
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn automated screening platform for improving the responsiveness of genetically encoded Ca2+ biosensors in mammalian cells†
Yufeng
Zhao
 *abc,
Yi
Shen
a,
Teodor
Veres
bd and
Robert E.
Campbell
*aef
*abc,
Yi
Shen
a,
Teodor
Veres
bd and
Robert E.
Campbell
*aef
aDepartment of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada
bCentre for Research and Applications in Fluidic Technologies, National Research Council of Canada, Toronto, Ontario M5S 3G8, Canada
cLeslie Dan Faculty of Pharmacy, University of Toronto, Toronto, Ontario M5S 3M2, Canada
dMedical Devices Research Center, Life Sciences Division, National Research Council of Canada, Boucherville, QC J4B 6Y4, Canada
eDepartment of Chemistry, Graduate School of Science, The University of Tokyo, Tokyo 113-0033, Japan
fCERVO Brain Research Center and Department of Biochemistry, Microbiology, and Bioinformatics, Université Laval, Québec, Québec G1V 0A6, Canada
First published on 18th July 2024
Abstract
Genetically-encoded, fluorescent protein (FP)-based biosensors are powerful tools for imaging dynamic cellular activities. Directed evolution is a highly effective method for developing enhanced versions of FP-based biosensors, but the screening process is laborious and time-consuming. Mammalian cell-based screening with electrical stimulation methods has been successful in accurately selecting variants of biosensors for imaging neuronal activities. We introduce an automated mammalian cell screening platform utilizing a fluorescence microscope and a liquid dispenser to enable the screening of biosensor responsiveness to chemical stimulation. We demonstrated the effectiveness of this platform in improving the response of a red fluorescent biosensor for Ca2+, K-GECO, for detection of histamine-induced changes in Ca2+ concentration. This method should be applicable to any FP-based biosensor that responds to pharmacological treatment or other exogenous chemical stimulation, simplifying efforts to develop biosensors tailored for specific applications in diverse biological contexts.
Introduction
Genetically-encoded, fluorescent protein (FP)-based biosensors are popular and powerful tools for monitoring or imaging of dynamic cellular activities.1 Compared to other types of techniques for analyzing cellular activities (e.g., synthetic chemosensors, biochemical assays, and mass spectrometry), they offer the distinctive advantages of low toxicity, subcellular targeting, and high temporal–spatial resolution. As the archetypical example, FP-based Ca2+ biosensors have revolutionized neuroscience research by enabling the in vivo recording of activities of dozens to hundreds of neurons in live mice and other model organisms.2–5 To develop FP-based biosensors with the requisite high-performance for in vivo imaging applications, directed protein evolution is widely used.6–8 While highly effective at producing enhanced versions of FP-based biosensors, directed evolution is an iterative method that necessitates many rounds of mutagenesis and screening, making the process laborious and time-consuming.The single most important aspect of a directed evolution strategy is a screening process that can effectively and reliably identify biosensor variants harboring beneficial mutations. A traditional approach, involving library expression in E. coli and subsequent screening in bacterial lysates, has been successfully used to develop various biosensors,4,7–9 but the throughput of this method is typically limited to at most a few 100 to 1000 variants per round. To achieve higher-throughput screening, researchers have previously used commercial or microfluidic fluorescence activated cell sorters (FACS).10–12 Nevertheless, the efficiency of those high throughput screening methods has been limited by issues of single-cell variability. Furthermore, since most biosensors find application in mammalian cell imaging, their performance in bacterial cells, or lysates, or buffers, may not accurately predict their performance in mammalian cells.
To address these challenges, methodologies have been successfully devised for screening biosensors directly within mammalian cells using automated fluorescence microscopy. For example, Wardill et al. reported a platform to screen the sensitivity of Ca2+ biosensors expressed in cultured rat neurons, yielding a series of ultrasensitive Ca2+ biosensors for recording neuronal activities.13 Villette et al. developed a screening platform to assess the sensitivity of voltage biosensors expressed in human embryonic kidney (HEK) cells.14 While these mammalian cell-based screening methods did not achieve higher throughput, their efficiency is significantly heightened due to their capacity to accurately select for variants that retain their performance in their intended end-use settings. Those platforms have a primary focus on enhancing biosensors for the detection of neuronal activities, employing electrical stimulation as a means to trigger the sensing action, but it is not readily apparent how this convenient triggering mechanism could be easily extended to evaluating other types of biosensors.
Here, we propose that chemical stimulations to induce analyte changes provide a versatile modality to screen libraries of biosensors expressed in mammalian cells for the purpose of directed evolution of higher performance variants. With this objective and drawing inspiration from prior platforms,15 we present an automated screening platform utilizing a fluorescence microscope and a liquid dispenser to enable the screening of biosensor responsiveness upon chemical stimulation. Specifically, we designed a screening assay by adapting the well-established method of histamine-induced intracellular Ca2+ change to our platform for evaluating the responsiveness of Ca2+ biosensors. We demonstrated the effectiveness of this platform for improving the response of a biosensor for the detection of histamine-induced Ca2+ activities. However, this method should be applicable to any FP-based biosensor that responds to a pharmacological treatment or other exogenous chemical stimulation.
