
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of structure on excited-state intramolecular proton transfer-based sensors for phosphonofluoridate G-series nerve agent vapour detection†
Shengqiang
Fan
,
Paul L.
Burn
 *,
Ian R.
Gentle
and
Paul E.
Shaw
*
*,
Ian R.
Gentle
and
Paul E.
Shaw
*
Centre for Organic Photonics & Electronics (COPE), School of Chemistry & Molecular Biosciences, University of Queensland, St. Lucia, QLD 4072, Australia. E-mail: p.burn2@uq.edu.au; p.shaw3@uq.edu.au
First published on 25th June 2024
Abstract
Excited-state intramolecular proton transfer (ESIPT) emitters are unique in that the emission is significantly red shifted relative to the absorption spectra. Herein we explore the effect of substituents on the ability of thin films of 2-[1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl]phenol-based ESIPT reporter compounds to detect hydrogen fluoride found in G-series nerve agents containing a phosphonofluoridate moiety. When the hydroxyl group of the 2-[1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl]phenol-based reporter compounds was protected as a silyl ether the photoluminescence emission spectra had vibrational structure and emission maxima at around 370 nm. The silyl protecting groups could be cleaved upon exposure to hydrogen fluoride in the G-series nerve agent simulant, di-iso-propyl fluorophosphate, leading to ESIPT emission with a peak maximum at around 470 nm, thus allowing identification of the presence of hydrogen fluoride. Films of the sensing materials with the different silyl protecting groups were found to have different stabilities to ambient conditions and reactivity with hydrogen fluoride, with the larger silyl ethers such as triethylsilyl and t-butyldimethyl silyl performing better overall when compared to the smaller trimethylsilyl ether. Steric encumberance or addition of polar solubilising groups was found to reduce the sensing capability. The optimal sensing material was lipophilic and contained a t-butyldimethyl silyl protecting group, with films capable of detecting hydrogen fluoride at a concentration of 0.1 ppm which, based on a sarin purity of 99%, would enable sarin to be detected at 1.2 ppm, which is below the LC50 five minute exposure limit for sarin of 1.6–3.2 ppm.
Introduction
Luminescence-based detection methods for chemical threats can be sensitive, rapid, and deployed in lightweight, low power, portable devices.1,2 However, selectivity of the response towards the desired analyte is often difficult to achieve. For example, while there is extensive literature on fluorescent materials that can detect nitro group containing explosives and taggants using a photoinduced electron transfer luminescence quenching mechanism,3–6 there are few reports of materials that are truly selective.7 In contrast to the detection of explosives there are comparatively fewer reports on using luminescence detection of chemical warfare agents, particularly nerve agents.8–10 The three classes of nerves agents are the V- and G-series,11 and the Novichoks (Fig. 1).12 Independent of class, nerve agents all have a phosphorous–oxygen double bond and are defined by the nature of the leaving group attached to the phosphorous atom.11 The electron affinities of nerve agents are not sufficiently high for photoinduced electron transfer (PET) luminescence quenching to be used for their detection. The main approaches investigated for the detection of nerve agents have utilised the fact that they contain an electrophilic phosphorous atom and a leaving group.13–16 The strategy has been to design luminescent (fluorescent) sensing materials that contain a nucleophilic group for attacking the phosphorous atom with subsequent displacement of the leaving group. While the approach is logical it suffers from several issues including achieving selectivity between different classes of nerve agents, low rates of reaction in the solid state, and the fact that not all nerve agents or their simulants are pure.8,17 For example, sarin is known to contain hydrogen fluoride, which is present post-synthesis and is also generated upon release into the atmosphere through hydrolysis by atmospheric water.12 Furthermore, while generally not reported, the simulants typically used for laboratory based sensing studies for the G-series nerve agents are unstable and if not prepared and stored correctly can contain acid.18 A consequence of using sensing materials with a nitrogen containing nucleophile (the common approach) has been that some reported sensing materials for the detection of nerve agents, particularly the G-series, have led to misleading results.8 For example, protonation of the nitrogen nucleophile can lead to the same luminescence response as the product expected from the nucleophile reacting with the nerve agent.17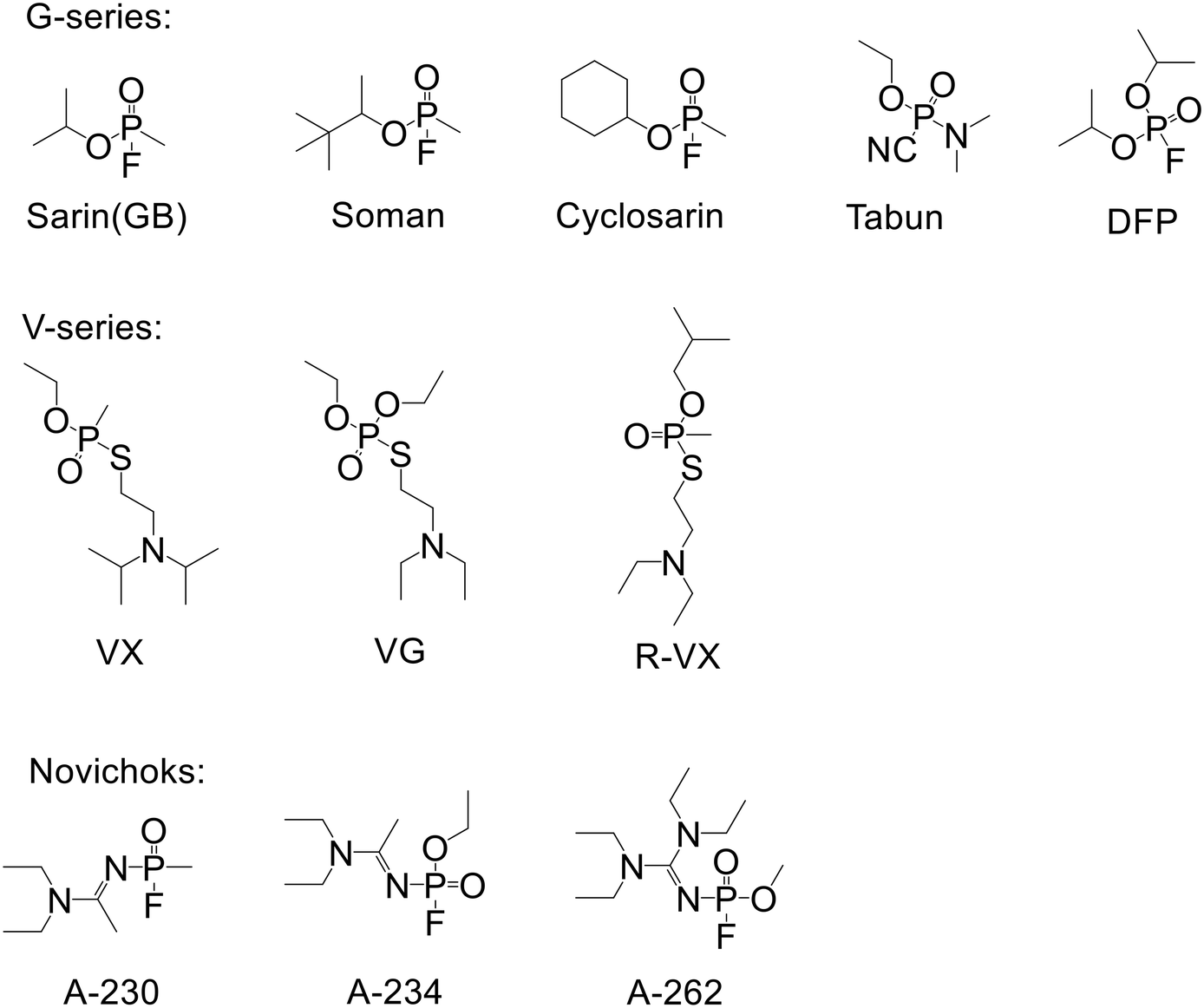 | ||
| Fig. 1 Structures of the different classes of nerve agent containing a phosphorous–oxygen double bond. The G-series agents other than tabun are generally defined by having a phosphonofluoridate moiety. DFP is a simulant used for the G-series nerve agents. | ||
Recognising that the G-series nerve agents composed of a phosphonofluoridate moiety always contain hydrogen fluoride (HF) we have pursued a different strategy. Our approach is to design hydroxyl group containing sensing materials that react with HF to give a luminescent reporter compound. However, a key issue of the deprotection approach is that simple silylated sensing materials and their deprotected derivatives are likely to have emission at similar wavelengths. To overcome this problem we chose a reporter compound chromophore that was capable of excited state intramolecular proton transfer (ESIPT) based emission. ESIPT emitters rely on the proton of a hydroxyl group to undergo the ESIPT process and they generally have a large shift between the emission peak and absorption onset (≈100 nm), which is significantly larger than the normal Stokes shift observed for fluorescent compounds. Thus, protecting the hydroxyl group with a silyl moiety stops the ESIPT process, with the sensing material simply being a fluorescent emitter. Therefore, the advantage of the ESIPT chromophore protection–deprotection strategy is that absorption and emission of the ESIPT reporter compound can be at longer wavelengths than the silylated sensing material, which emits via simple fluorescence. That is, the approach has the potential to overcome the problem of background luminescence from the sensing material that could reduce detection sensitivity.
