As a result, there was an urgent need for a commercially available diagnostic tool capable of providing rapid, high-medium accuracy results without requiring individuals to leave their homes or seek professional guidance. This demand aligned with the concept of point-of-care testing,13 leading to a resurgence in the popularity of LFAs In such emergencies, LFAs became indispensable as they offered a portable, affordable, and professional-free diagnostic solution (Scheme 1). LFAs struck a balance between turnaround time and accuracy, delivering results in under half an hour. In contrast, PCR testing required in-person sampling and took hours to days for results, posing additional cost burdens.14 Despite not being perfect, LFAs demonstrated sensitivity ranging from 37.7% to 99.2% and over 92% selectivity,15 surpassing self-diagnostics used at home. However, as LFAs gained popularity, concerns about sensitivity and selectivity grew.16 False negatives and false positives remained significant challenges,17–19 impacting human rights and endangering lives by affecting quarantine lengths and hospitalization decisions.
 | ||
| Scheme 1 Overview for the comparison of traditional RT-PCR with current LFAs aiming at enhanced sensitivity and selectivity. | ||
Continuous efforts are made to enhance the selectivity and sensitivity of LFAs, which can be categorized into internal and external modifications (Scheme 2). Internal modifications focus on improving chemical interactions within the LFA system, such as the use of polymer-based LFAs to facilitate protein grafting via the hydrophobic effect,20 the implementation of 3D bio-linkers to reduce steric hindrance and control fluid flow rate by increasing surface area,21 and the addition of extra conjugated pads to allow for multi-layer nanoparticle (NP) conjugation, thereby enhancing colorimetric signal strength.22 On the other hand, external modifications primarily target the expression of signals or readouts. Examples include: (i) adjusting the size of gold nanoparticles (AuNPs), (ii) optimizing the length of linkers, (iii) applying electroactive tags, and (iv) enhancing visual signals. The first two strategies focus on amplifying surface plasmon resonance (SPR), while the latter two aim to improve detection methods using electrical signals, chemiluminescence, colorimetry, or quantum dots.5,23 Additionally, magnetism can be utilized for sample localization and preconcentration.24 Despite the myriad of techniques available, these modifications often prioritize assay advancement and sample enrichment,25 potentially compromising the inherent advantages of LFAs relative to PCR, such as their short detection time, simple assay design, and minimal sample pretreatment requirements.23 Upon closer examination, it becomes apparent that the primary objective of conventional approaches is to improve binding efficiency, a consequence derived from achieving correct probe orientations.26,27
 | ||
| Scheme 2 Strategies of the modification of antibody orientation: membrane engineering and probe modification. | ||
Being conscious of the critical role of probe orientation, recent studies have placed significant emphasis on immobilization techniques. Immobilization, in essence, leverages (bio)chemistry to maximize the exposure of effective binding sites, thereby enhancing binding efficiency. Whether anchoring probes onto nanoparticles, specific analytes, or test and control lines, oriented immobilization ensures secure and precise bindings by maximizing the exposure of binding sites. In such scenarios, LFAs maintain their hallmark characteristics of short detection times, low complexity, and minimal sample pretreatment requirements. Additionally, higher sensitivity and/or selectivity are achieved as a result of an increased number of capture–analyte complexes.28 While the theory behind oriented immobilization appears straightforward, practical complexities often hinder scientists from implementing these techniques. Introducing immobilization methods that are easy to rationalize and operate, such as pH modulation and surface charge adaptation, may encounter obstacles, such as limited immobilization efficiency. Chemical bonds formed in these methods are often induced and therefore weak in energy, leading to potential challenges in achieving oriented immobilization.27,29 For methods with higher immobilization efficiency, costs may be reflected in terms of expense, time, and effort. Binding through chemically designed covalent bonds, for example, is relatively effective but requires additional steps and the risk of probe conformational changes must be considered.30 Incorporating proteins as assistants presents a double-edged sword. On one hand, proper proteins can guide probes directionally due to specific interactions in protein domains. On the other hand, as proteins are biologically active, factors such as environmental conditions, affinity to targets, and cross-activities to other functional groups must be carefully considered.21,31
It's undeniable that the existing methodologies aimed at improving the performance of LFAs still have room for enhancement. However, achieving high sensitivity and selectivity is imperative for realizing the concept of point-of-care diagnostics. Currently, oriented immobilization appears to offer the most promising solution. Generally, antibody immobilization methodologies can be categorized into two main types: non-covalent modification and covalent modification. Regardless of the principle employed, the primary goal is to maximize the exposure of binding sites to maximize sensitivity and selectivity in LFAs. When antibodies are attached to the membrane surfaces of LFAs, four types of arrangements may occur. Among these, the “end on” orientation is considered superior as it exposes the binding sites on Fab segments to a greater extent, significantly enhancing the likelihood of efficient bindings.32 Consequently, efforts focused on tuning antibodies to achieve an end-on orientation have become a focal point.
2. Manipulation of antibody orientation
Various methods exist for directing antibodies to achieve ideal orientation. Traditionally, physical adsorption is the most commonly used method due to its intuitive and easy operation. Physical adsorption involves immobilization approaches that do not significantly alter the electron structure of the molecules involved. Examples of non-covalent interactions utilized in physical adsorption include hydrophobic effects, hydrogen bonds, electrostatic interaction and van der Waals forces.33 Techniques categorized under physical adsorption typically leverage the surface properties of probes, nanoparticles, and platform materials themselves.34–36 For example, pH adjustment of the buffer to approach the isoelectric point of antibodies29 or replacing the material of the membrane strip with one that is more protein-reactive.37 However, since the chemical bonds formed in physical adsorption are induced, they are much weaker compared to covalent chemical bonds.38 Consequently, these methods are not as effective or oriented, resulting in random immobilizations.35,39To achieve more oriented and complete immobilization of antibodies, various modification methodologies have been developed. Oriented immobilizations can be categorized into two groups based on where the modifications occur: probe modification and membrane modification (Scheme 2). Probe modification involves both non-covalent and covalent methods depending on the type of chemical bonds formed. In contrast, membrane modification includes modification of test and control lines, as well as the substrate compositions of the test strips (Scheme 3).
 | ||
| Scheme 3 Tree diagram summarizing the strategies of antibody immobilization with ordered orientation. | ||
2.1. Modifications of probes
 | ||
| Scheme 4 Typical types of non-covalent bonding among proteins used in LFAs. | ||
2.1.1.1 Protein A and protein G. Protein A, protein G, and protein A/G are widely used proteins for immobilizing certain immunoglobulins. Although they possess differing domains, their specific binding affinities towards the Fc segments of immunoglobulins are consistent.43,44 To optimize binding capacity, careful selection of protein–immunoglobulin pairs is necessary, as affinity varies among immunoglobulin subclasses. Additionally, the pH of the buffer can impact binding efficiency.45 Despite the advantages of oriented immobilization techniques,46–48 protein A and protein G still have limitations. Firstly, there is a possibility that the segment on antibodies bound is Fab region rather than the hypothesized Fc, owing to cross-activity between protein domains.21,31,49 Secondly, the binding between protein A or protein G and antibodies is reported to be reversible,50,51 which can decrease the efficiency of oriented immobilization. In 2018, a novel and robust technology was introduced, leveraging the specific binding of the Fc region of antibodies with streptococcal protein G on the surface of probes, followed by covalent cross-linking at the binding sites to stabilize the interaction.52 This method significantly improved detection sensitivity, demonstrating that the proper orientation of antibodies on probe surfaces effectively enhances their immunological activity compared to random immobilization methods.
Recently, Sotnikov et al. developed an advanced secondary stage LFA based on the conventional type.53 By incorporating an immunoglobulin-binding protein, they amplified the oriented conjugate concentration, thereby significantly enhancing sensitivity (Fig. 1A(i)). The initial phase of immunochromatographic serodiagnosis was performed in its standard format, utilizing a conjugate of gold nanoparticles with staphylococcal immunoglobulin-binding protein A and an antigen immobilized on a working membrane. In the subsequent phase, a labeled immunoglobulin-binding protein was introduced, which intensified the coloration of the bound immune complexes. This two-step method—binding specific antibodies followed by enhancing the coloration of the complexes—significantly minimized the impact of non-specific immunoglobulins on the assay results (Fig. 1A(ii)). This approach was tested using a recombinant RBD protein of SARS-CoV-2, resulting in a more than two orders of magnitude increase in test zone coloration intensity, thereby markedly reducing false-negative outcomes. The diagnostic sensitivity of the LFA improved from 62.5% in the conventional format to 100% in the enhanced format. Similarly, in Kim's study, the oriented immobilization of antibodies on magnetic beads using protein G was compared to random immobilization with amine groups (Fig. 1B).54 Although the conjugate concentration was lower with oriented immobilization, a larger signal enhancement was observed at higher target concentrations. This indicates that using oriented immobilization techniques can indeed improve sensitivity. In addition to the direct application of protein A and protein G, crosslinking techniques can also be employed. Choi and his team genetically engineered protein G to include two cysteine residues at its C-terminus.55 This modification allows the thiol groups on cysteine to self-assemble onto a gold surface, greatly enhancing the fluorescence performance of the gold plate (Fig. 1C).
 | ||
| Fig. 1 Antibody oriented immobilization with the assistance with protein A or protein G. (A) Protein A facilitates secondary stage LFA: (i) scheme of the proposed enhanced serodiagnostic LFA and (ii) photographs of test strips after the common (left panel) and enhanced (right panel) LFA of samples containing 0 (1), 0.003 (2), 0.01 (3), 0.03 (4), 0.1 (5), 0.3 (6), 1 (7), 3 (8), and 10 (9) μg mL−1 of MAb RBD5313. (B) Immobilization of antibodies onto magnetic beads in random (left) and oriented (right) manners. (C-i) Schematic showing the IgG Fc-binding domain modification and IgG immobilization on gold surface by the cysteine-modified protein G. (C-ii) Fluorescence microscope analysis of Cy3-labeled antibodies immobilized on PBS-treated gold (left), wild-type protein-treated (middle), and cysteine-modified protein G-treated (right) gold surface. Reproduced with permission from ref. 53 and 55. | ||
2.1.1.2 Biotin–(strept)avidin interaction. Another commonly utilized protein pair consists of biotin and streptavidin (or its derivatives). Renowned for possessing one of the strongest non-covalent bonds (with a Kd value of ∼1014 M),56 biotin–streptavidin complexes exhibit exceptional stability across a wide range of pH levels, temperatures, and organic solvents. Furthermore, the binding ratio of biotin to streptavidin is high at 4
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1,57 making them highly suitable for biomolecule conjugation. It is important to note that despite sharing a common origin with “avidin,” streptavidin and avidin come from different biological sources.58 This results in differing affinities for biotin. Avidin, in particular, is reported to exhibit pseudocatalytic activity and nonspecific binding.59
:1,57 making them highly suitable for biomolecule conjugation. It is important to note that despite sharing a common origin with “avidin,” streptavidin and avidin come from different biological sources.58 This results in differing affinities for biotin. Avidin, in particular, is reported to exhibit pseudocatalytic activity and nonspecific binding.59
The pre-incubation of streptavidin with capture probes leads to the multi-oriented immobilization of streptavidin complexes, where the binding sites are then exposed for biotinylated detection probes. Although specific orientation was not realized through this interaction, the multivalent interaction of streptavidin with biotinylated antibodies usually lead to signal enhancement. Nichols and his colleagues successfully applied this method to achieve the detection of SARS-CoV-2, as shown in Fig. 2A.60 In another example, Wu's group devised BioAb/SA-BSA/MPA/AuNS/SPCE-based immunosensors specifically intended for detecting SARS-CoV-2 (Fig. 2B(i)).61 Specifically, this research developed label-free electrochemical impedance spectroscopy (EIS)-based immunosensors using gold nanostructured screen-printed carbon electrodes (AuNS/SPCEs) to detect the SARS-CoV-2 nucleocapsid protein (N-protein) in saliva. By utilizing short-chain 3-mercaptopropionic acid (MPA) as a linker to covalently bond streptavidin (SA) and bovine serum albumin (BSA) for controlling the oriented immobilization of the biotinylated anti-N-protein antibody (BioAb), the developed immunosensors exhibited improved sensitivity, a lower limit of detection (LOD), and better reproducibility compared to randomly immobilized antibody immunosensors and long-chain 11-mercaptoundecanoic acid (MUA)-modified immunosensors (BioAb/SA-BSA/MUA/AuNS/SPCEs). Additionally, the immunosensor displayed minimal cross-reactivity with other viral antigens, including MERS-CoV N-protein, influenza A N-protein, influenza B N-protein, and SARS-CoV-2 spike protein, underscoring the high specificity of the immunosensors (Fig. 2B(ii)). This illustrates the potential for improved virus detection through the use of BSA in immobilizing antibodies on various materials.
