
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid-state nanopore counting of amplicons from recombinase polymerase isothermal amplification†
Breeana
Elliott
 a,
Martin
Charron
a,
John
Pezacki
b,
Erin
McConnell
*c and
Vincent
Tabard-Cossa
*ab
a,
Martin
Charron
a,
John
Pezacki
b,
Erin
McConnell
*c and
Vincent
Tabard-Cossa
*ab
aDepartment of Physics, University of Ottawa, Ottawa, K1N 6N5, Ontario, Canada. E-mail: tcossa@uOttawa.ca
bDepartment of Chemistry and Biomolecular Sciences, University of Ottawa, Ottawa, K1N 6N5, Ontario, Canada
cDepartment of Chemistry, Carleton University, Ottawa, K1S 5B6, Ontario, Canada. E-mail: erin.mcconnell@carleton.ca
First published on 23rd August 2024
Abstract
Single-molecule detection methods based on electrical readout can transform disease diagnostics by miniaturizing the downstream sensor to enable sensitive and rapid biomarker quantification at the point-of-care. In particular, solid-state nanopores can be used as single-molecule electrical counters for a variety of biomedical applications, including biomarker detection. Integrating nanopores with efficient DNA amplification methods can improve upon sensitivity and accessibility concerns often present in disease detection. Here, we present nanopores as biosensors downstream of a reverse-transcription recombinase polymerase amplification (RT-RPA)-based assay targeting synthetic SARS-CoV-2 RNA. We demonstrate the efficacy of nanopore-integrated RT-RPA for the direct electrical detection of target amplicons, and discuss challenges from RPA-based assays and adaptations that facilitate solid-state nanopore readout.
Introduction
Over the past few years, the COVID-19 pandemic has prompted significant renewed interest in developing alternative disease diagnostics approaches, and in improving upon existing assays and detection methods.1–5 Currently, real-time reverse transcriptase-polymerase chain reaction (RT-qPCR) remains the gold standard for many molecular tests that require DNA amplification of a target RNA sequence, such as for SARS-CoV-2 diagnosis. The detection modality of RT-qPCR traditionally relies on the measurement of a fluorescent signal whose intensity is proportional to the presence of amplified nucleic acid products (amplicons) in the sample. Although highly sensitive, this technique has drawbacks for point-of-care applications since it involves repeated thermal cycling which can be time-consuming and requires more advanced equipment. The requirement for thermal and optical components (light source and detector) and the need for optical dyes can constrain the cost and size of instruments or reagents especially for decentralized and remote use. Consequently, there is a need for simpler amplification procedures and for new sensing modalities that can deliver a cost-effective and streamlined alternative method of testing without compromising on the speed, sensitivity and accuracy of the detection.6Nanopores can count single molecules purely electrically and without the use of labels, and as such provide an alternative strategy to detect and quantify amplified products.7–9 Nanopores are nanometer-sized holes in thin insulating membranes that partition reservoirs of electrolyte solution. The application of an electric potential difference across the membrane generates an electric field inside the pore that drives the flow of ions and can be used to capture charged polymers, like DNA, thus creating a steady ionic current that is modulated by the passage of individual biomolecules. The characteristics of the resulting electrical signatures (e.g., blockage depths and passage times) can be used to infer the identity of the translocating molecules. Having already transformed genomics by providing a low-cost and portable nucleic acid sequencing technology,10,11 nanopore-assisted counting of disease biomarkers, in particular amplicons from viral and bacterial pathogens, is therefore a promising alternative sensing method to optical sensors that can be developed into a rapid, inexpensive and ultra-sensitive diagnostic platform, compatible with decentralized deployment to enable point-of-need screening for infection.
In recent years, sensing schemes have been proposed to showcase how nanopores can be used to detect disease biomarkers.12–18 More specifically, solid-state nanopores fabricated from glass capillaries or in planar SiN membranes have shown promise for such diagnostics applications, as their dimensions can be tuned to match the target of interest as well as the requirements for different digital and multiplexing schemes.19,20 To provide specificity of nanopore signals from nucleic acid targets, these approaches made use of enzymes, labels, probes or DNA nanocarriers to accurately detect for example, the cystic-fibrosis mutation codon,21 the conserved sequences of the HIV RNA genome,17 the Zika virus gene, microRNA biomarker for human lung cancer,22–24 and circulating tumor DNA or microRNA biomarkers for prostate cancer.25,26
Enzymatic amplification strategies have also been used alongside nanopore detection to generate countable amplicon copies for the development of qualitative and quantitative nucleic acid tests. One of our recent studies employed a PCR-based assay to detect the presence of group A streptococcus (GAS) in throat swabs.4 Other studies have measured SARS-CoV-2 viral loads using RT-PCR or by simply using the reverse transcription in cases of high transcript concentrations,13,14 or have utilized loop-mediated isothermal amplification schemes.27 Common challenges in these studies included the need to tune the amplicon characteristics to distinguish the amplicon signal from the background and/or to clean up the proteins from the amplification mixture using purification kits and/or enzymatic digestion.4 Additionally, variations in pore characteristics that affect the molecular capture rate and limit the accuracy of quantitative measurements had to be addressed, either by performing calibration curves prior to sensing or making use of an internal standard for real-time calibration, as in Charron et al.28
Many isothermal amplification techniques have emerged as alternative methods29–31 to PCR that solve the need for a thermal cycler, thus reducing instrumentation complexity and improving upon the diagnostic turnaround time and cost. Recombinase polymerase amplification32–34 (RPA) is one such amplification method and involves recombinase proteins forming a complex with application-specific primers that amplify dsDNA template strands (see ESI† Fig. S0). When combined with reverse transcription (RT-RPA), RPA can be used to amplify complementary DNA (cDNA) strands from a viral RNA genome in one tube. The benefits of RPA for point-of-care or field use include: (i) a 37–42 °C operating range without the need for a tight temperature control; (ii) no initial denaturation steps to melt the dsDNA into single-strands; (iii) the use of only 2 primers per target; (iv) a limit of detection as low as 1–10 copies for certain targets/samples; (v) a rapid amplification in <45 minutes; and (vi) no refrigeration of reagents, which can instead be lyophilized.29–31 Particularly, we note that in contrast with loop-mediated isothermal amplification (LAMP), another popular isothermal amplification technique, RPA can be done at a lower temperature (37–42 °C compared to 60–65 °C) and requires fewer primers.35
Integrating RPA with nanopore-based single molecule detection holds promise in offering a simple and effective assay solution on a purely electrical platform well suited for performing decentralized molecular diagnostics. In this work, we demonstrate the counting of amplicons from RPA amplification of SARS-CoV-2 RNA with solid-state nanopores. We report on the assay optimization to produce relatively long amplicons (363 bp) well-suited for nanopore sensing and present how the presence of amplicons is detected by observing capture rate changes for the expected amplicon nanopore signals when compared to the blank sample (i.e. non-template control). We show that as low as 102 copies of the viral RNA can be detected following a simple dilution step of the assay reagents into the nanopore sensing buffer. Finally, we discuss challenges with performing quantitative measurements given the efficiency of the current assay and analog measurement mode despite counting single molecules.