In this study we chose 2-[1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl]phenol as the base ESIPT reporter19 chromophore to explore the effect of structural variations of the sensing material/reporter compound on the efficacy of HF detection. The structures of the sensing materials used in the current work are shown in Fig. 2 and include the choice of silyl protecting group, and the polarity (attachment of a glycol chain) or size (e.g., attachment of dendrons) of the ESIPT reporter chromophore. We describe their synthesis, photophysical properties, and capability to selectively detect HF over other acid vapours.
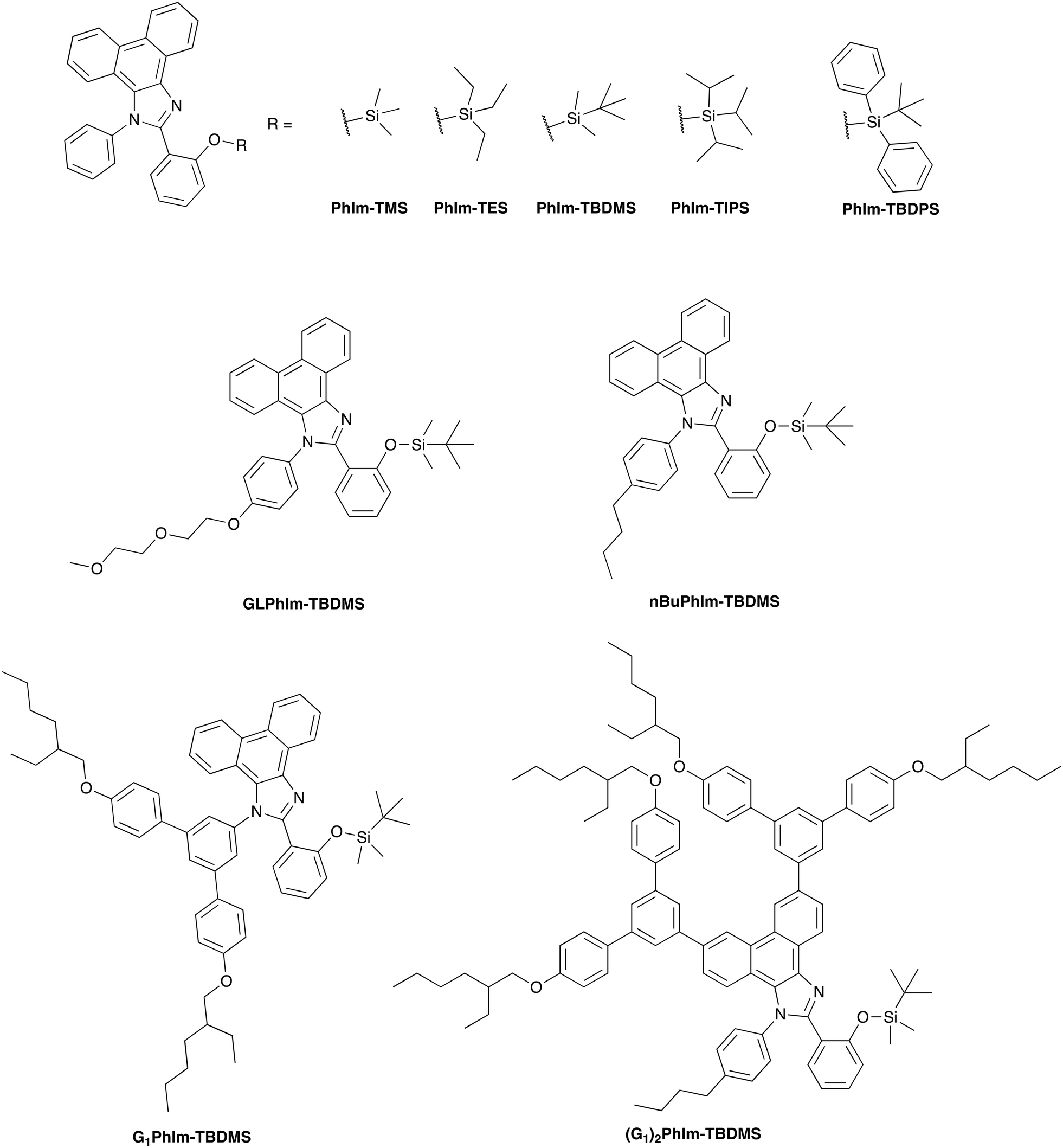 | ||
| Fig. 2 The silyl protected structural variants based on the basic ESIPT chromophore, 2-[1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl]phenol, showing the different silyl protecting groups, substituents on the N-phenyl ring (none, glycol chain, and n-butyl), and dendrimeric versions. | ||
Synthesis
Scheme 1 illustrates the synthetic routes to the different sensing materials, with the full experimental details given in the ESI.† In the first part of the study, we investigated the effect of the silyl protecting group on the sensing performance and for this we used the simple chromophore, PhIm-OH,20 as the reporter compound. The synthesis of different silylated materials was straightforward with PhIm-OH reacted with the requisite silyl chloride using imidazole as the catalyst. Under these conditions the silylated compounds PhIm-TMS, PhIm-TES, PhIm-TBDMS, PhIm-TIPS, and PhIm-TBDPS were isolated in yields between 57–77%. The synthesis of the n-butylphenyl derivative, nBuPhIm-TBDMS has been previously reported.21 The glycolated and N-dendronised derivatives were formed in similar two step reactions. In the first step the respective amines, 4-[2-(2-methoxyethoxy)ethoxy]phenylamine22 and 4,4′′-bis[(2-ethylhexyl)oxy]-[1,1′:3′,1′′-terphenyl]-5′-amine (G1-NH2) (see ESI† for the synthesis of G1-NH2), were condensed with 9,10-phenanthrenequinone and 2-hydroxybenzaldehyde in the presence of ammonium acetate and acetic acid to give GLPhIm-OH and G1PhIm-OH in yields of 40% and 35%, respectively. The two ESIPT reporter compounds were then reacted with tert-butyl dimethylsilyl chloride under the standard conditions to give GLPhIm-TBDMS (68%) and G1PhIm-TBDMS (52%). Finally, the doubly dendronised (G1)2PhIm-TBDMS was prepared in three steps: first, 4-n-butylaniline was condensed with 3,6-dibromophenanthrene-9,10-dione and 2-hydroxybenzaldehyde in the presence of ammonium acetate and acetic acid to give 2BrPhIm-OH (32%); second, 2BrPhIm-OH was reacted with [4,4′′-bis({2-ethylhexyl}oxy)-(1,1′:3′,1′′-terphenyl)-5′-yl]boronic acid23 under Suzuki coupling conditions to give the reporter compound, (G1)2PhIm-OH (71%); and finally, (G1)2PhIm-OH was converted to the tert-butyl dimethylsilyl ether, (G1)2PhIm-TBDMS (82%). The reason for only preparing the TBDMS silyl ethers of the latter materials will be discussed later. | ||
| Scheme 1 Reactions and conditions: i) trimethylsilyl chloride, triethylsilyl chloride, tert-butyl dimethylsilyl chloride, tri-iso-propylsilyl chloride, or tert-butyl diphenylsilyl chloride, imidazole, anhydrous N,N-dimethylformamide, Ar, rt or 40 °C, 16–48 h; ii) 2-hydroxybenzaldehyde, ammonium acetate, acetic acid, Ar, 110 °C, 16 h; iii) [4,4′′-bis({2-ethylhexyl}oxy)-(1,1′:3′,1′′-terphenyl)-5′-yl]boronic acid, Pd(PPh3)4, Na2CO3 (2 M, aq.), THF, Ar (g), 80 °C, 48 h. Yields: PhIm-TMS (67%), PhIm-TES (60%), PhIm-TBDMS (77%), PhIm-TIPS (65%), PhIm-TBDPS (57%), GLPhIm-OH (40%), GLPhIm-TBDMS (68%), G1PhIm-OH (35%), G1PhIm-TBDMS (52%), 2BrPhIm-OH (32%), (G1)2PhIm-OH (71%), (G1)2PhIm-TBDMS (82%). | ||
Photophysical properties
Fig. 3a illustrates the solution UV-vis absorption spectra of the different PhIm silyl ethers and reporter compounds in dichloromethane, with equivalent spectra for toluene and mixed methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dichloromethane solutions of the different PhIm-OH reported compounds shown in Fig. S1.† The solution absorption spectra of the differently silylated sensing materials based on PhIm-OH are identical as expected and are indicative that the differently sized silyl protecting groups do not cause twisting between the different aromatic units in the conjugated chromophore. Addition of the electron donating glycol unit did not red shift the onset of the absorption, which is due to the N-phenyl ring being twisted out of plane by the phenanthrene ring. Given the similarity of the absorption spectra it would be expected that the emission spectra of the silylated materials would also be the same and this was indeed found, with the vibrationally structured emission having peaks at around 365 nm and 375 nm (Fig. 3b). However, there are distinct differences in the solution UV-visible absorption and PL spectra between the silylated sensing materials and the ESIPT reporter compounds, PhIm-OH and