 | ||
| Fig. 2 Antibody oriented immobilization with the assistance from biotin–avidin interactions. (A-i) Development of a half-strip LFA with the introduction of biotin–avidin chemistry for the detection of SARS-CoV-2. (A-ii) Dose–response curve produced for the half-strip LFA by employing two commercially accessible SARS-CoV-2 nucleocapsid (N) proteins obtained from Genemedi and Genscript. (B-i) The steps showing the fabrication of the AuNS/SCPE-based immunosensor, including modification of AuNS/SPCE with MPA, activation with EDC/NHS, immobilization of SA-BSA, immobilization of BioAb, and finally, N-protein immunoreaction. (B-ii) Specificity performance of the BioAb/SA-BSA/MPA/SPCEs towards various analytes. Reproduced with permission from ref. 60 and 61. | ||
 | ||
| Scheme 5 The strategies of covalent bonding in tuning antibody orientation. (A) Antibody structure and functional Fc and Fab regions. The covalent bonding reactions between antibodies and probes, including (B) carboxylic acid–amine, (C) disulfide cleavage, and (D) carbohydrate reactions. | ||
2.1.2.1 Coupling between amine and carboxyl functional groups. Given that hydrophilic functional groups tend to become exposed during protein folding processes,64,65 traditional immobilization techniques often rely on the densely covered amine and carboxyl groups present on antibodies. The most commonly used method involves forming amide bonds through EDC/NHS coupling (Scheme 4B),66 while more sophisticated approaches include maleimide functionalization, which attaches targets to thiol/sulfhydryl groups on the surfaces of probes.67 Prof. Ding's and Tang's groups achieved successful coupling of antibodies with AIE luminogens (AIEgens) through EDC/NHS, enabling the visual detection of receptor binding domain (RBD) and N antigens of SARS-CoV-2 (Fig. 3A(i)).68 This method allows for a low limit of detection, as low as 6.9 ng mL−1 for RBD protein and 7.2 ng mL−1 for N protein with high specificity (Fig. 3A(ii)). The scope of targets that can be immobilized through amine and carboxyl coupling chemistries extends beyond antibodies. Nucleic acids69 and surfaces modifiable with reactive functional groups are also compatible,70,71 making amine/carboxyl modification widely applicable. However, the antibody orientation remains an issue when the Fab region reacts with the COOH groups on the reporter's surface, hindering interactions between the antibody and the target antigen. To address this issue, Chan's group used the two-step amine–carboxylic acid reactions with 4,7,10-trioxa-1,13-tridecanediamine, a flexible diamine-PEG linker, as the linker to couple polymers with antibodies (Fig. 3B).72 The study revealed negligible nonspecific adsorption, along with a significant increase in specific binding affinity, as depicted in Fig. 3B. These findings illustrate that the flexible diamine linker grants antibodies the freedom to orient themselves appropriately to align with the target antigen domains. However, it's important to note that amino groups are commonly found on antibodies, which can lead to random orientations during immobilization through amine and carboxyl groups.73 Additionally, the pH plays a crucial role because amino groups exhibit different pK values and thus buffers that are not sufficiently alkaline may result in some of them being protonated,66,74 thereby reducing modification efficiency.
 | ||
| Fig. 3 Modification of antibody orientation with the assistance from amine and carboxyl groups. (A-i) Schematic of the configuration and the detection mechanism of AIEgen-based test strips. (A-ii) Images of the test strips under 365 nm light irradiation for 0, 0.001, 0.01, 0.1, 1, 10, and 20 μg mL−1 RBD (left) and N (middle) proteins in PBS. Right panel: Images of the test strips under 365 nm light irradiation after reaction with samples containing different antigens (AFP, HCG, CEA, CA125, HSA, S2 protein, RBD protein, FBS, CRP, and N protein at 1 μg mL−1). (B-i) Schematic showing the traditional method for functionalization of antibodies on the probe surfaces. (B-ii) Modification of diamine as the linker between probe surfaces and antibodies. The bottom panels show the photographs of test strips after reaction with samples containing 0 or 5 ng mL−1 of PSA antigens. Reproduced with permission from ref. 68 and 72. | ||
2.1.2.2 Disulfide bond. Disulfide bonds present in the hinge regions of antibodies can also facilitate immobilization (Scheme 4C). Treating these disulfide bonds with reducing agents such as tris(2-carboxyethyl)phosphine (TCEP) or 2-mercaptoethylamine (2-MEA) results in half antibodies, which can then bind to exposed reduced thiols.75,76 Typically, these thiols can be coupled with probes such as Au nanoparticles, forming stable Au–S bonds.77,78 Conversely, thiols can also be modified on noble metal surfaces, allowing oriented antibody immobilization through linkages with various terminal functional groups.79 However, while disulfide modification enables site-directed immobilization, reported downsides include protein conformational changes and aggregation with nanoparticles, which can impede probe binding efficiency.80 Moreover, it's important to note that the term “disulfide bond” in antibodies refers to both interchain and intrachain types. When modifying antibodies, key factors to consider include the specific characteristics of different immunoglobulins, receptor affinity, chemical stability, and amino acid sequences.81 Jeong et al. accomplished approximately an eightfold increase in antibody binding on silicon nanoparticles through maleimide coupling, followed by azide–alkyne cycloaddition click chemistry. In the technique, the linker which included both maleimide and dibenzocyclooctyne groups as terminals was the key. The thiol groups from disulfide bonds on antibodies were covalently bond with one side of the maleimide, then, the dibenzocyclooctyne was coupled with the azide groups modified on silicon nanoparticles. This method contrasts with traditional EDC/NHS coupling, which typically results in random immobilization.82 The employment of thiol-immobilized antibodies demonstrated superior specific targeting ability due to the achieved appropriate orientation. For instance, a rapid detection platform for SARS-CoV-2 spike 1 (S1) antigen in saliva samples was developed using dual gold conjugates for signal enhancement.83 In this study, gold-labeled anti-S1 nanobodies (Nbs) served as the S1 detector conjugate, while gold-labeled angiotensin-converting enzyme 2 (ACE2) functioned as the S1 capturing conjugate (Fig. 4A). In early detection of positive samples with cycle threshold (Ct ≤ 30), Nbs-based LFT strips exhibited higher sensitivity (97.14%) and specificity (98.57%) compared to mAb-based strips (Fig. 4B), providing a sensitive diagnostic tool for rapid screening of SARS-CoV-2 S1 antigen in easily collected saliva samples.
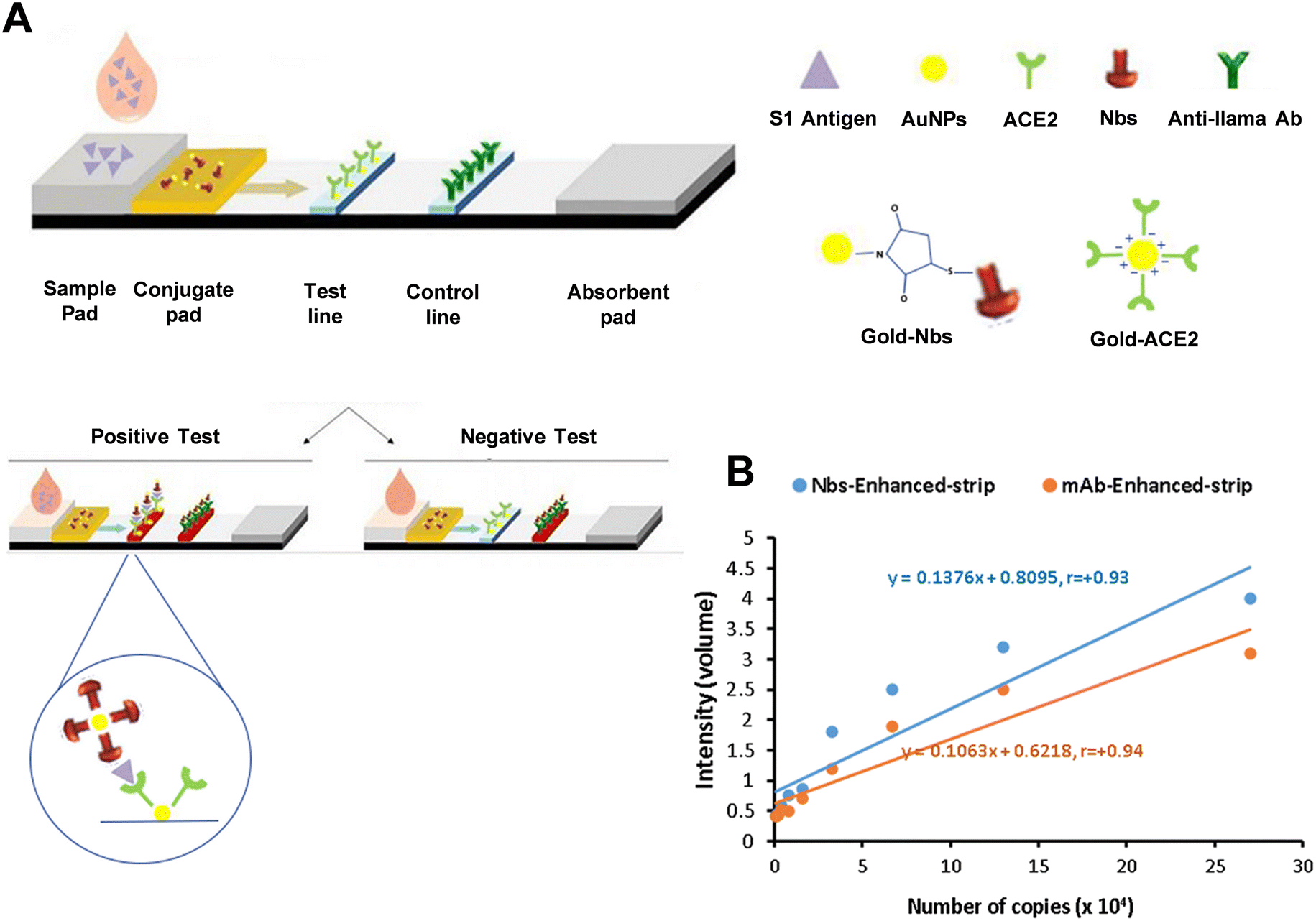 | ||
| Fig. 4 (A) Diagram illustrating the enhanced sandwich LFA for the detection of SARS-CoV-2 S1 antigen in saliva samples. (B) The relationships between the intensity of color in the enhanced LFA strip, using Nbs or mAbs, and the number of viral copies in saliva samples. Reproduced with permission from ref. 83. | ||
2.1.2.3 Carbohydrate. For immobilizing carbohydrates located on the Fc segments of antibodies, oxidation of diols to aldehydes is a viable method. Aldehyde groups are highly reactive and can couple with surfaces pre-functionalized with hydrazide or other amine groups.84–86 However, it is crucial to control the concentration of the oxidizing agent, processing time, and temperature, as overoxidation and cross-linking of antibodies have been reported as issues.87 Alternatively, treating cis diols on N-glycans with boronic acids under alkaline conditions forms boronate esters, a dehydration reaction, is much preferred immobilization method due to its single-step process.88 Additionally, enzymatic modification allows for oriented immobilization, as specific carbohydrate-reactive enzymes target the sugar monomers on Fc segments.89 Daniele's and Menegatti's groups achieved signal amplification through carbohydrate immobilization (Fig. 5A).90 In their study, they compared the efficiency of amine-based and carbohydrate-targeted conjugation chemistries in lateral flow assays. While amine condensation reactions using carbodiimide crosslinkers are common for antibody conjugation, they can lead to heterogeneous results and disrupt the Fab region of IgG, rendering the antibody ineffective for biosensing. Alternatively, conjugation through carbohydrate groups involves oxidizing glycosylated regions in the Fc part of the antibody. The resulting aldehydes react with amine groups in the detection molecule to form an imine, which is then reduced to a stable amine bond. This site-specific approach through Fc carbohydrate groups reduces conjugation heterogeneity and increases the likelihood of correctly oriented IgG-detector conjugates. Since the Fc region is far from the antibody's binding site, this method maintains more active binding sites, enhancing binding kinetics and increasing the number of binding events even at lower concentrations. Utilizing these selective conjugation methods in LFAs can improve detection limits, conserve reagents, and reduce detection time (Fig. 5B). The limits of detection for carbohydrate-conjugated and EDC-conjugated antibodies were reported as 10 pg mL−1 and 80 pg mL−1, respectively. Lin et al. fabricated a microarray platform using cyclic boronate diester to orientally bind with carbohydrates on capture antibodies for lectin detection (Fig. 5B(i)).91 The silver-enhanced image in Fig. 5B(ii) demonstrates that the antibody microarray immobilized in an oriented manner produced a well-defined and homogeneous array, unlike the weak signal observed with randomly immobilized antibodies. The darker spots in the oriented method likely result from increased capture of gold nanoparticles on the slide surface. This enhanced capture occurs because surface-conjugated boronic acids form cyclic boronate diesters with the diol groups of glycans in the Fc region of the antibody, keeping the Fab region exposed for better antigen capture and providing more gold seeds for nucleation. These findings highlight that the orientation of antibodies on the microarray significantly boosts detection sensitivity. The detection results were notably brighter compared to those obtained with random NHS chemistry immobilization, emphasizing the importance of directed antibody orientation.