Results and discussion
Optimization of nanopore sensing of RPA reaction
To investigate the feasibility of using RT-RPA to isothermally amplify target RNA prior to nanopore sensing (RT-RPA-NP), synthetic RNA fragments from SARS-CoV-2 were employed as proof-of-concept targets, from which various samples with predetermined template copy numbers were prepared (Fig. 1a). We first optimized the RPA reaction to produce amplicons from the N region encoding the nucleocapsid phosphoprotein of the synthetic SARS-CoV-2 fragmented genome (see Methods section). Optimization efforts focused on producing the longest possible amplicon to facilitate nanopore detection while maintaining a good amplification efficiency via minimization of non-specific background amplification and maximization of intended amplicon production. | ||
Fig. 1 Nanopore sensing of DNA control and amplified DNA. (a) Schematic representation of RPA-nanopore workflow. The RPA reaction is typically run at 42 °C for 45 minutes prior to nanopore sensing. (b) Agarose gel analysis of an RPA ran at 42 °C from 106 initial RNA template copies at different timepoints. Lanes from left to right: ladder; control held at 42 °C; aliquot removed after 15 minutes, 25 minutes, 35 minutes, and 45 minutes. (c) 2.5 second current trace for RT-RPA products amplified at 42 °C for 45 min, from 106 initial RNA copies. (d) Conductance blockage (maximum deviation) versus passage time for RPA products from amplification using 106 initial RNA template copies in a 50 μL reaction volume. Amplicons sensed on the nanopore over ∼30 minutes. Gaussian fits to the data show ![[small tau, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d4.gif) = 15 ± 4 μs and = 15 ± 4 μs and 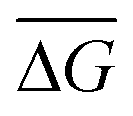 = 9.9 ± 0.7 nS. (e) 2.5 second current trace for 24 nM 400 bp dsDNA fragments. (f) Conductance blockage (maximum deviation) versus passage time for 24 nM 400 bp dsDNA fragments sensed on the nanopore over ∼10 minutes. Gaussian fits to the data show = 17 ± 4 μs and = 9.9 ± 0.7 nS. (e) 2.5 second current trace for 24 nM 400 bp dsDNA fragments. (f) Conductance blockage (maximum deviation) versus passage time for 24 nM 400 bp dsDNA fragments sensed on the nanopore over ∼10 minutes. Gaussian fits to the data show = 17 ± 4 μs and  = 10.2 ± 0.6 nS. Data was acquired on a 5.1 ± 0.5 nm nanopore at 200 mV in 3.6 M LiCl pH 8. Data sampled at 4.167 MS s−1 and low pass filtered at 400 kHz. = 10.2 ± 0.6 nS. Data was acquired on a 5.1 ± 0.5 nm nanopore at 200 mV in 3.6 M LiCl pH 8. Data sampled at 4.167 MS s−1 and low pass filtered at 400 kHz. | ||
Although shorter amplicons are known to be optimal for RPA reactions,34 our previous work4,28 suggests that amplicons <100 bp in length often translocate in <10 μs, which results in attenuated blockage signals or signals indistinguishable from the baseline current, thus limiting measurement accuracy. Furthermore, many background molecules (e.g., proteins, surfactants) found in amplification mixtures produce signals with durations in the 1–100 μs range.36 Signals from longer amplicons can thus be more clearly distinguished from background signals without prior purification steps. To this end, primer sets of various lengths were designed and screened according to the recommended reaction conditions. Considering these factors, alongside the practical limits of nucleic acid amplification (i.e. non-specific amplification more likely for longer target amplicons), we selected the primer pair that produced amplicon lengths of 363 bp, which we found maximizes length while producing minimal non-specific background amplification. See Tables S1, S2 and Fig. S0, S1 of the ESI† for a more detailed description of the optimization process.
Fig. 1b shows a representative agarose gel image following RT-RPA reactions at 42 °C from 106 initial RNA template copies for different reaction times. The RT-RPA reaction occurs efficiently in as little as 15 minutes with relatively low non-specific amplification when the copy number is high (>105, i.e., at a ∼fM levels from tens of μL volumes). At lower copy numbers (typically less than 104), although the amplicon band is observed, non-specific amplification is also present. This can be seen in the multiple faint bands at lengths other than the target amplicon (see ESI† Fig. S4 for reaction sensitivity at different initial template copies). The reaction temperature (up to 42°) was used to optimize reverse transcriptase activity whereas the reaction time (up to 45 min) was chosen to improve amplification efficiency and specificity (see ESI† Section S3).