GLPhIm-OH in dichloromethane. The absorption spectra of the two reporter compounds are identical but with the onset red shifted with respect to the silylated sensing materials (369 nm to 377 nm). Furthermore, there was a significant red shift in the PL emission for the reporter compounds relative to the silylated sensing materials (365 nm to 472 nm), with the emission from the reporter compounds lacking vibrational structure. The large red shift of the PL emission relative to the absorption spectra and lack of vibrational structure is consistent with the PL of PhIm-OH and GLPhIm-OH occurring via an ESIPT mechanism. The red shift in the onset of the absorption and PL emission for the reporter compounds is useful for sensing as the reporter compounds can be selectively excited in the presence of remaining sensing material, with emission being also selectively detected. This enables the background fluorescence from the sensing material to be avoided, which is important for improving the sensitivity of detection. Finally, comparison of the absorption and emission spectra of the reporter compounds showed that there was not a significant shift in the absorption or emission peaks (<10 nm) across different solvent polarities (Fig. S1†). However, in the presence of the protic solvent methanol, all the reporter compounds exhibited PL emission from the enol form (around 350–400 nm), which were at a similar wavelength and had a similar shape as the silylated sensing materials (Fig. 3a), in addition to the predominant ESIPT emission at ≈475 nm. The emergence of the enol emission in the presence of protic solvents has been reported before, and we attribute the presence of the enol emission to H-bonding of the methanol to the reporter compounds.19 It should be noted that (G1)2PhIm-OH displays both enol and keto form emission in all three solvents, suggesting that the substitution results in an equilibrium between the anti-conformer and the form that can undergo the ESIPT process.19
:dichloromethane solutions of the different PhIm-OH reported compounds shown in Fig. S1.† The solution absorption spectra of the differently silylated sensing materials based on PhIm-OH are identical as expected and are indicative that the differently sized silyl protecting groups do not cause twisting between the different aromatic units in the conjugated chromophore. Addition of the electron donating glycol unit did not red shift the onset of the absorption, which is due to the N-phenyl ring being twisted out of plane by the phenanthrene ring. Given the similarity of the absorption spectra it would be expected that the emission spectra of the silylated materials would also be the same and this was indeed found, with the vibrationally structured emission having peaks at around 365 nm and 375 nm (Fig. 3b). However, there are distinct differences in the solution UV-visible absorption and PL spectra between the silylated sensing materials and the ESIPT reporter compounds, PhIm-OH and GLPhIm-OH in dichloromethane. The absorption spectra of the two reporter compounds are identical but with the onset red shifted with respect to the silylated sensing materials (369 nm to 377 nm). Furthermore, there was a significant red shift in the PL emission for the reporter compounds relative to the silylated sensing materials (365 nm to 472 nm), with the emission from the reporter compounds lacking vibrational structure. The large red shift of the PL emission relative to the absorption spectra and lack of vibrational structure is consistent with the PL of PhIm-OH and GLPhIm-OH occurring via an ESIPT mechanism. The red shift in the onset of the absorption and PL emission for the reporter compounds is useful for sensing as the reporter compounds can be selectively excited in the presence of remaining sensing material, with emission being also selectively detected. This enables the background fluorescence from the sensing material to be avoided, which is important for improving the sensitivity of detection. Finally, comparison of the absorption and emission spectra of the reporter compounds showed that there was not a significant shift in the absorption or emission peaks (<10 nm) across different solvent polarities (Fig. S1†). However, in the presence of the protic solvent methanol, all the reporter compounds exhibited PL emission from the enol form (around 350–400 nm), which were at a similar wavelength and had a similar shape as the silylated sensing materials (Fig. 3a), in addition to the predominant ESIPT emission at ≈475 nm. The emergence of the enol emission in the presence of protic solvents has been reported before, and we attribute the presence of the enol emission to H-bonding of the methanol to the reporter compounds.19 It should be noted that (G1)2PhIm-OH displays both enol and keto form emission in all three solvents, suggesting that the substitution results in an equilibrium between the anti-conformer and the form that can undergo the ESIPT process.19
