
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magneto-plasmonic bionanocomposites for on-site SERS detection of water contaminants†
Sofia F.
Soares
 ,
Nuno M. A. S.
Silva
,
João
Brenheiro
,
Sara
Fateixa
,
Ana L.
Daniel-da-Silva
and
Tito
Trindade
*
,
Nuno M. A. S.
Silva
,
João
Brenheiro
,
Sara
Fateixa
,
Ana L.
Daniel-da-Silva
and
Tito
Trindade
*
CICECO-Aveiro Institute of Materials and Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal. E-mail: tito@ua.pt
First published on 31st May 2024
Abstract
Magneto-plasmonic nanosystems have emerged as important multifunctional structures for several sensing applications, including on-site water quality monitoring. In this scenario, these nanosystems can integrate magnetic assisted separation procedures associated with optical detection of water contaminants, by exploring the surface-enhanced Raman scattering effect (SERS). Among the several modalities proposed for such magneto-plasmonic nanosystems, bionanocomposite particles have not been explored in this context. Hence, this research introduces bionanocomposites comprising magnetite cores that have been coupled to Au nanoparticles (NPs) via an intermediate surface modification step using hybrid shells of trimethyl chitosan-SiO2. The magnetic bionanocomposites were decorated with Au NPs by exploring two methods: their assembly with pre-synthesized Au colloids and as heterogeneous substrates for the in situ synthesis of Au NPs. The resulting magneto-plasmonic nanosystems are responsive to an external magnetic gradient and show the localized surface plasmon resonance (LSPR) band ascribed to the Au NPs. Therefore, such multifunctionality was explored here by assessing the SERS performance of the magneto-plasmonic substrates after their use as magnetic nanosorbents for the uptake of organic dyes, specifically methylene blue (MB) and rose bengal (RB), as water contaminant models. The results showed that both types of substrates are effective, though the ex situ bionanocomposites have shown better SERS activity. As such, the latter have been selected to further demonstrate the versatility of the bionanocomposites for the SERS detection of other types of water contaminants, such as salicylic acid (SA), a pharmaceutical compound that is classified as a teratogen substance. Overall, these findings indicate that magneto-plasmonic bionanocomposites, indeed can be explored as more sustainable platforms for analytical purposes, combining the ability for magnetic separation and SERS trace detection.
Introduction
Water pollution is a major global issue that poses considerable threat to human health and ecological environments.1,2 This issue encompasses a variety of water pollutants known as contaminants of emerging concern (CEC), which include vestiges of pharmaceutical compounds, pesticides and textile dyes, all of which can have detrimental effects on human health and various ecosystems.2 Furthermore, less than 1% of the surface water available on Earth is accessible for safe drinking; the scarcity of potable water makes of critical importance methods not only for efficiently treating wastewater but also for monitoring its quality.1,3 Unfortunately, conventional wastewater treatment methods for CEC often prove ineffective, resulting in the release of treated yet still contaminated effluents into the environment.4,5 Additionally, preventive measures should be firstly considered in relation to water remediation strategies. Addressing this challenge demands urgent development of efficient, environmentally friendly, and reliable sensing technologies capable of detecting trace pollutants in water, ensuring safe water supplies. Indeed, accurate analysis of water pollutants is essential for effective environmental safety monitoring.Surface-enhanced Raman scattering (SERS) has emerged as an attractive spectroscopic tool for detecting trace pollutants in water, owing to its distinctive molecular fingerprint, high sensitivity, and selectivity.6,7 The performance of SERS sensors depends upon several factors, with the characteristics of the analytical substrate being of paramount importance. Hence, extensive research has been conducted on metal surfaces featuring plasmonic nanoparticles with size- and shape-tuned properties.8–10 These metal nanostructures show localized surface plasmon resonances (LSPR) in the visible region of the spectrum, which contribute significantly for the observation of SERS spectra of molecular adsorbates when probed using a laser line with wavelength in that spectral window.11 Regions of the substrate in which the local electromagnetic field is strongly intensified are called hotspots, which have been described as occurring on the apexes of anisotropic nanometals or at the interfaces of clustered plasmonic nanoparticles.8,9 In parallel, the development of functional materials integrating such plasmonic nanoparticles has become an active field of research, namely for expanding the applicability of SERS sensors in a variety of practical contexts.12–14 In line with these endeavours, we have been engaged in fabricating SERS substrates for water pollutants detection, encompassing diverse metal-loaded nanocomposites, including textile fibres,15,16 filter membranes,17 paper18 and 3D scaffold composites.19 More recently, SERS substrates containing magnetic nanoparticles (MNPs) have also been explored as multifunctional magneto-plasmonic materials, namely as nanosorbents for water quality monitoring sensors.20–24
A magneto-plasmonic nanosorbent takes advantage of both functionalities conferred by its compositional phases, such as SERS activity due to plasmonic nanometals and the responsiveness to an external magnetic field due to magnetic nanoparticles.25,26 Furthermore, magneto-plasmonic materials are of great interest because the intertwining of magnetic and plasmonic behaviour can expand their effectiveness in new sensing applications.25,27,28 In particular, magneto-plasmonic materials are of great interest as nanosorbents for on-site water contaminants' detection since they are easily separated from the water sample, with the possibility of SERS analysis using Raman portable equipment. Moreover, these materials open an avenue for new analytical protocols that take advantage of magnetic separation and the sorbents' reuse. The presence of an external magnetic gradient can also be explored to increase the number of hotspots during the extraction and post-separation process, which can further improve the SERS sensitivity.20,21
Several studies have explored magneto-plasmonic nanostructures of different types, typically including plasmonic Ag/Au NPs and magnetic iron oxides, with different shapes, such as core–shell, core–satellite, non-spherical and multi-functionalized MNPs.20,21,29,30 Silver nanostructures might exhibit better SERS sensitivity and are more affordable,31,32 but in comparison to Au NPs, they are not so chemically stable and biocompatible. This is of relevance when considering applications that require analytical-kits with longer shelf-life time, such as on site analysis in remote regions of the globe.31,33 Moreover, by adjusting the size, shape, and concentration of Au NPs on the surfaces of magnetic-based nanocomposites, it is possible to create new materials with unique properties for SERS applications.33 The assembly of the magneto-plasmonic heterostructures has been mediated by chemical modification at the particles' surfaces, which might involve inorganic shells or organic coatings.20,30 The quest for eco-friendly and low-cost effective materials has prompted recent developments of biopolymer-based materials and their application in water decontamination. However, in this context, there are no reports of SERS studies employing magneto-plasmonic nanostructures comprising hybrid surfaces of biopolymeric origin, which is an important field that remains largely unexplored. This research explores for the first time, the application of magneto-plasmonic nanosorbents based on a biopolymer of natural origin (chitosan), as the surface modifier of magnetite cores upon which Au NPs have been attached. The hybrid nanosorbents were then employed as SERS substrates for detecting organic molecules dissolved in water, such as the organic dyes methylene blue (MB) and rose bengal (RB), and the pharmaceutical compound salicylic acid (SA). We also note that these bionanocomposites have been developed for SERS detection, which might integrate analytical kits that require just minor amounts of Au NPs, thus contributing to materials sustainability.