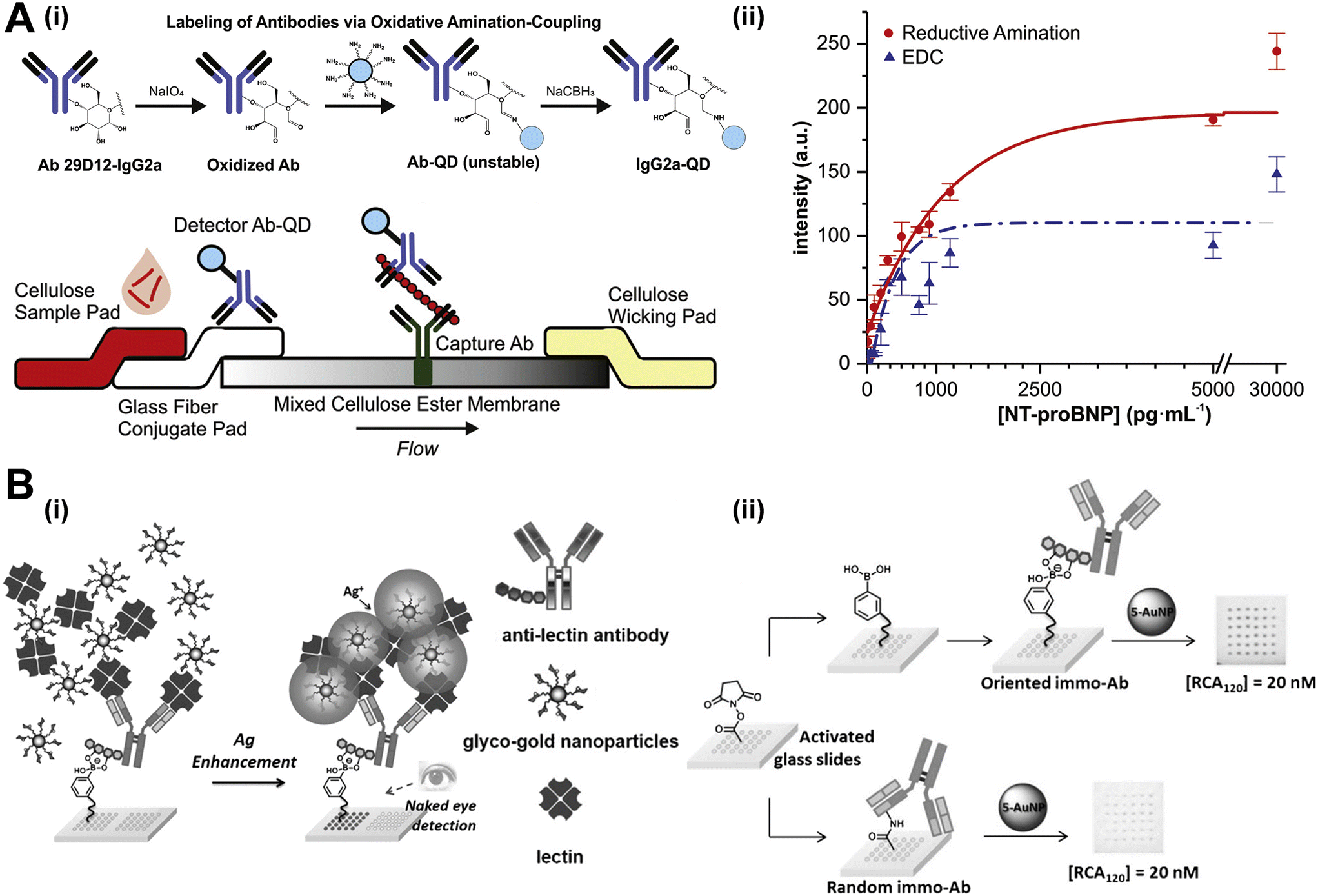 | ||
| Fig. 5 Antibody oriented immobilization with the assistance from carbohydrate. (A-i) Schematic diagram showing the lateral flow sandwich immunoassay with quantum dots as the signal reporters. Conjugation between antibody and quantum dot through carbohydrate groups is achieved by oxidation of glycosylated regions in the Fc region of the antibody. (A-ii) Comparison of signal response to analytes between common EDC coupling and carbohydrate oxidation followed by reductive amination. (B-i) Diagrammatic sketch of a glyconanoparticle-based oriented antibody microarray for lectin sensing assay. (B-ii) The comparison of the detection performance between the capture antibody fabricated by oriented immobilization through boronate formation and by random amide bond formation. Reproduced with permission from ref. 90 and 91. | ||
So far, the most common and practical oriented immobilization methodologies have been introduced. For more comprehensive or advanced techniques that can be applied to LFAs, several reviews have discussed them in detail.51,92
2.2. Modifications of test strips
In addition to probe modifications for antibody orientation, the membrane facilitating the capillary effect is often overlooked compared to probes. The structure of the LFA membrane includes the nitrocellulose membrane and the coated test/control lines (Scheme 6) in which both compositions can also undergo oriented modifications. Techniques in this category include well-established protein precoating and newly developed cellulose materials. | ||
| Scheme 6 The strategies of membrane modifications in tuning antibody orientation. | ||
2.2.1.1 Protein A and protein G. In addition to conjugation with antibody probes, protein A and protein G can be used for modifying test and control (T/C) lines. By precoating these lines with protein A or protein G, which specifically bind to the Fc region of antibodies, the binding sites are oriented outward for optimal antigen capture. Notably, the binding efficiencies of protein A and protein G with antibodies vary depending on the immunoglobulin subclasses and their donor species. For instance, protein G can bind effectively with IgG1, IgG2, IgG3, and IgG4, while protein A shows little affinity for IgG3. Despite its broader binding range, protein G has been reported to bind with the Fab region of antibodies (the antigen-binding site) and albumin (commonly found in serum samples), which can reduce specific binding.93–95 Additionally, protein A has five binding sites, compared to only two effective domains in protein G.96 Antibodies immobilized using this protein method demonstrate a 25-fold smaller dissociation constant compared to those attached via physical adsorption-based nanoprobes, indicating a superior antigen-binding rate due to their spatially accessible orientation.97 Cai et al. precoated the test line with protein G, enabling the oriented capture of anti-rSjSAP4 antibody complexes when the antigen is present (Fig. 6A).98 The device demonstrated a satisfactory detection limit, maintaining R values above 1 even at 320-fold sample dilution (Fig. 6B). However, at higher serum sample concentrations (1
:5 and 1:10 dilutions), nonspecific antibody bindings were observed, leading to lower R values. Since most probes and serum used in LFAs are protein-based, cross-reactivity among proteins can often lead to false detection results. In such cases, coating the test and control lines with protein A and protein G may not be ideal; instead, using antigens as capture probes is recommended. This is due to the specific interaction between antigens and their corresponding immunoglobulins, which reduces the likelihood of non-specific binding. Additionally, selecting the appropriate serum and performing dilution steps are crucial. Serum with fewer interfering proteins minimizes the risk of binding with protein A or protein G and the immobilized capture probes, thereby improving sensitivity and selectivity.99 In another example, to fulfill the diagnosis of visceral leishmaniasis (VL), the Anfossi's team not only used protein A to coat the control line but also made gold nanoparticle signal reporters which labelled with protein A (GNP-pA).100 As for the material of test line, it was composed of recombinant chimeric antigen (rCAg) which can target VL specifically. It is worthwhile noticing that VL is often linked to hypergammaglobulinemia, where high levels of gamma globulins can saturate the probe's binding capacity, preventing it from reacting with the immunoglobulins at the control line. When the sample is loaded, fluid containing only generic canine IgG will be captured by GNP-pA and subsequently by the protein A-coated control line, resulting in a negative outcome. However, if anti-leishmanial antibodies are present, the GNP-pA will bind with both, producing two red lines (a positive result) from the rCAg-coated test line that target anti-leishmanial antibodies, and the protein A-coated control line which detect generic canine IgG.
 | ||
| Fig. 6 (A) Test line modification with pre-coated recombinant protein G for oriented antibody immobilization. (B) Detection limit of dilutions range from 1:5 to 1:40690 by introducing Kato Katz-positive participant's pooled serum sample. Detection limit of 1:20480 was reported. C: control line; T: test line; NC, a pooled serum sample from non-endemic controls tested at a dilution of 1:5. Reproduced with permission from ref. 98. | ||
2.2.1.2 Biotin–(strept)avidin. Similarly, directly coating streptavidin or biotin on detection lines as capture probes has proven effective, leading to the development of numerous LFAs leveraging streptavidin–biotin interactions.101–104 These interactions provide several advantages over traditional LFAs, including longer shelf life, enhanced signal, high potential for chemical engineering, and the ability to recognize DNA and RNA sequences. Even more, detection of protein biomarkers such as calprotectin and PTP1B were also realized recently.105,106
In addition to conventional capture substrates like antibodies and RNA or DNA sequences, streptavidin-precoated strip lines enable the oriented immobilization of biotinylated nanobodies. Nanobodies (Nbs), as fragments of antibodies, consist of a single recombinant variable domain of the heavy chain with a dissociation constant ranging from 10−6–10−11 M.107,108 This characteristic makes Nbs significantly smaller (∼15 kDa) compared to conventional antibodies (∼150 kDa), reducing steric hindrance during deposition. Furthermore, the absence of light chains eliminates a large number of reactive amine groups, decreasing the likelihood of unoriented binding. Additionally, their higher resistance to thermal and chemical denaturation compared to conventional antibodies further enhances the shelf life of LFAs.
Magez's group designed LFAs utilizing the Nb44/Nb42 pair to specifically target T. congolense (Fig. 7A).107 During optimization, they discovered that biotinylated Nb42 conjugated with streptavidin-coated test lines outperformed directly deposited Nb42, achieving a LOD of 0.22 TcoPYK μg ml−1 compared to 0.88 TcoPYK μg ml−1 for the latter (Fig. 7B). Magez et al. attributed this improvement to the enhanced orientation and immobilization efficiency provided by BSA pairs. Furthermore, they optimized the assay buffer, using 1% methylcellulose and 1% Tween20 in PBS, to increase sample viscosity and reduce plasma matrix effects. This adjustment lowered the LOD to 0.014 TcoPYK μg ml−1 (in naïve mouse serum), compared to 0.110 TcoPYK μg ml−1 with the untreated buffer (Fig. 7C), highlighting the importance of factors beyond merely capture probes and targets. In detection tests for T. congolense, mice and cattle served as subjects. In the mice “test-of-cure” experiments, infected subjects were divided into three groups: untreated, treated with berenil at 6 days post-infection, and treated at 14 days post-infection. The performances of Nb44/Nb42 LFAs were compared with ELISA and microscopy results. Kappa coefficient analysis yielded values of 0.746 ± 0.211, 0.653 ± 0.221, and 0.764 ± 0.195 for microscopy-ELISA, microscopy-LFA, and ELISA-LFA, respectively. The authors noted a higher false negative rate for the LFAs, attributing it to the technical limitations of LFA, such as insufficient incubation time and the absence of an enzyme-mediated signal amplification step. In cattle tests, the sensitivity was 79.17% and the selectivity was 91.89%, with a visual LOD approximately 60 times lower (0.88 TcoPYK μg ml−1) than in mice tests (0.11 TcoPYK μg ml−1). The authors suggested that the difference was due to variations in plasma composition between animals. They proposed using a bivalent capture nanobody with AuNPs (Nb44–Nb44–AuNPs) to enhance affinity and signal amplification, potentially overcoming these limitations.
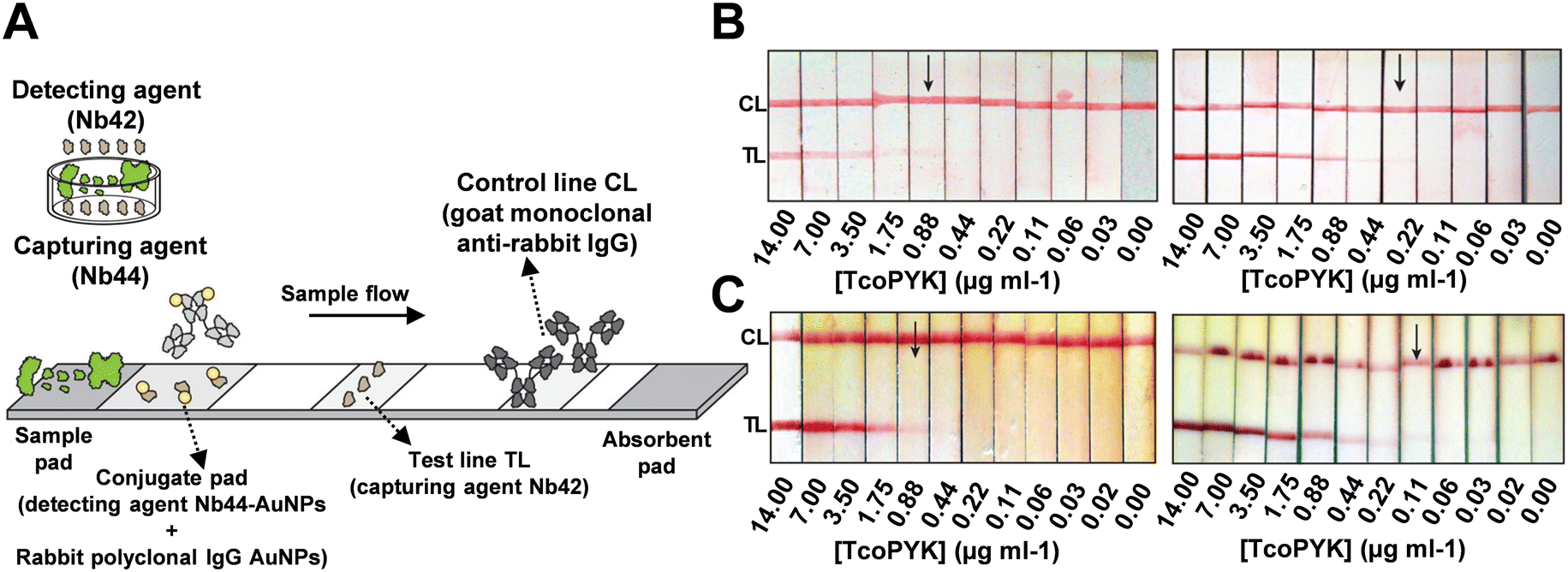 | ||
| Fig. 7 (A) Design of Nb-based LFA that targets T. congolense. (B) Detection limit of PBS spiked with a dilution series of TcoPYK: Nb42-coated test line (0.88 μg ml−1, left) and biotinylated Nb42-coated test line (0.22 μg ml−1, right). (C) Detection limit of naive cattle serum spiked with a dilution series of TcoPYK: utilizing monovalent Nb44–AuNPs (0.88 μg ml−1, left) and bivalent Nb44–Nb44–AuNPs (0.11 μg ml−1, right) as capture antibodies. Reproduced with permission from ref. 107. | ||
2.2.1.3 Nucleic acid. Typically, the immobilization of nucleic acids, whether DNA or RNA, involves using proteins like biotin, streptavidin, or protein A and protein G. These indirect methods generally require additional pre-incubation steps, making them less cost-effective and time-efficient as POC tools. However, a technique using UV light has been developed to form covalent bonds between nucleic acids and nitrocellulose or nylon membranes.109,110 Introduced in 2013, this method involves UV cross-linking non-labeled oligonucleotides to attach the test and control lines onto the nitrocellulose membrane.110 During UV exposure, thymine becomes highly reactive and forms covalent bonds with amine groups on the membrane surface, creating a stable bond. This allows for stringent assay conditions without losing substrate molecules, reducing the fabrication time and cost of test strips. The UV-assisted DNA immobilization process takes only 120 seconds and is five times cheaper than biotin-labeled LFAs while maintaining comparable sensitivity and specificity for HIV-1.