As discussed, accurate nanopore detection of RPA reaction amplicons requires the electrical identification of amplicon population from the background molecules present in solution. This background can include, proteins, recombinase and polymerase enzymes, as well as interfering DNA fragments such as primer–dimer complexes, RNA–DNA hybrids, non-specifically amplified DNA, and other reagents used in the commercial RPA mixture. In contrast to PCR or LAMP the individual reagents for RPA are not available off the shelf, and thus optimization of the reaction mixture composition for nanopore sensing is not readily available as in our previous study.4 Nonetheless, the nanopore characteristics and operating conditions can be optimized to reduce the observation of these background molecules while maximizing the signal amplitude from the amplicon (see ESI† Fig. S11 and S12 for nanopore optimization tests). Here, following King et al.,4 we used nanopores 5 nm in diameter in thin 12 nm SiN membranes, operated in 3.6 M LiCl pH 8 at 200 mV.
To develop an understanding of the electrical signature (i.e., types of signals) generated by the passage of RPA amplicons through nanopores, PCR-purification kits were used to filter the amplification products (see Methods) from 106 initial template copies (∼30 fM initial target concentration in a 50 μL sensing volume) to remove excess nanopore background signals (see Fig. S1† for non-specific amplification from RPA positive control reactions). A representative ionic current trace of the amplicon translocation events is shown in Fig. 1c, where single transient blockades are observed. Fig. 1d illustrates the purified RPA product translocation characteristics, by displaying the conductance blockage depth, ΔG, measured as the maximum conductance deviation from the baseline during the event, versus the translocation time, τ, of each individual event. The population is centered at a mean translocation time of = 15 ± 4 μs and a blockage depth tightly distributed around a mean of 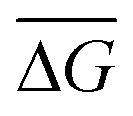 = 9.9 ± 0.7 nS. Note that some events are also observed <10 μs.
= 9.9 ± 0.7 nS. Note that some events are also observed <10 μs.
Control experiments were performed with dsDNA to help confirm the identity of the populations resulting from the passage of purified RPA products. A representative ionic current trace of these events is shown in Fig. 1e. Fig. 1f shows a scatterplot of conductance blockage versus translocation time of 400 bp dsDNA in the same pore. A population is observed centered around a mean translocation time of = 17 ± 4 μs and a mean blockage depth of  = 10.2 ± 0.6 nS. This blockage level corresponds to the expected value from the single-file passage of dsDNA under these operating conditions. This then suggests the population observed in Fig. 1d is the single-file RPA amplicon population. Lastly, we argue that the shallower and faster events <10 μs in Fig. 1d is a result of remaining primers and/or non-specific amplification producing shorter amplicons. ESI† Fig. S15 provides some evidence to support this by showing the results of a control experiment of translocating primers through a similarly sized pore.
= 10.2 ± 0.6 nS. This blockage level corresponds to the expected value from the single-file passage of dsDNA under these operating conditions. This then suggests the population observed in Fig. 1d is the single-file RPA amplicon population. Lastly, we argue that the shallower and faster events <10 μs in Fig. 1d is a result of remaining primers and/or non-specific amplification producing shorter amplicons. ESI† Fig. S15 provides some evidence to support this by showing the results of a control experiment of translocating primers through a similarly sized pore.
Given that both the translocation time and conductance blockage distributions of purified RPA amplicon signals are well described by Gaussian distributions centered around and  , with widths of στ and σΔG, respectively, we define an elliptical event classification filter in ΔG vs. τ scatterplots which retains most of the amplicon population as in King et al.4
, with widths of στ and σΔG, respectively, we define an elliptical event classification filter in ΔG vs. τ scatterplots which retains most of the amplicon population as in King et al.4
 | (1) |
Nanopore detection of unpurified RPA products
An ideal RT-RPA-NP assay scheme would involve only two steps to streamline the assay: (i) one-tube reverse transcription and amplification, and (ii) direct nanopore sensing of RT-RPA products without any cleanup steps. Omitting a purification stage prior to nanopore sensing means that in addition to amplicons, background molecules such as excess primers, enzymes and other proteins involved in the reaction mixture will be present in the sample, which will make it more challenging to accurately identify and quantify amplicons. To investigate this, we now show the response of a nanopore sensor to unpurified RPA products simply diluted in a 3.6 M LiCl pH 8 sensing buffer.Fig. 2a–d respectively depict the blockage depths ΔG and translocation times of individual nanopore signals from the master mix (MM), i.e. the RPA reaction mixture with no RNA template, from the non-template control (NTC), i.e. the master mix sample incubated at 42 °C for 45 minutes, and from the RPA products resulting from the amplification of 105 copies and 102 copies of RNA templates. Fig. 2c shows that signals from the RPA products of the 105 copy number sample are more tightly distributed than the MM and NTC samples, and that parameters for an elliptical event classification filter could be determined (eqn (1)) from its distributions in order to classify RPA amplicons of interest. To quantify how much the background molecules overlap with the RPA amplicons, this same filter was applied to the MM and NTC samples, as per Fig. 2a–c which highlight, in blue, events that fall within the classification filter, and in gray those that do not.