Experimental
Hardware of the platform
Cell imaging was conducted using a Zeiss Axiovert 200 fluorescence microscope equipped with a 20× objective and a motorized X–Y–Z positioning control system. Fluorescence signals from the green and red Ca2+ biosensors were collected using a GFP filter set (470/40 nm excitation and 525/50 nm emission) and an RFP filter set (535/50 nm excitation and 609/57 nm emission), respectively. Digital signal input/output was facilitated by a NI USB-6501 (National Instruments) data acquisition board. A Microlab 600 dispenser (Hamilton) equipped with PTFE tubings was employed to withdraw histamine solution from the reservoir and inject it into the wells to stimulate intracellular Ca2+ changes in HeLa cells.Software for screening and data analysis
The fluorescence microscope was operated through MetaMorph (Molecular Devices), while the Microlab 600 dispenser was controlled by a LabVIEW (National Instruments) program adapted from the manufacturer's original design to enable initiation upon receiving a digital signal. Image processing was accomplished using a customized macro program in ImageJ to measure the fluorescence trace of individual cells over time in the images. Subsequently, the average fluorescence response of each biosensor variant was calculated and statistical analysis was conducted using MATLAB (MathWorks).Library construction
Libraries of K-GECO1.3 variants in the pcDNA or pBAD vector at single positions were constructed through site-directed mutagenesis using the QuikChange II site-directed mutagenesis kit (Agilent) and a primer with the degenerate codon NNK. For random mutagenesis, libraries were generated via error-prone PCR using the GeneMorph II – random mutagenesis kit (Agilent). The resulting amplified products were transformed into E. coli DH10B electrocompetent cells (Thermo Scientific™) through electroporation. These transformed cells were then plated on LB agar plates supplemented with 100 μg mL−1 ampicillin and 0.2% arabinose for an overnight culture in a 37 °C incubator. On the following day, individual colonies were selected for either plasmid purification or protein characterization.HeLa cell culture and transfection
HeLa cells (ATCC CCL-2) were cultured and maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 1% penicillin–streptomycin and 10% fetal bovine serum (FBS). The cells were seeded in black/glass-bottom 96-well plates at approximately 90% confluency and immediately transfected with K-GECO plasmids using TurboFect Transfection Reagent (Thermo Scientific™) following the manufacturer's protocol. Approximately 2 hours post-transfection, the transfection medium was replaced with fresh cell culture media. The transfected cells were then cultured in a 5% CO2 incubator at 37 °C and subjected to imaging 24 to 48 hours after transfection.Screening process
Prior to screening, the cell culture medium was substituted with HEPES-buffered Hank's Balanced Salt Solution (HBSS) solution for imaging, with each well containing 100 μL of the imaging buffer. The histamine stock solution was initially diluted into 50 mL of HEPES-buffered HBSS, achieving a concentration of 20 μM. The inlet tubing was immersed in the histamine solution, while the outlet tubing was positioned above the microwell plate. To ensure prompt delivery of histamine to the first well, the dispenser tubing was preloaded with the solution. Subsequently, 100 μL of histamine-containing buffer was injected into each well for 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixing with the imaging buffer, resulting in a final concentration of 10 μM histamine. The objective was initially centered below well A1, and cells were manually focused in the bright field. The X, Y, and Z positions after focusing were recorded as the initial reference point for configuring the positions of the remaining wells. The 96-well plate was systematically scanned from the top left (A1) to the bottom right (H12) using the multi-dimensional acquisition (MDA) mode in MetaMorph.
:1 mixing with the imaging buffer, resulting in a final concentration of 10 μM histamine. The objective was initially centered below well A1, and cells were manually focused in the bright field. The X, Y, and Z positions after focusing were recorded as the initial reference point for configuring the positions of the remaining wells. The 96-well plate was systematically scanned from the top left (A1) to the bottom right (H12) using the multi-dimensional acquisition (MDA) mode in MetaMorph.
Purification of K-GECOs
The gene of K-GECO variants was cloned into the pBAD His-tag backbone for expression in E. coli cells and subsequent purification. Initially, the assembled plasmid was transformed into DH10B electrocompetent cells, which were then plated on LB agar supplemented with ampicillin and arabinose for an overnight culture at 37 °C. Colonies were selected for inoculation in liquid LB medium and incubated in a shaker incubator at 37 °C. Afterward, cells were collected for plasmid miniprep, and the sequence was confirmed through Sanger sequencing.For protein purification, a single E. coli colony was chosen for inoculation in 100 mL LB medium and shaken at 37 °C for 18 hours. The following day, cells were harvested by centrifugation, resuspended in Tris-buffered saline (TBS), and lysed using sonication. The lysates were then centrifuged at 4 °C for 30 minutes, and the supernatant was transferred to a 50 mL centrifuge tube. Subsequently, Ni-NTA beads slurry was added to the supernatant, and the tube was shaken on ice for 2 hours. The mixture was transferred to a Poly-Prep® chromatography column (Bio-Rad) and drained under vacuum. The Ni-NTA column underwent washing with TBS buffer containing 10 mM imidazole, and the proteins were eluted using TBS buffer with 200 mM imidazole. The purified proteins were transferred to an Amicon ultrafiltration tube (MilliporeSigma) for buffer exchange to remove excess imidazole. The final purified proteins were stored at 4 °C.
In vitro characterization of K-GECOs
In vitro characterization was conducted using a Biotek Synergy Neo2 plate reader. The protocol is similar to ones that have been previously reported.4 Proteins were diluted into buffers and transferred to clear-bottomed 96 or 384 well plates for measurement. Excitation and emission spectra were recorded for K-GECO variants in 30 mM MOPS, 100 mM KCl, with either 10 mM EGTA or 10 mM EGTA-Ca2+. The spectra for each variant were normalized to those in the Ca2+-bound state. To determine the extinction coefficient, proteins were initially denatured in 1 M NaOH for absorbance measurements, enabling the calculation of concentration based on the assumed extinction coefficient of the denatured chromophore at 44000 M−1 cm−1 at 446 nm. Subsequently, this concentration was utilized to calculate the extinction coefficient of the chromophore in its native state, obtained by dividing the peak absorbance by the concentration.
To determine the quantum yield, K-GECO variants were initially diluted across a range of factors for the measurement of their fluorescence and absorbance. The quantum yield of K-GECO1 in the Ca2+-bound state was employed as the reference standard. The total integrated fluorescence versus the absorbance peak for each variant was plotted, and the slope was calculated through linear fitting. The quantum yield of each variant was then determined by comparing its slope with that of K-GECO1 in the Ca2+-bound state.
The Kd of the K-GECO variants was determined by measuring the fluorescence of diluted proteins in a series of calibration buffers. These buffers were prepared by mixing Ca2+-free buffer (30 mM MOPS 100 mM KCl with 10 mM EGTA) and Ca2+ buffer (30 mM MOPS 100 mM KCl with 10 mM EGTA-Ca2+) in various ratios. The concentration of free Ca2+ in the buffer was calculated using an online calculation tool (https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/CaEGTA-TS.htm). The fluorescence plotted against the concentration of free Ca2+ was then fitted to the Hill equation to determine the Kd.