In Bruylants' dipstick assay,111 the immobilization of p14 peptide aptamers on test lines as capture probes was achieved via BSA interaction, targeting the cancer biomarker Mdm2. The system achieved a detection limit of 2 nM using AgNPls-X4–p53 as a colorimetric detector, outperforming commercially available anti-Mdm2 polyclonal antibodies. Interestingly, it is suggested that the presence of five arginine residues on each p14 peptide may cause electrostatic attraction to streptavidin and AgNPls-X4–p53, leading to false positives. To demonstrate the value of the peptide aptamer-based system, the experimental group named p14 BSA was compared with the traditional antibody system, anti-Mdm2 pAB. When exposed to 8 nM Mdm2, only the assays containing peptide aptamers produced visible test lines, regardless of whether the detector reagent was AgNPls-X4–p53 or AgNPls-X4–anti-Mdm2 pAB. Additionally, the shelf-life test showed that the peptide aptamer-based system could reliably detect Mdm2 even after one year of storage at room temperature, in contrast to conventional antibody-based assays, which lost function after a few weeks at 4 °C.
To address the limitation of recognition probes, self-assembled tetrahedral DNA nanostructures (TDNs) were developed. Through a series of processes including size control, ssDNA hybridization, Au–S bond self-assembly, and capture probe modification, four ssDNA oligonucleotides are formed into a 3D structure specifically immobilized on a gold surface. This structure provides high mechanical strength and modifiability. The rigid frame of TDNs allows for easier tuning of orientation and density, reducing steric hindrance commonly encountered in LFAs. The versatile DNA aptamers enable targeting a wide range of nucleic acids and proteins, making the TDNs-based biosensing platform universal. This platform can target DNAs,112 microRNAs,113 small molecules,114 and protein biomarkers.115 Furthermore, due to TDNs' adaptability to various capture probes, multiplex bioanalysis is possible.116,117 However, despite their many advantages, the time-consuming and delicate experimental procedures are significant drawbacks that need to be addressed.118,119
The work of Zuo et al. clearly demonstrates the significance of oriented antibodies and the spacing between capture probes.120 Using TDNs, the team developed a gold electrode platform covered with a TDN monolayer through Au–S chemistry. For comparison, they also constructed a monolayer of thiolated double-strand DNA (ds-DNA). Both layers were incubated overnight to fully deposit on the gold electrodes. To achieve oriented immobilization of antibodies, linkers with alkyne groups were paired complementarily with the ds-DNA and TDNs, allowing for covalent bonding of capture antibodies with the alkyne groups. This ensured a well-ordered orientation of the antibodies. Upon treatment with PSA, the tetrahedron-based monolayer achieved a detection limit of 500 pg ml−1, while the ds-DNA-based monolayer had a detection limit of only 10 ng ml−1. Zuo's group attributed this significant difference to the dense packing caused by ds-DNAs, resulting in 2.4 nm spacing. In contrast, the TDN-based structure allowed for a 5.0 nm spacing, comparable to the size of the capture antibody. This increased spacing is crucial for oriented immobilization, as it reduces steric hindrance, minimizes unordered physical adsorption, and exposes more Fab regions on the antibodies for detection. To further enhance the detection of extremely low concentrations of PSA, AuNPs were used to amplify the signal, resulting in detection limits of 1 pg ml−1 and 50 pg ml−1 for the TDN and ds-DNA-based gold electrodes, respectively. In selectivity tests, the TDN-based electrodes with PSA capture probes showed minimal signals even when exposed to 10 μg ml−1 of CEA and AFP. Practical serum tests from 11 patients corresponded well with actual PSA concentrations. Additionally, the platform demonstrated programmability by successfully detecting CEA and AFP through the modification of capture antibodies.
The implementation of TDNs on lateral flow test strips was accomplished by Yu and Zha's team.121 Their design involved the use of barcode TDNs on the test lines (tDTs) and biotinylated TDNs on the control lines (cDTs), which were applied using a dispenser. For the ratiometric visual detection of exosomal miRNA-150-5p, the recognition substrates were designed to hybridize with hairpin 1, which carries a sequence complementary to the capture probe on the tDTs, and hairpin 2, which has a biotin label on its 5′ end to bind with streptavidin-modified AuNPs. This setup ensures that any H1/H2-streptavidin labeled AuNP complexes not captured by the tDTs will anchor to the cDTs through B-SA interaction (Fig. 8A). The competitive binding of tDTs and cDTs with the recognition hybrids creates a ratiometric effect, allowing T/C ratios to serve as indicators in sensitivity and selectivity tests. When exposed to varying concentrations of miRNA-150-5p ranging from 10−8 to 10−13 M, a linear calibration curve with a correlation coefficient of 0.9921 was obtained, with a detection limit calculated at 58.60 fM (Fig. 8B). The strip's high selectivity was demonstrated by testing with four other miRNAs, where only the miRNA-150-5p group exhibited a high T/C ratio (Fig. 8C). In the clinical utility experiment, the ratiometric strip measured miRNA-150-5p at 1.43 pM, compared to 1.72 pM obtained via real-time fluorescence PCR, demonstrating a high degree of accuracy and suggesting the potential for TDNs in diagnostic applications. However, for the still-developing TDNs technology, the key challenges remain to simplify probe fabrication procedures and reduce detection time (optimized to 50 min in this study) to achieve a true POC tool.119,122
 | ||
| Fig. 8 (A) Design of ratiometric LFA based on DNA tetrahedron-coated strips that target exosomal microRNA-150-5p. (B) Sensitivity test by applying 0, 10−13, 10−12, 10−11, 10−10, 10−9, and 10−8 M of miR-150-5p (left). LOD of 0.9921 was yielded built on the data from sensitivity test (right). (C) Selectivity test by introducing miR-150-5p, single-base mismatched miR-150-5p, two-base mismatched miR-150-5p, random miRNAs, and miR-21 (group b–f, accordingly. With group a as blank) (left). The ratiometric device performed significantly larger T/C ratios when treated with miR-150-5p, group b (right). Reproduced with permission from ref. 121. | ||
2.2.1.4 Amphiphilic hydrophobin (HFBI) protein. The bioactivity of antibodies immobilized on the test line of a porous nitrocellulose membrane is crucial for determining analytical sensitivity. Traditionally, antibodies are deposited on the test line by spraying, which often results in random orientation and multilayer superposition, leaving the Fab regions unexposed and causing a loss of bioactivity. Additionally, dispensers are frequently used to apply antibodies, but this method carries the risk of aggregation or conformational changes which can reduce the proper orientation of the antibodies.123,124 In a study by Zhang et al., amphiphilic hydrophobin (HFBI), a fungal protein, was used to facilitate the ordered self-assembly of PSA antibodies on the test line (Fig. 9A).125 Various surface characterization techniques confirmed the self-assembling performance of HFBI on the membrane and the “standup” orientation of immobilized antibodies facilitated by HFBI adsorption. This method achieved a detection limit as low as 0.06 ng mL−1-two orders of magnitude lower than conventional test strips. Additionally, a calibration curve with a coefficient of determination of 0.965 was obtained for PSA detection (Fig. 9B). For specificity testing, the assay was challenged with four other analytes (CEA, CA153, HCG, and AFP), and distinguished fluorescence signals were only observed in the PSA-treated group. These results were further validated by the T/C and S/N signal ratios. The accuracy of the HFBI-modified LFA was also demonstrated by testing 150 clinical serum samples, with detection results aligning with those obtained from electrochemiluminescence immunoassay.
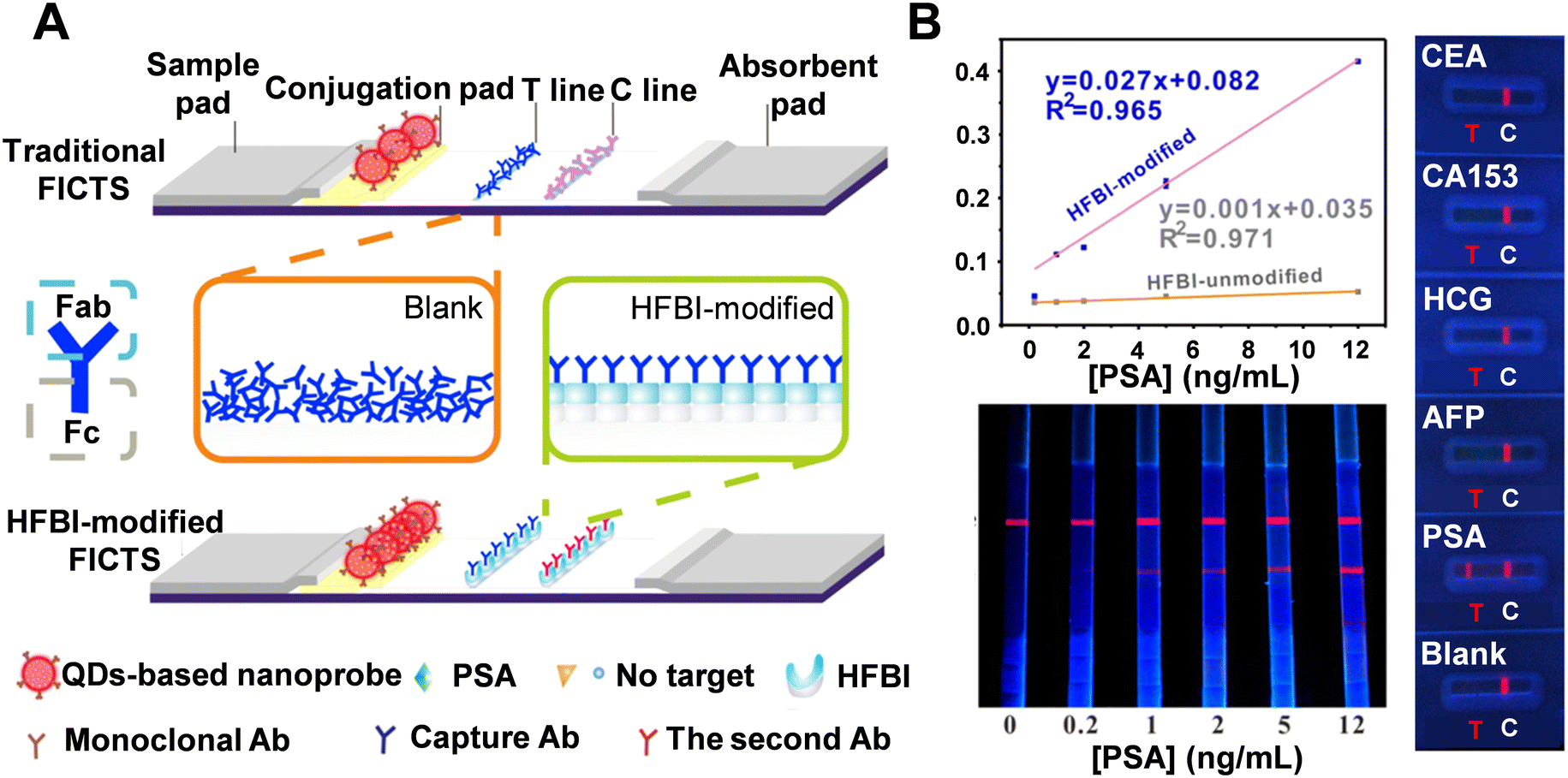 | ||
| Fig. 9 (A) Schematic comparison of traditional and HFBI-modified LFA. (B) LODs for HFBI-modified and unmodified FICTS for detection of PSA targets (top left). Sensitivity test (bottom left): visual readout on HFBI-modified FICTS by treating PSA targets of 0, 0.2, 1, 2, 5, and 12 ng mL−1, respectively. Selectivity test (right): HFBI-modified LFA after reacting with different antigens. Reproduced with permission from ref. 125. | ||
2.2.2.1 Cellulose-based membranes. To improve orientation control, cellulose-based LFAs have been introduced. Unlike the widely used nitrocellulose (NC) membrane, which is made of nitrated glucose, cellulose-based materials consist solely of carbohydrate polymers. These glucose monomers form cellulose, which is more affordable, biocompatible, and biodegradable. From a commercial perspective, cellulose-based materials offer cost advantages. In the context of LFAs, cellulose membranes facilitate more organized flow migration of immune complexes through capillary action due to the numerous hydroxyl groups on their surface. This contrasts with the disorganized migration caused by weak physical adsorption on NC membranes. Moreover, research has indicated that non-nitrated cellulose membranes surpass nitrocellulose membranes in mechanical strength and hydrophilicity, making them a compelling alternative to traditional NC membranes.126,127 Comparisons between cellulosic substrates (filter paper, cotton fiber, and cellulose fibers, for instance) and other commonly used materials in LFAs have been extensively studied; as used as sample and conjugate pads, cellulosic substrates offer higher bed volume and greater resistance to non-specific bindings, leading to increased sensitivity. As a stacking pad, cellulosic substrates outperform polyester and glass fiber, producing the most intense test line. However, despite its numerous proven advantages, practical applications in POC tools have been rare and primarily limited to cellulose fiber materials. Additionally, research in this area has been conducted by only a few groups.128–133
As a relatively new method for creating well-oriented POC devices, cellulose-based membranes are now being integrated with various domains found in carbohydrate-active enzymes.134 This fusion is then anchored with selected proteins capable of specifically binding antibodies, thereby achieving oriented antibody immobilization. For example, carbohydrate-binding modules (CBMs) are distinct protein segments found in many carbohydrate-active enzymes.135,136 Their capacity to bind a variety of carbohydrates, ranging from disaccharides and oligosaccharides to polysaccharides like cellulose, with high selectivity and specificity has led to numerous biotechnological applications. In essence, CBM-fused antibodies exploit the natural affinity of CBMs for cellulose, combined with specific domain or fragment binding steps. This innovative approach leverages the inherent properties of CBMs to facilitate the precise orientation of antibodies on cellulose-based membranes. The initial layer of fusion between CBMs and the cellulose membrane is achieved using biomolecular engineering, employing carbohydrate-active enzymes like cellulosomal-scaffolding protein A from Clostridium thermocellum (CBM3). The second layer involves anchoring CBMs to biorecognition proteins through specific domains. Several well-known CBM–protein pairs have been discovered and studied, including FLAG tag-IgG, avidin–biotin, PDZ domain-peptide, and zz-IgG. The third layer focuses on immobilizing antibodies via their Fc regions to expose the Fab portions, or alternatively, attaching oligonucleotides to the biorecognition proteins.131 When antibodies are used, biorecognition proteins are required to bind to the Fc region, ensuring the Fab region is exposed for target interaction. In contrast, if oligonucleotides are employed, terminal modifications are necessary to enable covalent bonding with recombinant CBM fusion proteins.131,132 This is accomplished through specific combinations of domains on the proteins and their corresponding substrates. These layers of fusion enable the development of oriented CBM-assisted LFAs on cellulose-based membranes.