 | ||
| Fig. 2 Nanopore sensing of RT-RPA amplicons without purification. Scatterplot of the conductance blockage (max deviation) as a function passage time for all passage events collected over 1800 seconds on the same nanopore for (a) master mix prior to heating (n = 1363 total, n = 58 classified events by selection filter) with a representative passage event trace, (b) non-template control (NTC) (n = 990 total, n = 188 classified events) with a representative passage event trace, (c) amplification using 105 initial RNA template copies – in 50 μL, equivalent to a ∼3 fM initial concentration (n = 1261 total, n = 443 classified events) with representative amplicon-classified event traces (blue) and background event traces (grey), (d) amplification using 102 initial RNA template copies in 50 μl, equivalent to a ∼3 aM initial concentration (n = 1247 total, n = 311 classified events). Amplicon-classified passage events are shown in blue. (e) RT-RPA product samples on a 2% agarose gel at 70 V for 100 minutes. Nanopore data was acquired on a 5.3 ± 0.5 nm nanopore at 200 mV in 3.6 M LiCl 10 mM HEPES pH 8 and any other reagents contained in the TwistAmp Basic RPA kit from TwistDx. Each RPA reaction was run for 45 minutes at 42 °C prior to sensing. | ||
The master mix sample exhibits some amplicon-classified events, but the corresponding detection rate is low at only 0.03 ± 0.01 Hz (58 for 1363 total events). The non-template control sample contains a somewhat larger number of events in the region expected for the amplicon, with a capture rate of 0.10 ± 0.01 Hz (188 for 990 total events). In contrast, when looking at the 105 copy number sample, we observe a significant increase in signal rate in comparison to the non-template control. The amplicon-classified molecules exhibit a capture rate of 0.24 ± 0.03 Hz (443 for 1261 total events), corresponding to a +0.14 Hz increase.
As shown in Fig. 2d, despite the large number of background molecules present in the unpurified solution, we were able to discern the 102-copy number sample from the non-template control as its amplicon-classified events display a capture rate of 0.17 ± 0.02 Hz (311 for 1247 total events). However, amplification and sensing at low copy numbers was inconsistent (see ESI† Sections S3 and S10) as amplification did not always occur consistently and efficiently for reactions from <104 initial RNA template copies (i.e., sub-fM initial concentration in a 50 μL volume).
To understand the signals observed in the NTC sample, all samples were also examined on a 2% agarose gel as shown in Fig. 2e. Multiple bands are present in the NTC lane, indicating the presence of several DNA lengths or complexes in the samples. Such streaking is also present in the RNA-positive samples, but in contrast their lanes contain a bright band corresponding the correctly amplified 363 bp target amplicon. Notably, at low copy numbers (<104) non-specific amplification and amplification artifacts are more likely, as illustrated by one of the separate samples of 102 RNA copy numbers exhibiting split amplicon bands. Consistent with our observations, amplification artifacts produced during RPA have been found to exhibit lengths shorter or longer than the intended amplicon. The short artifacts are often primer–dimer complexes that result from homology between primer sequences, or primers that partially hybridize non-specifically with enough stability to allow for polymerization.37 Note that nanopores cannot differentiate polymer length varying by less than a few hundred bp due to the dynamics of the translocation process leading to wide distribution of passage times for identical molecules.38,39 Therefore, the different bands seen in the gel overlap in the nanopore scatter plots.
The observed background signals from the unpurified NTC sample (i.e., the blank) have a substantial impact on the limit of detection of this RT-RPA-NP assay. Nanopores can provide precise and sensitive quantification of molecules of known length by controlled counting or capture rate measurements, provided that the target can be distinguished from other molecules in solution. In this case, since there are no characteristic structures in the events (e.g., sub-levels) and the passage times of similarly sized DNA polymers coincide significantly, the background and amplicon populations can overlap, which complicates classification and thus quantification at low copy numbers. As observed in Fig. 2d however, despite the presence of the non-specific background molecules, using 102 initial RNA copies is enough to produce sufficient specific amplification to detect a signal rate increase of the amplicon population on the nanopore (>2 s.d. above the limit of blank40). We note that RT-RPA assays designed specifically for COVID-19 detection typically use <200 bp amplicons,41–43 and have been shown to detect <101 total RNA template copies, on par with detection limits from RT-qPCR. The fact that this RT-RPA-NP assay relies on a longer amplicon length (363 bp) could explain the inconsistent amplification results, as shorter amplicons result in more efficient amplification.
It should be noted that while it was possible to obtain statistically relevant numbers of translocation events (which we define here as >100 events) when running unpurified samples to estimate the capture rate, we observed an increase in the likelihood of clogging and a decrease in pore longevity (see ESI† Fig. S13) compared to running purified samples. Out of 10 bare SiN pores tested with unpurified samples, 6 clogged prior to running all control experiments. To investigate the ability of this RT-RPA-NP assay to quantify RNA viral load while using the same amplicon length, we next move to analyzing purified RPA samples to better isolate the specific DNA amplicon prior to nanopore sensing.