Experimental section
Materials
The following chemicals were used as purchased from Sigma-Aldrich: ferrous sulfate heptahydrate (FeSO4·7H2O, >99%), potassium nitrate (KNO3, >99%), sodium citrate tribasic dihydrate (Na3C6H5O7·2H2O, 99%), tetrachloroauric(III) acid trihydrate (HAuCl4·3H2O, >99.9%), tetraethyl orthosilicate (Si(OC2H5)4, TEOS, >99%) 3-(triethoxysilyl)propyl isocyanate ((C2H5O)3Si(CH2)3NCO, ICPTES, 95%) and rose bengal (RB, C20H2Cl4I4Na2O5, 95%). Potassium hydroxide (KOH, >86%) was supplied by LabChem. N,N-dimethylformamide (HCON(CH3)2, >99%) and ethanol (CH3CH2OH, >99%) were obtained from Carlo Erba Reagents. Methanol (CH3OH, >99%), ammonia solution (NH3, 25%) and hydrochloric acid (HCl, 37%) were acquired from VWR. Trimethyl chitosan (TMC, degree of quaternization 15.3%) was acquired from Henan Tianfu Chemical Co., Ltd (China). Methylene blue (MB, C16H18ClN3S) was purchased from Riedel-de-Häen. Salicylic acid (SA, HOC6H4COOH, >98%) was obtained from Fluka Chemika. All the aqueous solutions were freshly prepared using ultrapure water obtained from the Synergy equipment (18.2 MΩ·cm, Milli-Q, Millipore).Synthesis of magnetic bionanocomposites
The magnetic bionanocomposites were synthesized using a procedure comprising two-steps. First, spheroidal magnetite (Fe3O4) NPs (50 nm average diameter) were prepared via the oxidative hydrolysis of a Fe(II) salt in alkaline aqueous medium.34 Then, the magnetite cores were subsequently coated with biopolymer/siliceous hybrid shells using sol–gel chemistry.35,36 For the synthesis of the Fe3O4 NPs, KOH 1.90 g of and 1.52 g of KNO3 were added to 25 mL of deoxygenated water. The resulting mixture was heated to 60 °C under N2 stream and mechanical stirring (500 rpm). After complete dissolution, 25 mL of an aqueous solution containing 4.75 g FeSO4·7H2O was added dropwise to the heated solution, and the stirring speed was increased to 700 rpm. The reaction was left to proceed for 30 min. Then, the mixture was heated to 90 °C for 4 h, without stirring. The resulting black powder was magnetically collected and then washed with deoxygenated water and ethanol, followed by drying at room temperature in a desiccator containing silica gel.The Fe3O4 NPs were coated with hybrid shells through alkaline hydrolysis and condensation of a mixture of TEOS and chemically modified trimethyl chitosan (TMC). The biopolymer TMC was modified via reaction with the alkoxysilane coupling agent 3-(triethoxysilyl)propyl isocyanate (ICPTES). Initially, 1 g of dry TMC was dispersed in 13 mL of N,N-dimethylformamide and the mixture was then heated to 100 °C in a N2 atmosphere, while stirring (500 rpm). Then, 1.3 mL of ICPTES was added to the mixture and the reaction was left to proceed for 24 h. Afterwards, the resulting modified TMC precursor (SiTMC) was thoroughly washed with dry methanol and dry ethanol, and dried by solvent evaporation. For the coating step, an ethanolic suspension containing 40 mg of Fe3O4 NPs in 38 mL was prepared under sonication, using a horn Sonics-Vibracell, for 15 min while the reaction flask was kept in an ice bath. Then, 0.406 mL of TEOS, 0.3 g of SiTMC and 2.4 mL of ammonia solution were added to the mixture. The reaction was allowed to proceed for 2 h under sonication and in an ice bath. The resulting magnetic bionanocomposites (Fe3O4@SiO2/SiTMC) were magnetically separated from the solution using a NdFeB magnet, and then washed with ethanol and subsequently dried at room temperature.
Synthesis of magneto-plasmonic bionanocomposites
The magneto-plasmonic bionanocomposites were prepared through two distinct approaches. In the ex situ method, pre-synthesized colloidal Au NPs were assembled onto the magnetic bionanocomposites, while in the in situ method, the Au colloids have been prepared in the presence of such bionanocomposites. The Au colloids were previously prepared by the reduction of [AuCl4]− using sodium citrate in aqueous solution.37 Briefly, 50 mL of a solution of HAuCl4·3H2O (1 mM) in ultra-pure water was heated for 10 min at 95 °C under reflux conditions with vigorous stirring (500 rpm). Then, 5 mL of sodium citrate (Na3C6H5O7·2H2O) aqueous solution (38.8 mM) was added. The reaction was left to proceed for 1 h and afterwards was gradually cooled to room temperature. For the ex situ method, an aqueous mixture containing 10 mg of Fe3O4@SiO2/SiTMC NPs was mechanically stirred (300 rpm) with 10 mL of citrated coated Au colloid, at room temperature for 24 h and in the dark. Subsequently, the resulting bionanocomposites (Fe3O4@SiO2/SiTMC/Auex situ) were separated magnetically from the solution using a NdFeB magnet, and dried before characterization. The in situ synthesis followed identical experimental conditions to those used in the synthesis of the Au colloid, as described above, but in the presence of the magnetic bionanocomposites. Typically, 50 mg of Fe3O4@SiO2/SiTMC were added to the initial HAuCl4·3H2O (1 mM) aqueous solution (50 mL), with mechanically stirring (500 rpm) for 10 min. Then, the mixture was heated to 95 °C under reflux conditions and stirred (500 rpm). Finally, 5 mL of a sodium citrate solution (38.8 M) was added. The reaction proceeded over 1 h before cooling down to room temperature. The magneto-plasmonic bionanocomposites (Fe3O4@SiO2/SiTMC/Auin situ) were then collected from the solution using a magnet NdBFe, and subsequently dried.Instrumentation and methods
The UV-VIS spectra of samples' solutions were obtained using quartz cuvettes placed in a GBC Cintra 303 UV-visible spectrophotometer. After the sorption experiments, the concentration of analyte in the supernatant solution was determined spectrophotometrically by monitoring the absorbance at 663 nm (MB), 549 nm (RB) and 230 nm (SA). The calibration curves were obtained by the best linear fitting applied to experimental data obtained for solutions of known concentration in the respective analyte (Fig. S1, ESI†). For each nanosorbent, the adsorption capacity was calculated using eqn (1), where C0 and Ct (mg L−1) are the concentration in solution at time t = 0 and t, respectively: | (1) |
Zeta potential and dynamic light scattering (DLS) measurements were recorded using a Malvern Zetasizer Nano ZS equipped with a standard 633 nm laser.