Model to estimate library coverage
A previously described statistical model16 was used to describe the relation of sampling number vs. library coverage:| T = −Vln(1 − Pi) |
Results
Design of the screening platform
The screening platform comprises a fluorescence microscope utilized for image acquisition and a liquid dispenser for solution injection, both under the control of a computer (Fig. 1b and S1†). To ensure synchronization between liquid injection and image acquisition, a data acquisition board is used to manage the input and output of digital signals. Optical mounting posts are employed to position the injection tubing, with its tip placed approximately 1 cm above the multi-well plate. Mammalian cells expressing a specific biosensor variant are cultured in each well (Fig. 1a). The multi-well plate is scanned well by well, using customized settings in the multi-dimensional acquisition mode of MetaMorph. A singular field of view is chosen for image recording in each well, necessitating a high cell confluency to ensure an ample cell count for reliable measurements.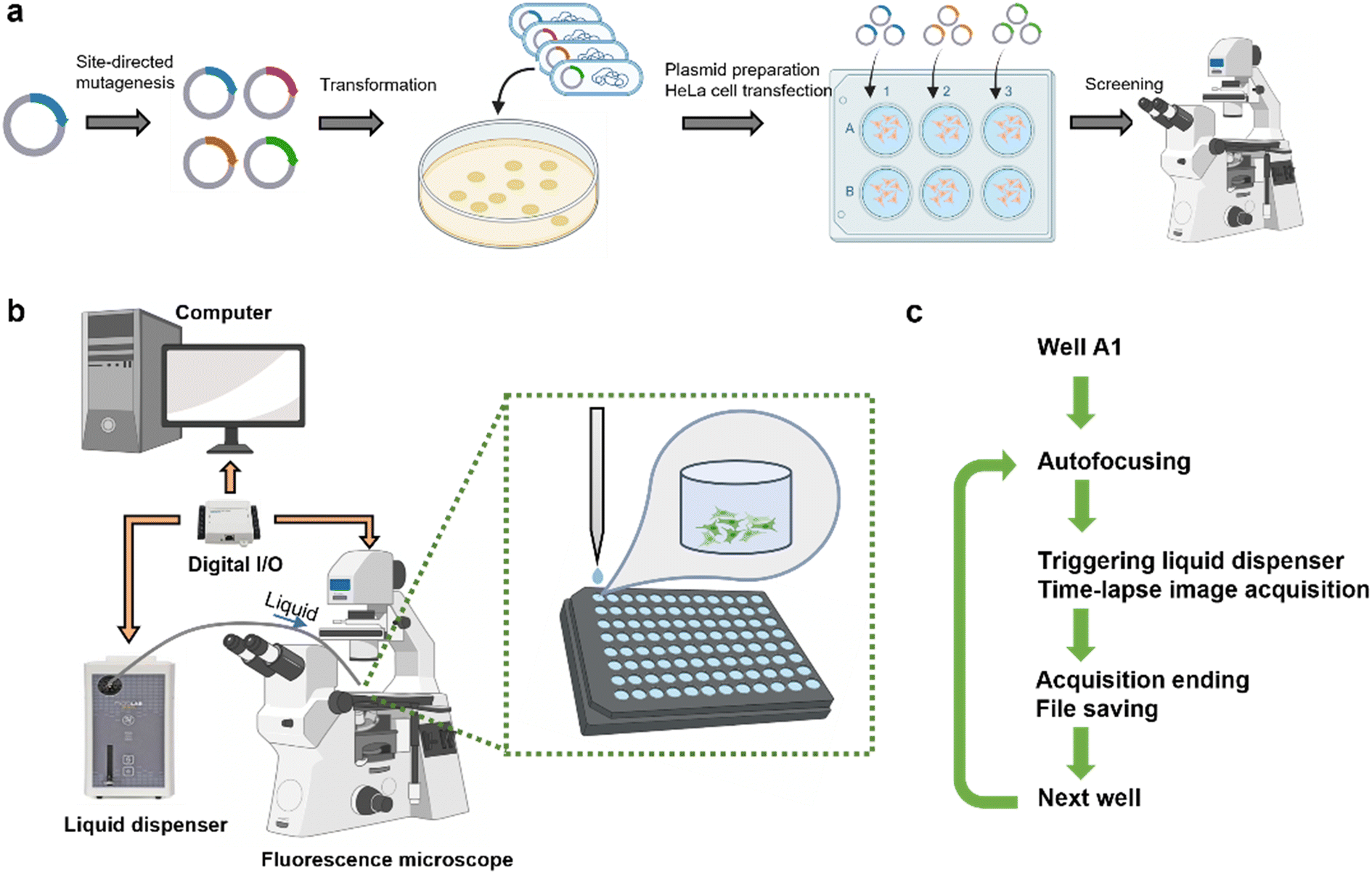 | ||
| Fig. 1 Schematic workflow and hardware of the automated screening platform. (a) Overall workflow from generation of the plasmid of biosensor variants, cell culture, variant expression, and screening. (b) Hardware setup includes a fluorescence microscope, a liquid dispenser, a computer and a data acquisition board. (c) Automated iterative screening steps of a multi-well plate on a microscope. | ||
The automated screening workflow is illustrated in Fig. 1c. For each well, a z-scale autofocusing is first applied, followed by the initiation of time-lapse recording and a simultaneous digital signal output to trigger the liquid dispenser. Upon the completion of the recording for each well, the resulting image stack is saved before proceeding to the subsequent well. To effectively screen for the response of a biosensor to the change of its analyte, an appropriate recording duration is set to capture the signal change, facilitating a quantitative evaluation of the responsiveness of a variant. Each variant was expressed and measured in triplicate wells, allowing for the evaluation of 31 variants versus one benchmark variant in one 96-well plate.
Image analysis is conducted using a customized ImageJ macro program (Fig. S2†). From the image stack, an image with the fluorescence of the cell population nearing the peak is chosen to generate a binary mask for automated cell segregation. Subsequently, the intensity of each segregated cell is measured throughout the entire time-lapse image stack. The results are exported and processed in Matlab calculation of the responsiveness of the variant in cells. The fluorescence response, (Fmax − Fmin)/Fmin, was computed for each individual cell and subsequently averaged across the entire population within each well. The response of each variant was then derived by averaging the results from the triplicate wells. Apart from manual examination of the data, cells exhibiting a response of (Fmax − Fmin)/Fmin > 20 were automatically excluded from the analysis, as this was likely indicative of an artificial situation, such as the presence of a floating cell entering the field of view.