Nanopore quantification of purified RPA products
One way of reducing the background signals to improve detection sensitivity and to prolong the lifetime of the nanopore sensor (without chemically functionalizing the SiN surface) is to filter the RPA samples with a purification kit prior to sensing. Cleaned up samples can facilitate the use of a single pore in generating calibration curves of increasing template concentrations and in testing samples of interest of unknown concentrations. Here, to address the above, we used a PCR-purification kit (see Methods section) to remove residual reaction enzymes, primers, nucleotides and short DNA molecules less than 100 bp.Fig. 3 shows the results of nanopore detection of different RPA reactions following a purification step with a PCR-purification kit, sensed by the same 5.3 nm nanopore at 200 mV in pH 8 3.6 M LiCl for 30 minutes for each sample. Fig. 3a depicts the conductance blockage versus translocation times for all events for the non-template control and RNA-positive samples ranging from 103 to 106 initial copies. The amplicon translocation events are highlighted on the scatterplots and were identified using an elliptical filter as defined above (eqn (1)) with parameters obtained from fitting the 106 sample, as it presents the most clearly separated population from the background signals. All samples were also spiked with a 2 kbp dsDNA internal control to monitor pore behaviour fluctuations between each experiment, as demonstrated more extensively in Fig. S14 of the ESI.†
 | ||
| Fig. 3 Nanopore identification of RT-RPA amplicons after PCR purification kit. (a) Scatterplot of the conductance blockage (max deviation) vs. passage time for all passage events (grey) and amplicon-classified events (color) collected over 1800 seconds on the same nanopore for RT-RPA product samples (from left to right): non-template control (n = 524 total, n = 95 classified events), amplification from 103 initial RNA copies – in 50 μL, equivalent to a ∼30 aM initial concentration (n = 780 total, n = 128 classified events), amplification from 104 initial RNA copies – in 50 μL, equivalent to a ∼0.3 fM initial concentration (n = 519 total, n = 179 classified events), amplification from 105 initial RNA copies – in 50 μL, equivalent to a ∼3 fM initial concentration (n = 561 total, n = 237 classified events), and amplification from 106 initial RNA copies – in 50 μL, equivalent to a 30 fM initial concentration (n = 870 total, n = 581 classified events). The capture rate of each sample is shown in the top-right corner of each panel. (b) Distribution of the log of inter-event times for the amplicon-classified events from each sample. (c) Nanopore extracted amplicon signal rate (circles) compared with amplicon band intensity extracted from a gel (triangles) for each sample. (d) RT-RPA product samples ran on a 2% agarose gel at 100 V for 60 minutes. Nanopore data was acquired on a 5.3 ± 0.5 nm nanopore at 200 mV in 3.6 M LiCl 10 mM HEPES pH 8. Data sampled at 4.167 MS s−1 and low pass filtered at 400 kHz. Band intensities were determined using GelAnalyzer 19.1 analysis software. | ||
Fig. 3b illustrates the inter-event delay histograms from which the amplicon capture rate of each sample was calculated.28Fig. 3c illustrates a clear correlation between the signal rate and the initial copy number of the RNA template, showing an increase in amplicon signal rate as the initial copy number rises. However, over the range of copy numbers tested, this assay exhibits a subpar calibration sensitivity as demonstrated by the weak sub-linear dependence of capture rate on copy number which limits the quantification precision of the assay.
As shown in Fig. 3a, the presence of background molecules (grey events) is reduced for samples with more than 104 initial template copies, which helps separate amplicon populations from background signals. In the samples with only 103 initial copies, amplification did not occur as efficiently as can be seen by the 0.08 ± 0.01 Hz capture rate and the high number of background events (429 unclassified out of 524 total events). This sample maintained a high concentration of non-specifically amplified DNA as can be seen by the elevated capture of short molecules (shown in grey, <10 μs). In comparison, the sample with 106 initial RNA copies has a significantly higher amplicon capture rate of 0.35 ± 0.03 Hz and a much lower background count (289 unclassified out of 870 total events).
The poor quantification sensitivity of this RT-RPA-NP assay can be linked to the performance our implementation of the RPA amplification step. This is illustrated in Fig. 3d, which shows the migration of the same RPA products on a 2% agarose gel. A strong band is present at the amplicon length in the samples with greater than 104 initial RNA template copies. Conversely, the amplicon band appears faint in the 103 initial RNA template copy sample, and akin to the NTC samples, shorter RPA products are also visible. We can see that this particular amplification reaction performed more poorly at low template copy numbers (under 103 copies) in comparison to the gel in Fig. 2e, which highlights the amplification challenges and the variability of the RPA process in our hands.
Fig. 3c compares the capture rate of the RPA samples on the nanopore (circles) to their relative band intensity on an agarose gel (squares). The gel results depict a similar lack of separation of the amplified DNA and the presence of a non-specific amplicon population, particularly at lower copy numbers, which agree with the results of nanopore detection scheme. The nanopore is able to detect a small increase in capture rate for the 103 sample in comparison to the NTC (0.08 ± 0.01 Hz vs. 0.05 ± 0.01 Hz, respectively) while there is no visible difference in their lanes on the gel. We thus relate the difficulty in obtaining a precise calibration curve to the challenges with the RPA isothermal amplification process. Unlike with PCR assays, where background signals can be reduced by custom master mix formulation,4 there are limitations on the development of custom RPA assays and amplification formulation as commercial RPA kits are only commonly available from one source. More importantly, RPA uses a time threshold instead of a cycle threshold, which is inherently dependent on the reaction kinetics. This time threshold is not only controlled by the initial template concentration, but also by the temperature, reagent concentration, and mixing step of the reaction. Therefore, further optimization of the RPA reaction to improve the amplification efficiency for low copy numbers is needed to improve the sensitivity and quantification accuracy of this RT-RPA-NP assay.
Conclusion
We have demonstrated the feasibility of detecting amplicons produced from RT-RPA amplification of SARS-CoV-2 RNA using solid-state nanopores. We showed that as low as 102 copies of RNA can be detected using our RPA implementation with 363 bp amplicons. Furthermore, we discussed the challenges associated with accurately classifying the amplicon population and performing quantitative measurements due to the limited amplification efficiency of the current RPA assay leading to poor calibration sensitivity.Additional optimization of the amplicon length could facilitate quantification of the initial copy number by minimizing potential overlap with the non-specifically amplified DNA. As it is, the amplicon does not produce a very specific electrical signal and is indistinguishable from any other DNA of similar length present in solution. Increasing the amplicon length closer to the 1000 bp upper limit for RPA would further separate the amplicon's dwell time from the background fragments and facilitate the analysis for low-copy numbers. Other isothermal amplification methods, such as RT-LAMP, might produce less non-specific fragments, but their amplicon is often size-limited to <300 bp which may not be compatible with nanopore sensors since translocations could be too fast (<10 μs) to be reliably detected. An alternative approach would be to design a digital assay scheme integrated with isothermal amplification instead of only relying on an analog capture rate signal. This could diminish the effects of the non-specific fragments by generating distinct features in the blockage patterns of the amplicons, permitting digital quantification of the amplicons as opposed to absolute measurements and thus removing instrumentation noise from the quantification.44–46
The results presented in this study demonstrate that RPA is compatible with nanopore detection. Given its simplicity and the fact that amplification requires only a relatively low and constant temperature, this technique continues to hold promise for future applications in disease diagnostics, for which point-of-care/need use is an attribute of ever-increasing importance.