Transmission electron microscopy (TEM) was carried out using a JEOL 2200FS instrument operating at 200 kV, equipped with Oxford energy-dispersive X-ray spectroscopy (EDS) detector. Samples for TEM were prepared by placing an aliquot of an aqueous suspension of the bionanocomposites onto the surface of a Cu grid coated with amorphous carbon film and then let the solvent to evaporate. The powder X-ray diffraction (XRD) patterns of the crystalline materials were obtained using a PANalytical Empyrean X-ray diffractometer equipped with a Cu-Kα monochromatic radiation source at 45 kV/40 mA.
Fourier transform infrared (FTIR) spectra of the samples were measured using the solid state. The FTIR spectra of the materials were collected using a Bruker Optics Tensor 27 spectrometer coupled to a horizontal attenuated total reflectance (ATR) cell, using 256 scans at a resolution of 4 cm−1. The elemental analysis of carbon, nitrogen, hydrogen, and sulfur was obtained using a Leco Truspec-Micro CHNS 630-200-200.
The Raman studies were carried out using a confocal Raman-AFM-SNOM microscope WiTec alpha300 RAS+ using a He:Ne laser operating at 633 nm (0.1–0.2 mW). Confocal Raman microscopy was employed in all the Raman imaging experiments. High-resolution Raman images were obtained for selected Fe3O4@SiO2/SiTMC/Au samples treated either with MB or RB solutions. The Raman images were obtained by recording 150 × 150 Raman spectra (in total 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 spectra) in an area of 30 μm × 30 μm, with the integration time for each spectrum set as 0.05 s for MB and 0.1 s for RB (0.1 mW of laser power, 100× objective). Raman images were obtained by the integration of the absolute area of the MB Raman band at 1621 cm−1 (ascribed to the νin plane(CC) + δ(CH) (ring), ν(CN) + ν(CC)) and RB Raman band at 1619 cm−1 (ascribed to νsym(C
500 spectra) in an area of 30 μm × 30 μm, with the integration time for each spectrum set as 0.05 s for MB and 0.1 s for RB (0.1 mW of laser power, 100× objective). Raman images were obtained by the integration of the absolute area of the MB Raman band at 1621 cm−1 (ascribed to the νin plane(CC) + δ(CH) (ring), ν(CN) + ν(CC)) and RB Raman band at 1619 cm−1 (ascribed to νsym(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) (ring)) using WITec software. The portable Raman SERS experiments were performed using the portable Raman spectroscopic module C13560 from Hamamatsu Photon Business with the sample excitation source operating at 785 nm (5 mW). Each Raman spectrum was acquired with ten acquisitions, 1000 ms each acquisition. For the calculation of the enhancement factors (EF), we have employed the Raman band at 1621 cm−1 (MB) and 1619 cm−1 (RB), using the following equation:38
C) (ring)) using WITec software. The portable Raman SERS experiments were performed using the portable Raman spectroscopic module C13560 from Hamamatsu Photon Business with the sample excitation source operating at 785 nm (5 mW). Each Raman spectrum was acquired with ten acquisitions, 1000 ms each acquisition. For the calculation of the enhancement factors (EF), we have employed the Raman band at 1621 cm−1 (MB) and 1619 cm−1 (RB), using the following equation:38
 | (2) |
SERS studies using magneto-plasmonic bionanocomposites as substrates
The magneto-plasmonic bionanocomposites were investigated as SERS substrates for detecting MB, RB, and SA dissolved in water. The SERS experiments were conducted using as the substrates, the magneto-plasmonic bionanocomposites magnetically collected after the treatment of aqueous solutions containing the above analytes. For all SERS measurements, control experiments have been carried out by using as substrates the magnetic bionanocomposites, i.e. the samples without plasmonic Au NPs present. In a typical sample preparation experiment, 5 mg of the bionanocomposites were dispersed in an aqueous solution (10 mL, pH = 6) of the selected analyte; the concentration of the analyte varied in the range 1 × 10−5–1 × 10−9 M for MB, 5 × 10−5–5 × 10−9 M for RB, and 1 × 10−3 M for SA. The aqueous mixtures containing both the analyte and the nanosorbents were incubated over 4 h at 25 °C ± 1 °C, using a vertical rotating mixer (30 rpm). After this period, the bionanocomposites were separated magnetically from the test solution using a NdFeB magnet and were then washed with ultrapure water. Finally, an aliquot of 10 μL of an aqueous suspension of the bionanocomposites was deposited onto glass slides and allowed to air-dry, at room temperature. The Raman spectra of samples prepared as above were acquired by recording five spectra (10 acquisitions, 2 s each) on each sample, and an average Raman spectrum was obtained using the WITec software (Project 5.3+).Results and discussion
Materials characterization
We report the first example of nanosystems comprising magneto-plasmonic bionanocomposites for SERS detection. Two distinct chemical routes have been investigated for their preparation, which are termed here as ex situ and in situ methods (Fig. 1). In both cases, the bionanocomposites share similar qualitative composition, specifically, they comprise plasmonic Au NPs, ferrimagnetic Fe3O4 NPs and, hybrid shells of SiO2 and trimethyl chitosan. This polysaccharide contains cationic trimethylammonium groups that are expected to promote the electrostatic interactions between the coated magnetic particles to the anionic citrate coated colloidal Au NPs, thus conferring to the final bionancomposites Au surfaces for Raman signal enhancement. | ||
| Fig. 1 Scheme illustrating of the preparation of the magneto-plasmonic bionanocomposites using the ex situ and in situ methods. | ||
Firstly, the Fe3O4 cores were prepared by oxidative hydrolysis of Fe(II) in an alkaline medium,34 which resulted in spheroidal shaped ferrimagnetic particles with an average size of 56 ± 11 nm (Fig. 2a) and 83 emu g−1 of saturation magnetization.39 The Fe3O4 cores were then coated with hybrid siliceous shells enriched in TMC, resulting in core/shell structures whose TEM images are shown in Fig. 2b. The presence of organic moieties in the shells was confirmed by elemental analysis of the carbon and nitrogen content (Table 1). Negligible carbon and nitrogen content (<0.1%) were detected in the non-modified Fe3O4 NPs, and an increase in the carbon (33.3%) and nitrogen (6.3%) content was observed in the Fe3O4@SiO2/SiTMC bionanocomposites. These observations are in line with our earlier syntheses on several magnetic bionanocomposites containing silicious shells and distinct polysaccharides.35,36,39
 | ||
| Fig. 2 TEM images of a) Fe3O4 and b) Fe3O4@SiO2/SiTMC NPs. | ||
| Sample | C (%) | H (%) | N (%) |
|---|---|---|---|
| TMC | 36.96 | 6.63 | 6.55 |
| SiTMC | 36.91 | 6.55 | 6.60 |
| Fe3O4 | 0.08 | 0.25 | 0.08 |
| Fe3O4@SiO2/SiTMC | 33.34 | 5.59 | 6.31 |
As illustrated in Fig. 1, the magnetic bionanocomposites were used as substrates to promote the electrostatic attachment of previously prepared colloidal AuNPs (ex situ method) or the growth of Au NPs (in situ method). In both strategies, the Au NPs were obtained by colloid synthesis employing sodium citrate as the reducing agent and HAuCl4 as the Au(III) precursor.37 This method has been extensively used for preparing citrate coated Au nanospheres, which in this case showed 12.2 ± 1.2 nm in average diameter and a LSPR band in the visible spectrum peaked at 520 nm (Fig. S2, ESI†). The zeta potential was determined as −37.1 ± 1.5 mV (pH = 5.9), which is due to the negatively charged coating composed of citrate anions.