Screening assay
To demonstrate the efficacy of the screening platform, we chose to screen Ca2+ biosensors to improve their sensitivity in response to histamine-induced intracellular Ca2+ change in HeLa cells. Broadly recognized as an activator of Ca2+ signals, histamine stimulates the histamine H1 receptor, eventually leading to the release of Ca2+ from endoplasmic reticulum pools into the cytosol through the mediation of inositol 1,4,5-trisphosphate (IP3).17 The histamine assay serves as a well-established method for the preliminary evaluation of genetically encoded Ca2+ biosensor performance in mammalian cell culture,6,18,19 providing a foundational step prior to advancing into more specialized biological applications. Adapting this assay to our platform, we determined that a recording duration of 30 seconds per well is necessary to capture the initial predominant Ca2+ signal wave (Fig. 2). In this experimental setup, a 96-well plate containing cells expressing the biosensor can be systematically scanned in approximately 1.5 hours. | ||
| Fig. 2 Characterization of the screening platform with Ca2+ biosensors. (a and c) Fluorescence trace over time of individual cells expressing GCaMP6s (a) and K-GECO1 (c). (b) Fluorescence microscopic images of cells expressing GCaMP6s at different time points marked by the grey dashed lines in (a). (d) Average response of the biosensors in response to histamine induced Ca2+ release. Each variant was expressed and measured in three independent wells for replicates. Error bars represent standard deviation. Two-tailed t test. n.s. (not significant) P > 0.05. *P < 0.05. **P < 0.005. | ||
Characterization of the platform
We characterized our screening platform using several widely used Ca2+ biosensors including a green FP-based biosensor (GCaMP6s2), and three red FP-based biosensors (R-GECO1,6 jR-GECO1 (ref. 20) and K-GECO1 (ref. 4)). The representative traces of individual cells expressing the same biosensor in response to histamine stimulated Ca2+ release was illustrated in Fig. 2a–c. Since all these biosensors manifest a direct response to changes in Ca2+ levels, specifically an increase in fluorescence in response to elevated Ca2+ concentration, they were initially dimly fluorescent when cells were in a resting state with a low cytoplasmic Ca2+ concentration. The addition of histamine for stimulation induced the release of Ca2+ from intracellular Ca2+ stores into the cytosol, leading to an increase in fluorescence signals.Each biosensor was measured in three replicated wells. Among the four biosensors, GCaMP6s exhibited the highest fluorescence response (∼11 fold) to the stimulation (Fig. 2d), consistent with previous characterizations.21 While jR-GECO1a and K-GECO1 was reported to outperform R-GECO1 in response to action potentials in dissociated neurons, R-GECO1 exhibited larger responsiveness in our assay and platform. A possible reason is that, compared to jR-GECO1a and K-GECO1, the higher Kd of R-GECO1 makes it more sensitive for the histamine stimulated Ca2+ concentration change (from ∼100 nM to ∼5 μM (ref. 22 and 23)) in HeLa cells. The relative standard deviation varies from 5% (R-GECO1) to 24% (GCaMP6s and K-GECO1). Identification of a significantly improved variant with a 95% confidence interval requires an improvement of twice the standard deviation compared to the template (e.g. 48% higher (Fmax − Fmin)/Fmin for a K-GECO variant vs. K-GECO1 in this experiment). Our results demonstrated that the screening platform generated robust measurement of the responsiveness of the Ca2+ biosensors and can be utilized for screening variants of these Ca2+ biosensors.
Screening and evolution of K-GECO1
K-GECO1 has been reported to exhibit comparable sensitivity to jR-GECO1a in terms of fluorescence response to single actional potentials, and has the advantage of less photoactivation and aggregation in cells.4 We reasoned that using our screening platform could further optimize the biosensor for sensitive detection of intracellular Ca2+ activities. Prior to screening, we first introduced the mutation C47A because we considered that the thiol or thiolate form of C47 could be serving as a potential excited state proton donor or acceptor, respectively, during the photoactivation of K-GECO1 in the Ca2+-apo state by blue light (Fig. S3†).24,25 This C47A variant suffered from low brightness, which was recovered by two rounds of random mutagenesis with traditional colony screening for brightness on agar plates and lysate screening for responsiveness in a plate reader. The product bearing six new mutations with recovered brightness but approximately 50% diminished dynamic range in bacterial lysates, was designated K-GECO1.3 and used as the template for further evolution using our new automated screening platform. Note that it remains to be determined whether K-GECO1.3 and its variants exhibit blue-light photoactivation under live-cell imaging conditions.Inspired by the structure guided mutagenesis rationale that has been used to guide the development of the GCaMP series,2 we reasoned that mutations at the interface of Ca2+ binding domain, CaM, and its interacting peptide, ckkap, can potentially lead to a more profound conformational change upon Ca2+ binding. The conformational change will be transduced to the linker residues in vicinity of the chromophore for modulation of its extinction coefficient or quantum yield, eventually improving the fluorescence response. Therefore, 19 positions at the interface of CaM and ckkap were selected for site-directed mutagenesis using the degenerate codon NNK at single positions (Fig. 3a). Libraries of variants in pcDNA vectors were used to transform bacterial cells from which single colonies were picked. For each of the 19 positions, approximately 15–30 colonies were randomly picked, the plasmid DNA was isolated, and the variants were expressed in HeLa cells for screening. In our screening, up to three top variants exhibiting at least 50% higher responsiveness and more than twice the standard deviation of K-GECO1.3 from each plate were selected for DNA recovery. These variants were then validated on subsequent days using the same assay with triplicate wells. The top three variants identified from the screening were I14S, M374V, and E379Q, exhibiting over 70% higher response to histamine stimulated Ca2+ elevation (Fig. 3b), and 2–3 fold higher dynamic range but lower affinity in bacterial lysates compared to their progenitor K-GECO1.3 (Fig. S4†).
 | ||
| Fig. 3 Design and screening of K-GECO1.3 libraries. (a) Crystal structure of K-GECO1 with highlighted positions selected for mutagenesis. (b) Summary of the first round of screening as the responsiveness of K-GECO1.3 variants relative to K-GECO1.3. Relative responsiveness = 1 represents equal performance to K-GECO1.3. Average value of each position is marked by the black solid line. (c) Summary of the second round of screening as the responsiveness of K-GECO1.3 variants relative to its template (K-GECO1.3 M374V or E379Q). | ||
We next designed a library on the basis of the three mutants for the second round of screening. We re-screened one winner position on top of another (e.g. M374X on E379Q), or screened a new position (L304X, Q306X, M409X and M410X) on the winner E379Q. However, no variant with further improved responsiveness was identified (Fig. 3c).