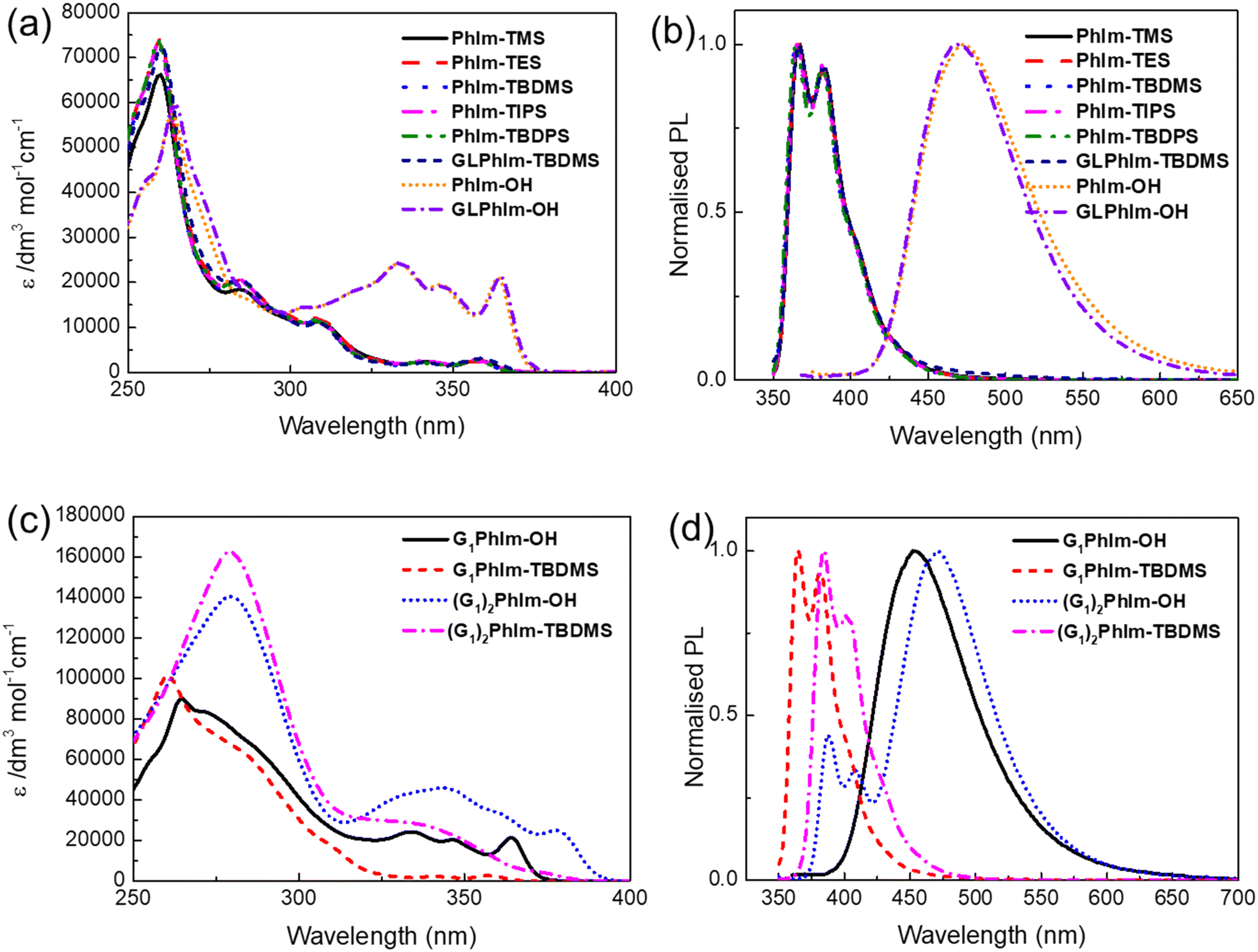 | ||
| Fig. 3 Solution (dichloromethane) (a) UV-vis absorption and (b) PL spectra of PhIm-OH, PhIm-Silyl ether, GLPhIm-OH and GLPhIm-TBDMS, and (c) UV-vis absorption and (d) PL spectra of G1PhIm-OH, G1PhIm-TBDMS, (G1)2PhIm-OH and (G1)2PhIm-TBDMS. | ||
Fig. 3c shows the solution UV-vis spectra of the two dendronised sensing materials and their corresponding reporter compounds. When the dendron was attached to the imidazole nitrogen (G1PhIm-TBDMS and G1PhIm-OH) the absorption onset was at a similar wavelength to the non-dendronised materials. In contrast, the onset to the absorption was red shifted for (G1)2PhIm-TBDMS and (G1)2PhIm-OH, reflecting the fact that the branching phenyl rings of the dendrons when attached to the PhIm unit extends the conjugation of the chromophores. A key difference in the absorption spectra of the dendronised sensing and reporter materials was that the molar extinction coefficient at short wavelengths (≈265 nm) was larger than that of the non-dendronised materials. The larger molar extinction coefficient arises from the biphenyl chromophores in the dendrons, with the doubly dendronised (G1)2PhIm-TBDMS and (G1)2PhIm-OH having the largest short wavelength molar extinction coefficients due to them having the largest number of biphenyl chromophores. The PL emission of G1PhIm-TBDMS occurred at similar wavelengths as the non-dendronised sensing materials whilst the emission from (G1)2PhIm-TBDMS was red shifted, which is consistent with the changes in the absorption onset (Fig. 3d). Both reporter compounds G1PhIm-OH and (G1)2PhIm-OH) were found to have ESIPT based emission although (G1)2PhIm-OH was also found to have a component of the emission associated with normal S1 to S0 decay, reminiscent of the (G1)2PhIm-TBDMS. The fact that the emission from (G1)2PhIm-OH has two components could arise from the bulk of the two dendrons slowing the structural rearrangement required for the ESIPT mechanism to occur. Given that the materials are intended to be used in the solid state for sensing we measured the film photoluminescence quantum yields (PLQYs) of the reporter compounds. The reporter compounds were found to be sufficiently luminescent to be used in sensing, with PLQYs of PhIm-OH (27%), GLPhIm-OH (24%), nBuPhIm-OH (31%), G1PhIm-OH (9%), and (G1)2PhIm-OH (26%).
Detection properties of the phenanthro[9,10-d]imidazole-based silyl ethers
For the sensing measurements we used di-iso-propyl fluorophosphate (DFP), which is an established simulant for G-series agents containing a phosphonofluoridate group.8 As commented on earlier, G-series nerve agents are not pure and contain HF. For example, the sarin used in the Tokyo attack was determined to have a purity of only 25%.24 In order to evaluate the efficacy of our sensing materials for dilute concentrations of HF, we used DFP with relatively high purity levels (95% and 99% as determined using 31P NMR).In the first part of the detection study we investigated the effect of the silyl protecting group on the ability of the sensing materials to detect HF in DFP, with the changes in PL shown in Fig. 4. The films were left for two hours under ambient conditions before the detection measurement was undertaken. The films were excited at 365 nm and ESIPT emission (peak at around 475 nm) was observed in all cases. It is important to note that all the sensing films had some ESIPT emission after two hours, indicating that under the ambient storage conditions some deprotection had occurred due to the moisture in the environment. However, the amount of deprotection of the sensing materials under ambient conditions was not the same, with the order being PhIm-TMS ≈ -TES > -TIPS ≈ -TBDMS > -TBDPS. This order reflects the relative steric hindrance of the silyl ethers with TMS being the smallest and most reactive and TBDPS providing the most steric encumbrance and being the least reactive. The amount of deprotection under ambient conditions is an important factor when considering deployment of the sensor in the field. If the sensing material is too reactive then it could be consumed before a threat event is detected. The films were then exposed to vapour pulses of DFP with the PL spectra after a single pulse shown in Fig. 4 and the time dependent changes in PL intensity shown in Fig. 5. The PL spectra shown in Fig. 4 are for a single DFP exposure in which the DFP was injected over a period of approximately 10 seconds, with the spectra collected once the PL response had equilibrated after a further 10 seconds. It can be seen that the magnitude of the PL response to the HF in the DFP was in the order PhIm-TMS > -TES ≈ -TIPS ≈ -TBDMS > -TBDPS. That is, the least stable PhIm-TMS gives the largest response and the most stable PhIm-TBDPS the smallest. Thus, while the TMS ether had the largest response under laboratory conditions we concluded that it did not possess sufficient stability for practical long-term use in the field.