Fig. 3 shows the TEM images of bionanocomposites synthesized using both methods: ex situ (sample Fe3O4@SiO2/SiTMC/Auex situ) and in situ (sample Fe3O4@SiO2/SiTMC/Auin situ). The TEM images of the final bionanocomposites show the magnetic hybrid substrates decorated with AuNPs (Fig. 3). The latter are clearly distinguished by their average size (12.2 ± 1.2 nm) in the ex situ nanocomposites (Fig. 3a), though particle agglomerates are also observed. For the in situ nanocomposites (Fig. 3b), the TEM images showed Au nanospheres with comparable average diameter (13.1 ± 4.8 nm) but more dispersed over the samples.
 | ||
| Fig. 3 TEM images of the magneto-plasmonic bionanocomposites prepared by the two distinct routes: a) Fe3O4@SiO2/SiTMC/Auex situ and b) Fe3O4@SiO2/SiTMC/Auin situ. | ||
Also note that the TEM images of the nanocomposites produced in situ show morphological alterations on the outer shells, suggesting a smoother coating after thermal treatment (95 °C) during the synthesis of the Au NPs, as compared to the non-decorated bionanocomposites (Fig. 2b). The spatial distribution of the nanophases over the ex situ and in situ nanocomposites was investigated through EDS mapping analysis (Fig. 4) and is detailed in the EDS spectra provided in Fig. S3 (ESI†). The EDS maps clearly show that the magnetic particles are present in the bionanocomposites cores, while the Au NPs are mostly located at the outer regions delimited by the hybrid silicious shells.
 | ||
| Fig. 4 TEM images and EDS maps of the Fe3O4@SiO2/SiTMC/Auex situ and Fe3O4@SiO2/SiTMC/Auin situ bionanonanocomposites. | ||
The XRD patterns of the powdered samples also confirmed the presence of the crystalline phases (Fe3O4 and Au) in the nanocomposites. Hence, Fig. S4 (ESI†) shows the reflections (220), (311), (400), (422), (511), and (440) for the Fe3O4@SiO2/SiTMC/Auex situ and Fe3O4@SiO2/SiTMC/Auin situ samples, that match the reported diffraction patterns of magnetite (Fe3O4) with cubic inverse spinel structure (JCPDS Card No. 19-0629).40 Additionally, the three additional reflections that appeared in the diffractogram of the ex situ and in situ nanocomposites are ascribed to the (111), (200) and (220) reflections of the Au face-centered cubic (fcc) crystalline structure (JCPDS Card No. 04-0784).41
The surface charge of the magnetic nanocomposites was evaluated through zeta potential measurements. Fig. 5 shows the zeta potential results conducted at pH range 5 to 8. The original positively charged Fe3O4@SiO2/SiTMC particles became less positive upon electrostatic attachment of citrate coated AuNPs, which have a negative surface charge (−37.1 ± 1.6 mV). Regardless of the pH range tested, we consistently observed that the surface charge of the Fe3O4@SiO2/SiTMC/Auin situ sample was lower compared to the ex situ nanocomposites. We interpret this observation as consequence of the partial loss of the hybrid coating due to the thermal treatment employed in the in situ synthesis.
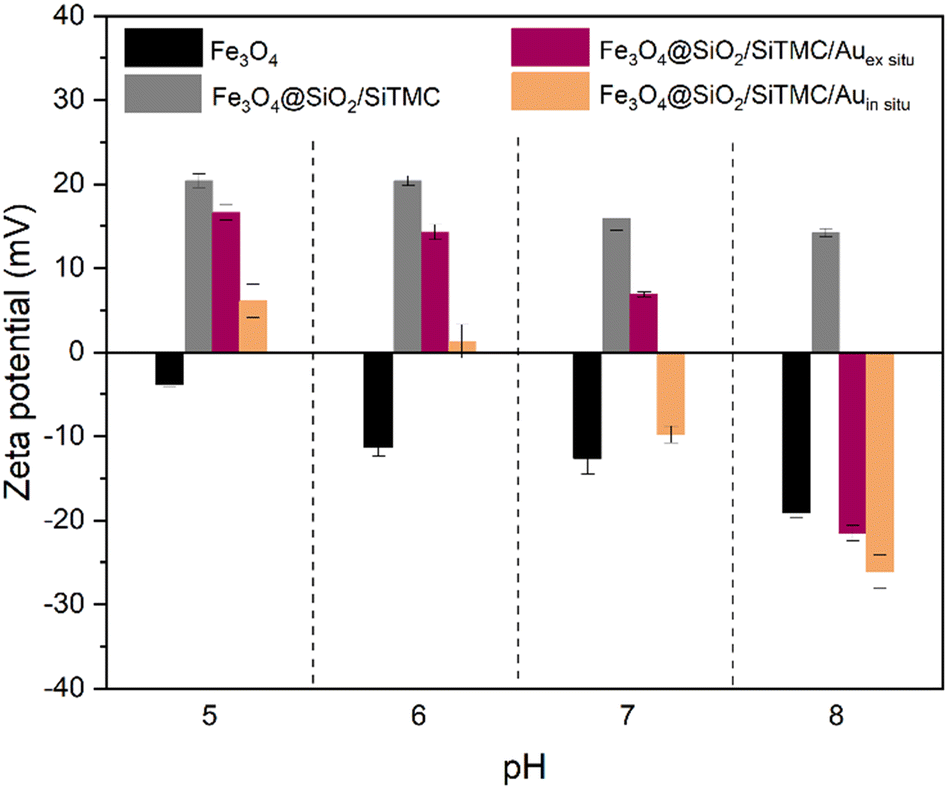 | ||
| Fig. 5 Results of zeta potential measurements performed on colloidal samples of Fe3O4, Fe3O4@SiO2/SiTMC, Fe3O4@SiO2/SiTMC/Auex situ and Fe3O4@SiO2/SiTMC/Auin situ, in the pH range 5 to 8. | ||
Fig. 6 shows the ATR-FTIR spectra of Fe3O4@SiO2/SiTMC modified with AuNPs (both ex situ and in situ methods), highlighting the wavenumbers of characteristic IR bands. The spectra of commercial TMC, SiTMC, Fe3O4, and the Fe3O4@SiO2/SiTMC particles are also shown for comparative purposes. The FTIR spectra of Fe3O4@SiO2/SiTMC/Auex situ and Fe3O4@SiO2/SiTMC/Auin situ show a vibrational band at 557 cm−1, which is attributed to the Fe–O lattice vibration.34 In addition, the vibrational bands characteristic of TMC moieties are observed within the range 1000 to 1633 cm−1,36,42 which include the CO stretching (amide I) and N–H bending (amide II) vibrations at 1628 and 1525 cm−1, respectively.36,42 A less intense band is observed at 1403 cm−1 which might be assigned to the deformation vibration of CH3 group present in TMC.35,36,42 Moreover, the bands between 1000 and 1200 cm−1 were indicative of C–O vibrations of the TMC backbone, while the band at 2887 cm−1 is characteristic of asymmetrical stretching of C–H bonds and the band at 3303 cm−1 is associated with O–H bonds.42 The silicious domains in the hybrid shells is also confirmed by the observation of vibrational bands at 436 cm−1 and 1046 cm−1, assigned to the O–Si–O deformation mode and Si-O-Si asymmetric stretching, respectively.35,36 Notably, the latter overlaps with the most intense bands of TMC.