Characterization of K-GECO variants
We next purified the three variants with the best performance, and K-GECO1, for in vitro characterization. The excitation and emission spectra of the variants in the Ca2+-apo and Ca2+-bound states are essentially identical to that of K-GECO1 (Fig. 4a–d). All three variants exhibit reduced Ca2+ affinity (higher Kd) than K-GECO1 (Fig. 4e). K-GECO1.3 I14S has the highest Kd (1130 nM), followed by K-GECO1.3 E379Q (811 nM) and K-GECO1.3 M374V (467 nM). K-GECO1.3 I14S and M374V have substantially improved molecular brightness at the Ca2+-bound state than K-GECO1, largely attributed to their higher extinction coefficient (Table 1). While the molecular brightness of K-GECO1.3 E379Q is slightly lower than K-GECO1, its dynamic range ((Fmax − Fmin)/Fmin) is the highest, followed by I14S, M374V and K-GECO1.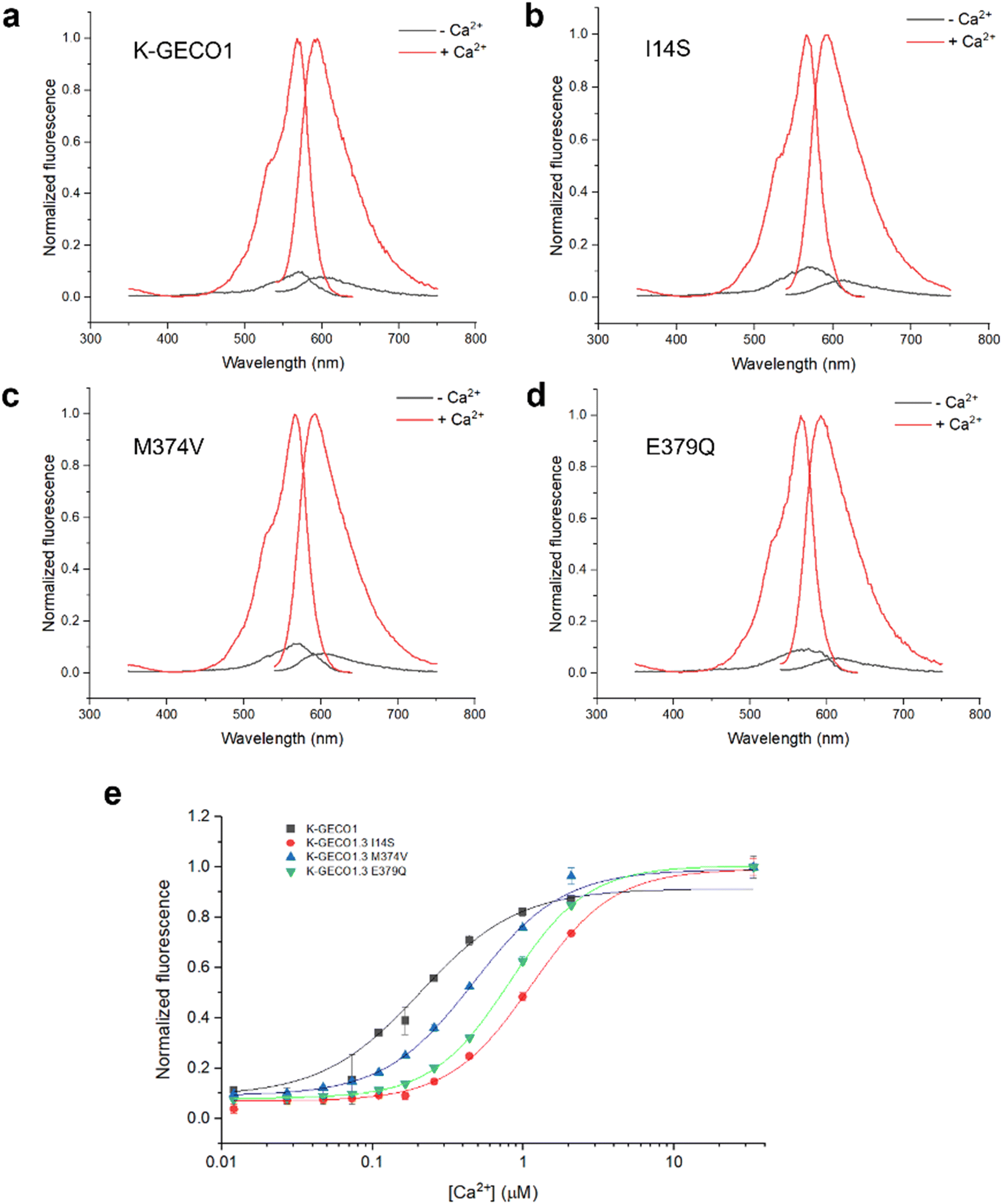 | ||
| Fig. 4 In vitro characterization of K-GECO variants. (a)–(d) Normalized excitation and emission spectra of K-GECO variants in the Ca2+-apo and Ca2+-bound states. (e) Ca2+ titration of K-GECO variants. | ||
| Variant | (Fmax − Fmin)/Fmin | K d/nM | Extinction coefficient ε (M−1 cm−1/1000) | Quantum yield ϕ | Molecular brightness ε × ϕ |
|---|---|---|---|---|---|
| K-GECO1 | 11.6 | 199 | 15 (− Ca2+) | 0.12 | 1.8 |
| 56 (+ Ca2+) | 0.45 | 25.2 | |||
| K-GECO1.3 I14S | 14.3 | 1130 | 26 (− Ca2+) | 0.09 | 2.3 |
| 70 (+ Ca2+) | 0.49 | 34.3 | |||
| K-GECO1.3 M374V | 12.3 | 467 | 25 (− Ca2+) | 0.12 | 3.0 |
| 83 (+ Ca2+) | 0.43 | 35.7 | |||
| K-GECO1.3 E379Q | 15.9 | 811 | 23 (− Ca2+) | 0.07 | 1.6 |
| 62 (+ Ca2+) | 0.39 | 24.2 |
Discussion
In this study, we devised an automated platform for screening the responsiveness of genetically encoded Ca2+ biosensors in the context of histamine-stimulated Ca2+ activities. While our demonstration focuses on screening Ca2+ biosensors, the approach can be readily generalized to enhance other biosensors either expressed intracellularly (e.g., PyronicSF26 for pyruvate and Citron1 (ref. 7) for citrate) or displayed on the membrane (e.g., iGluSnFR27 for glutamate and the GRAB family of GPCR-based biosensors28–31). To apply the screening platform to other types of FP-based biosensors, a major challenge is the selection of an appropriate stimulus to rapidly elevate the concentration of the intracellular or extracellular analyte to trigger the biosensor response while having minimal effect on cell viability. Additionally, a modified image analysis program needs to be developed for non-cytoplasmic expressed biosensors to effectively segregate and measure the signals.The throughput of our screening platform is 32 variants per 1.5 hours on a 96-well plate with triplicate wells for each variant, comparable to the 96-well neuron-based screening platform. With this throughput, up to 300 variants (10 plates) can be screened in one week when the entire workflow, from library construction to screening, is considered. While others have demonstrated a semi-automated screening platform for a genetically encoded voltage indicator (GEVI) using a 384-well plate,14 thereby increasing throughput, the GEVI was stimulated through a different modality involving an electroporation process. In our platform, employing a 384-well plate would present technical challenges in liquid injection and mixing due to the smaller well size. Although the traditional method screens biosensor variants in bacterial lysates at a higher throughput (>1000 variants per week), the larger dynamic range observed in the bacterial system may not translate well to the mammalian system, thereby compromising its screening efficiency. For mammalian cell-based screening, a major limiting factor is the manual processing involved in cloning, plasmid purification, and cell culture. To advance this platform towards greater automation and reduced dependence on manual labor, the incorporation of a liquid handling robot is imperative to automate as many steps as possible, especially the tedious plasmid purification and mammalian cell transfection in our workflow. The utilization of purified linear PCR products for transfection can further simplify the process by eliminating the need for cloning and plasmid preparation.14
The optimization of the degenerate codon utilized in the library holds the potential to enhance screening efficiency. Although we employed the conventional NNK codon for its ease of use, it exhibits considerable redundancy, often necessitating oversampling to achieve a high percentage of library coverage. In our specific context, sampling 15–30 colonies at a single position is estimated to yield a coverage of 38–60% based on the model proposed by Reetz et al.16 (Fig. S5†). Under a circumstance with certain resources, we opted to explore more positions rather than striving for full coverage at a single position, aiming for a broader coverage of variants. For instance, supplementing the sampling of 30 colonies at a specific position with an additional 30 colonies can elevate the coverage from 60% to 85%, yielding a low marginal effect. Conversely, sampling 30 more colonies at a different unexplored position results in an additional 60% in coverage, offering a more efficient strategy.