 | ||
| Fig. 4 PL spectra of PhIm- (a) TMS, (b) TES, (c) TBDMS, (d) TIPS, and (e) TBDPS silyl ethers after 2 hours under ambient conditions (background) and then exposure to 95% DFP headspace vapours. Saturated DFP vapours (3 mL) were injected at 20 mL min−1 into a nitrogen flow (200 mL min−1) and the spectra were collected 10 seconds after completion of the injection. The films were excited at 365 nm using an LED light source. | ||
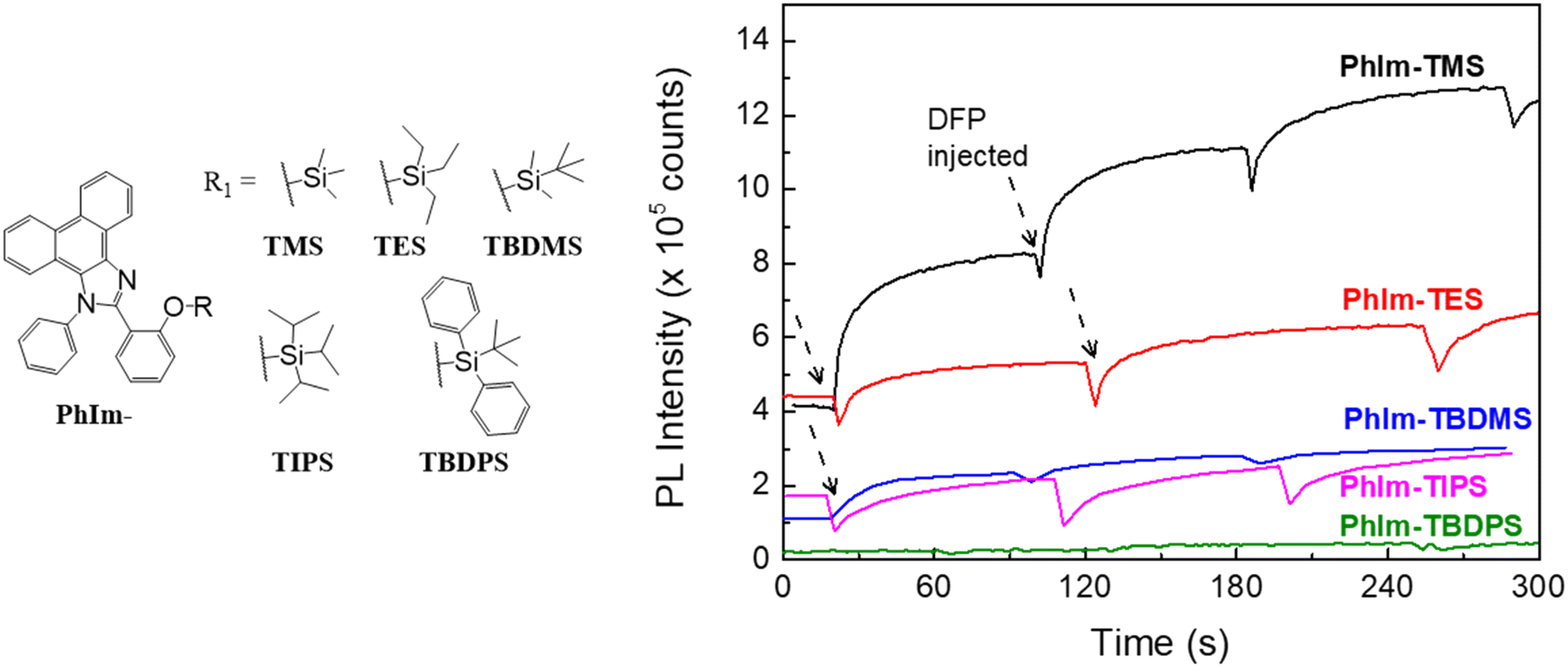 | ||
| Fig. 5 Change in PL intensity versus time for sequential DFP injections for films of PhIm-TMS, PhIm-TES, PhIm-TBDMS, PhIm-TIPS, and PhIm-TBDPS. The DFP purity was 95% and saturated DFP vapour (1 mL) pulses were injected each time at 20 mL min−1 into a nitrogen flow (200 mL min−1). The films were excited at 365 nm using an LED light source. | ||
The reactivity of the different silyl protecting groups was reflected in the time-dependent data shown in Fig. 5. It should be noted that the amount of DFP vapour injected was a third of that shown in Fig. 4 and hence the magnitude of the PL responses were smaller. The data showed an initial rapid decrease in the PL intensity, which we ascribe to a change in the optical path as the DFP is injected, followed in all cases, except PhIm-TBDPS, by an increase in the PL intensity at the emission wavelength of the reporter compound, PhIm-OH. PhIm-TMS was found to have the largest response due to it being the least sterically hindered and most reactive. Given that the same amount of HF is injected for each experiment, the smaller responses for the PhIm-TES, PhIm-TBDMS and PhIm-TIPS films could arise from the HF diffusing into the film to a lesser extent than in the case of PhIm-TMS and/or the silyl protecting groups being less reactive. It is not possible to differentiate these two factors but the trends in Fig. 4 suggest that the reactivity of the silyl protecting groups plays an important role in the response of the sensing materials to HF. A final point to note from the time-dependent measurements is that for the films that showed a response to HF, the PL intensity was found to increase upon further exposure to HF. This is important for practical deployment as a single film can be used until there is no sensing material left, that is, only the reporter compound is present. Balancing the current costs (TMS < TBDMS < TES < TBDPS < TIPS) of the chloride reagents used to form the sensing materials, relative stability to ambient conditions (PhIm-TMS ≈ -TES < -TIPS ≈ -TBDMS < -TBDPS) and response to HF (PhIm-TMS > -TES ≈ -TIPS ≈ -TBDMS > -TBDPS) led us to choose the TBDMS silyl ether for the study that investigated how changes to the emissive chromophore structure affected the ability to detect HF.