 | ||
| Fig. 6 ATR-FTIR spectra of TMC, SiTMC, Fe3O4, Fe3O4@SiO2/SiTMC, Fe3O4@SiO2/SiTMC/Auex situ and Fe3O4@SiO2/SiTMC/Auin situ. | ||
SERS studies using bionanocomposites as substrates
The magneto-plasmonic bionanocomposites (Fe3O4@SiO2/SiTMC/Auex situ and Fe3O4@SiO2/SiTMC/Auin situ) described above were investigated as multifunctional nanosorbents for SERS detection of selected pollutants dissolved in water. Firstly, the SERS activity and adsorption capabilities of the nanocomposites were initially assessed using two dyes as model analytes, specifically MB and RB. MB is a cationic dye commonly used as a colorant and is used in methemoglobinemia treatment, while RB serves as a fluorophore with relevant applications in biology and medicine; both dyes have applications as photosensitizing agents in photodynamic therapies.43,44 Additionally, these organic dyes occur as distinctly charged molecular species in aqueous solutions at pH 6, which was set as the working pH value. Hence, in aqueous solution at pH 6, RB is predominantly present in the form of anions (pKα1 = 1.9 and pKα2 = 3.9).45,46 The UV-VIS spectra of MB and RB aqueous solutions are shown in Fig. 7a and b, respectively. The uptake of the organic dyes by the dispersed nanosorbents was assessed by monitoring the absorbance in the electronic spectra of post-treated solutions containing the dye. It was observed that both ex situ and in situ nanocomposites exhibited greater affinity for RB (69–73%) as compared to MB (<1%) (Fig. S5, ESI†). This outcome was expected because a solution at pH 6 renders the surface of the colloidal nanocomposite particles positively charged (+14 mV and +1 mV, for ex situ and in situ, respectively). | ||
| Fig. 7 Electronic spectra of a) methylene blue and b) rose bengal; average SERS spectra of c) methylene blue (1 × 10−5 M) and d) rose bengal (5 × 10−5 M) using Fe3O4@SiO2/SiTMC, Fe3O4@SiO2/SiTMC/Auex situ and Fe3O4@SiO2/SiTMC/Auin situ nanocomposites as substrates, under 633 nm laser excitation. | ||
The Raman bands of the neat SERS substrates, i.e. prior adsorption of the analytes, and the respective band assignments are shown in Fig. S6 and Table S1 (ESI†).15,47,48 The laser power used for conducting the SERS studies was set at a low level (0.1 mW) to avoid photodegradation of the organic dyes (MB and RB). At such low laser power settings, we have observed the enhanced Raman bands of the molecular analytes, but not the Raman bands of the underlying substrates. Fig. 7c and d show the SERS spectra of MB and RB, using as substrates the magneto-plasmonic bionanocomposites described above. In contrast, Fe3O4@SiO2/SiTMC particles without Au NPs, which were used as a substrate in control experiments, did not result in Raman signal for MB or RB. Both the ex situ and in situ nanocomposites exhibited SERS activity in the detection of MB at a concentration of 1 × 10−5 M in the sample solution (Fig. 7c). Note that the SERS signal for MB was observed even though the extent of its adsorption on the Au containing bionanocomposites was minimal (less than 1% of the MB concentration). This suggests that MB molecules exhibit a preferential adsorption tendency toward the surface of gold nanoparticles.49 Furthermore, because in this case the excitation line (633 nm) overlaps the absorption band of MB peaked at 663 nm, there might be additional band enhancement due to surface-enhanced resonance Raman scattering (SERRS). In fact, characteristic Raman bands of MB are clearly observed in the respective spectrum (Fig. 7c), including those at 448 cm−1 attributed to the skeletal deformation (CN, CS and CH3), 480 cm−1 associated with δin-plane thiazine ring, 1396 cm−1 attributed to νsym(CN) (lateral and centre) + δin-plane (CH) ring + δout-plane (CH), δ(CH3), ν(CN) + ν(CC), νasym(CN) and 1621 cm−1 associated with ν(CC) + δin-plane (CH) (ring), ν(CN) + ν(CC).15,47 However, upper closer examination of the MB Raman spectra obtained using both nanocomposites, differences in the relative intensity of the bands at 448 and 480 cm−1 become apparent. These vibrational bands are indicative of the presence of monomer and dimer forms of MB. Specifically, the band at 480 cm−1 (associated with the thiazine group) is attributed to the monomer form, while the band at 448 cm−1 (corresponding to CN skeletal deformation) is associated with MB dimers.50,51 The MB Raman spectrum obtained using the Fe3O4@SiO2/SiTMC/Auin situ nanocomposite exhibited both bands (448 and 480 cm−1), indicating a mixture of monomer and dimer (orange line, Fig. 7c). Conversely, the Raman spectrum of MB on the Fe3O4@SiO2/SiTMC/Auex situ substrates shows a strong band at 480 cm−1, which indicates the predominance of the monomer form (purple line, Fig. 7c). These findings suggest that the method used to fabricate the bionanocomposites influence the experimental SERRS spectrum, which is consistent with distinct surface effects caused by the differences observed in their morphological characteristics.