In recent years, codons for saturation mutagenesis have undergone significant optimization to minimize redundancy. For example, the NDT codon, which encodes only twelve different amino acids, has proven highly efficient. This codon efficiently covers the main amino acids with a variety of physicochemical properties and eliminates the three stop codons.16 The 22c-trick, a design involving the use of three primers with a total of 22 different codons to encompass all 20 amino acids, has successfully reduced bias and redundancy, thereby minimizing the need for oversampling.32 With the decreasing cost of DNA synthesis, a library with deterministic codons becomes an option, alleviating the challenge of oversampling, although more efforts are unavoidable in the cloning and DNA purification steps.
We have identified three new variants demonstrating enhanced responsiveness in cells compared to its progenitor K-GECO1.3. All these variants exhibit larger dynamic range and lower Ca2+ affinities in vitro. Indeed, an optimal Kd at the one-micromolar level maximizes the response to histamine-induced cytoplasmic Ca2+ changes, while a Kd around 100 nanomolar is conducive to the sensitive detection of neuronal Ca2+ activities. However, combining two beneficial mutations did not further improve its performance in cells, possibly due to instability of the protein or a shift of Kd towards the suboptimal millimolar range.
Directed evolution of FP-based biosensors is a protracted process that often requires the utilization of different screening technologies at various stages. Typically, screening in bacterial lysates is a common practice during the initial stages of biosensor development, especially for the first generation, which typically exhibits a dynamic range of 5-fold or higher signal change. This is unless the biosensor cannot be tested in bacterial lysates, such as GPCR-based biosensors.28–31,33 As the screening in bacterial lysates reaches its maximum efficacy, often constrained by throughput limitations and the capacity to predict the transferability of a variant, a more robust or specialized screening technique becomes imperative. For instance, the adaptation of a biosensor to different cell types or organelles may require adjustments to the Kd and pH resistance of the biosensor.34,35 Consequently, the ideal development strategy involves screening in the actual application scenario, as exemplified by the successful development of GCaMP6 and its derivatives through neuronal-based screening for neuroscience applications. The implementation of our designed automated screening platform, coupled with a chemical stimulation modality, will establish a foundation, simplifying efforts in the development of biosensors tailored for specific applications in diverse biological contexts.
Data availability
The data in this study are available from the authors upon request.Author contributions
Y. Z. designed the screening platform, screened libraries of K-GECO1.3, and characterized the final three variants. Y. S. performed the initial evolution of K-GECO and developed K-GECO1.3. T. V. and R. E. C. supervised the research. Y. Z. and R. E. C. wrote the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by grants from the Canadian Institutes of Health Research (CIHR, FS-154310 to REC) and the Natural Sciences and Engineering Research Council of Canada (NSERC, RGPIN 2018-04364 to REC). REC is currently supported by a Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research S (KAKENHI) (19H05633). The authors thank Prof. Keith Pardee at the University of Toronto for facility access.References
- E. C. Greenwald, S. Mehta and J. Zhang, Genetically Encoded Fluorescent Biosensors Illuminate the Spatiotemporal Regulation of Signaling Networks, Chem. Rev., 2018, 118, 11707–11794 CrossRef CAS PubMed.
- T. W. Chen, T. J. Wardill, Y. Sun, S. R. Pulver, S. L. Renninger, A. Baohan, E. R. Schreiter, R. A. Kerr, M. B. Orger, V. Jayaraman, L. L. Looger, K. Svoboda and D. S. Kim, Ultrasensitive Fluorescent Proteins for Imaging Neuronal Activity, Nature, 2013, 499, 295–300 CrossRef CAS PubMed.
- H. Dana, Y. Sun, B. Mohar, B. K. Hulse, A. M. Kerlin, J. P. Hasseman, G. Tsegaye, A. Tsang, A. Wong, R. Patel, J. J. Macklin, Y. Chen, A. Konnerth, V. Jayaraman, L. L. Looger, E. R. Schreiter, K. Svoboda and D. S. Kim, High-Performance Calcium Sensors for Imaging Activity in Neuronal Populations and Microcompartments, Nat. Methods, 2019, 16, 649–657 CrossRef CAS PubMed.
- Y. Shen, H. Dana, A. S. Abdelfattah, R. Patel, J. Shea, R. S. Molina, B. Rawal, V. Rancic, Y. F. Chang, L. Wu, Y. Chen, Y. Qian, M. D. Wiens, N. Hambleton, K. Ballanyi, T. E. Hughes, M. Drobizhev, D. S. Kim, M. Koyama, E. R. Schreiter and R. E. Campbell, A Genetically Encoded Ca2+ Indicator Based on Circularly Permutated Sea Anemone Red Fluorescent Protein EqFP578, BMC Biol., 2018, 16, 1–16 CrossRef PubMed.