Methods
Gel benchmarking
2% agarose (w/v) gels were made in 1× TE buffer with either GelRed stain (MilliporeSigma) or with SYBR safe DNA gel stain (Thermo Fisher). Sample aliquots (2 μL) were diluted in 6× DNA gel loading dye (Thermo Fisher) with water to 12 μL. Aliquots (6 μL) of the GeneRuler ready-to-use low range DNA ladder (Thermo Fisher) or the Invitrogen 1 Kb Plus DNA ladder (Thermo Fisher) were used as a benchmark. Gels were run at 70 V or 100 V until bands were visibly separated. Gels were imaged using an AzureC150 Gel imaging system (Azure Biosystems) using the default settings for ethidium bromide staining. Band intensities were determined using GelAnalyzer 19.1 analysis software.Nanopore-optimized RPA
Recombinase polymerase amplification was conducted using the TwistDx TwistAmp® basic kit following the outlined protocol. Briefly, RPA master mix was prepared (to a total volume of 47.5 μL) using 25 μL of 2× reaction buffer, 2.25 μL dNTPs, 5 μL 10× basic E-mix, and 5.45 μL of water. 2.4 μL of each primer at a concentration of 10 μM were added to 0.2 mL PCR tubes. 2.5 μL of 20× core reaction mix, 1 μL of reverse transcriptase, and 1 μL of RNA template were added to the PCR tube, along with 41.7 μL of the master mix. To begin the reaction, 2.5 μL of 280 mM MgOAc was added to the tube. The samples were incubated at 42 °C for 45 minutes.Nanopore fabrication
Nanopores were fabricated using controlled dielectric breakdown (CBD) in 12 nm thick SiN membranes purchased from Norcada Inc. (NXDB-50B405A122) using the Spark-E2 nanopore fabrication device (Northern Nanopore Instruments). To reduce capacitive noise, chips were painted in polydimethylsiloxane (PDMS) prior to fabrication. Pores were fabricated using a linear voltage ramp in 1 M KCl pH 8 buffered with 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), in custom 3D-printed millifluidic flow cells made of Tough 1500 resin. Pores were enlarged by applying AC square pulses of ±3 V in 3.6 M LiCl buffered with 10 mM HEPES pH 8 until a low noise pore, with a diameter of ∼5 nm ± 0.5 nm was reached. In-depth protocols can be found in Waugh et al.47RPA product purification
For the indicated experiments, RPA products were purified using a QIAquick® PCR purification kit (Qiagen) prior to nanopore sensing to remove primers, nucleotides, and enzymes from the sample and prevent clogging of the nanopore. This kit purifies DNA ranging from 100 bp to 10 kbp. Samples were purified using the protocols outlined by the QIAGEN QIAquick spin handbook, with 10 μL of the original RPA sample diluted 5× in buffer prior to centrifuge processing and DNA elution.Nanopore sensing and data analysis
Prior to nanopore sensing, 2.5 μL aliquots of the RPA products were diluted 100× in 3.6 M LiCl buffered with 10 mM HEPES pH 8, and 50 μL of the 100×-diluted sensing mixtures were used to perform the nanopore experiments. For samples cleaned with a PCR-purification kit, 5 μL of the purified sample and 1 μL of 0.38 μM 2 kbp DNA (Thermo Fisher Scientific) were diluted with 74 μL of 3.6 M LiCl 10 mM HEPES pH 8. Nanopore experiments were conducted in 3.6 M LiCl pH 8, 10 mM HEPES using a −200 mV voltage bias. Data was acquired using the VC100 amplifier (Chimera Instruments) sampled at 4.17 MHz and low-pass filtered at 400 kHz prior to analysis (or at 200 kHz in some of the ESI† sections). Events were fitted using custom CUSUM+ algorithm.48Data availability
Event trace data used in the main text figures for this article is accessible via Federated Research Data Repository: https://doi.org/10.20383/103.01023. Any other data including raw current trace data and data from ESI† figures are available from the corresponding author upon request.Author contributions
Breeana Elliott: investigation, formal analysis, writing – original draft. Martin Charron: formal analysis, writing – review and editing. John Pezacki: resources, conceptualization. Erin McConnell: investigation, conceptualization, writing – review and editing. Vincent Tabard-Cossa: supervision, conceptualization, writing – review and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
M. C. acknowledges support from the Ontario Graduate Scholarship (OGS). We thank David Prescott for isolation and characterization of mRNA samples used for preliminary work. All authors would like to acknowledge the support of the Natural Sciences and Engineering Research Council of Canada (NSERC), [funding reference number ALLRP 555057-2020].References
- M. Alafeef and D. Pan, Diagnostic Approaches for COVID-19: Lessons Learned and the Path Forward, ACS Nano, 2022, 16, 11545–11576 CrossRef CAS PubMed.
- Z. Li, X. Xu, D. Wang and X. Jiang, Recent advancements in nucleic acid detection with microfluidic chip for molecular diagnostics, TrAC, Trends Anal. Chem., 2023, 158, 116871 CrossRef CAS PubMed.
- X. Chen, S. Zhou, Y. Wang, L. Zheng, S. Guan and D. Wang, et al., Nanopore single-molecule analysis of biomarkers: Providing possible clues to disease diagnosis, TrAC, Trends Anal. Chem., 2023, 162, 117060 CrossRef CAS PubMed.
- S. King, K. Briggs, R. Slinger and V. Tabard-Cossa, Screening for Group A Streptococcal Disease via Solid-State Nanopore Detection of PCR Amplicons, ACS Sens., 2022, 7(1), 207–214 CrossRef CAS PubMed.