In the next part of the study we compared the impact of hydrophobicity and steric bulk on the ability of the sensing materials to detect HF (see Fig. 6). The measurements were carried out in the same way as those that gave the data in Fig. 4 to allow for direct comparison. Fig. 6a shows the results for the sensing material where the N-phenyl was substituted in the para position by the lipophilic n-butyl group. The key improvement observed for nBuPhIm-TBDMS over PhIm-TBDMS was that it was more stable under ambient conditions. In contrast, while GLPhIm-TBDMS showed a good PL response to HF it was less stable under ambient conditions when compared to either nBuPhIm-TBDMS or PhIm-TBDMS. We suspect that the glycol units encourage water from the environment to diffuse into the film and react with the TBDMS group. Addition of the first generation dendron to the imidazole nitrogen did not improve the ambient stability relative to the simple PhIm-TBDMS sensing material nor did it enhance the response to HF. Furthermore, doubly dendronising the PhIm unit to give (G1)2PhIm-TBDMS also did not improve the stability towards environmental moisture relative to nBuPhIm-TBDMS and in addition, it suppressed the ability to detect HF. In the case of (G1)2PhIm-TBDMS the poor performance may be in part due to the dendrons restricting the ability of the chromophore to adopt the conformation required for ESIPT emission, noting that both ESIPT and S1 to S0 emission was observed in solution. It would be anticipated that such an effect would be exacerbated in the solid state.
 | ||
| Fig. 6 PL spectra of (a) n-butyl, (b) glycol or (c and d) dendron-containing PhIm silyl ethers after 2 hours under ambient conditions (background) and then exposure to DFP of 95% purity. Saturated DFP vapours (3 mL) were injected at 20 mL min−1 into a nitrogen gas flow (200 mL min−1). The films were excited at 365 nm using an LED light source. | ||
From the results presented on the relative stability to ambient conditions and PL response to HF we concluded that nBuPhIm-TBDMS was on balance the best performing of the materials reported thus far. To show that it would not give false positive responses to common acids we carried out sensing measurements with a range of acids (Fig. 7). Critically, we did not observe a PL turn-on response similar to that of HF at the emission peak of the ESIPT reporter compound, even after five minutes exposure. To understand the selectivity, we conducted 1H NMR measurements in which a solution of nBuPhIm-TBDMS was exposed to two acids that might be found in the domestic arena, hydrochloric acid and acetic acid (Fig. S2 and S3†). It should be noted that the experimental conditions used for the NMR experiments were harsher than those encountered in the film measurements, where films of the sensing material are exposed to vapours of the acids. Fig. S3a and b† shows that there was no change in the 1H NMR spectrum of nBuPhIm-TBDMS upon exposure to an excess of acetic acid, even after 21 hours. In contrast, the 1H NMR shows that nBuPhIm-TBDMS rapidly protonates in the presence of hydrochloric acid and that after 2 hours desilylation begins to occur (Fig. S3c and d†). The fact that desilylation occurs with hydrochloric acid in solution after 2 hours is not an issue for in-field detection, with the detection of HF vapour by a thin film of the sensing material achieved within seconds of exposure (Fig. 5).
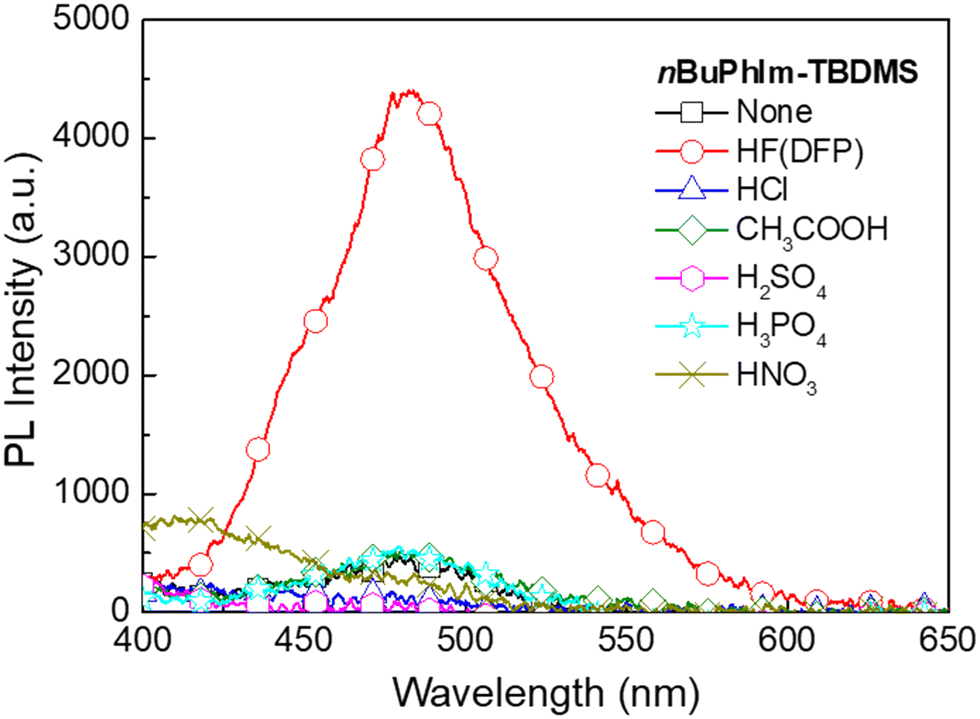 | ||
| Fig. 7 PL spectra of nBuPhIm-TBDMS films before and after exposure to various acid vapours. Before exposure to the acids, the films were kept under ambient conditions for 2 hours. For each acid, 10 μL of the respective acid solution was added to a container (200 mL plastic bottle for DFP/HF and 250 mL conical flask for all other acids) and allowed to sit for 30 minutes. Then the sensing films were exposed to the acid vapours for 5 minutes and the PL spectra were recorded. The acids used for exposure included DFP/HF (DFP purity 95%), hydrochloric acid (32%), acetic acid (100%), sulfuric acid (98%), phosphoric acid (85%), and nitric acid (70%). | ||
In the final part of the study we were interested in determining the limit of detection (LoD) of the basic PhIm-TBDMS to HF as well as the best sensing material that has the n-butyl solubilising group, nBuPhIm-TBDMS, and whether they were both below the LC50 for sarin. The LC50 is the concentration that can kill 50% of test animals within a predetermined observation period, which for sarin is 1.6–3.2 ppm over a five-minute exposure.25 Our approach to identifying sarin by detecting the hydrogen fluoride it contains poses challenges in determining the LoD for the agent itself. This difficulty arises because the quantity of acid present is contingent upon the purity of the agent. To estimate the LoD for sarin, we employed DFP containing 1% HF, with the concentration of HF determined using 19F NMR spectroscopy. Fig. 8a and b illustrate the dependence of PL response on HF vapour concentration for PhIm-TBDMS and nBuPhIm-TBDMS, respectively. For both sensing materials, an increase in HF vapor concentration resulted in a larger change in PL intensity. From this, we established the effective LoD of HF to be approximately 0.2 ppm for PhIm-TBDMS and around 0.1 ppm for nBuPhIm-TBDMS following a 5-minute exposure. Since hydrogen fluoride is more volatile than DFP, it is the primary constituent of the headspace vapour despite being an impurity in the liquid. This corresponds to an estimated LoD of DFP containing 1% HF being around 0.8 ppm for PhIm-TBDMS and about 0.3 ppm for nBuPhIm-TBDMS. The enhanced LoD for nBuPhIm-TBDMS is attributed to its higher stability under ambient conditions, as evidenced from the changes in PL intensity that occurred without exposure to HF (Fig. 8a and b). Given that sarin is four times more volatile than DFP,26 and assuming the same concentration of HF, the data suggests that PhIm-TBDMS and nBuPhIm-TBDMS can effectively detect sarin at approximately 3.2 and 1.2 ppm, respectively. The latter is lower than the LC50 value (1.6–3.2 ppm for 5-minute exposure) and hence is a promising material to use for the detection of sarin.