- M. Inoue, A. Takeuchi, S. Manita, S. i. Horigane, M. Sakamoto, R. Kawakami, K. Yamaguchi, K. Otomo, H. Yokoyama, R. Kim, T. Yokoyama, S. Takemoto-Kimura, M. Abe, M. Okamura, Y. Kondo, S. Quirin, C. Ramakrishnan, T. Imamura, K. Sakimura, T. Nemoto, M. Kano, H. Fujii, K. Deisseroth, K. Kitamura and H. Bito, Rational Engineering of XCaMPs, a Multicolor GECI Suite for In Vivo Imaging of Complex Brain Circuit Dynamics, Cell, 2019, 177, 1346–1360 CrossRef CAS PubMed.
- Y. Zhao, S. Araki, J. Wu, T. Teramoto, Y. F. Chang, M. Nakano, A. S. Abdelfattah, M. Fujiwara, T. Ishihara, T. Nagai and R. E. Campbell, An Expanded Palette of Genetically Encoded Ca2+ Indicators, Science, 2011, 333, 1888–1891 CrossRef CAS PubMed.
- Y. Zhao, Y. Shen, Y. Wen and R. E. Campbell, High-Performance Intensiometric Direct- And Inverse-Response Genetically Encoded Biosensors for Citrate, ACS Cent. Sci., 2020, 6, 1441–1450 CrossRef CAS PubMed.
- Y. Nasu, C. Murphy-Royal, Y. Wen, J. N. Haidey, R. S. Molina, A. Aggarwal, S. Zhang, Y. Kamijo, M. E. Paquet, K. Podgorski, M. Drobizhev, J. S. Bains, M. J. Lemieux, G. R. Gordon and R. E. Campbell, A Genetically Encoded Fluorescent Biosensor for Extracellular L-Lactate, Nat. Commun., 2021, 12, 7058 CrossRef CAS PubMed.
- T. Wu, M. Kumar, J. Zhang, S. Zhao, M. Drobizhev, M. McCollum, C. T. Anderson, Y. Wang, A. Pokorny, X. Tian, Y. Zhang, T. Tzounopoulos and H. W. Ai, A Genetically Encoded Far-Red Fluorescent Indicator for Imaging Synaptically Released Zn2+, Sci. Adv., 2023, 9, eadd2058 CrossRef CAS PubMed.
- B. L. Fiedler, S. Van Buskirk, K. P. Carter, Y. Qin, M. C. Carpenter, A. E. Palmer and R. Jimenez, Droplet Microfluidic Flow Cytometer for Sorting on Transient Cellular Responses of Genetically-Encoded Sensors, Anal. Chem., 2017, 89, 711–719 CrossRef CAS PubMed.
- Y. Zhao, W. Zhang, Y. Zhao, R. E. Campbell and D. J. Harrison, A Single-Phase Flow Microfluidic Cell Sorter for Multiparameter Screening to Assist the Directed Evolution of Ca2+ Sensors, Lab Chip, 2019, 19, 3880–3887 RSC.
- L. M. Davis, J. L. Lubbeck, K. M. Dean, A. E. Palmer and R. Jimenez, Microfluidic Cell Sorter for Use in Developing Red Fluorescent Proteins with Improved Photostability, Lab Chip, 2013, 13, 2320–2327 RSC.
- T. J. Wardill, T. W. Chen, E. R. Schreiter, J. P. Hasseman, G. Tsegaye, B. F. Fosque, R. Behnam, B. C. Shields, M. Ramirez, B. E. Kimmel, R. A. Kerr, V. Jayaraman, L. L. Looger, K. Svoboda and D. S. Kim, A Neuron-Based Screening Platform for Optimizing Genetically-Encoded Calcium Indicators, PLoS One, 2013, 8, e77728 CrossRef CAS PubMed.
- V. Villette, M. Chavarha, I. K. Dimov, J. Bradley, L. Pradhan, B. Mathieu, S. W. Evans, S. Chamberland, D. Shi, R. Yang, B. B. Kim, A. Ayon, A. Jalil, F. St-Pierre, M. J. Schnitzer, G. Bi, K. Toth, J. Ding, S. Dieudonné and M. Z. Lin, Ultrafast Two-Photon Imaging of a High-Gain Voltage Indicator in Awake Behaving Mice, Cell, 2019, 179, 1590–1608 CrossRef CAS PubMed.
- E. Greenwald, C. Posner, A. Bharath, A. Lyons, C. Salmerón, K. Sriram, S. Z. Wiley, P. A. Insel and J. Zhang, GPCR Signaling Measurement and Drug Profiling with an Automated Live-Cell Microscopy System, ACS Sens., 2023, 8, 19–27 CrossRef CAS PubMed.
- M. T. Reetz, D. Kahakeaw and R. Lohmer, Addressing the Numbers Problem in Directed Evolution, ChemBioChem, 2008, 9, 1797–1804 CrossRef CAS PubMed.
- M. Montero, C. D. Lobatón, S. Gutierrez-Fernández, A. Moreno and J. Alvarez, Modulation of Histamine-Induced Ca2+ Release by Protein Kinase C: Effects on Cytosolic and Mitochondrial [Ca2+] PEAKS, J. Biol. Chem., 2003, 278, 49972–49979 CrossRef CAS PubMed.
- Y. Zhao, D. Bushey, Y. Zhao, E. R. Schreiter, D. J. Harrison, A. M. Wong and R. E. Campbell, Inverse-Response Ca2+ Indicators for Optogenetic Visualization of Neuronal Inhibition, Sci. Rep., 2018, 8, 11758 CrossRef PubMed.
- I. Farhana, M. N. Hossain, K. Suzuki, T. Matsuda and T. Nagai, Genetically Encoded Fluorescence/Bioluminescence Bimodal Indicators for Ca2+ Imaging, ACS Sens., 2019, 4, 1825–1834 CrossRef CAS PubMed.
- H. Dana, B. Mohar, Y. Sun, S. Narayan, A. Gordus, J. P. Hasseman, G. Tsegaye, G. T. Holt, A. Hu, D. Walpita, R. Patel, J. J. Macklin, C. I. Bargmann, M. B. Ahrens, E. R. Schreiter, V. Jayaraman, L. L. Looger, K. Svoboda and D. S. Kim, Sensitive Red Protein Calcium Indicators for Imaging Neural Activity, eLife, 2016, 5, e12727 CrossRef PubMed.