- S. Chandra and T. Hu, From Prevention to Therapy: A Roadmap of Nanotechnologies to Stay Ahead of Future Pandemics, ACS Nano, 2022, 16, 9985–9993 CrossRef CAS PubMed.
- C. D. Flynn, D. Chang, A. Mahmud, H. Yousefi, J. Das and K. T. Riordan, et al., Biomolecular sensors for advanced physiological monitoring, Nat. Rev. Bioeng., 2023, 1(8), 560–575 CrossRef PubMed.
- L. Xue, H. Yamazaki, R. Ren, M. Wanunu, A. P. Ivanov and J. B. Edel, Solid-state nanopore sensors, Nat. Rev. Mater., 2020, 5(12), 931–951 CrossRef CAS.
- J. A. Alfaro, P. Bohländer, M. Dai, M. Filius, C. J. Howard and X. F. van Kooten, et al., The emerging landscape of single-molecule protein sequencing technologies, Nat. Methods, 2021, 18(6), 604–617 CrossRef CAS PubMed.
- Y. L. Ying, Z. L. Hu, S. Zhang, Y. Qing, A. Fragasso and G. Maglia, et al., Nanopore-based technologies beyond DNA sequencing, Nat. Nanotechnol., 2022, 17(11), 1136–1146 CrossRef CAS PubMed.
- D. Deamer, M. Akeson and D. Branton, Three decades of nanopore sequencing, Nat. Biotechnol., 2016, 34(5), 518–524 CrossRef CAS PubMed.
- S. Lindsay, The promises and challenges of solid-state sequencing, Nat. Nanotechnol., 2016, 11(2), 109–111 CrossRef CAS PubMed.
- R. Ren, S. Cai, X. Fang, X. Wang, Z. Zhang and M. Damiani, et al., Multiplexed detection of viral antigen and RNA using nanopore sensing and encoded molecular probes, Nat. Commun., 2023, 14, 7362 CrossRef CAS PubMed.
- X. F. van Kooten, Y. Rozevsky, Y. Marom, E. Ben Sadeh and A. Meller, Purely electrical SARS-CoV-2 sensing based on single-molecule counting, Nanoscale, 2022, 14(13), 4977–4986 RSC.
- U. Stein, A. Meller, Y. Rozevsky, T. Gilboa, X. F. Van Kooten and D. Kobelt, et al., Quantification of mRNA expression using single-molecule nanopore sensing, ACS Nano, 2020, 14(10), 13964–13974 CrossRef PubMed.
- F. Bošković and U. F. Keyser, Nanopore microscope identifies RNA isoforms with structural colours, Nat. Chem., 2022, 14(11), 1258–1264 CrossRef PubMed.
- J. Zhu, R. Tivony, F. Bošković, J. Pereira-Dias, S. E. Sandler and S. Baker, et al., Multiplexed Nanopore-Based Nucleic Acid Sensing and Bacterial Identification Using DNA Dumbbell Nanoswitches, J. Am. Chem. Soc., 2023, 145(22), 12115–12123 CrossRef CAS PubMed.
- K. Sethi, G. P. Dailey, O. K. Zahid, E. W. Taylor, J. A. Ruzicka and A. R. Hall, Direct Detection of Conserved Viral Sequences and Other Nucleic Acid Motifs with Solid-State Nanopores, ACS Nano, 2021, 15(5), 8474–8483 CrossRef CAS PubMed.
- O. K. Zahid, F. Rivas, F. Wang, K. Sethi, K. Reiss and S. Bearden, et al., Solid-state nanopore analysis of human genomic DNA shows unaltered global 5-hydroxymethylcytosine content associated with early-stage breast cancer, Nanomed.: Nanotechnol., Biol. Med., 2021, 35, 102407 CrossRef CAS PubMed.
- E. Beamish, H. Kwok, V. Tabard-Cossa and M. Godin, Precise control of the size and noise of solid-state nanopores using high electric fields, Nanotechnology, 2012, 23(40), 405301 CrossRef PubMed.
- A. J. Storm, J. H. Chen, X. S. Ling, H. W. Zandbergen and C. Dekker, Fabrication of solid-state nanopores with single-nanometre precision, Nat. Mater., 2003, 2(8), 537–540 CrossRef CAS PubMed.
- T. J. Morin, T. Shropshire, X. Liu, K. Briggs, C. Huynh, V. Tabard-Cossa and M. Wanunu, et al., Nanopore-Based Target Sequence Detection, PLoS One, 2016, 11(5), e0154426 CrossRef PubMed.
- E. Beamish, V. Tabard-Cossa and M. Godin, Digital counting of nucleic acid targets using solid-state nanopores, Nanoscale, 2020, 12(34), 17833–17840 RSC.
- E. Beamish, V. Tabard-Cossa and M. Godin, Programmable DNA Nanoswitch Sensing with Solid-State Nanopores, ACS Sens., 2019, 4(9), 2458–2464 CrossRef CAS PubMed.
- A. H. Squires, E. Atas and A. Meller, Genomic pathogen typing using solid-state nanopores, PLoS One, 2015, 10(11), 1–16 CrossRef PubMed.
- S. Cai, T. Pataillot-Meakin, A. Shibakawa, R. Ren, C. L. Bevan and S. Ladame, et al., Single-molecule amplification-free multiplexed detection of circulating microRNA cancer biomarkers from serum, Nat. Commun., 2021, 12(1), 1–12 CrossRef PubMed.
- N. Burck, T. Gilboa, A. Gadi, M. Patkin Nehrer, R. J. Schneider and A. Meller, Nanopore Identification of Single Nucleotide Mutations in Circulating Tumor DNA by Multiplexed Ligation, Clin. Chem., 2021, 67(5), 753–762 CrossRef PubMed.