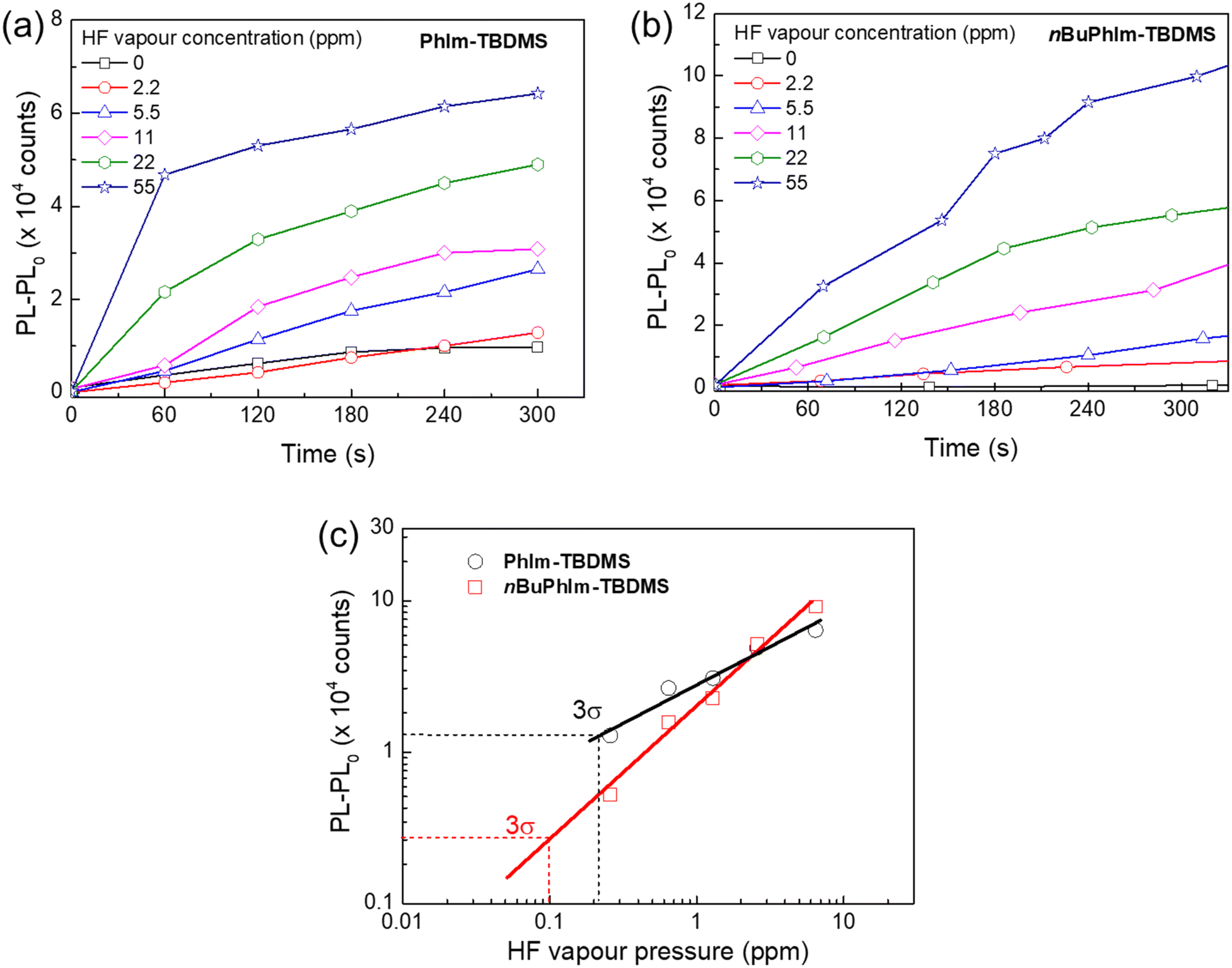 | ||
| Fig. 8 PL responses of (a) PhIm and (b) nBuPhIm-based sensing materials against HF vapour concentration, and (c) estimation of the LoD using three times the standard deviation (3σ) of the change in PL intensity in air. Saturated DFP vapour was continuously injected at a rate of 0–5 mL min−1 into a nitrogen flow (200 mL min−1). The DFP purity was 99%. The films were excited at 365 nm using an LED light source. | ||
Conclusion
In conclusion, utilization of ESIPT emissive 2-[1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl]phenol-based compounds has proven to be an effective strategy for the vapour phase detection of hydrogen fluoride in G-series nerve agents containing a phosphonofluoridate moiety. Exposure of films of the silylated sensing material to vapours of a G-series nerve agent simulant, di-iso-propyl fluorophosphate (which like sarin contains hydrogen fluoride as an impurity), led to the cleavage of the silyl protecting group and triggering of ESIPT emission from the different reporter compounds with a distinctive peak maximum at longer wavelengths than the corresponding silylated sensing materials. The increase in emission intensity allowed for the rapid identification of the presence of hydrogen fluoride. It was found that the structural variant, 2-[2-({tert-butyldimethylsilyl}oxy)phenyl]-1-[4-n-butylphenyl]-1H-phenanthro[9,10-d]imidazole, with a lipophilic n-butyl group attached to the N-phenyl ring, demonstrated the best sensing capabilities, enabling the detection of hydrogen fluoride after a few seconds of exposure. This research not only contributes to the understanding of ESIPT-based sensing mechanisms but also holds promise for practical applications in the rapid and sensitive detection of phosphonofluoridate-based chemical warfare agents.Experimental
Sensing film preparation
The sensing compounds were dissolved in toluene (10 mg mL−1). Thin films were prepared on fused silica substrates by spin-coating at 2000 rpm for 60 s to give thicknesses of 20–40 nm.Vapour generation and sensing measurement
The work on DFP was conducted in a fumehood by a trained professional wearing appropriate personal protective equipment. Fig. 9 shows the setup for generation of the DFP vapours and injection of pulses onto the film. Caution: di-iso-propyl fluorophosphate is a highly toxic compound and a comprehensive risk assessment should be undertaken before its synthesis, purification, use and disposal. To generate the saturated headspace vapour 2 μL of DFP was added to a 10 mL plastic syringe and kept for 30 min to allow the analyte evaporation at 20–22 °C. The vapour was then injected via syringe pump at a flow rate between 0 and 20 mL min−1 into a nitrogen flow (200 mL min−1) and the mixed gas was introduced into the optical chamber for the sensing measurement. The nitrogen stream was eventually passed through a scrubbing solution (20 wt% sodium hydroxide in water) to break down the excess simulant.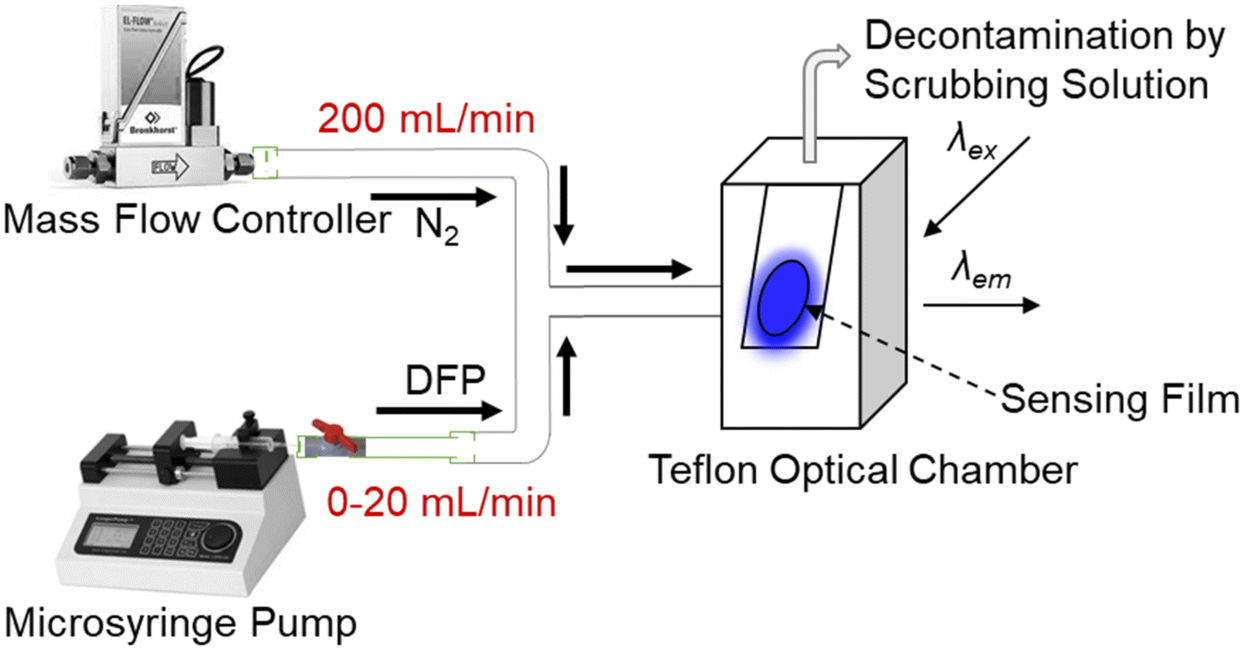 | ||
| Fig. 9 Setup for the sensing measurements using diluted DFP vapour. DFP vapour was diluted by mixing with a nitrogen flow and introduced to the optical chamber where the PL of the sensing film was recorded. | ||
The sensing film samples on fused silica substrates were mounted in a closed sample chamber. The sample chamber featured three optical windows to allow for excitation of the films and subsequent detection of the film PL at right angles to the excitation. The sample chamber was connected via optical fibre to the optical components. An LED light source (365 nm, OceanOptics) was used for the excitation and the film. PL spectra before and after exposure to analyte and the PL kinetics at the emissive peak were recorded using OceanView software (OceanOptics).