To assess whether CBM–cellulose fused LFAs are more effective in terms of sensitivity and selectivity compared to NC-based LFAs, several experiments were conducted. França Prazeres's group first prepared a traditional LFA format, which relies on the random adsorption of capture antibodies on the test lines of an NC membrane, to compare with an LFA modified by ZZ-CBM3 fusions (Fig. 10A).132 With the exception of the dilution test (ZZ-CBM3: capture antibody, not shown here), all other experiments involved first dispensing ZZ-CBM3 over the strips, followed by the addition of capture antibodies. In traditional NC membranes, controlling the orientation and accessibility of antibodies is challenging, as they can adopt various spatial positions after immobilization, resulting in less effective analyte capture. The study compared an NC membrane that physically adsorbed ZZ-CBM3 fusions with a strip coated with cellulose and chemically bound to ZZ-CBM3. By introducing a cellulose layer at the active test line of LFA strips, ZZ-CBM3 fusions could properly anchor and orient the capture antibodies (CBM binds to cellulose, ZZ captures antibodies via the Fc portion). To evaluate the impact of the cellulose layer on fluorescence signals, a control system (biotin–BSA:Alexa-streptavidin) was used. Biotin–BSA lines were dispensed on NC and NC + cellulose strips, and LFA cartridges were assembled. Buffer samples containing either 1 ng mL−1 or 2 ng mL−1 of Alexa-labeled streptavidin were then run. Results indicated that when cellulose was used as a coating, the fluorescence lines were generally thicker, more intense (Fig. 10B(i)), and displayed a larger pixel volume. This increased line thickness was attributed to enhanced lateral diffusion of the biotin–BSA solution on the cellulose layer, confirming that the cellulose coat effectively serves as an anchor point for ZZ-CBM3 fusions. Another experiment compared the capture ability of Alexa-labeled antibodies by protein A and ZZ-CBM3 fusions dispensed on LFA test lines (Fig. 10B(ii)). Protein A has five antibody-binding domains (A–E), while ZZ-CBM3 has two Z domains.31 The D and E domains of protein A have an affinity for the Fab region of antibodies, leading to partially random orientation of the capture antibodies. Consequently, the higher intensity observed with ZZ-CBM3 fusions in NC + cellulose strips, compared to protein A, can be attributed to the more favorable orientation provided by the ZZ domain in the fusion.
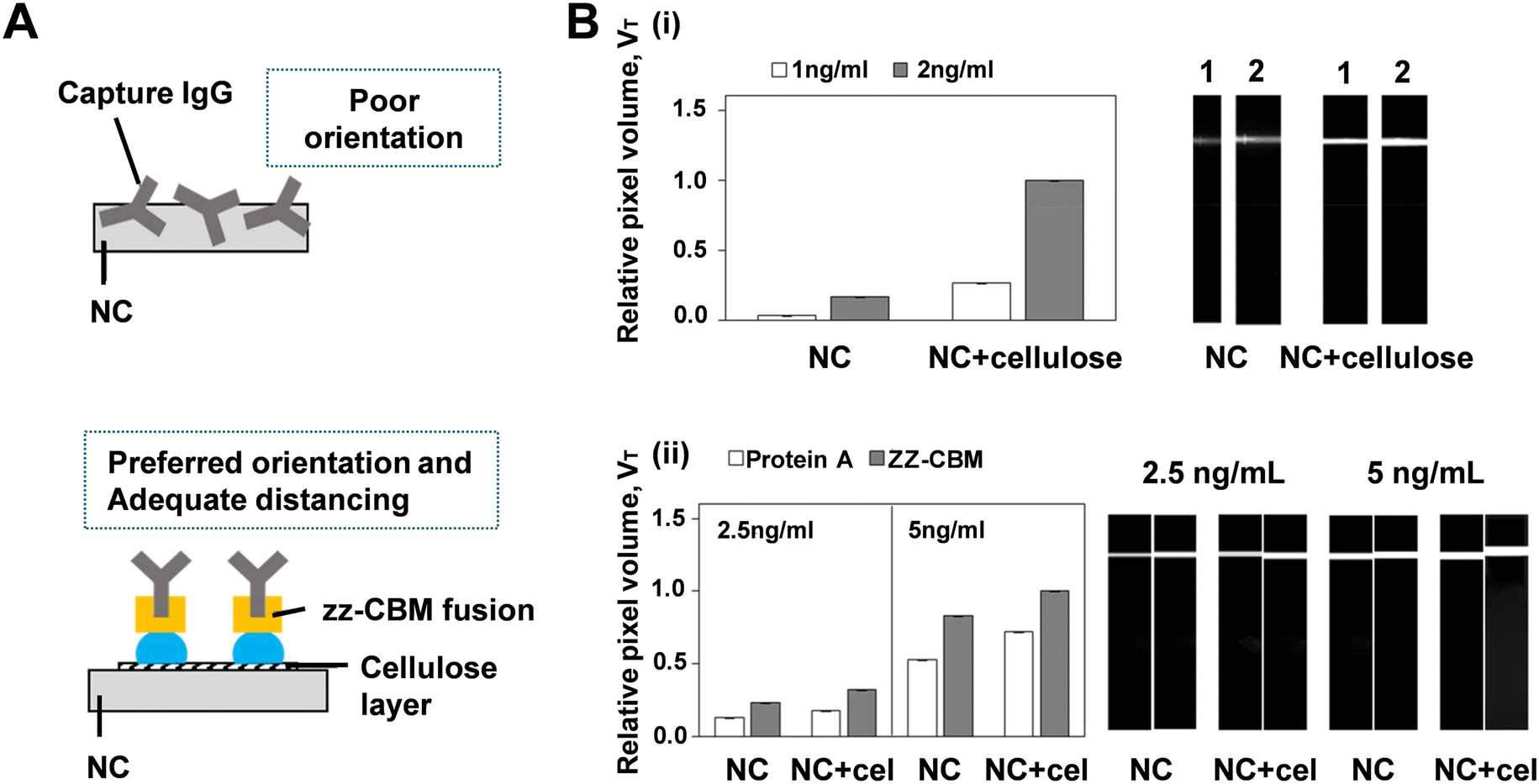 | ||
| Fig. 10 (A) Comparison between traditional NC membranes and membranes coated with ZZ-CBM fusions in LFA. (B-i) Effect of cellulose coating on NC strips on the fluorescence intensity of generated signals. (B-ii) Comparative capture efficiency of Alexa-labeled antibodies by protein A and ZZ-CBM3 fusions on LFA test lines. Experiments utilized NC strips and NC strips with an added cellulose layer (NC + cel) on the test line. Reproduced with permission from ref. 132. | ||
Similarly, Kolmar and Schwall et al. developed genetic fusions of single-chain variable fragments (scFv) or full-length antibodies (IgG) with the CBM3a domain from the cellulosomal scaffold of Clostridium thermocellum. This approach significantly improved cellulose-binding capacity, leading to enhanced sensitivity and overall performance of cellulose-based lateral flow devices compared to those using bare scFvs or IgG (Fig. 11A).133 To verify the broad applicability of CBM-fused detection antibodies, they created pregnancy LFA devices using both CBM-anti-hCG scFv and solitary anti-hCG scFv. The CBM-assisted LFAs exhibited increased sensitivity compared to devices functionalized with the solitary scFv. In another set of experiments, LFAs were designed to detect SARS-CoV-2-specific antibodies using CBM-fused antibodies in two different setups (Fig. 11B). The first batch was functionalized with either CBM-anti-Fc scFv or solitary anti-Fc scFv. A second batch was functionalized with full-length IgG multiclonal antibody constructs, either anti-human IgG or the IgG-CBM fusion variant. The CBM-scFv-functionalized COVID-19 antibody test showed sensitive detection of 125 ng of SARS-CoV-2-specific antibody, and the strips functionalized with anti-hIgG-CBM demonstrated comparable sensitivity. In contrast, LFAs functionalized with solitary scFv and sole anti-hIgG showed faint test lines, barely discernible to the naked eye. This work underscores the significant advantages of using cellulose-based papers over nitrocellulose in serologic LFA devices.
 | ||
| Fig. 11 (A) Cellulose-based LFAs for hCG detection using cellulose paper with a capillary flow rate of approximately 60 s/4 cm (C60). Test lines were modified with either CBM-anti-hCG scFv or solitary anti-hCG scFv. (B) Cellulose-based LFAs for detecting SARS-CoV-2-specific antibodies in serum samples, with test lines coated with CBM-anti-Fc scFv, anti-hIgG-CBM, anti-Fc scFv, or anti-hIgG as detection antibodies. Reproduced with permission from ref. 133. | ||
In another example, a CBP31-BC linker was created by genetically fusing cellulose-binding modules from family 3 (CBM3) and family 1 (CBM1) with the antibody-binding B and C domains of protein A (Fig. 12A).128 Analysis showed that the addition of these CBMs resulted in high cellulose-binding capacity for CBP31-BC. This strong binding allowed the BC domain to be efficiently exposed, achieving about six times higher antibody-binding efficiency (∼32.5%) compared to the BC domain alone (∼5.5%). The effectiveness of the CBP31-BC linker in LFIA was evaluated by comparing it with conventional LFIAs that use physical adsorption of antibodies, using prostate-specific antigen (PSA) as the target analyte (Fig. 12B). The CBP31-BC-based LFIA detected PSA at levels as low as 0.25 ng mL−1 within 20 minutes, which is approximately 10 times more sensitive than conventional LFIAs. Additionally, the CBP31-BC-based LFIA demonstrated a lower detection range of 0.25–2.5 ng mL−1 compared to traditional methods. These findings highlight the significant advantage of using the CBP31-BC linker for the efficient and oriented immobilization of capture antibodies on cellulose membranes.