- Y. Zhao, A. S. Abdelfattah, Y. Zhao, A. Ruangkittisakul, K. Ballanyi, R. E. Campbell and D. J. Harrison, Microfluidic Cell Sorter-Aided Directed Evolution of a Protein-Based Calcium Ion Indicator with an Inverted Fluorescent Response, Integr. Biol., 2014, 6, 714–725 CrossRef CAS PubMed.
- A. E. Palmer and R. Y. Tsien, Measuring Calcium Signaling Using Genetically Targetable Fluorescent Indicators, Nat. Protoc., 2006, 1, 1057–1065 CrossRef CAS PubMed.
- A. Miyawaki, O. Griesbeck, R. Heim and R. Y. Tsien, Dynamic and Quantitative Ca2+ Measurements Using Improved Cameleons, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2135–2140 CrossRef CAS PubMed.
- Q. Wang, L. J. Byrnes, B. Shui, U. F. Röhrig, A. Singh, D. M. Chudakov, S. Lukyanov, W. R. Zipfel, M. I. Kotlikoff and H. Sondermann, Molecular Mechanism of a Green-Shifted, PH-Dependent Red Fluorescent Protein MKate Variant, PLoS One, 2011, 6, e23513 CrossRef CAS PubMed.
- M. Z. Lin, M. R. McKeown, H. L. Ng, T. A. Aguilera, N. C. Shaner, R. E. Campbell, S. R. Adams, L. A. Gross, W. Ma, T. Alber and R. Y. Tsien, Autofluorescent Proteins with Excitation in the Optical Window for Intravital Imaging in Mammals, Chem. Biol., 2009, 16, 1169–1179 CrossRef CAS PubMed.
- R. Arce-Molina, F. Cortés-Molina, P. Y. Sandoval, A. Galaz, K. Alegría, S. Schirmeier, L. F. Barros and A. S. Martín, A Highly Responsive Pyruvate Sensor Reveals Pathway-Regulatory Role of the Mitochondrial Pyruvate Carrier MPC, eLife, 2020, 9, e53917 CrossRef PubMed.
- A. Aggarwal, R. Liu, Y. Chen, A. J. Ralowicz, S. J. Bergerson, F. Tomaska, B. Mohar, T. L. Hanson, J. P. Hasseman, D. Reep, G. Tsegaye, P. Yao, X. Ji, M. Kloos, D. Walpita, R. Patel, M. A. Mohr, P. W. Tillberg, L. L. Looger, J. S. Marvin, M. B. Hoppa, A. Konnerth, D. Kleinfeld, E. R. Schreiter and K. Podgorski, Glutamate Indicators with Improved Activation Kinetics and Localization for Imaging Synaptic Transmission, Nat. Methods, 2023, 20, 925–934 CrossRef CAS PubMed.
- F. Sun, J. Zeng, M. Jing, J. Zhou, J. Feng, S. F. Owen, Y. Luo, F. Li, H. Wang, T. Yamaguchi, Z. Yong, Y. Gao, W. Peng, L. Wang, S. Zhang, J. Du, D. Lin, M. Xu, A. C. Kreitzer, G. Cui and Y. Li, A Genetically Encoded Fluorescent Sensor Enables Rapid and Specific Detection of Dopamine in Flies, Fish, and Mice, Cell, 2018, 174, 481–496 CrossRef CAS PubMed.
- T. Patriarchi, J. R. Cho, K. Merten, M. W. Howe, A. Marley, W. H. Xiong, R. W. Folk, G. J. Broussard, R. Liang, M. J. Jang, H. Zhong, D. Dombeck, M. von Zastrow, A. Nimmerjahn, V. Gradinaru, J. T. Williams and L. Tian, Ultrafast Neuronal Imaging of Dopamine Dynamics with Designed Genetically Encoded Sensors, Science, 2018, 360, eaat4422 CrossRef PubMed.
- J. Wan, W. Peng, X. Li, T. Qian, K. Song, J. Zeng, F. Deng, S. Hao, J. Feng, P. Zhang, Y. Zhang, J. Zou, S. Pan, M. Shin, B. J. Venton, J. J. Zhu, M. Jing, M. Xu and Y. Li, A Genetically Encoded Sensor for Measuring Serotonin Dynamics, Nat. Neurosci., 2021, 24, 746–752 CrossRef CAS PubMed.
- M. Jing, Y. Li, J. Zeng, P. Huang, M. Skirzewski, O. Kljakic, W. Peng, T. Qian, K. Tan, J. Zou, S. Trinh, R. Wu, S. Zhang, S. Pan, S. A. Hires, M. Xu, H. Li, L. M. Saksida, V. F. Prado, T. J. Bussey, M. A. M. Prado, L. Chen, H. Cheng and Y. Li, An Optimized Acetylcholine Sensor for Monitoring in Vivo Cholinergic Activity, Nat. Methods, 2020, 17, 1139–1146 CrossRef CAS PubMed.
- S. Kille, C. G. Acevedo-Rocha, L. P. Parra, Z. G. Zhang, D. J. Opperman, M. T. Reetz and J. P. Acevedo, Reducing Codon Redundancy and Screening Effort of Combinatorial Protein Libraries Created by Saturation Mutagenesis, ACS Synth. Biol., 2013, 2, 83–92 CrossRef CAS PubMed.
- T. Patriarchi, A. Mohebi, J. Sun, A. Marley, R. Liang, C. Dong, K. Puhger, G. O. Mizuno, C. M. Davis, B. Wiltgen, M. von Zastrow, J. D. Berke and L. Tian, An Expanded Palette of Dopamine Sensors for Multiplex Imaging in Vivo, Nat. Methods, 2020, 17, 1147–1155 CrossRef CAS PubMed.
- M. J. Henderson, H. A. Baldwin, C. A. Werley, S. Boccardo, L. R. Whitaker, X. Yan, G. T. Holt, E. R. Schreiter, L. L. Looger, A. E. Cohen, D. S. Kim and B. K. Harvey, A Low Affinity GCaMP3 Variant (GCaMPer) for Imaging the Endoplasmic Reticulum Calcium Store, PLoS One, 2015, 10, e0139273 CrossRef PubMed.
- H. Shinoda, M. Shannon and T. Nagai, Fluorescent Proteins for Investigating Biological Events in Acidic Environments, Int. J. Mol. Sci., 2018, 19, 1548 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00138a |
| This journal is © The Royal Society of Chemistry 2024 |