Data availability
The data supporting this article have been included as part of the ESI.† Further data is available upon reasonable request from the authors.Conflicts of interest
The authors (SF, PLB and PES) have filed a provisional patent relating to the results in the manuscript.Acknowledgements
This research was supported by funding from the Australian Research Council under the Discovery Program (DP170102072).References
- H. Peng, L. Ding and Y. Fang, J. Phys. Chem. Lett., 2024, 15, 849–862 CrossRef CAS PubMed.
- P. E. Shaw and P. L. Burn, Phys. Chem. Chem. Phys., 2017, 19, 29714–29730 RSC.
- Y. J. Jang, K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2015, 115, PR1–PR76 CrossRef CAS PubMed.
- X. Sun, Y. Wang and Y. Lei, Chem. Soc. Rev., 2015, 44, 8019–8061 RSC.
- Y. Salinas, R. Martínez-Máñez, M. D. Marcos, F. Sancenón, A. M. Costero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261–1296 RSC.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed.
- Y. Geng, M. A. Ali, A. J. Clulow, S. Fan, P. L. Burn, I. R. Gentle, P. Meredith and P. E. Shaw, Nat. Commun., 2015, 6, 8240 CrossRef CAS PubMed.
- S. Fan, G. Zhang, G. H. Dennison, N. FitzGerald, P. L. Burn, I. R. Gentle and P. E. Shaw, Adv. Mater., 2020, 32, 1905785 CrossRef CAS PubMed.
- V. Kumar, H. Kim, B. Pandey, T. D. James, J. Yoon and E. V. Anslyn, Chem. Soc. Rev., 2023, 52, 663–704 RSC.
- E. Butera, A. Zammataro, P. A. Pappalardo and G. T. Sfrazzetto, ChemPlusChem, 2021, 86, 681–695 CrossRef PubMed.
- S. Costanzi, J.-H. Machado and M. Mitchell, ACS Chem. Neurosci., 2018, 9, 873–885 CrossRef CAS PubMed.
- T. Franca, D. Kitagawa, S. Cavalcante, J. da Silva, E. Nepovimova and K. Kuca, Int. J. Mol. Sci., 2019, 20, 1222 CrossRef CAS PubMed.
- B. Zhu, R. Sheng, T. Chen, J. Rodrigues, Q.-H. Song, X. Hu and L. Zeng, Coord. Chem. Rev., 2022, 463, 214527 CrossRef CAS.
- W.-Q. Meng, A. C. Sedgwick, N. Kwon, M. Sun, K. Xiao, X.-P. He, E. V. Anslyn, T. D. James and J. Yoon, Chem. Soc. Rev., 2023, 52, 601–662 RSC.
- Q. Chen, J. Liu, S. Liu, J. Zhang, L. He, R. Liu, H. Jiang, X. Han and K. Zhang, Anal. Chem., 2023, 95, 4390–4394 CrossRef CAS PubMed.
- L. Chen, D. Wu and J. Yoon, ACS Sens., 2018, 3, 27–43 CrossRef CAS PubMed.
- S. Fan, G. H. Dennison, N. FitzGerald, P. L. Burn, I. R. Gentle and P. E. Shaw, Commun. Chem., 2021, 4, 45 CrossRef CAS PubMed.
- D. R. Heiss, D. W. Zehnder, D. A. Jett, G. E. Platoff, D. T. Yeung and B. N. Brewer, J. Chem., 2016, 3190891 Search PubMed.
- K. Skonieczny, A. I. Ciuciu, E. M. Nichols, V. Hugues, M. Blanchard-Desce, L. Flamigni and D. T. Gryko, J. Mater. Chem., 2012, 22, 20649–20664 RSC.
- K. Wang, F. Zhao, C. Wang, S. Chen, D. Chen, H. Zhang, Y. Liu, D. Ma and Y. Wang, Adv. Funct. Mater., 2013, 23, 2672–2680 CrossRef CAS.
- S. Fan, A. S. Loch, K. Vongsanga, G. H. Dennison, P. L. Burn, I. R. Gentle and P. E. Shaw, Small Methods, 2024, 8, 2301048 CrossRef CAS PubMed.
- M. Stein, S. J. Middendorp, V. Carta, E. Pejo, D. E. Raines, S. A. Forman, E. Sigel and D. Trauner, Angew. Chem., Int. Ed., 2012, 51, 10500–10504 CrossRef CAS PubMed.
- Y.-J. Pu, R. E. Harding, S. G. Stevenson, E. B. Namdas, C. Tedeschi, J. P. J. Markham, R. J. Rummings, P. L. Burn and I. D. W. Samuel, J. Mater. Chem., 2007, 17, 4255–4264 RSC.
- T. T. Tu, J. Mass Spectrom. Soc. Jpn., 1996, 44, 293–320 CrossRef.
- S. W. Wiener and R. S. Hoffman, J. Intensive Care Med., 2004, 19, 22–37 CrossRef PubMed.
- D. H. Rosenblatt, M. J. Small, T. A. Kimmell and A. W. Anderson, Background chemistry for chemical warfare agents and decontamination processes in support of delisting waste streams at the U.S. Army Dugway Proving Ground, Utah, OSTI, TN, USA, 1996, DOI:10.2172/258187.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00120f |
| This journal is © The Royal Society of Chemistry 2024 |