 | ||
| Fig. 12 (A) Schematic illustration of the paper-based LFA platform utilizing the CBP31-BC linker, an engineered protein that combines cellulose-binding modules with antibody-binding domains. (B) Photographic comparison of LFAs featuring the CBP31-BC fusion on the test line (left panel) versus conventional LFAs (right panel) at PSA concentrations of 0, 0.25, 0.5, 2.5, 5, 10, and 20 ng mL−1. The normalized test line intensities corresponding to these PSA concentrations are displayed below. Reproduced with permission from ref. 128. | ||
In 2023, Kim's group developed a colorimetric LFA platform for early SARS-CoV-2 detection using a bifunctional fusion linker, CBP31-BC.129 This linker combines cellulose-binding and antibody-binding domains to orient antibodies on a cellulose membrane. As shown in Fig. 13A, the test zone featured CBP31-BC preincubated with an anti-SARS-CoV-2 RBD antibody at the test line and CBP31-BC alone at the control line. The optimal molar ratio of CBP31-BC to the capture antibody was determined to be 1:20 to minimize nonspecific adsorption of the detection antibody on gold nanoparticles. The CBP31-BC-based LFA demonstrated high specificity for the SARS-CoV-2 antigen using S1 antigens from SARS-CoV, MERS-CoV, and CoV-H229E (Fig. 13B). The platform's practical capability was assessed with cultured SARS-CoV-2 samples, showing visual sensitivity and a LOD of 5 × 105 copies per mL (Fig. 13C). Additionally, the LFA's performance was confirmed using nasopharyngeal swab samples from COVID-19 patients (n = 16) and healthy subjects (n = 3), displaying high concordance with RT-qPCR results (Fig. 13D).
 | ||
| Fig. 13 (A) Schematic of the CBP31-BC-based LFA for detecting SARS-CoV-2. (B) Normalized test line intensity of the LFIA for SARS-CoV-2 RBD, SARS-CoV S1, MERS-CoV S1, and CoV-H229E S1 antigens, with the dotted line indicating no sample. (C) Photographic images and normalized test line intensity of the LFA strip for various cultured SARS-CoV-2 concentrations, with the black dotted line indicating the cutoff value (mean of 3 blank samples + 3 standard deviations). (D) Comparison of LFA performance using nasopharyngeal swab samples from COVID-19 patients versus PCR results. Reproduced with permission from ref. 129. | ||
While the concept of “immobilization leads to well-orientation” is a major advantage promoted for CBM–cellulose fusion, practical challenges have emerged. A significant issue is the reduction in oriented immobilization caused by cross-reactivity between proteins. Biorecognition protein domains may bind to other antibody segments or react with non-target analytes, resulting in decreased sensitivity and selectivity.132,133 This cross-reactivity is particularly problematic when dealing with biological fluid samples, such as serum or urine, which contain numerous non-target proteins, leading to false negatives and false positives.129 To address these challenges, various solutions have been proposed, ranging from simple sample dilution to advanced antibody editing, which removes interfering segments using specific enzymes. Additionally, site-directed covalent protein–cellulose conjugation presents a promising solution.132 Besides, cellulose-based membranes face several challenges, including the spreading of fluorescence on test and control lines due to cellulose's high adsorption properties.129,130 Additionally, the inability to plot reliable quantitative calibration curves arises because the fused proteins often possess multiple binding sites.132 Another significant challenge is simplifying the biomolecular engineering processes involved. These issues remain to be resolved.
2.2.2.2 Cellulose nanofiber-modified lines. In recent years, the addition of a cellulose nanofiber (CNF) layer to LFAs has become a mature technique aimed at enhancing sensitivity. The porosity of the paper is crucial for the sensitivity of the strips. Smaller pores lead to higher sensitivity; however, this can impede sample flow, increase assay time, and raise the likelihood of flow stops or membrane defects, resulting in low reproducibility. CNFs, with their smaller pore size, enhance porosity, surface area, and the number of exposed hydroxyl groups.137,138 This increases adsorption capacity and, indirectly, sensitivity. However, this method primarily relies on physical adsorption, which is less effective for antibody immobilization compared to the CBM fusion strategy. Additionally, the dense surface created by CNFs can hinder sample migration, thereby reducing reproducibility.139 Merkoçi's group found that CNF gel increases the density of antibodies near the paper surface, thereby enhancing the signal by retaining more reporting probes on positive samples.137,138 CNFs were used to penetrate the pores of nitrocellulose paper, reducing pore size only in the test area (Fig. 14A). As shown in Fig. 14B, this modification keeps antibodies close to the strip surface, increasing the density of selectively attached gold nanoparticles in the test line area and boosting sensitivity by an average of 36.6%. The authors demonstrated that this modification is simple, cost-effective, and can be easily applied to any type of LFA strip, making it suitable for point-of-care applications.
 | ||
| Fig. 14 (A) Schematic representation of a positive test line not modified (upper panel) and modified with cellulose nanofibers dispensed (bottom panel). Prof. Merkoçi's LFAs by introducing cellulose nanofiber. (B) Photographic images (up) of LFAs with and without the CNF applied on the test lines with HIgG of 0, 0.01, 0.05, 0.10, 0.25, 0.50, 0.75, and 1.00 μg ml−1. Bar chart (below) was made for the comparison of percentage of signal enhancement. Reproduced with permission from ref. 140. | ||
3. Conclusion
The critical importance of sensitivity and selectivity in LFAs is underscored by the evolution of various modifications. From external strategies aimed at amplifying signals and readouts to internal optimizations of chemical interactions within the devices, the primary challenge remains the orientation of antibodies. Here, we have discussed several approaches: (i) physical adjustment. (ii) Covalent bond formation. (iii) Non-covalent bond formation. (iv) Cellulose-based membranes. (v) Cellulose nanofiber coatings. Conventional techniques such as (i) and (ii) often suffer from weak interactions, operational complexities, and unsatisfactory affinity. Additionally, these methods require multiple device or sample pretreatments, which are time-consuming. The (iii) approach, while benefiting from biochemical specificity, encounters issues like protein cross-reactivity and difficulties in finding appropriate conjugate pairs. The relatively new methodology (iv), involving biorecognition led by CBMs, also risks protein cross-reactivity and entails complicated pre-coating steps. Method (v), which uses denser cellulose coatings, does enhance signal strength. However, challenges such as line spreading and unoriented immobilization persist. The advantages and disadvantages of these bond formation techniques are summarized in Table 1. For newer modifications involving probes, test/control lines, or membrane compositions, the reported effectiveness is listed in Table 2.| Bond(s) type | Utilization | Advantage(s) | Limitation(s) reported |
|---|---|---|---|
| Non-covalent | Physical adsorption | Simple operation | Unoriented immobilization35,38,39 |
| Straightforward theory | |||
| Protein A, protein G | Oriented immobilization46–48 | Protein cross-activity21,31,49 | |
| Reversible reaction50,51 | |||
| Extra pretreatment | |||
| Biotin–(strept)avidin | Oriented immobilization60,61 | Protein cross-activity59 | |
| High B-SA binding ratio57 | Extra pretreatment | ||
| High condition-tolerance57 | |||
| Covalent | Amine, carboxyl group | Wide availability | Unoriented immobilization73 |
| pH dependence66,74 | |||
| Extra pretreatment | |||
| Disulfide bond | Oriented immobilization75–79,82,83 | Conformational change80 | |
| Ab-AuNP aggregation80 | |||
| Extra pretreatment | |||
| Carbohydrate moiety | Oriented immobilization84–86,88–91 | Overoxidation87 | |
| Antibody cross-linking87 | |||
| Extra pretreatment |
| Method | Medium | Device | Target | LOD | Sensitivity (in fold(s) of enhancement or %) | Ref. |
|---|---|---|---|---|---|---|
| Probe modification | Protein A | LFA | Pathogen-specific antibodies | 300 ng mL−1 | 30 folds | 53 |
| Protein G | Gold plate | Rabbit IgG | N/A | 11 folds | 55 | |
| BSA | LFA | SARS-CoV-2 | 0.65 ng mL−1 (Genemedi N protein) | N/A | 60 | |
| N-protein (Genemedi N and Genscript N protein) | 3.03 ng mL−1 (Genscript N protein) | |||||
| BSA | AuNS/SPCEs | Mouse monoclonal anti-SARS-CoV-2 | 6 pg mL−1 | 2.9 folds | 61 | |
| N-protein | ||||||
| Amine/carboxyl groups | LFA | SARS-CoV-2 | 6.9 ng mL−1 (RBD protein) | N/A | 68 | |
| RBD protein and N protein | 7.2 ng mL−1 (N protein) | |||||
| Amine/carboxy groups | LFA | Pathogen-specific antibodies | 1.07 pg mL−1 | N/A | 72 | |
| Disulfide bond | LFA | SARS-CoV-2 S1 antigen | 0.4 × 104 copies per ml | 97.14% (relative to 90.04% of unmodified device, Ct ≤ 30 (%)) | 83 | |
| Carbohydrate | LFA | N-terminal | 10 pg mL−1 | N/A | 90 | |
| Pro-B-type | ||||||
| Natriuretic peptide | ||||||
| Carbohydrate | Microarray | Lectins (RCA120, ConA, and WGA) | 3 pM (RCA120) 16 pM (ConA) 12 pM (WGA) | 2.2 × 103 folds 9.7 × 102 folds same | 91 | |
| T/C lines modification | Protein G | GICA | S. japonicum saposin protein | 1:20480 diluted sample |
95% (non-endemic group) | 98 |
| 85% (KK (−) and F_ddPCR (−) group) | ||||||
| BSA | LFA | Salmonella | 1 × 10−13 M | N/A | 141 | |
| Nucleic acid | LFA | HIV-1 RNA | 10.5log10 copies (batch 1) |
N/A | 109 | |
| 9.5log10 copies (batch 2) |
||||||
| Hydrophobin | FICTS | Pathogen-specific antibodies | 0.06 ng mL−1 | 3 folds | 125 | |
| Membrane composition | Cellulose membrane | LFA | Cystatin C | N/A | Compatible with the clinical diagnostic range | 132 |
| Cellulose membrane | LFA | SARS-CoV-2 specific antibodies | 125 ng per 150 μL | 4.6 folds | 133 | |
| Cellulose membrane | LFA | Pathogen-specific antibodies | 0.25 ng mL−1 | 10 folds more | 128 | |
| Cellulose membrane | LFA | Inactivated SARS-CoV-2 and RBD protein | 0.63 ng mL−1 (RBD protein) | N/A | 129 | |
| 5 × 104 copies per mL (inactivated SARS-CoV-2) | ||||||
| Cellulose nanofiber T/C lines | LFs | Human IgG | 0.01 μg mL−1 | 36.6% signal enhancement | 140 |
In addition to the conventional and novel immobilization techniques discussed, the methods for attaching, depositing, or coating capture probes onto T/C lines are often overlooked. The spraying process relies on passive bond formation, including hydrophobic interactions and electrostatic attraction, leading to random immobilization.123,124 This review aims to address and improve this random immobilization. If the detection lines themselves are not properly oriented in their compositions, the subsequent layers of capture and recognition probes will also lack proper orientation.
In conclusion, despite the numerous techniques developed thus far, none have fully achieved the ideal POC standards. LFAs still have significant room for improvement in both sensitivity and selectivity. Additionally, cost and global availability are crucial factors that need attention to reduce economic and personnel burdens. Therefore, advancements in chemical engineering, fusion techniques, and biomolecular exploration are areas where further efforts and research are needed.
Data availability
All data created or analyzed in this study are available on request from the corresponding author.Conflicts of interest
There are no conflicts to declare.References
- B. G. Andryukov, AIMS Microbiol., 2020, 6, 280–304 CAS.
- B. O'Farrell, Evolution in Lateral Flow–Based Immunoassay Systems, Humana Press, 2009 Search PubMed.
- G. A. Bonwick and C. J. Smith, Int. J. Front. Sci. Technol., 2004, 39, 817–827 CAS.
- S. Ahmed, J. Ning, D. Peng, T. Chen, I. Ahmad, A. Ali, Z. Lei, M. Abu bakr Shabbir, G. Cheng and Z. Yuan, Food Agric. Immunol., 2020, 31, 268–290 CrossRef CAS.
- V. Gubala, L. F. Harris, A. J. Ricco, M. X. Tan and D. E. Williams, Anal. Chem., 2012, 84, 487–515 CrossRef CAS PubMed.
- S. Deepak, K. Kottapalli, R. Rakwal, G. Oros, K. Rangappa, H. Iwahashi, Y. Masuo and G. Agrawal, Curr. Genomics, 2007, 8, 234–251 CrossRef CAS PubMed.
- I. M. Artika, Y. P. Dewi, I. M. Nainggolan, J. E. Siregar and U. Antonjaya, Genes, 2022, 13, 2387 CrossRef CAS PubMed.
- P. Kralik and M. Ricchi, Front. Microbiol., 2017, 8, 108 Search PubMed.
- D. Wang, Y. Chen, S. Xiang, H. Hu, Y. Zhan, Y. Yu, J. Zhang, P. Wu, F. Y. Liu, T. Kai and P. Ding, Front. Cell. Infect. Microbiol., 2023, 12, 1040248 CrossRef PubMed.