- Z. Tang, G. Choi, R. Nouri and W. Guan, Loop-Mediated Isothermal Amplification-Coupled Glass Nanopore Counting Toward Sensitive and Specific Nucleic Acid Testing, Nano Lett., 2019, 19(11), 7927–7934 CrossRef CAS PubMed.
- M. Charron, K. Briggs, S. King, M. Waugh and V. Tabard-Cossa, Precise DNA Concentration Measurements with Nanopores by Controlled Counting, Anal. Chem., 2019, 91(19), 12228–12237 CrossRef CAS PubMed.
- Y. Zhao, F. Chen, Q. Li, L. Wang and C. Fan, Isothermal Amplification of Nucleic Acids, Chem. Rev., 2015, 115(22), 12491–12545 CrossRef CAS PubMed.
- J. Glökler, T. S. Lim, J. Ida and M. Frohme, Isothermal amplifications–a comprehensive review on current methods, Crit. Rev. Biochem. Mol. Biol., 2021, 56(6), 543–586 CrossRef PubMed.
- P. Craw and W. Balachandran, Isothermal nucleic acid amplification technologies for point-of-care diagnostics: A critical review, Lab Chip, 2012, 12(14), 2469–2486 RSC.
- I. M. Lobato and C. K. O'Sullivan, Recombinase polymerase amplification: Basics, applications and recent advances, TrAC, Trends Anal. Chem., 2018, 98, 19–35 CrossRef CAS PubMed.
- J. D. Sherwood, M. Mao and S. Ghosal, Electroosmosis in a finite cylindrical pore: Simple models of end effects, Langmuir, 2014, 30(31), 9261–9272 CrossRef CAS PubMed.
- R. K. Daher, G. Stewart, M. Boissinot and M. G. Bergeron, Recombinase polymerase amplification for diagnostic applications, Clin. Chem., 2016, 62(7), 947–958 CrossRef CAS PubMed.
- M. Tan, C. Liao, L. Liang, X. Yi, Z. Zhou and G. Wei, Recent advances in recombinase polymerase amplification: Principle, advantages, disadvantages and applications, Front. Cell. Infect. Microbiol., 2022, 12, 1–13 Search PubMed.
- C. Plesa, S. W. Kowalczyk, R. Zinsmeester, A. Y. Grosberg, Y. Rabin and C. Dekker, Fast translocation of proteins through solid state nanopores, Nano Lett., 2013, 13(2), 658–663 CrossRef CAS PubMed.
- J. Qian, S. A. Boswell, C. Chidley, Z. X. Lu, M. E. Pettit and B. L. Gaudio, et al., An enhanced isothermal amplification assay for viral detection, Nat. Commun., 2020, 11(1), 1–10 CrossRef PubMed.
- S. Carson, J. Wilson, A. Aksimentiev and M. Wanunu, Smooth DNA Transport through a Narrowed Pore Geometry, Biophys. J., 2014, 107(10), 2381–2393 CrossRef CAS PubMed.
- M. Charron, L. Philipp, L. He and V. Tabard-Cossa, Elucidating the dynamics of polymer transport through nanopores using asymmetric salt concentrations, Nano Res., 2022, 15, 9943–9953 CrossRef CAS.
- D. A. Armbruster and T. Pry, Limit of blank, limit of detection and limit of quantitation, Clin. Biochem. Rev., 2008, 29(Suppl 1), S49–S52 Search PubMed.
- Y. Sun, L. Yu, C. Liu, S. Ye, W. Chen and D. Li, et al., One-tube SARS-CoV-2 detection platform based on RT-RPA and CRISPR/Cas12a, J. Transl. Med., 2021, 19(1), 1–10 CrossRef PubMed.
- Y. Song, P. Huang, M. Yu, Y. Li, H. Jin and J. Qiu, et al., Rapid and Visual Detection of SARS-CoV-2 RNA Based on Reverse Transcription-Recombinase Polymerase Amplification with Closed Vertical Flow Visualization Strip Assay, Microbiol. Spectrum, 2023, 11(1), 1–10 Search PubMed.
- J. L. Malaga, M. J. Pajuelo, M. Okamoto, E. K. Tsinda, K. Otani and P. Tsukayama, et al., Rapid Detection of SARS-CoV-2 RNA Using Reverse Transcription Recombinase Polymerase Amplification (RT-RPA) with Lateral Flow for N-Protein Gene and Variant-Specific Deletion–Insertion Mutation in S-Protein Gene, Viruses, 2023, 15, 1254 CrossRef CAS PubMed.
- L. He, D. R. Tessier, K. Briggs, M. Tsangaris, M. Charron and E. M. McConnell, et al., Digital immunoassay for biomarker concentration quantification using solid-state nanopores, Nat. Commun., 2021, 12, 5348 CrossRef CAS PubMed.
- L. Cohen and D. R. Walt, Single-Molecule Arrays for Protein and Nucleic Acid Analysis, Annu. Rev. Anal. Chem., 2017, 10(1), 1–19 CrossRef PubMed.
- D. M. Rissin, C. W. Kan, T. G. Campbell, S. C. Howes, D. R. Fournier and L. Song, et al., Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations, Nat. Biotechnol., 2010, 28(6), 595–599 CrossRef CAS PubMed.
- M. Waugh, K. Briggs, D. Gunn, M. Gibeault, S. King and Q. Ingram, et al. Solid-state nanopore fabrication by automated controlled breakdown, Nat. Protoc., 2020, 15(1), 122–143 CrossRef CAS PubMed.
- J. H. Forstater, K. Briggs, J. W. F. Robertson, J. Ettedgui, O. Marie-Rose and C. Vaz, et al., MOSAIC: A modular single-molecule analysis interface for decoding multistate nanopore data, Anal. Chem., 2016, 88(23), 11900–11907 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00159a |
| This journal is © The Royal Society of Chemistry 2024 |