The Raman spectra presented in Fig. 7d demonstrate the successful SERS detection of RB molecules (5 × 10−5 M) using both bionanocomposites as substrates. Specifically, the spectra show the three major Raman bands located at 614 cm−1, 1488 cm−1 and 1619 cm−1, which were not detected when using the Fe3O4@SiO2/SiTMC sample, as a control substrate. These characteristic peaks are associated with the (C–I) stretching mode and, the asymmetric and symmetric stretching of (CC) bonds within the aromatic rings of RB, respectively.44,48,52 The enhancement of these Raman bands suggests that RB molecules are interacting with Au surface in such way that the –O– and –COO– groups of RB molecules are oriented towards the –N+(CH3)3 group of the TMC. Similar findings have been previously reported for RB molecules adsorbed on the surface of Au nanorods, Au nanospheres and Ag films.48,53,54
A comparative analysis of the SERS performance of the two bionanocomposite substrates, while indicating that both magneto-plasmonic nanostructures can be explored as Raman amplifiers for sensing devices, the ex situ sample showed better performance (see Fig. S7†). As such, the latter has been selected in our subsequent experiments, including to further investigate the spatial distribution and spectral information of the dye molecules onto Fe3O4@SiO2/SiTMC/Auex situ composites using high-resolution Raman imaging. One plausible reason for the differences observed in the SERS performance between the two samples may result from the spatial arrangement of the Au nanophases within them, as revealed by EDS mapping analysis (Fig. 4 and S3†). The EDS maps show a more uniform distribution of the Au nanophases over the ex situ bionanocomposite, characterized by extensive regions having interparticle gaps to interact with the analyte molecules. In contrast, the in situ sample exhibits a clustering of Au NPs primarily concentrated in certain surface regions. Fig. 8 presents the optical images with the scanned area marked in red, together with the respective 2D Raman images, showing the spatial distribution of MB (1 × 10−5 M) and RB (5 × 10−5 M) molecules over the Fe3O4@SiO2/SiTMC/Auex situ bionanocomposites. The absolute area underneath the selected Raman bands of the organic dyes, 1621 cm−1 for MB (ascribed to the νin plane(CC) + δ(CH) (ring), ν(CN) + ν(CC)) and 1619 cm−1 for RB (ascribed to νsym(CC) (ring)) were integrated and used to establish the colour intensity scale in the Raman image (Fig. 8). The brighter colour in the Raman image indicates a stronger SERS signal for the molecular analyte under analysis. This not only probes the MB or RB molecules adsorbed on the bionanocomposites but also provides information about the distribution of the Au nanophases on the substrates.
 | ||
| Fig. 8 Optical images (left) and Raman images (right) obtained by integrating the Raman intensity of a) the MB's band at 1621 cm−1 and b) RB's band at 1619 cm−1 recorded using the Fe3O4@SiO2/SiTMC/Auex situ composites as substrates (excitation at 633 nm, 0.1 mW laser power). The vertical bar shows the colour profile in each image, with the relative intensity scale. | ||
To further evaluate the SERS sensitivity of the Fe3O4@SiO2/SiTMC/Auex situ bionanocomposites in detecting MB and RB, these substrates were treated with aqueous solutions with varying concentration in the analyte, respectively for MB (1 × 10−5 M to 1 × 10−9 M) and RB (5 × 10−5 M to 5 × 10−9 M). Fig. 9a and b show the respective Raman spectra, which provide information concerning the concentration detection limits in these experimental conditions. Remarkably, the characteristic Raman peaks of MB can still be observed in solutions at the concentration of 1 × 10−9 M. This demonstrates the high sensitivity of the ex situ composites, enabling them to achieve trace-level detection of MB by SERRS. In the case of RB, Raman characteristic peaks of RB could still be observed when the concentration was as low as 5 × 10−7 M. We have also calculated the EF for MB (10−9 M) and RB (5 × 10−7 M), giving values of 1.7 × 106 for MB and 3.8 × 103 for RB. These EF values are comparable to data published in the literature for magneto-plasmonic particles and dyes.55,56
 | ||
| Fig. 9 Average SERS spectra of a) methylene blue and b) rose bengal with different concentrations enhanced by Fe3O4@SiO2/SiTMC/Auex situ nanocomposites under 633 nm laser excitation. | ||
SERS sensors enable fast on-site detection of water pollutants using currently affordable portable Raman instruments, thereby reducing the time needed for sample transportation and analysis. As a proof of concept, Fe3O4@SiO2/SiTMC/Auex situ bionanocomposites were utilized as substrates to detect MB and RB using a portable Raman device. Fig. 10 shows the SERS spectra of MB (1 × 10−4 M and 1 × 10−5 M) and RB (5 × 10−5 M) solutions deposited on Fe3O4@SiO2/SiTMC/Auex situ substrates. Note that the characteristic Raman features of MB and RB were distinctly observed, consistent with those shown in Fig. 9.
 | ||
| Fig. 10 SERS spectra of methylene blue (1 × 10−4 M and 1 × 10−5 M) and rose bengal (5 × 10−5 M) at variable concentrations using the Fe3O4@SiO2/SiTMC/Auex situ nanocomposites in conjunction with a portable Raman device. | ||
The long-term SERS sensitivity of the Fe3O4@SiO2/SiTMC/Auex situ bionanocomposites was investigated over a period of 6 months, keeping the MB and RB as molecular probes. Despite the observed decrease in the Raman intensities (Fig. 11), these results indicate that the bionanocomposites still present SERS activity after a period of 6 months, which is crucial for ensuring practical detection applications on the site in remote areas.
 | ||
| Fig. 11 Average SERS spectra of a) methylene blue and b) rose bengal using Fe3O4@SiO2/SiTMC/Auex situ nanocomposites as synthesized and after 6 months of sensor storage. | ||
To expand the applicability of our SERS substrates, their effectiveness in detecting salicylic acid (SA) was also investigated. Salicylic acid is a contaminant that might be found in wastewater effluents and potable water, and the conventional water purification methods often prove insufficient in removing this pollutant.57 Moreover, salicylic acid has demonstrated its SERS-activity when interacting with metal nanoparticles.58 The UV-VIS spectrum of SA, along with its chemical structure, is depicted in Fig. 12a. Both ex situ and in situ nanocomposites showed affinity to remove SA (36–28% removal, Fig. S5, ESI†). The SA adsorption can be ascribed to the electrostatic attraction between deprotonated SA molecules (pKα = 2.7)59 and the positively charged surface of the bionanocomposites (at pH 6). Fig. 12b shows the Raman and SERS spectra of SA using these substrates. Firstly, it should be noted that no Raman signal was detected when using a SA solution at a concentration of 1 × 10−3 M. Likewise, no Raman signal was observed using the non-plasmonic Fe3O4@SiO2/SiTMC substrates used as control. However, the Raman spectrum probed at the surface of Fe3O4@SiO2/SiTMC/Auex situ substrates previously treated with a SA solution (1 × 10−3 M), originate enhanced Raman bands in the spectrum (Fig. 12b). Specifically, the SERS spectrum shows three major bands located at 1625 cm−1 (CO stretching mode) and the vibrational modes of the benzene ring at 1583 cm−1 and 1390 cm−1.58,60 These results confirm our findings above, that ex situ bionanocomposites have potential for developing magneto-plasmonic sensors for routine environmental monitoring of certain water contaminants.
 | ||
| Fig. 12 Electronic spectrum of a) salicylic acid solution and b) Raman and SERS spectra of salicylic acid (1 × 10−3 M) using Fe3O4@SiO2/SiTMC, Fe3O4@SiO2/SiTMC/Auex situ and Fe3O4@SiO2/SiTMC/Auin situ nanocomposites as substrates under 633 nm laser excitation. | ||
Conclusions
This study focused on the preparation and evaluation of magneto-plasmonic bionanocomposites, synthesized via ex situ and in situ methods, as nanosorbents for optical detection of selected water pollutants. The bionanocomposites reported here take advantage of the properties of each component, resulting in multifunctional nanosorbents for SERS analysis. Hence, the magnetite cores facilitate the rapid and efficient separation of the nanosorbents from treated aqueous samples, while the hybrid shells enhance the chemical stability of the particles and promote the attachment of Au nanophases, which significantly enhanced the Raman signals of molecular analytes adsorbed near the metal surfaces. In addition, the functional groups of the biopolymer can contribute to capturing the target analyte near the surface of the plasmonic nanoparticles, thereby enhancing sensing capabilities. Moreover, it was demonstrated that the ex situ nanocomposites showed superior performance in the SERS detection of several water pollutants, including methylene blue, rose bengal and salicylic acid, even after a period of 6 month. We anticipate that the approach reported here paves the way to develop magneto-plasmonic sensors based on other biocomposites, which hold promise for SERS applications applied to water quality monitoring using on site analysis protocols.Author contributions
Conceptualization: TT; methodology: SFS and TT; formal analysis and investigation: SFS, NMASS, JB, SF, ALD-d-S and TT; writing – original draft preparation: SFS and SF; writing – review and editing: SFS, SF, ALD-d-S and TT; funding acquisition: ALD-d-S and TT; resources: ALD-d-S and TT; supervision: SFS and TT.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is financed by Portugal 2020 through European Regional Development Fund (ERDF) in the frame of CENTRO2020 in the scope of the project BIOMAG, CENTRO-01-0145-FEDER-181268 and was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 (DOI: https://doi.org/10.54499/UIDB/50011/2020), UIDP/50011/2020 (DOI: https://doi.org/10.54499/UIDP/50011/2020) & LA/P/0006/2020 (DOI: https://doi.org/10.54499/LA/P/0006/2020), financed by national funds through the FCT/MCTES (PIDDAC). S.F. thanks FCT for her research contract (REF-069-88-ARH-2018), which is funded by national funds (OE) through FCT-Fundação para a Ciência e a Tecnologia, I.P., in the scope of the framework contract foreseen in the numbers 4, 5, and 6 of the article 23, of the Decree-Law 57/2016, of August 29, changed by Law 57/2017, of July 19. ALD-d-S acknowledges FCT for funding (CEECIND/03075/2018/CP1559/CT0020; DOI: https://doi.org/10.54499/CEECIND/03075/2018/CP1559/CT0020).References
- J. K. Bediako, Y. El Ouardi, E. S. Massima Mouele, B. Mensah and E. Repo, Chemosphere, 2023, 325, 138418 CrossRef CAS PubMed.
- S. Shukla, R. Khan and A. Daverey, Environ. Technol. Innovation, 2021, 24, 101924 CrossRef CAS.
- N. B. Singh, G. Nagpal, S. Agrawal and Rachna, Environ. Technol. Innovation, 2018, 11, 187–240 CrossRef.
- M. L. Sikosana, K. Sikhwivhilu, R. Moutloali and D. M. Madyira, Procedia Manuf., 2019, 35, 1018–1024 CrossRef.
- J. You, L. Wang, Y. Zhao and W. Bao, J. Cleaner Prod., 2021, 281, 124668 CrossRef CAS.
- S. Fateixa, H. I. S. Nogueira and T. Trindade, Phys. Chem. Chem. Phys., 2015, 17, 21046–21071 RSC.
- P. C. Pinheiro, A. L. Daniel-da-Silva, H. I. S. Nogueira and T. Trindade, Eur. J. Inorg. Chem., 2018, 2018, 3443–3461 CrossRef CAS.
- Y. Zhou, Y. Lu, Y. Liu, X. Hu and H. Chen, Biosens. Bioelectron., 2023, 228, 115231 CrossRef CAS PubMed.
- L. Wang, M. Hasanzadeh Kafshgari and M. Meunier, Adv. Funct. Mater., 2020, 30, 2005400 CrossRef CAS.
- J. Krajczewski, K. Kołątaj and A. Kudelski, RSC Adv., 2017, 7, 17559–17576 RSC.
- E. C. Le Ru and P. G. Etchego, in Principles of surface-enhanced Raman spectroscopy, Elsevier, New Zealand, 2009 Search PubMed.
- A. Szaniawska and A. Kudelski, Front. Chem., 2021, 9, 664134 CrossRef CAS PubMed.
- U. Tamer, H. Torul, Ü. Doğan, M. Eryılmaz, A. Gümüştaş, İ. H. Boyacı, S. A. Özkan and B. Uslu, in Nanotechnology Applications in Health and Environmental Sciences, ed. N. Saglam, F. Korkusuz and R. Prasad, Springer Cham, 2021, pp. 197–236 Search PubMed.
- C. Li, Y. Huang, X. Li, Y. Zhang, Q. Chen, Z. Ye, Z. Alqarni, S. E. J. Bell and Y. Xu, J. Mater. Chem. C, 2021, 9, 11517–11552 RSC.
- S. Fateixa, M. Wilhelm, H. I. S. Nogueira and T. Trindade, J. Raman Spectrosc., 2016, 47, 1239–1246 CrossRef CAS.
- S. Fateixa, M. Raposo, H. I. S. Nogueira and T. Trindade, Talanta, 2018, 182, 558–566 CrossRef CAS PubMed.
- S. Fateixa, H. I. S. Nogueira and T. Trindade, ACS Omega, 2018, 3, 4331–4341 CrossRef CAS PubMed.
- N. C. T. Martins, S. Fateixa, T. Fernandes, H. I. S. Nogueira and T. Trindade, ACS Appl. Nano Mater., 2021, 4, 4484–4495 CrossRef CAS.
- S. Fateixa, M. Landauer, J. Schneider, S. Kumar and R. Böhm, Macromol. Mater. Eng., 2023, 308, 2300060 CrossRef CAS.
- H. Huang, Z. Zhang and G. Li, Biosensors, 2022, 13, 30 CrossRef PubMed.
- D. Song, R. Yang, F. Long and A. Zhu, J. Environ. Sci., 2019, 80, 14–34 CrossRef CAS PubMed.
- P. Pinheiro, S. Fateixa and T. Trindade, Magnetochemistry, 2017, 3, 32 CrossRef.
- P. Pinheiro, S. Fateixa, H. Nogueira and T. Trindade, Nanomaterials, 2018, 9, 31 CrossRef PubMed.
- P. C. Pinheiro, S. Fateixa, A. L. Daniel-da-Silva and T. Trindade, Sci. Rep., 2019, 9, 19647 CrossRef CAS PubMed.
- G. Sirgedaite, M. Talaikis, G. Niaura and L. Mikoliunaite, New J. Chem., 2023, 47, 402–411 RSC.
- C. de Julián Fernández and F. Pineider, in New Trends in Nanoparticle Magnetism, ed. D. Peddis, S. Laureti and D. Fiorani, Springer, Cham, 2021, vol. 308, pp. 107–136 Search PubMed.
- A. Guadagnini, S. Agnoli, D. Badocco, P. Pastore, D. Coral, M. B. Fernàndez van Raap, D. Forrer and V. Amendola, J. Colloid Interface Sci., 2021, 585, 267–275 CrossRef CAS PubMed.
- S. Scaramuzza, D. Badocco, P. Pastore, D. F. Coral, M. B. Fernández van Raap and V. Amendola, ChemPhysChem, 2017, 18, 1026–1034 CrossRef CAS PubMed.
- L. B. Berganza, L. Litti, M. Meneghetti, S. Lanceros-Méndez and J. Reguera, ACS Omega, 2022, 7, 45493–45503 CrossRef CAS PubMed.
- H. Lai, F. Xu and L. Wang, J. Mater. Sci., 2018, 53, 8677–8698 CrossRef CAS.
- S. B. Ikramova, Z. N. Utegulov, K. K. Dikhanbayev, A. E. Gaipov, R. R. Nemkayeva, V. G. Yakunin, V. P. Savinov and V. Y. Timoshenko, Int. J. Mol. Sci., 2022, 23, 2590 CrossRef CAS PubMed.
- P. Pal, A. Bonyár, M. Veres, L. Himics, L. Balázs, L. Juhász and I. Csarnovics, Sens. Actuators, A, 2020, 314, 112225 CrossRef CAS.
- Á. I. López-Lorente, Anal. Chim. Acta, 2021, 1168, 338474 CrossRef PubMed.
- R. Oliveira-Silva, J. Pinto da Costa, R. Vitorino and A. L. Daniel-da-Silva, J. Mater. Chem. B, 2015, 3, 238–249 RSC.
- S. F. Soares, C. O. Amorim, J. S. Amaral, T. Trindade and A. L. Daniel-da-Silva, J. Environ. Chem. Eng., 2021, 9, 105189 CrossRef CAS.
- S. F. Soares, T. Fernandes, T. Trindade and A. L. Daniel-da-Silva, Molecules, 2019, 24, 1958 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- S. Fateixa, M. Raposo, H. I. S. Nogueira and T. Trindade, Talanta, 2018, 182, 558–566 CrossRef CAS PubMed.
- S. F. Soares, M. J. Rocha, M. Ferro, C. O. Amorim, J. S. Amaral, T. Trindade and A. L. Daniel-da-Silva, Int. J. Biol. Macromol., 2019, 139, 827–841 CrossRef CAS PubMed.
- Joint Committee on Powder Diffraction Standards (JCPDS), card no. 19-0629.
- Joint Committee on Powder Diffraction Standards (JCPDS), card no. 04-0784.
- C. Chen, S. Tao, X. Qiu, X. Ren and S. Hu, Carbohydr. Polym., 2013, 91, 269–276 CrossRef CAS PubMed.
- M. Q. Doan, N. H. Anh, N. X. Quang, N. X. Dinh, D. Q. Tri, T. Q. Huy and A.-T. Le, J. Electron. Mater., 2022, 51, 150–162 CrossRef CAS.
- Y. Xia, P. Padmanabhan, S. Sarangapani, B. Gulyás and M. Vadakke Matham, Sci. Rep., 2019, 9, 8497 CrossRef PubMed.
- P. C. D. S. Pereira, P. F. D. A. Costa, D. S. Pellosi, I. R. Calori, B. H. Vilsinski, B. M. Estevão, N. Hioka and W. Caetano, J. Mol. Liq., 2017, 230, 674–685 CrossRef CAS.
- T. M. Eldeeb, U. O. Aigbe, K. E. Ukhurebor, R. B. Onyancha, M. A. El-Nemr, M. A. Hassaan, S. Ragab, O. A. Osibote and A. El Nemr, Biomass Convers. Biorefin., 2024, 14, 9361–9383 CrossRef CAS.
- G.-N. Xiao and S.-Q. Man, Chem. Phys. Lett., 2007, 447, 305–309 CrossRef CAS.
- A. M. Gabudean, M. Focsan and S. Astilean, J. Phys. Chem. C, 2012, 116, 12240–12249 CrossRef CAS.
- S. Dutta Roy, M. Ghosh and J. Chowdhury, J. Phys. Chem. C, 2018, 122, 10981–10991 CrossRef CAS.
- S. Fateixa, M. Wilhelm, A. M. Jorge, H. I. S. Nogueira and T. Trindade, J. Raman Spectrosc., 2017, 48, 795–802 CrossRef CAS.
- P. H. B. Aoki, P. Alessio, A. Riul, J. A. De Saja Saez and C. J. L. Constantino, Anal. Chem., 2010, 82, 3537–3546 CrossRef CAS PubMed.
- T. Vosgröne and A. J. Meixner, ChemPhysChem, 2005, 6, 154–163 CrossRef PubMed.
- J. Kneipp, H. Kneipp, A. Rajadurai, R. W. Redmond and K. Kneipp, J. Raman Spectrosc., 2009, 40, 1–5 CrossRef CAS.
- C. Farcău, J. Mol. Struct., 2022, 1250, 131583 CrossRef.
- A. Shumskaya, I. Korolkov, A. Rogachev, Zh. Ignatovich, A. Kozlovskiy, M. Zdorovets, M. Anisovich, M. Bashouti, A. Shalabny, R. Busool, S. Khubezhov, D. Yakimchuk, V. Bundyukova, L. V. Panina and E. Kaniukov, Colloids Surf., A, 2021, 626, 127077 CrossRef CAS.
- M. Ye, Z. Wei, F. Hu, J. Wang, G. Ge, Z. Hu, M. Shao, S.-T. Lee and J. Liu, Nanoscale, 2015, 7, 13427–13437 RSC.
- C. Escapa, R. N. Coimbra, S. Paniagua, A. I. García and M. Otero, J. Environ. Manage., 2017, 203, 799–806 CrossRef CAS PubMed.
- S. Adomavičiūtė-Grabusovė, S. Ramanavičius, A. Popov, V. Šablinskas, O. Gogotsi and A. Ramanavičius, Chemosensors, 2021, 9, 223 CrossRef.
- V. Bernal, L. Giraldo and J. C. Moreno-Piraján, Adsorpt. Sci. Technol., 2018, 36, 833–850 CrossRef CAS.
- J. L. Castro, J. F. Arenas, M. R. López-Ramírez, D. Peláez and J. C. Otero, J. Colloid Interface Sci., 2009, 332, 130–135 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00049h |
| This journal is © The Royal Society of Chemistry 2024 |