- H. Onyeaka, C. K. Anumudu, Z. T. Al-Sharify, E. Egele-Godswill and P. Mbaegbu, Sci. Prog., 2021, 104, 00368504211019854 CAS.
- P. Korneta and M. Chmiel, Int. J. Environ. Res. Public Health, 2022, 19, 14827 CrossRef PubMed.
- S. Sun, Z. Xie, K. Yu, B. Jiang, S. Zheng and X. Pan, Glob. Health, 2021, 17, 14 CrossRef PubMed.
- B. Heidt, W. F. Siqueira, K. Eersels, H. Diliën, B. van Grinsven, R. T. Fujiwara and T. J. Cleij, Biosensors, 2020, 10, 133 CrossRef PubMed.
- A. Taylor, R. Calvez, M. Atkins and C. G. Fink, Health Sci. Rep., 2022, 5, e811 CrossRef PubMed.
- D. A. Mistry, J. Y. Wang, M.-E. Moeser, T. Starkey and L. Y. W. Lee, BMC Infect. Dis., 2021, 21, 828 CrossRef CAS PubMed.
- B. A. Trombetta, S. E. Kandigian, R. R. Kitchen, K. Grauwet, P. K. Webb, G. A. Miller, C. G. Jennings, S. Jain, S. Miller, Y. Kuo, T. Sweeney, T. Gilboa, M. Norman, D. P. Simmons, C. E. Ramirez, M. Bedard, C. Fink, J. Ko, E. J. De León Peralta, G. Watts, E. Gomez-Rivas, V. Davis, R. M. Barilla, J. Wang, P. Cunin, S. Bates, C. Morrison-Smith, B. Nicholson, E. Wong, L. El-Mufti, M. Kann, A. Bolling, B. Fortin, H. Ventresca, W. Zhou, S. Pardo, M. Kwock, A. Hazra, L. Cheng, Q. R. Ahmad, J. A. Toombs, R. Larson, H. Pleskow, N. M. Luo, C. Samaha, U. M. Pandya, P. De Silva, S. Zhou, Z. Ganhadeiro, S. Yohannes, R. Gay, J. Slavik, S. S. Mukerji, P. Jarolim, D. R. Walt, B. C. Carlyle, L. L. Ritterhouse and S. Suliman, BMC Infect. Dis., 2021, 21, 580 CrossRef CAS PubMed.
- Y. Galipeau, A. Xavier, A. Dyks, C. Cooper and M.-A. Langlois, Front. Public Health, 2023, 11, 1240308 CrossRef PubMed.
- Y. Lei, X. Lu, D. Mou, Q. Du, G. Wang and Q. Wang, Open Life Sci., 2022, 17, 1487–1496 CrossRef CAS PubMed.
- R. Y. T. Chiu, N. Kojima, G. L. Mosley, K. K. Cheng, D. Y. Pereira, M. Brobeck, T. L. Chan, J. S.-T. Zee, H. Kittur, C. Y. T. Chung, E. Tsang, K. Maran, R. W.-H. Yung, A. C.-P. Leung, R. H.-P. Siu, J. P.-L. Ng, T. H. Choi, M. W. Fung, W. S. Chan, H. Y. Lam, K. H. Lee, S. Parkin, F. C. Chao, S. K.-N. Ho, D. R. Marshak, E. S.-K. Ma and J. D. Klausner, Microbiol. Spectrum, 2021, 9, 10 CrossRef PubMed.
- J. C. Linnes, N. M. Rodriguez, L. Liu and C. M. Klapperich, Biomed. Microdevices, 2016, 18, 30 CrossRef CAS PubMed.
- Y. Liu and J. Yu, Microchim. Acta, 2016, 183, 1–19 CrossRef CAS.
- M. Ishii, P. Preechakasedkit, K. Yamada, O. Chailapakul, K. Suzuki and D. Citterio, Anal. Sci., 2018, 34, 51–56 CrossRef CAS PubMed.
- Y. Liu, L. Zhan, Z. Qin, J. Sackrison and J. C. Bischof, ACS Nano, 2021, 15, 3593–3611 CrossRef CAS PubMed.
- Y. Zhao, J. Sang, Y. Fu, J. Guo and J. Guo, Analyst, 2023, 148, 3418–3431 RSC.
- S. Nayak, N. R. Blumenfeld, T. Laksanasopin and S. K. Sia, Anal. Chem., 2017, 89, 102–123 CrossRef CAS PubMed.
- Y. Jung, J. Y. Jeong and B. H. Chung, Analyst, 2008, 133, 697–701 RSC.
- N. G. Welch, J. A. Scoble, B. W. Muir and P. J. Pigram, Biointerphases, 2017, 12, 02D301/313 CrossRef PubMed.
- A. Trilling, J. Beekwilder and H. Zuilhof, Analyst, 2013, 138, 1619–1627 RSC.
- G. Ruiz, K. Tripathi, S. Okyem and J. D. Driskell, Bioconjugate Chem., 2019, 30, 1182–1191 CrossRef CAS PubMed.
- S. Gao, J. M. Guisán and J. Rocha-Martin, Anal. Chim. Acta, 2022, 1189, 338907 CrossRef CAS PubMed.
- B. Jansson, M. Uhlén and P.-Å. Nygren, FEMS Immunol. Med. Microbiol., 1998, 20, 69–78 CrossRef CAS PubMed.
- V. Susini, C. Sanguinetti, S. Ursino, L. Caponi and M. Franzini, Antibody-Antigen Binding Events: The Effects of Antibody Orientation and Antigen Properties on the Immunoassay Sensitivity, IntechOpen, 2023 Search PubMed.
- N. Aljamali, R. Khdur and I. Alfatlawi, International Journal of Thermodynamics and Chemical Kinetics, 2021, 7, 1–8 Search PubMed.
- C. Parolo, A. Sena, J. F. Bergua Canudo, E. Calucho, C. Fuentes Chust, L. Hu, L. Rivas, R. Alvarez Diduk, E. Nguyen, S. Cinti, D. Quesada and A. Merkoçi, Nat. Protoc., 2020, 15, 3788–3816 CrossRef CAS PubMed.
- F. Di Nardo, S. Cavalera, C. Baggiani, C. Giovannoli and L. Anfossi, ACS Appl. Mater. Interfaces, 2019, 11, 32758–32768 CrossRef CAS PubMed.
- J. Saidykhan, L. Selevic, S. Cinti, J. E. May and A. J. Killard, Anal. Chem., 2021, 93, 14007–14013 CrossRef CAS PubMed.
- T.-T. Tsai, T.-H. Huang, C.-A. Chen, N. Y.-J. Ho, Y.-J. Chou and C.-F. Chen, Sci. Rep., 2018, 8, 17319 CrossRef PubMed.
- E. Müller, Ind. Eng. Chem. Res., 2001, 40, 2193–2211 CrossRef.
- M. Park, BioChip J., 2019, 13, 82–94 CrossRef CAS.
- J. E. Lee, J. H. Seo, C. S. Kim, Y. Kwon, J. H. Ha, S. S. Choi and H. J. Cha, Korean J. Chem. Eng., 2013, 30, 1934–1938 CrossRef CAS.
- H. Miyao, Y. Ikeda, A. Shiraishi, Y. Kawakami and S. Sueda, Anal. Biochem., 2015, 484, 113–121 CrossRef CAS PubMed.
- T. Lu, X. Chen, Q. Shi, Y. Wang, P. Zhang and X. Jing, Acta Biomater., 2008, 4, 1770–1777 CrossRef CAS PubMed.
- W. Choe, T. A. Durgannavar and S. J. Chung, Materials, 2016, 9, 994 CrossRef PubMed.
- H. Yang, P. V. Gurgel, D. K. Williams, Jr., B. G. Bobay, J. Cavanagh, D. C. Muddiman and R. G. Carbonell, J. Mol. Recognit., 2010, 23, 271–282 CrossRef CAS PubMed.
- J. B. Fishman and E. A. Berg, Cold Spring Harb. Protoc., 2019 DOI:10.1101/pdb.top099101.
- D. V. Sotnikov, N. A. Byzova, A. V. Zherdev, Y. Xu and B. B. Dzantiev, Biosensors, 2023, 13, 750 CrossRef CAS PubMed.
- M. Manasa, P. Revathi, M. P. Chand, V. Maroudam, P. Navaneetha, G. D. Raj, P. B. K. Kishor, B. De and P. Rathnagiri, J. Immunoassay Immunochem., 2019, 40, 149–158 CrossRef CAS PubMed.
- G. P. Anderson, M. A. Jacoby, F. S. Ligler and K. D. King, Biosens. Bioelectron., 1997, 12, 329–336 CrossRef CAS PubMed.
- R. O'Kennedy, C. Murphy and T. Lysakova-Devine, Antib. Technol. J., 2016, 6, 17–32 CrossRef.
- A. C. Grodzki and E. Berenstein, Antibody Purification: Affinity Chromatography – Protein A and Protein G Sepharose, Humana Press, 2010 Search PubMed.
- S. Gao, J. M. Guisán and J. Rocha-Martin, Anal. Chim. Acta, 2022, 1189, 338907 CrossRef CAS PubMed.
- D. Lou, L. Fan, Y. Cui, Y. Zhu, N. Gu and Y. Zhang, Anal. Chem., 2018, 90, 6502–6508 CrossRef CAS PubMed.
- D. V. Sotnikov, N. A. Byzova, A. V. Zherdev, Y. Xu and B. B. Dzantiev, Biosensors, 2022, 12, 434 CrossRef CAS PubMed.
- Y. Ryu, Z. Jin, M. S. Kang and H.-S. Kim, BioChip J., 2011, 5, 193–198 CrossRef CAS.
- H. Kim, D.-Y. Kang, H.-J. Goh, B.-K. Oh, R. P. Singh, S.-M. Oh and J.-W. Choi, Ultramicroscopy, 2008, 108, 1152–1156 CrossRef CAS PubMed.
- L. Deng, E. N. Kitova and J. S. Klassen, J. Am. Soc. Mass Spectrom., 2013, 24, 49–56 CrossRef CAS PubMed.
- J. P. Dhandhukia, D. A. Brill, A. Kouhi, M. K. Pastuszka and J. A. MacKay, Protein Sci., 2017, 26, 1785–1795 CrossRef CAS PubMed.
- A. Jain, A. Barve, Z. Zhao, W. Jin and K. Cheng, Mol. Pharmaceutics, 2017, 14, 1517–1527 CrossRef CAS PubMed.
- J. H. T. Luong and S. K. Vashist, ACS Omega, 2020, 5, 10–18 CrossRef CAS PubMed.
- B. D. Grant, C. E. Anderson, J. R. Williford, L. F. Alonzo, V. A. Glukhova, D. S. Boyle, B. H. Weigl and K. P. Nichols, Anal. Chem., 2020, 92, 11305–11309 CrossRef CAS PubMed.
- C.-C. Wu, Y.-H. Chiang and H.-Y. Chiang, Biosensors, 2022, 12, 265 CrossRef CAS PubMed.
- F. Rusmini, Z. Zhong and J. Feijen, Biomacromolecules, 2007, 8, 1775–1789 CrossRef CAS PubMed.
- E. Steen Redeker, D. T. Ta, D. Cortens, B. Billen, W. Guedens and P. Adriaensens, Bioconjugate Chem., 2013, 24, 1761–1777 CrossRef CAS PubMed.
- H. J. Dyson, P. E. Wright and H. A. Scheraga, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13057–13061 CrossRef CAS PubMed.
- Y.-K. Cheng and P. J. Rossky, Nature, 1998, 392, 696–699 CrossRef CAS PubMed.
- S. Puertas, M. Moros, R. Fernández-Pacheco, M. R. Ibarra, V. Grazú and J. M. de la Fuente, J. Phys. D: Appl. Phys., 2010, 43, 474012 CrossRef.
- L. Zhang, Y. Mazouzi, M. Salmain, B. Liedberg and S. Boujday, Biosens. Bioelectron., 2020, 165, 112370 CrossRef CAS PubMed.
- G.-Q. Zhang, Z. Gao, J. Zhang, H. Ou, H. Gao, R. T. K. Kwok, D. Ding and B. Z. Tang, Cell Rep. Phys. Sci., 2022, 3, 100740 CrossRef CAS PubMed.
- M. McKenna, F. Soberon, A. J. Ricco, S. Daniels and S. M. Kelleher, Sens. Actuators, B, 2016, 236, 286–293 CrossRef CAS.
- S. Udomsom, U. Mankong, P. Paengnakorn and N. Theera-Umpon, Coatings, 2021, 11, 595 CrossRef CAS.
- S. Smith, K. Goodge, M. Delaney, A. Struzyk, N. Tansey and M. Frey, Nanomaterials, 2020, 10, 2142 CrossRef CAS PubMed.
- P.-Y. You, F.-C. Li, M.-H. Liu and Y.-H. Chan, ACS Appl. Mater. Interfaces, 2019, 11, 9841–9849 CrossRef CAS PubMed.
- S. Sharma, H. Byrne and R. O'Kennedy, Essays Biochem., 2016, 60, 9–18 CrossRef PubMed.
- S. Gao, F. Rojas-Vega, J. Rocha-Martin and J. M. Guisán, Int. J. Biol. Macromol., 2021, 177, 19–28 CrossRef CAS PubMed.
- A. Makaraviciute, C. D. Jackson, P. A. Millner and A. Ramanaviciene, J. Immunol. Methods, 2016, 429, 50–56 CrossRef CAS PubMed.
- A. J. Counsell, S. J. Walsh, N. S. Robertson, H. F. Sore and D. R. Spring, Org. Biomol. Chem., 2020, 18, 4739–4743 RSC.
- A. A. Karyakin, G. V. Presnova, M. Y. Rubtsova and A. M. Egorov, Anal. Chem., 2000, 72, 3805–3811 CrossRef CAS PubMed.
- A. Makaraviciute, T. Ruzgas, A. Ramanavicius and A. Ramanaviciene, Anal. Methods, 2014, 6, 2134–2140 RSC.
- M. Yüce and H. Kurt, RSC Adv., 2017, 7, 49386–49403 RSC.
- K. Yoshimoto, M. Nishio, H. Sugasawa and Y. Nagasaki, J. Am. Chem. Soc., 2010, 132, 7982–7989 CrossRef CAS PubMed.
- B. Moritz and J. O. Stracke, Electrophoresis, 2017, 38, 769–785 CrossRef CAS PubMed.
- S. Jeong, J. Y. Park, M. G. Cha, H. Chang, Y.-i. Kim, H.-M. Kim, B.-H. Jun, D. S. Lee, Y.-S. Lee, J. M. Jeong, Y.-S. Lee and D. H. Jeong, Nanoscale, 2017, 9, 2548–2555 RSC.
- S. Maher, M. Kamel, Z. Demerdash, H. El Baz, O. Sayyouh, A. Saad, N. Ali, F. Salah and S. Atta, Sci. Rep., 2023, 13, 10643 CrossRef CAS PubMed.
- X. Hu, M. J. Hortigüela, S. Robin, H. Lin, Y. Li, A. P. Moran, W. Wang and J. G. Wall, Biomacromolecules, 2013, 14, 153–159 CrossRef CAS PubMed.
- J. Hu, S. Zhou, L. Zeng, Q. Chen, H. Duan, X. Chen, X. Li and Y. Xiong, Talanta, 2021, 223, 121723 CrossRef CAS PubMed.
- E. L. Gulyak, V. A. Alferova, V. A. Korshun and K. A. Sapozhnikova, Molecules, 2023, 28, 7890 CrossRef CAS PubMed.
- C. A. C. Wolfe and D. S. Hage, Anal. Biochem., 1995, 231, 123–130 CrossRef CAS PubMed.
- F. Duval, T. A. van Beek and H. Zuilhof, Analyst, 2015, 140, 6467–6472 RSC.
- Q. Chao, Y. Ding, Z.-H. Chen, M.-H. Xiang, N. Wang and X.-D. Gao, Front. Chem., 2020, 8, 513 CrossRef CAS PubMed.
- M. D. Wilkins, B. L. Turner, K. R. Rivera, S. Menegatti and M. Daniele, Sens. Bio-Sens. Res., 2018, 21, 46–53 CrossRef.
- L.-D. Huang, A. K. Adak, C.-C. Yu, W.-C. Hsiao, H.-J. Lin, M.-L. Chen and C.-C. Lin, Chem. – Eur. J., 2015, 21, 3956–3967 CrossRef CAS PubMed.
- S. K. Vashist, E. Lam, S. Hrapovic, K. B. Male and J. H. Luong, Chem. Rev., 2014, 114, 11083–11130 CrossRef CAS PubMed.
- Z. Peng, F. Estelle, R. Simons and A. B. Becker, J. Immunol. Methods, 1991, 145, 255–258 CrossRef CAS PubMed.
- W. Choe, T. A. Durgannavar and S. J. Chung, Materials, 2016, 9, 994 CrossRef PubMed.
- R. Ollier, P. Wassmann, T. Monney, C. Ries Fecourt, S. Gn, V. C A, D. Ayoub, C. Stutz, G. S. Gudi and S. Blein, mAbs, 2019, 11, 1464–1478 CrossRef CAS PubMed.
- S. Liu, E. Haller, J. Horak, M. Brandstetter, T. Heuser and M. Lämmerhofer, Talanta, 2019, 194, 664–672 CrossRef CAS PubMed.
- Y. Li, Z. Zhu, W. Qu, Q. Yang, Y. Liu, Q. Wang, S. Duan, J. Wu, Z. Gong and L. Xu, Food Qual. Saf., 2023, 7, 1–8 Search PubMed.
- Y. Mu, D. P. McManus, C. A. Gordon, H. You, A. G. Ross, R. M. Olveda and P. Cai, Front. Immunol., 2023, 14, 1165480 CrossRef CAS PubMed.
- G. Li, Q. Li, X. Wang, X. Liu, Y. Zhang, R. Li, J. Guo and G. Zhang, Int. J. Biol. Macromol., 2023, 242, 125186 CrossRef CAS PubMed.
- L. Anfossi, F. Di Nardo, M. Profiti, C. Nogarol, S. Cavalera, C. Baggiani, C. Giovannoli, G. Spano, E. Ferroglio, W. Mignone and S. Rosati, Anal. Bioanal. Chem., 2018, 410, 4123–4134 CrossRef CAS PubMed.
- Z. Ali, E. Sánchez, M. Tehseen, A. Mahas, T. Marsic, R. Aman, G. Sivakrishna Rao, F. S. Alhamlan, M. S. Alsanea, A. A. Al-Qahtani, S. Hamdan and M. Mahfouz, ACS Synth. Biol., 2022, 11, 406–419 CrossRef CAS PubMed.
- Y. Wang, C. Fill and S. R. Nugen, Biosensors, 2012, 2, 32–42 CrossRef CAS PubMed.
- S. Dalirirad, D. Han and A. J. Steckl, ACS Omega, 2020, 5, 32890–32898 CrossRef CAS PubMed.
- S. Pavagada, R. B. Channon, J. Y. H. Chang, S. H. Kim, D. MacIntyre, P. R. Bennett, V. Terzidou and S. Ladame, Chem. Commun., 2019, 55, 13470 RSC.
- C. Díaz-Perlas, B. Ricken, L. Farrera-Soler, D. Guschin, F. Pojer, K. Lau, C.-B. Gerhold and C. Heinis, Nat. Commun., 2023, 14, 2774 CrossRef PubMed.
- X. Li, Q. Zhu, F. Xu, M. Jian, C. Yao, H. Zhang and Z. Wang, Anal. Biochem., 2022, 648, 114671 CrossRef CAS PubMed.
- J. E. Pinto Torres, J. Goossens, J. Ding, Z. Li, S. Lu, D. Vertommen, P. Naniima, R. Chen, S. Muyldermans, Y. G. J. Sterckx and S. Magez, Sci. Rep., 2018, 8, 9019 CrossRef PubMed.
- T. He, J. Zhu, Y. Nie, R. Hu, T. Wang, P. Li, Q. Zhang and Y. Yang, Toxins, 2018, 10, 180 CrossRef PubMed.
- B. A. Rohrman, V. Leautaud, E. Molyneux and R. R. Richards-Kortum, PLoS One, 2012, 7, e45611 CrossRef CAS PubMed.
- M. Pongsuchart, A. Sereemaspun and K. Ruxrungtham, J. Life Sci., 2013, 1, 172–175 Search PubMed.
- B. Gosselin, M. Retout, I. Jabin and G. Bruylants, Sens. Diagn., 2024, 3, 248–255 RSC.
- M. Lin, J. Wang, G. Zhou, J. Wang, N. Wu, J. Lu, J. Gao, X. Chen, J. Shi, X. Zuo and C. Fan, Angew. Chem., Int. Ed., 2015, 54, 2151–2155 CrossRef CAS PubMed.
- H. Wei, F. Li, T. Xue, H. Wang, E. Ju, M. Li and Y. Tao, Bioact. Mater., 2023, 28, 50–60 CAS.
- B. Dai, Y. Hu, J. Duan and X. D. Yang, Onco Targets Ther, 2016, 7, 38257–38269 Search PubMed.
- D. Feng, J. Su, Y. Xu, G. He, C. Wang, X. Wang, T. Pan, X. Ding and X. Mi, Microsyst. Nanoeng., 2021, 7, 33 CrossRef CAS PubMed.
- Z. Li, B. Zhao, D. Wang, Y. Wen, G. Liu, H. Dong, S. Song and C. Fan, ACS Appl. Mater. Interfaces, 2014, 6, 17944–17953 CrossRef CAS PubMed.
- Z. Zhou, Y. S. Sohn, R. Nechushtai and I. Willner, ACS Nano, 2020, 14, 9021–9031 CrossRef CAS PubMed.
- D. X. Wang, J. Wang, Y. X. Wang, Y. C. Du, Y. Huang, A. N. Tang, Y. X. Cui and D. M. Kong, Chem. Sci., 2021, 12, 7602–7622 RSC.
- M. Lin, P. Song, G. Zhou, X. Zuo, A. Aldalbahi, X. Lou, J. Shi and C. Fan, Nat. Protoc., 2016, 11, 1244–1263 CrossRef CAS PubMed.
- X. Chen, G. Zhou, P. Song, J. Wang, J. Gao, J. Lu, C. Fan and X. Zuo, Anal. Chem., 2014, 86, 7337–7342 CrossRef CAS PubMed.
- Y. Xu, J. Da, Q. Lan, J. Luo, Z. Lu, R. Peng, F. Yu and Y. Zha, Sens. Actuators, B, 2023, 393, 134266 CrossRef CAS.
- L. Yu, S. Yang, Z. Liu, X. Qiu, X. Tang, S. Zhao, H. Xu, M. Gao, J. Bao, L. Zhang, D. Luo, K. Chang and M. Chen, Mater. Today Bio, 2022, 15, 100276 CrossRef CAS PubMed.
- A. Rathore, V. Joshi and N. Yadav, BioPharm Int., 2013, 26, 40–45 CAS.
- A. P. H. Le, Q. L. Nguyen, B. H. Pham, T. H. M. Cao, T. V. Vo, K. Huynh and H. T. T. Ha, HardwareX, 2023, 15, e00455 CrossRef PubMed.
- B. Zhang, W. Gao, J. Piao, Y. Xiao, B. Wang, W. Peng, X. Gong, Z. Wang, H. Yang and J. Chang, ACS Appl. Mater. Interfaces, 2018, 10, 14549–14558 CrossRef CAS PubMed.
- M. M. Bordbar, A. Sheini, P. Hashemi, A. Hajian and H. Bagheri, Biosensors, 2021, 11, 316 CrossRef CAS PubMed.
- R. Tang, M. Xie, X. Yan, L. Qian, J. P. Giesy and Y. Xie, Cellulose, 2023, 30, 6457–6469 CrossRef CAS PubMed.
- J. M. Yang, K. R. Kim, S. Jeon, H. J. Cha and C. S. Kim, Sens. Actuators, B, 2021, 329, 129099 CrossRef CAS.
- A. S. Lee, S. M. Kim, K. R. Kim, C. Park, D. G. Lee, H. R. Heo, H. J. Cha and C. S. Kim, Sens. Actuators, B, 2023, 379, 133245 CrossRef CAS PubMed.
- S. Natarajan, J. Jayaraj and D. M. F. Prazeres, Biosensors, 2021, 11, 49 CrossRef CAS PubMed.
- M. Barbosa, H. Simões and D. M. F. Prazeres, Materials, 2021, 14, 3175 CrossRef CAS PubMed.
- S. Natarajan, J. Joseph and D. M. França Prazeres, Sci. Rep., 2022, 12, 5478 CrossRef CAS PubMed.
- A. Elter, T. Bock, D. Spiehl, G. Russo, S. C. Hinz, S. Bitsch, E. Baum, M. Langhans, T. Meckel, E. Dörsam, H. Kolmar and G. Schwall, Sci. Rep., 2021, 11, 7880 CrossRef CAS PubMed.
- A. B. Boraston, D. N. Bolam, H. J. Gilbert and G. J. Davies, Biochem. J., 2004, 382, 769–781 CrossRef CAS PubMed.
- O. Shoseyov, Z. Shani and I. Levy, Microbiol. Mol. Biol. Rev., 2006, 70, 283–295 CrossRef CAS PubMed.
- C. Oliveira, V. Carvalho, L. Domingues and F. M. Gama, Biotechnol. Adv., 2015, 33, 358–369 CrossRef CAS PubMed.
- D. Quesada-González, C. Stefani, I. González, A. de la Escosura-Muñiz, N. Domingo, P. Mutjé and A. Merkoçi, Biosens. Bioelectron., 2019, 141, 111407 CrossRef PubMed.
- R. H. Tang, L. N. Liu, S. F. Zhang, A. Li and Z. Li, Microchim. Acta, 2019, 186, 831 CrossRef CAS PubMed.
- N. Alam, L. Tong, Z. He, R. Tang, L. Ahsan and Y. Ni, Cellulose, 2021, 28, 8641–8651 CrossRef CAS PubMed.
- D. Quesada-González, C. Stefani, I. González, A. de la Escosura-Muñiz, N. Domingo, P. Mutjé and A. Merkoçi, Biosens. Bioelectron., 2019, 141, 111407 CrossRef PubMed.
- F. Feng, Q. Fu, F. Cao, Y. Yuan, R. Kong, D. Ji and H. Liu, ChemBioChem, 2024, 25, e202300575 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |