
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The integrated on-chip isolation and detection of circulating tumour cells
Sophia M.
Abusamra†
 a,
Robert
Barber†
b,
Mohamed
Sharafeldin
c,
Claire M.
Edwards
ad and
Jason J.
Davis
*b
a,
Robert
Barber†
b,
Mohamed
Sharafeldin
c,
Claire M.
Edwards
ad and
Jason J.
Davis
*b
aNuffield Department of Surgical Sciences, University of Oxford, Oxford, OX3 9DU, UK
bDepartment of Chemistry, University of Oxford, Oxford, OX1 3QZ, UK. E-mail: jason.davis@chem.ox.ac.uk
cDepartment of Chemistry, University of Otago, Dunedin, 9016, New Zealand
dNuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Systems, University of Oxford, Oxford, UK
First published on 26th March 2024
Abstract
Circulating tumour cells (CTCs) are cancer cells shed from a primary tumour which intravasate into the blood stream and have the potential to extravasate into distant tissues, seeding metastatic lesions. As such, they can offer important insight into cancer progression with their presence generally associated with a poor prognosis. The detection and enumeration of CTCs is, therefore, critical to guiding clinical decisions during treatment and providing information on disease state. CTC isolation has been investigated using a plethora of methodologies, of which immunomagnetic capture and microfluidic size-based filtration are the most impactful to date. However, the isolation and detection of CTCs from whole blood comes with many technical barriers, such as those presented by the phenotypic heterogeneity of cell surface markers, with morphological similarity to healthy blood cells, and their low relative abundance (∼1 CTC/1 billion blood cells). At present, the majority of reported methods dissociate CTC isolation from detection, a workflow which undoubtedly contributes to loss from an already sparse population. This review focuses on developments wherein isolation and detection have been integrated into a single-step, microfluidic configuration, reducing CTC loss, increasing throughput, and enabling an on-chip CTC analysis with minimal operator intervention. Particular attention is given to immune-affinity, microfluidic CTC isolation, coupled to optical, physical, and electrochemical CTC detection (quantitative or otherwise).
1 Introduction
Cancer cells that detach from a primary tumour or metastatic site and circulate though the bloodstream are known as circulating tumour cells (CTCs), and represent a promising target for non-invasive tumour sampling. Despite an established clinical and biological relevance, a cost effective, robust, and rapid method of assaying these with sufficient sensitivity has yet to materialise. CTCs are highly associated with disease metastasis, yet the first FDA-approved method, CellSearch®, has been shown to detect them in only 57% of patients with metastatic disease, with a prognostic cut-off of >5 CTCs/7.5 mL of blood.1 Therefore, substantial and impactful improvements to address both practical applicability (cost, blood volumes etc., see below), and the high proportion of patients with metastatic disease who are likely below this threshold, are needed.CTC analysis is comprised of two crucial steps: isolation (specific capture from within an enormous excess of blood cells), and subsequent detection (quantification and/or identification of captured cells). Nearly all reported platforms, including CellSearch®, separate these two processes, despite it being known that sample transfer between dissociated isolation and detection contributes to cell loss and increased cost, time, and labour.2–5 As such, there is significant interest in integrated platforms that enable a rapid, sensitive, and streamlined analysis from low blood volumes. This review will focus on the state of the art on-chip technologies that integrate CTC isolation and detection into a single, and potentially highly scalable, microfluidic device.
1.1 Biological significance and clinical utility of CTCs
The metastasis of cancer from primary tumour to distal tissues is associated with approximately 90% of cancer deaths, of which there were almost 10 million in 2020.6 The lethality of metastasis is grimly indicated by the drastic difference in five-year survival rates between patients with localised and distant disease (99% versus 27%).7 Thus, understanding how cancer cells transform between primary and metastatic sites is crucial if one seeks to prevent disease progression and guide treatment decisions.8 At present, it is known that CTCs are shed from the primary tumour and intravasate into circulation, where they can potentially extravasate into distant tissues and seed secondary sites of disease (Fig. 1). However, further understanding is required to elucidate disease dissemination and metastasis mechanisms; understanding which could be provided through improved CTC analyses.9–12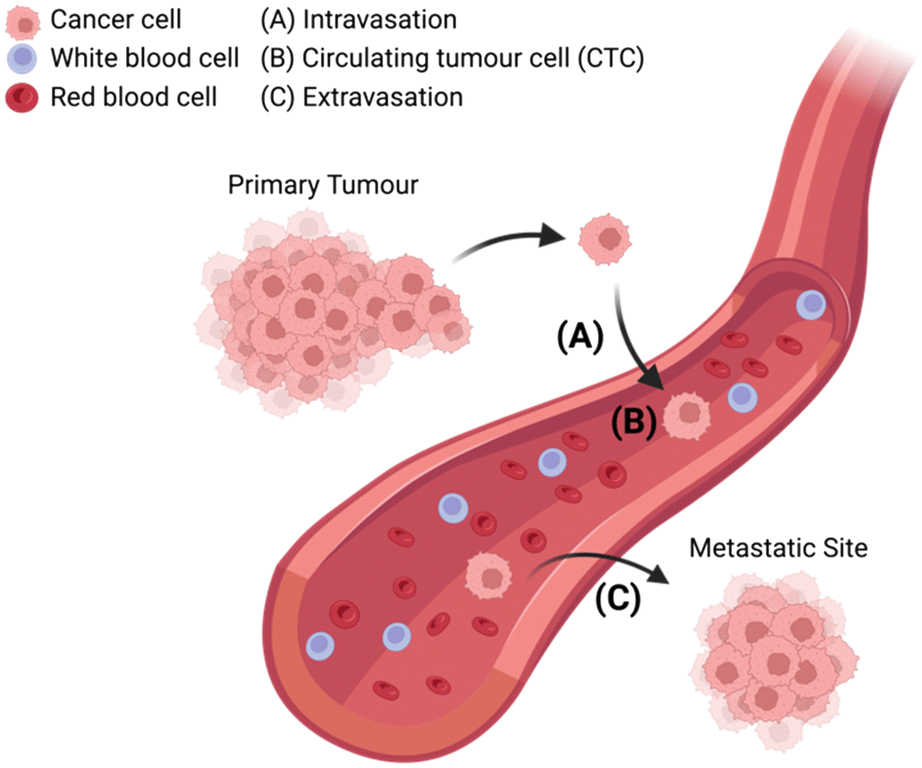 | ||
| Fig. 1 Schematic representation of circulating tumour cells (CTCs) breaking off from the primary tumour site and (A) intravasating into circulation, (B) traveling through the bloodstream among red blood cells and white blood cells, (C) extravasating from circulation with the potential to seed sites of cancer metastasis in distant tissue. | ||
The isolation of CTCs from whole blood is a form of liquid biopsy, and as such offers great clinical utility, as it is less invasive (accessible via peripheral venous phlebotomy) and less expensive than solid tissue biopsies (performed by highly trained professionals and may require radiographic guidance).13,14 Furthermore, solid tissue biopsies can easily miss small cancerous lesions (especially in early-stage disease), and may fail to capture intra-tumour heterogeneity and intra-patient tumour evolution with time.15–17 CTCs have also been shown to change in a more dynamic way than other serum cancer biomarkers such as PSA, AFP, CA-199, and CEA.18,19 They provide, then, a more accurate and up to date clinical picture of disease, in capturing a ‘snapshot’ of the tumour condition,9,20 representing a validated prognostic marker for numerous malignancies.21–26 While other methods of liquid biopsy including circulating tumour DNA/cell-free DNA (ctDNA/cfDNA) and exosomes have been described, CTCs offer innate advantages with the opportunity to study whole cells, as they can be cultured for in vivo and functional studies.27–29 Furthermore, CTCs allow for protein- and RNA-based molecular profiling and are certain to be of tumour origin, unlike ctDNA/cfDNA, wherein DNA analyses can be confounded by clonal haematopoietic mutations of indeterminate potential (CHIP).27–32
The biological significance of CTCs is most clearly demonstrated by the very direct association of increased CTC load (higher haematological abundance) with poor prognosis and reduced overall disease-free survival.33–38 In addition to enumeration, these cells hold a wealth of biological information that can be unlocked via genetic profiling. Recent studies have focused on genomic sequencing of CTCs to reveal their neoplastic origin, elucidating clonal evolution and offering insight into metastatic pathways.39–41 This downstream genetic profiling also has the potential to predict response to therapy and to risk stratify patients, guiding treatment decisions.42–50
1.2 Microfluidics (μFs) for CTC isolation & detection
Microfluidics (μFs) is a multidisciplinary field that enables the complex manipulation of microlitre volumes of reagents and as such, has demonstrated applicability to numerous areas of research, including drug discovery, cell culture, and personalised medicine.51,52 The microchannel configurations of microfluidic chips can be designed with diverse geometries to achieve a range of tasks including reagent delivery, mixing, or extraction of specific targets.36 Historically, formative manufacturing has been the default microfluidic fabrication technique, wherein a liquid polymer precursor (e.g., polydimethylsiloxane, PDMS) is cast and cured into a photolithographically fabricated mould (Fig. 2A). Conversely, subtractive manufacturing involves the selective removal of substrate material via laser ablation or computer numerical control (CNC) milling to form a channel (Fig. 2B). Recently, the decreasing cost of high-resolution desktop 3D printers using stereolithography (SLA) through point-by-point laser scanning or digital light processing (DLP) techniques have enabled in-house additive manufacturing of microfluidic channels with features down to 25 μm (Fig. 2C). These methods use a laser or UV light source to selectively polymerise a liquid resin into the desired solid geometry at very low cost (around £0.03–0.15 per cm3). 3D printing with inherently non-cytotoxic materials or the post-cure application of a protective coating (e.g., bovine serum albumin, BSA) has allowed for the fabrication of biocompatible microfluidic chips.53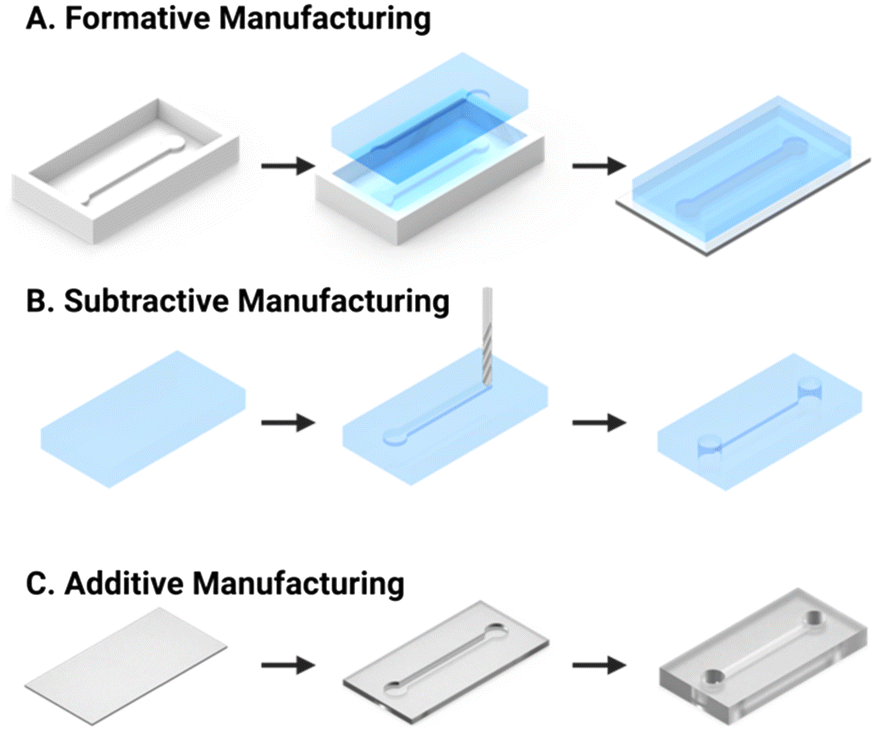 | ||
| Fig. 2 Schematic representation of the most common microfluidic fabrication strategies. (A) Formative manufacture is achieved by casting of optically transparent PDMS into a mould. The chip is then formed by mounting the resultant PDMS cast piece onto a glass slide. (B) Subtractive manufacture is performed via computer numerical control (CNC) milling of a solid substrate. (C) Additive manufacture utilises layer by layer selective curing of transparent resin to assemble the chip and internal channel (e.g., SLA or DLP 3D printing). | ||
Microfluidic platforms, or “chips” have been extensively applied to the challenge of CTC isolation, using both immunoaffinity and physical methods, as discussed in sections 2.1 and 2.2, respectively. They present an opportunity for a simplified and miniaturised workflow, requiring less time (<5 hours) and sample volume (sub mL)54–56 than the CellSearch® commercialised standard for CTC enrichment (7.5 mL).57–60 The detection of CTCs in microfluidic devices has most frequently been achieved using optical microscopy methods (section 3.2), mandating good optical transparency of the platform. For this reason, transparent PDMS substrates, and photo-curing resins, are ubiquitous.
1.3 On-chip integration of CTC isolation and detection
As mentioned above, CTCs analyses include two critical steps: isolation and detection. Isolation is the separation of tumour cells from blood, with detection being either a subsequent quantification (i.e., electrochemical or optical enumeration) or identification (i.e., confirmation that captured cells are CTCs and not an erroneously captured cell).The majority of reported methods of CTC isolation, including CellSearch®, necessitate the elution and manual transfer of isolated cells to a separate module or device for detection (schematised in Fig. 3A).61–65 CTCs can be lost during this transfer via adhesion to pipette tips and sample tubes across experimental steps, or simple retention within the device or chamber.2,3 Such manual transfer increases time and labour, introduces inter-test variability, adversely affects assay accuracy, and can inflict cell damage.4,5 The latter arises because human cells are mechanically fragile, deformable, and sensitive to environmental change (and thus transport/manipulation can affect sample quality).66,67 As CTCs need to be in optimal condition for potential downstream analysis, such as genomic interrogation or culture, each damaged cell represents the loss or tarnishing of potentially valuable information. In response to such issues, a low but growing number of reports have integrated CTC isolation and detection into a single microfluidic chip, as summarised in Table 1 and described as “integrated” methods from hereon. These methods require less time to operate, reducing assay cost, curtailing opportunities for cell loss, and necessitating notably lower sample volumes.52,68,69 They facilitate convenient sample processing wherein multiple experimental steps can be completed on-chip without manual transfer.70 Such integrated approaches have been shown to support a robust sensitivity (detection limit of <10 cells per mL) in a scalable, fast (<1 hour per sample) system capable of analysing CTCs in blood volumes as low as 1 mL.71–73
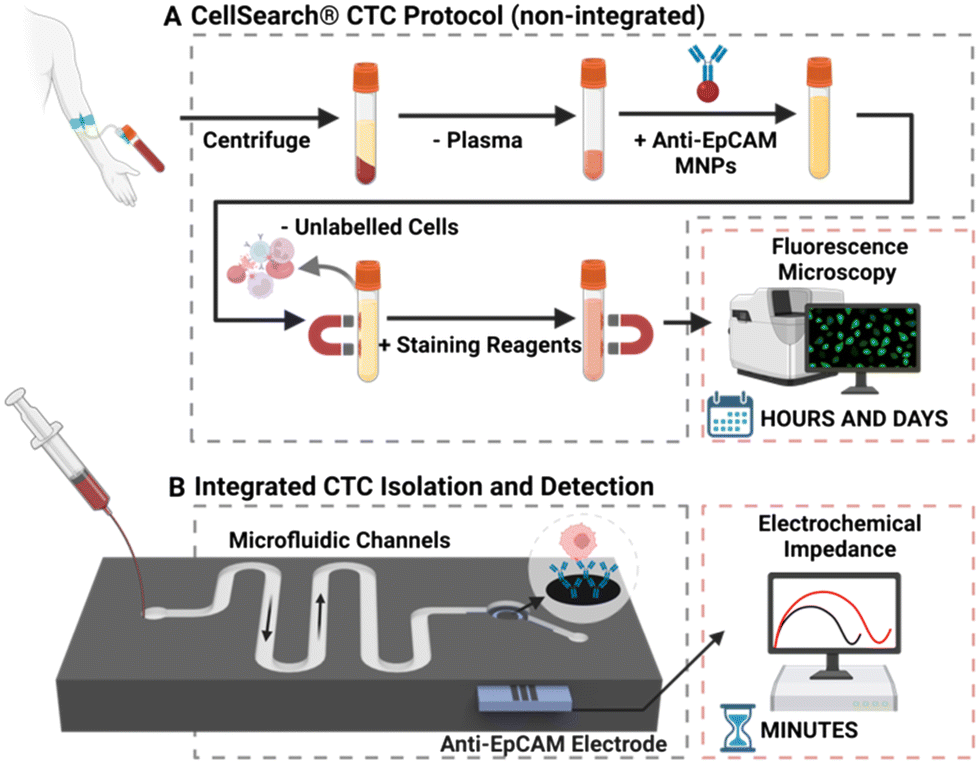 | ||
| Fig. 3 (A) Schematic representation of the CellSearch® platform. Firstly, some 7.5 ml of whole blood is centrifuged to remove plasma, the residual volume is then mixed with anti-EpCAM modified magnetic nanoparticles (MNPs) to separate CTCs from healthy cells. The isolated cells are then enumerated using immunofluorescence microscopy. (B) Representative schematic of a configuration that facilitates CTC isolation and electrochemical (e.g., impedance) detection within a single microfluidic chip. | ||
| Integrated detection method | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Electrochemical detection | Optical detection | Physical and optical detection | |||||||||||
| Amperometry | Impedance | Voltammetric pulse | UCL imaging | RIDA | In situ Raman barcoding | Brightfield microscopy | Immuno-blotting | IF | Deformability cytometry | Cavitation inception pressure | |||
| DEP, dielectrophoresis; NP, nanoparticle; Ab, antibody; UCL, up-conversion luminescence; RIDA, rapid isothermal nucleic acid detection assay; IF, immunofluorescence.a Platforms that use multiple detection methods.b Platforms that use multiple isolation methods. | |||||||||||||
| Integrated isolation method | Antigen-independent (physical) isolation | Size-based exclusion with mesh or microfilter | 41b | 137 | |||||||||
| Size-based sorting in trap channel or hole array | 113b | 142b | 55 | 138 | |||||||||
| 139 | |||||||||||||
| DEP | 54 | 146b | 142b | ||||||||||
| Hydrodynamic separation with chip geometry | 146b | 120b | 41b | ||||||||||
| Hydrodynamic cavitation | 14 | ||||||||||||
| Antigen-dependent isolation | Immobilized Ab on-chip | 112a | 114 | 116 | |||||||||
| 119a | 112a | 119a | |||||||||||
| 56 | |||||||||||||
| Off-chip incubation with Ab-conjugated NPs | 117 | 118 | 120b | 121 | |||||||||
| 115 | |||||||||||||
| 122 | |||||||||||||
| Ab-conjugated Raman active nanoprobes | 113b | ||||||||||||
Within integrated platforms, CTCs can either be isolated and detected on the same surface (e.g., isolation of CTCs using a filter mesh followed by immunofluorescence staining on the same mesh), or flow within the same chip from isolation to detection regions (e.g., CTC isolation on channel walls with immobilised antibodies, followed by direct flow of captured cells to an electrochemical detection module), as schematised in Fig. 3B. Within this review, we provide an overview of common approaches for CTC isolation and elaborate on the implementation of such methods within integrated systems. Subsequently, strategies for CTC detection are summarised and the coupling of such methods on integrated platforms is detailed.
2 CTC isolation
As discussed, integrated platforms for CTC analysis (summarised in Table 1) both isolate and detect these rare cells on a single microfluidic chip. Isolation from whole blood is challenging, with the most significant barriers being the low abundance,74,75 morphological similarity to white blood cells (WBCs),76,77 and phenotypic heterogeneity.78 Nonetheless, CTC isolation has been demonstrated using many proof of principle methods, broadly divisible into two approaches: antigen-dependent (immunoaffinity; e.g., antibody-loaded magnetic nanoparticles or other solid supports), and antigen-independent (i.e., separation through physical properties).37,76,79 Section 2 will overview current immunoaffinity and physical approaches to CTC isolation (see section 2.1 and section 2.2, respectively) and detail how these approaches are applied within integrated platforms (see section 2.1.1 and section 2.2.1).To evaluate the performance of CTC isolation techniques, the following parameters are considered: purity, enrichment or capture efficiency, and throughput (or turnaround time/TAT).33 Purity describes the specific capture of CTCs from a heterogeneous background of other cells, while enrichment refers to the ability to increase the proportion of CTCs within a given sample. Capture efficiency is a metric presented to summarise the number of CTCs captured out of a known population (as a percentage), and the amount (in volume or number of samples) that can be processed per a given amount of time is known as throughput/TAT.33
2.1 Antigen-dependant isolation
Antigen-dependent (immunoaffinity-based) methods of isolation utilise antigen recognition to select for or against cells based on markers present on the cell membrane, achieved by immobilising a complimentary antibody or aptamer on a supporting structure.80,81 Positive enrichment selects for CTCs; conversely, negative enrichment excludes non-CTCs.12 These strategies often target epithelial cell adhesion molecule (EpCAM/CD326), as a high proportion (up to 90%) of cancers are of epithelial origin and thus express this marker.12,82–84As an exemplar, the CellSearch® platform mentioned in section 1 uses anti-EpCAM conjugated magnetic nanoparticles (MNPs) to positively select for EpCAMpositive cells (Fig. 3A). The protocol results in a low purity output (60–70%) due to a high residual leucocyte population of ∼1000–3000 cells from the 7.5 mL sample.85–87 This background noise necessitates the use of immunofluorescent (IF) imaging of cytoplasmic and cell surface markers to detect and enumerate CTCs, resulting in a high instrument cost (∼£200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), with cost per test ∼£800 and TAT ∼5 hours.88–90
000), with cost per test ∼£800 and TAT ∼5 hours.88–90
Relying on positive enrichment alone may erroneously include non-tumour EpCAMpositive cells and fail to address the dynamic expression of cell surface antigens.39,91–93 CTCs are known to undergo an epithelial-to-mesenchymal transition (EMT) while in circulation, during which they acquire the migratory properties typical of mesenchymal cells, lose their epithelial characteristics and downregulate EpCAM expression.79,94 Attempts to mitigate this downregulation have sought to diversify the surface antigens used in antigen-dependant CTC capture by including mesenchymal (e.g., vimentinpositive) and pseudo-endothelial (e.g., dual EpCAMpositive and CD31positive) markers.39,68,95,96 Targeting of malignancy specific markers has also been investigated to address this challenge, including positive selection for prostate specific membrane antigen (PSMA) in prostate cancer and for human epidermal growth factor 2 (HER2) in breast and gastric cancers.22,97
The inverse immune-affinity approach, negative enrichment, makes use of antibody-coated solid supports to target cell surface antigens such as CD45 (specific to white blood cells) to deplete healthy blood cells from a sample.98,99 A key advantage of negative enrichment is that intact (label-free) CTCs are obtained independent of their specific antigen expression, and thus, a heterogenous population of CTCs can be isolated (i.e., including low-EpCAM-expressing CTCs).100,101 This approach does not, however, afford a tumour-specific selection and thus typically results in a low purity (as exemplified by the low and inconsistent purities across different systems varying between 0.97–10%, going up to a maximum of 34.8 ± 14%).102–111
Integrated platforms using Ab-functionalised microfluidic channels capture CTCs on the channel walls by flowing a cell-spiked matrix through the device, while endogenous or exogenous healthy cells continued unimpeded to the channel outlet.56,112,114,116,119 These reports have demonstrated capture efficiencies of 72–88% from whole blood, and purities of 85–99.6%.56,116,119 Both IF staining56,116 and electrochemical detection methods (based on impedance or voltammetry112,114,119) have been effectively integrated with upstream channel-wall-immobilised antibody methods of isolation, either by directly imaging the captured CTCs within the channels or probing perturbations to electrode impedance induced by CTC/antibody binding.
One such integrated platform described a dislocated herringbone microfluidic channel modified with anti-EpCAM antibodies.116 This channel geometry caused increased turbulence in the flowing sample which increased the likelihood for CTC immunocapture. The authors reported a capture efficiency of 87% using a lung cancer CTC model (H1975) in whole blood, without the addition of any nanoparticle species. The H1975 cells were subsequently enumerated by conducting IF staining and imaging for CK18positive and CD45 (leukocyte marker)negative directly on the chip (with captured and stained cells inside).
Importantly, a controlled release of CTCs following antibody-based capture is critical for downstream analysis. However, the release of viable cells is a challenge for antigen-dependent isolation methods. Pahattuge et al. demonstrated the successful CTC release from an isolation channel on a SMART-chip (Fig. 4B).119 This was achieved by exposure of the transparent channel to blue light (400–450 nm), photocleaving a coumarin-based linker which was used to immobilise antibodies onto the channel walls, achieving a 90% release. The controlled elution enabled captured CTCs to proceed for viability analysis (Fig. 4C) and immunofluorescent staining (Fig. 4D) within the respective downstream modules, a significant achievement in an 80 mm platform.
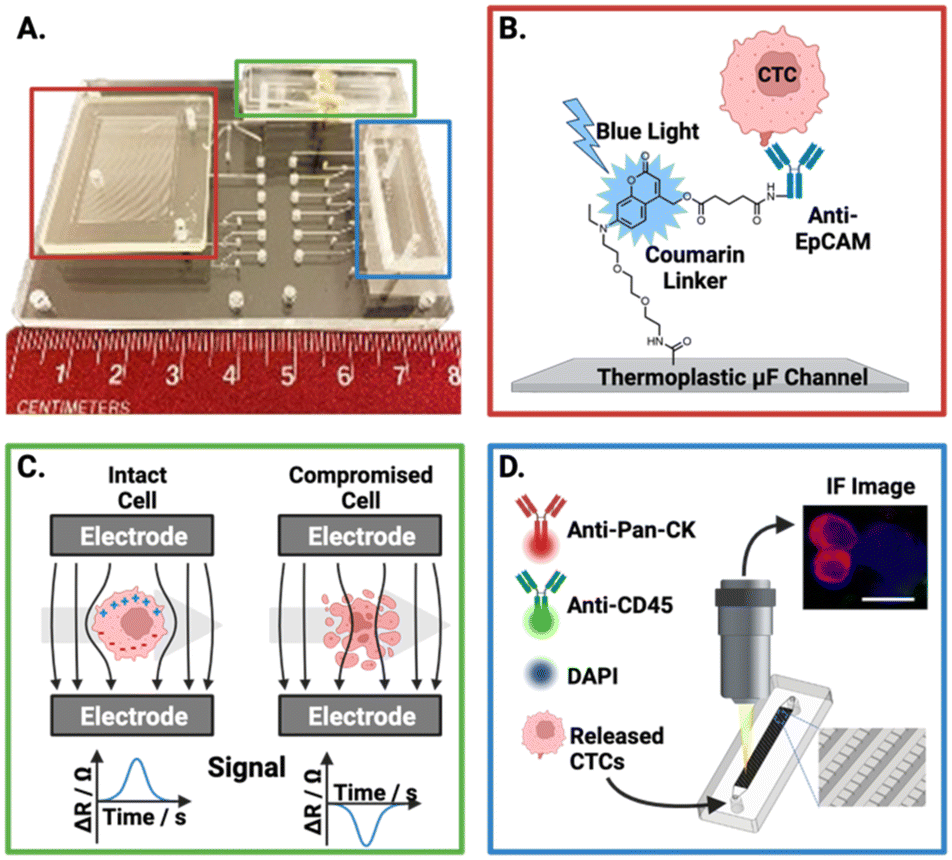 | ||
| Fig. 4 Representative example of an affinity-based CTC isolation and detection system, wherein the principles behind the three SMART-chip modules are schematised. (A) Image of the SMART-chip with each module highlighted. (B) CTC selection module; CTCs are enriched from a flowing blood sample by anti-EpCAM antibodies tethered to a microfluidic channel, the channel is then flushed with buffer to remove unwanted cell background. Isolated CTCs are then released by cleaving the coumarin-based linker with blue light. (C) Impedance module; isolated CTCs proceed to an impedance-based cell enumeration channel to confirm viability. This module had Pt electrodes situation orthogonal to the fluidic channel with ∼50 μm distance. Cells with intact membranes have a higher resistance than the buffer (ΔR) and yield positive polarity signals, with dead cells apparently yielding negative signals. (D) Imaging module; the enriched cell population is stained and imaged using a selection of fluorescent antibodies. Reproduced with permission.119 | ||
One unique report of an affinity-based CTC isolation in an integrated device uses neither channel-immobilised Abs nor Ab-conjugated MNPs, but rather Ab-conjugated Raman active nanoprobes to capture CTCs.113 In this work, Cho et al. triple functionalised a suite of gold nanoparticles (Au-NPs) for CTC isolation and detection. The bare Au-NPs were modified with biotinylated dsDNA (enabling their capture on the streptavidin pillars), unique Raman reporters (later used for CTC detection, as discussed in section 3.2.1) and a corresponding CTC specific antibody (CD133, HER2, EGFR, EpCAM, or MUC1), forming 5 subsets of labelled Au-NPs. The group described a microfluidic chip with 5 μm-wide micro-pillars to complete size-based exclusion of RBCs and WBCs. A turbulent state of sample flow within the chip facilitated cell–particle mixing. The streptavidin-rich pillar surfaces passively recruited the biotinylated, Raman barcoded CTCs, capturing them within the chip. While this method was not applied to cancer patient blood samples, it was reported to achieve 90% capture efficiency of model CTCs spiked in 4 mL of healthy human blood. However, this required significant pre-treatment of the samples to first isolate mononuclear cells using a density gradient Ficoll-Paque protocol involving dilution, centrifugation, washing and separation.
While the chemical (biotin–streptavidin) mediated capture of CTCs within the chip in the above report was reversible by enzymatic cleaving of a dsDNA linker, Au-NPs cannot be magnetically manipulated. An alternative, would be, of course, to employ antibody-MNPs. CTC–MNP complexes can be simply isolated and released from the running solution by application or removal of a permanent or modulating electromagnetic field. This enables the controlled retention of CTCs within the channel, while other contaminants are eluted from the device and eventually released into a clean running buffer, as required.117,118
These Ab-modified MNP approaches have reported capture efficiencies of 65–99.9% from whole blood. To do so, microfluidic platforms have utilised various channel designs to achieve passive mixing and thereby, maximum capture.115,117,118,120–122 These include serpentine and herringbone geometries, both of which have been shown to disturb laminar flow within the channel to improve mixing efficiency beyond what would be possible by diffusion alone.123 One study noted ∼80 WBCs captured along with CTCs from 1 mL of whole blood, a considerable reduction from ∼10000 WBCs per 7.5 mL of blood recorded for the isolation method used by CellSearch®.117
While most reports using Ab-conjugated MNPs utilise a constant magnetic field and a single molecular target,117,118,121 recent fully integrated platforms have included progressively more sophisticated techniques, selecting for multiple cell surface markers and ranking captured cells based on magnetic gradients. An example which exploits the power of full integration is one that achieved CTC isolation and ranked these based on surface antigen expression. Poudineh and colleagues, demonstrated their magnetic ranking cytometry (MagRC) microfluidic chip, featuring a magnetic field gradient along a channel to sort cells based on magnetisation. The amount of nanoparticle loading on cells was, specifically, reflective of the degree of expression of the target antigen, which allowed for instantaneous on-chip CTC profiling. The group reported a capture efficiency of 90–97% and an integrated IF staining based detection limit of 1–10 cells per mL.115
A similar demonstration of this graduation strategy by Lee et al. employed MNPs conjugated to anti-HER2 antibodies to target breast cancer cells; once captured from culture media (capture efficiency >99%), CTCs were sorted based on a magnetic gradient into ‘HER2 positive’ and ‘HER2 negative’ regions (Fig. 5A), allowing near instantaneous grading and readout of CTC phenotype based on the expression of the oestrogen receptor (OR) and progesterone receptor (PR). This is particularly impactful as these biomarkers can aid in the prediction of tumour metastasis (Fig. 5B).122 Both of these integrated ranking reports utilised the variance in magnetic properties in MNP–CTC complexes to gain insight into phenotypic profiles of the captured CTC population and fully integrated isolation and IF staining. To date, there is only one report which employed an electrochemical method of CTC detection after upstream Ab-MNP assisted isolation.117 It can be conjectured that this is because IF can be conducted on captured CTCs without removal of MNPs, whereas introducing MNPs to one of the alternative detection methods discussed in section 3.1 potentially requires CTC separation from MNPs prior to quantification.
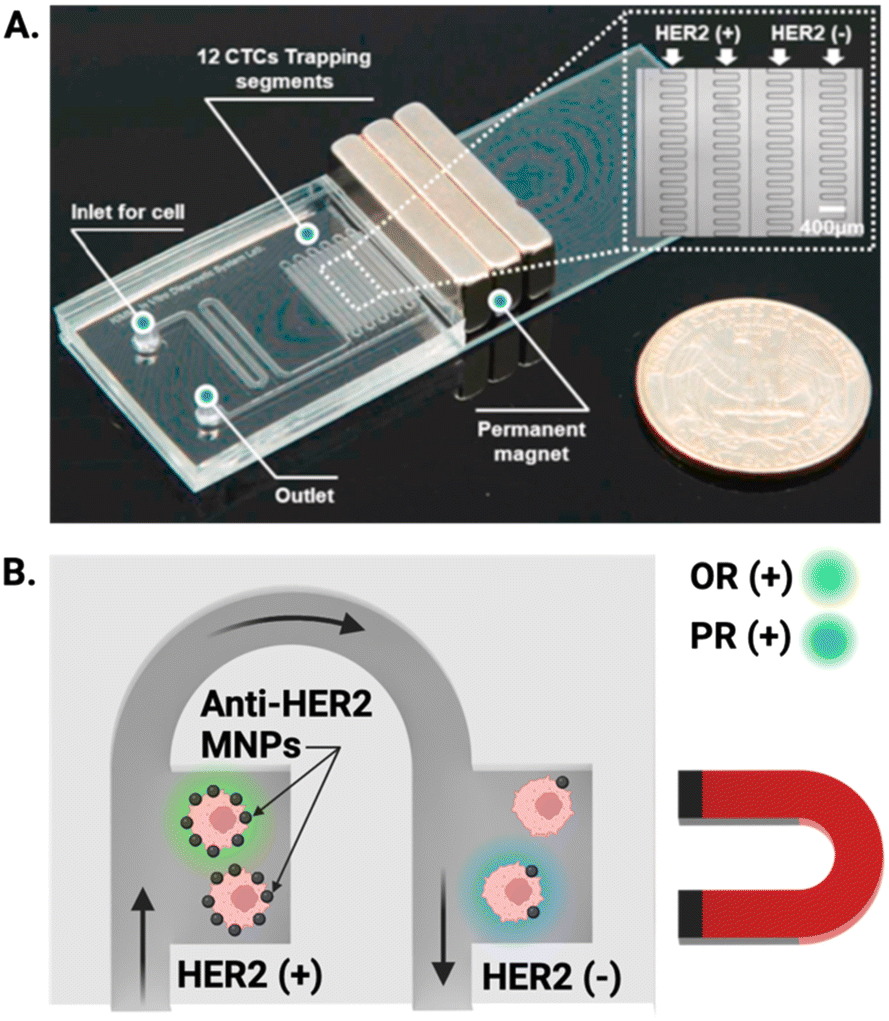 | ||
| Fig. 5 Representative example of MNP assisted isolation of CTCs in an integrated microfluidic chip. (A) Image of a PDMS microfluidic cell sorter which enabled the specific labelling of HER2 receptors with anti-HER2 coated MNPs under the influence of an external magnetic field. (B) Schematic showing magnetically trapped CTCs IF visualised on chip by flowing reagents through the channel for the oestrogen (OR) and progesterone (PR) receptors. Reproduced with permission.122 | ||
While the release of CTCs from Ab-functionalised MNPs presents a challenge, it has been documented, for example, by Wang and colleagues who released 80% of captured CTCs from their anti-EpCAM modified MNPs and showed successful proliferation for 7 days in culture.118 Similarly, Poudineh et al. achieved a 92% release efficiency, of which 98% of the cells were viable.115 Meanwhile, the above discussed report by Pahattuge et al. achieved a release of 90% with their photocleavable linker.
In summary, antigen-dependent methods (where capture antibodies are immobilised on NPs or microfluidic channels) have been able to achieve successful CTC isolation from low mL volumes of patient blood samples with capture efficiencies spanning 65–99.9% and 72–88% respectively. Methods utilising MNPs have thus far utilised off-chip incubation of the sample (tens of minutes to a few hours) and then complete magnetic isolation on-chip. Channel-immobilised methods allow for direct injection of the sample into the antibody-modified channel. To date, as noted, the majority of platforms completing CTC isolation by antigen dependant methods have used downstream immunofluorescence imaging as the assaying method.
2.2 Physical isolation
Physical, or “antigen-independent” methods isolate CTCs based on differences in their physical properties from healthy blood cells (independent of antigen expression) and are thus less susceptible to issues associated with phenotypic heterogeneity, as discussed in section 2.1.124 Common physical differences leveraged for such methods include size, density, deformability, or charge.125 Size-based physical isolation techniques use filtering apparatus to (rather crudely) exploit the geometric difference in size between CTCs (mean diameter 15.6 μm), healthy red blood cells (RBCs) (7.5–8.7 μm) and WBCs (12–15 μm).126–128 However, a subpopulation of WBCs known as monocytes (15–18 μm) pose a significant problem.129 A more sophisticated method of antigen-independent CTC isolation can be achieved by microfluidic hydrodynamic separation which is achieved by judicious selection of channel geometry and can be assisted by application of an acoustic waveform.130 The profound variances in density and deformability of CTCs affects their loci within a channel due to enforced flow through the channel geometry. CTCs can also be physically isolated using dielectrophoresis, an approach which exploits the dielectric characteristics which differ from healthy cells. This is achieved by flowing samples through a channel under the influence of an external, non-uniform electrical field, thereby inducing directional movement of cells to facilitate isolation.125,131Issues associated with these physical methods, such as size overlap, progressive membrane obstruction, and lack of tumour-specificity can result in antigen-independent isolation methods recovering a lower number of CTCs than the molecular enrichment techniques discussed earlier, and with a notably low purity (e.g., 1–3%).125,132–136 However, their relative simplicity and straightforward integration to a microfluidic configuration justifies their inclusion as a viable CTC isolation tool.
Size-based isolation on integrated devices has taken the form of either micromachined ‘hole’ or ‘trap’ arrays within the microfluidic channel system, or the use of meshes or microfilters. Mesh or microfilter approaches have achieved capture efficiencies of 87–99% from whole blood, 90% CTC purity or a 99.9% removal of WBCs.41,137 The simplest example of size-based filtration is presented by the use of CellSieve™ microfilters, where blood is drawn through a polymer film with an array of 7 μm diameter pores.137 CTCs (captured at >90% efficiency) could then be easily fixed, stained, and imaged directly on the filter. Physical isolation of CTCs has also been achieved in integrated platforms using trap channels and hole arrays, which complete a size-based cell sorting. In these reports, samples are flowed through microfluidic chips and CTCs are captured in vortices generated by chamber geometries,138 or by settling into hole arrays.55,139 These integrated methods have achieved capture efficiencies of 25–99% from a few mL of whole blood and obtained purities of 35–92%, with a WBC depletion of up to 98.7%.55,138–140 A recent study utilized a nanostructured array to isolate individual cells within nanodroplets, functioning as individual reactors for evaluating the secretion of matrix metalloproteinase 9 (MMP6), an enzyme crucial in the EMT process of cancer cells. The methodology involved the use of a specially designed FRET probe to assess enzyme activity without affecting cell integrity or contents, thus enabling the recovery of individual cells for subsequent analyses, as the method maintains the integrity of cells without requiring any surface modifications.141
Combining multiple antigen-independent isolation techniques within a chip (including those integrated with downstream analysis) has been shown to boost the achievable capture efficiency. In one report of such multiplexed isolation, Farasat et al. detailed a transparent (enabling CTC detection by brightfield microscopy) PDMS porous membrane, placed above microfabricated gold electrodes to achieve isolation driven by dielectrophoretic force. The PDMS microfluidic chip featured an array of 20–30 μm pores patterned by soft lithography. By applying a non-uniform electric field to the embedded electrodes, model CTCs migrated downwards and settled into the pores. This achieved isolation based on both dielectric properties and size.142 Unfortunately, this report did not include any detail on parameters of capture efficiency or purity.
Another group have detailed two integrated approaches to separate CTCs from whole blood by selectively increasing retention time within a microfluidic channel (Fig. 6A).54 Firstly, an electrostatic enrichment was achieved by modifying a channel with a polymer/lipid layer. As cancer cells are known to have an increased expression of anion transporters on their surface,143,144 CTCs flowing through the channel were observed to have a temporary but increased electrostatic interaction with the positively charged lipid layer than healthy cells.54 The second design aspect capitalised on the larger size and dielectrophoretic properties of these CTCs. The application of an alternating current waveform to electrodes integrated within the walls of a microfluidic channel induced small oscillating movements of cells, perpendicular to the direction of flow (i.e., towards and away from the channel walls). CTCs were shown to have slower oscillations due to their larger size, resulting in a longer retention time in the channel, thereby enabling physical separation from the smaller cellular components of whole blood. This bimodal, particle and antibody free, CTC isolation was followed by an electrochemical assay and IF imaging, both conducted directly on the chip.
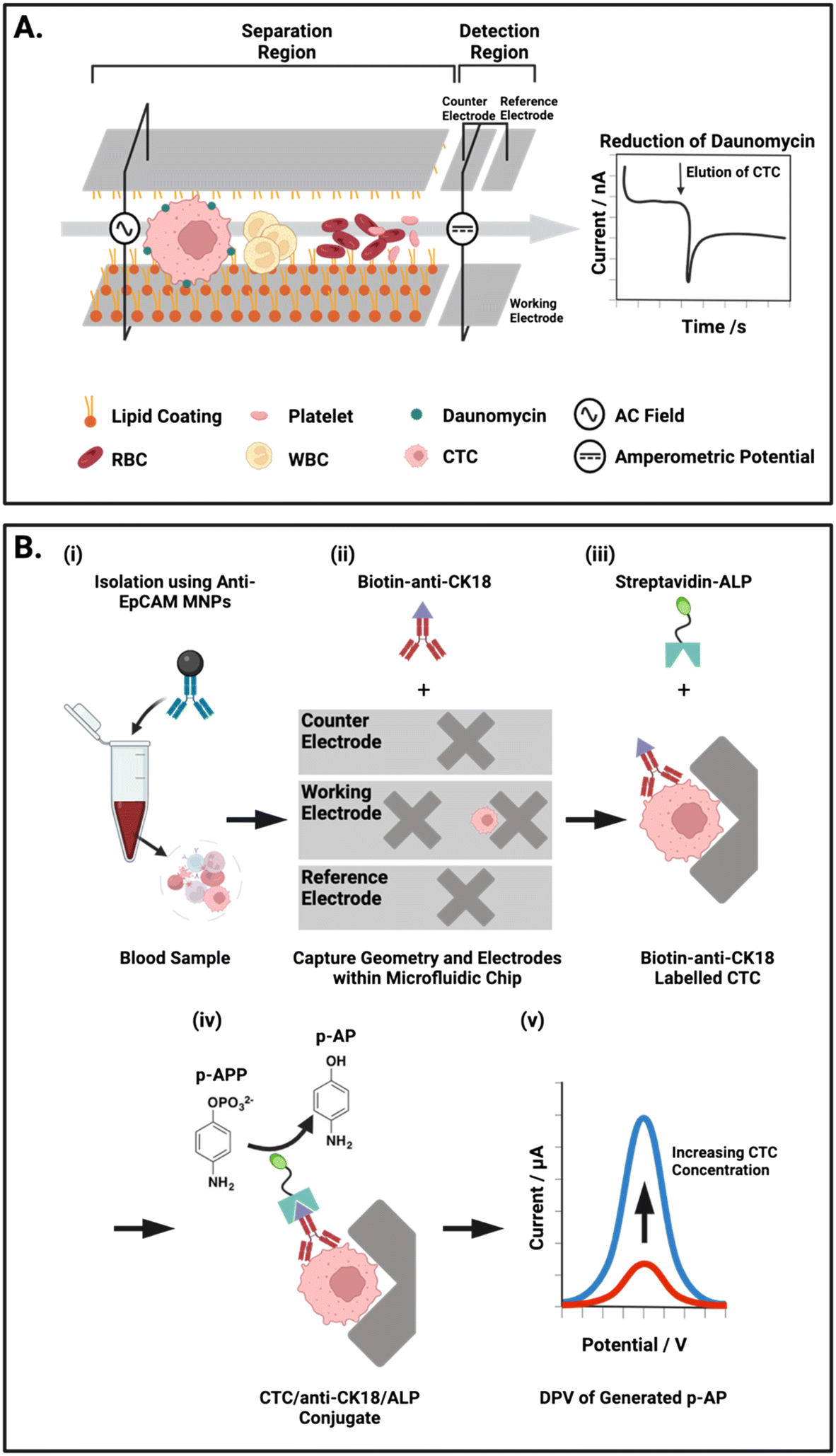 | ||
| Fig. 6 Representative examples of integrated on-chip physical CTC isolation and subsequent electrochemical detection. (A) Schematic representation of the microfluidic channel described by Gurudatt et al.54 An AC potential is applied to the carbon/polymer printed microfluidic channels to physically isolate CTCs from blood using a combination of lipid–lipid and electrostatic interactions. The reduction of daunomycin (redox-active cancer drug) labelled CTCs at the detection electrodes at the channel terminus enabled the chronoamperometric quantification of CTCs. (B) Electrochemical ELISA for the detection of CTCs from human blood. (i) Whole blood mixed with anti-EpCAM MNPs off-chip. (ii) Transfer of captured cells to the microfluidic chip where the low flow regions facilitate isolation and subsequent tagging of CTCs with biotin anti-CK18 antibodies. (iii) Labelled CTCs are exposed to a solution of alkaline phosphatase (ALP) enzyme tagged with streptavidin, forming a complex with the biotin-CK18. (iv) p-Aminophenyl phosphate (p-APP) is flowed in excess through the chip were ALP converts it to the electroactive p-aminophenol (p-AP). (v) Differential pulse voltammetry (DPV) is used to oxidise p-AP to generate a signal corresponding to the concentration of CTCs within the sample. | ||
Another report by Abdulla et al. integrated both size and hydrodynamic physical isolation within a PDMS chip mounted on a glass slide. The group sought to reduce opportunities for filter blocking through the addition of a winding channel geometry to separate CTCs from whole blood prior to sample reaching a filter membrane.41 By adjusting the flow rate within the separation channel, they demonstrated that larger CTCs were located proximal to the channel midline, with smaller cells were pushed out distally. The relatively fast flow rate (1.4 mL min−1) maintained laminar flow (Reynolds number: 29.6, cf. <2000 is deemed laminar) and induced spatial segregation in the channel between cell types. They specifically reported the separation of white blood cells from MCF-7 cells (as a breast cancer CTC model) suspended in 0.9% saline at a purity of 70% after hydrodynamic separation alone. The enriched CTCs then continued to a filter membrane for a second stage physical exclusion before IF analysis of protein cargo by cell lysis on the chip itself (Fig. 7). Although the 70% purity obtained by this physical approach is lower than that achievable by an antigen-dependent method, achieving this degree of separation without an immunorecognition event is significant.125,132–136
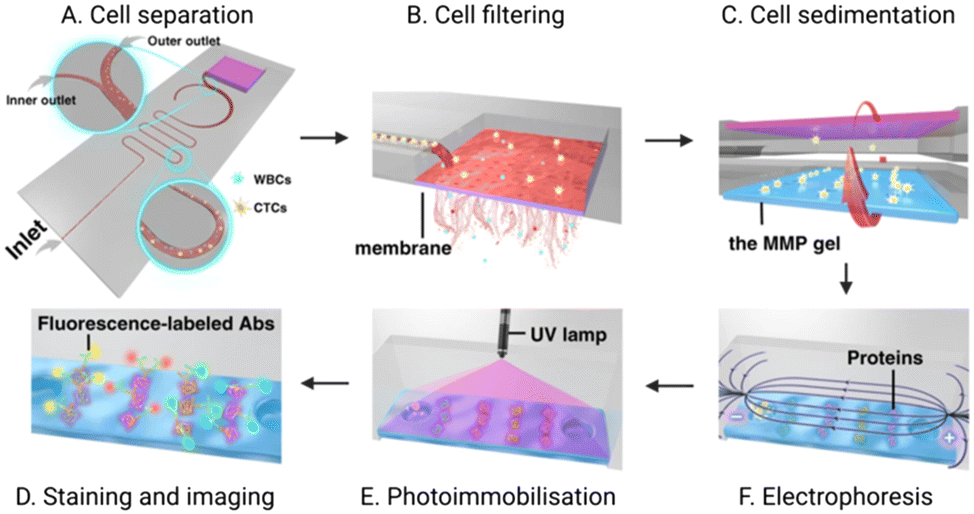 | ||
| Fig. 7 Schematic of an integrated microfluidic platform completing CTC isolation and detection. (A) CTCs are first hydrodynamically separated from WBCs; (B) the enriched CTC population is then purified via an in situ membrane filter. (C) Inversion of the chip enables CTCs to transition to the polyacrylamide gel where they are (D) chemically lysed to enable electrophoretic separation of the resultant proteins. (E) an ultraviolet (UV) light source immobilises these proteins for (F) subsequent fluorescent immunoblotting. Reproduced with permission.41 | ||
It could be expected that technologies which combine size-based filtration to compliment antibody mediated CTC isolation should achieve an enriched CTC population with superior purity to using either one alone.131,145 Several proof of principle integrated platforms that demonstrate bimodal isolation strategies exist in the literature. One such report used an initial antibody-mediated bead-based capture, followed by hydrodynamic separation of the CTC–bead conjugates from the complex matrix into micromachined chip geometries with two “fences” with diameters of 9 μm and 15 μm, in which the 23 μm microspheres could be captured. The approach achieved a high capture efficiency (>85%), but a low CTC purity (20–40%) was reported, making downstream analysis more difficult.120
Clearly, antibody independent methods of CTC isolation warrant further investigation as recent reports which employ them in integrated isolation/detection platforms have claimed very high capture efficiencies (80–99%).41,54,55,120,137,139,146 Furthermore, some reports herein discussed have combined antigen-dependent and physical means of isolation (i.e., antibody-functionalized chip with inertial microflow147 or antibody-functionalised nanoparticles injected into a microfluidic chip148), achieving high capture efficiencies of 90–94%. However, sample input across these works has not been consistent, with some groups completing their enrichment on unmodified patient samples,137,139 whilst other studies utilised pre-processing, including RBC lysis, various washing steps,41 and PBS dilution of blood.55,138 As with integrated platforms applying antigen-dependent isolation, the vast majority of physical methods utilised within integrated formats have been paired with optical detection methods,41,55,137–140,142,149 with only two reports of electrochemical means of detection.54,146
3 Detection of CTCs
Once CTCs have been isolated on-chip using the antibody-mediated or physical methods discussed in sections 2.1 and 2.2, subsequent detection is required (i.e., quantitation and/or identification). As mentioned in the previous examples, this can involve either quantification (i.e., enumeration), or optical or physical confirmation that captured cells are CTCs. Methods available can be broadly categorised, then, as those which are optical, electrochemical or physical.Optical methods are most commonly fulfilled through well-established immunofluorescence (IF) imaging, using multichannel fluorescence staining of cellular markers.55,56,115,116,119,121,122,137,139,140,149 CTCs are typically detected based on CK (cytokeratin)positive, EpCAMpositive, nuclear DAPIpositive, and CD45negative signatures.69 However, IF requires cell fixation, numerous manual transfer steps and ultimately, interpretation by a clinical cytologist or pathologist. The laborious and time consuming (typically ∼5 active working hours) nature of the method, may prohibit the translation of IF-based CTC screening platforms into routine clinical practice. Additionally, relying on this approach reduces achievable throughput, introduces multiple opportunities for systematic or random errors, and ultimately presents an increased cost per test.20,150
Furthermore, a perfectly specific positive marker for CTC identification has not been identified, as EpCAM can be, as noted earlier, downregulated during the EMT and CK is not universally expressed across all tumours. This results in CKnegative and EpCAMnegative CTCs evading detection when relying on these markers. This is concerning as CTCs with this signature have already been identified in peripheral blood of patients with breast cancer.93
The enzyme-linked immunosorbent assay (ELISA) workflow, typically used for high-throughput protein biomarker assays, is another optical technique applicable to CTC detection, whereby primary and secondary antibodies can be used to first capture, and then generate a quantifying signal. This format can be adapted to produce an electrochemical output by exchanging the optically responsive label/substrate for a redox-active species produced by the enzyme-conjugated secondary antibody. Using voltammetry or amperometry to assay the production of such, the CTC concentration in the sample can be inferred. Label-free electrochemical methods are also applicable to CTC detection in integrated platforms as techniques. This includes electrochemical impedance spectroscopy (EIS) which enables the direct assessment of an interaction between CTCs and an electrode surface. Electrochemical diagnostics are already known to have sufficient sensitivity to assay metabolites and biomarkers in human samples (glucose, lactate, pH, pO2 and pCO2).112,151–155
Additionally, the cost and footprint of electrochemical instruments has been decreasing over the last decade, with a 10-gram potentiostat recently demonstrating similar results in amperometric virus detection, for example, when compared to an instrument 50 times its size, with no statistically significant difference in the signal obtained (p < 0.05).156 A miniaturised electrochemical means of CTC analysis integrated with on-chip isolation presents a real opportunity for an automated (or semi-automated) configuration with a high potential for clinical impact.
Detection can also be achieved by using physical differences between CTCs and other cell populations.94,157 These do not employ immunorecognition agents or staining protocols and therefore have a rapid TAT when compared to methods that do (i.e., immediate detection with high-speed camera or pressure gauge).138,158 Physical methods of CTC detection have been less frequently reported (integrated or otherwise), but one non-integrated example utilised specialised software to confirm CTC identity following isolation with CellSearch® based on size, nuclear–cytoplasmic ratio, and elongation (i.e., cell morphology).94 These technologies have not yet been widely adopted, however a few examples of physical CTC detection on integrated platforms are discussed below (section 3.2.3).
3.1 Integrated on-chip CTC detection
Optical, electrochemical and physical methods of CTC detection have been employed in integrated on-chip platforms where the channels have been patterned in a PDMS substrate or otherwise microfabricated by lithography. The detection methods are summarised in Table 1 and Fig. 8 and the sections below provide a description of their operational principles and methods of integration.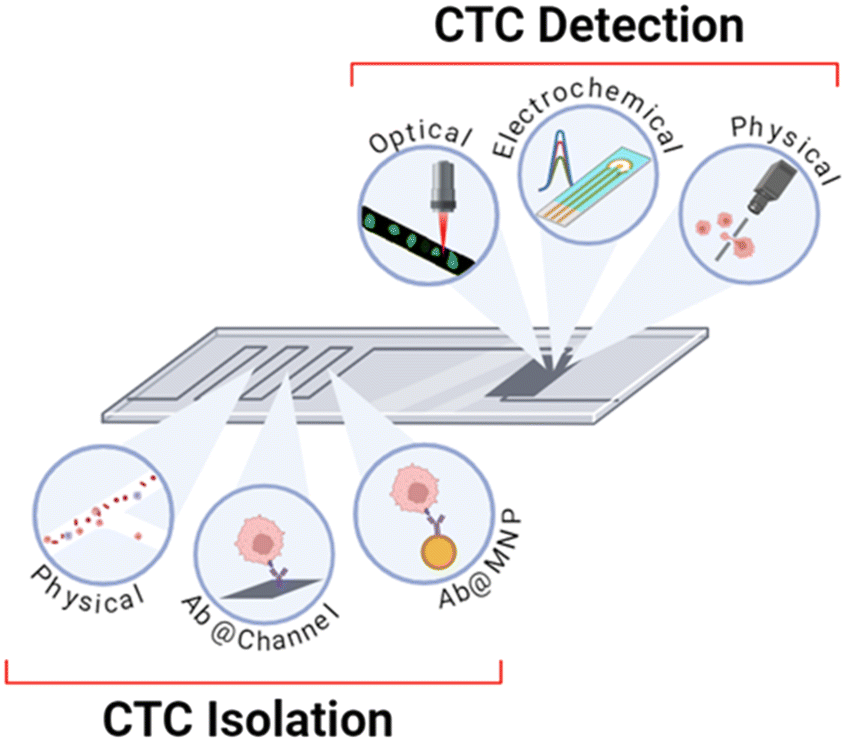 | ||
| Fig. 8 Schematic summary of the methods of isolation and detection that are utilised within integrated platforms discussed herein. Isolation can be completed using physical methods, antibody-modified channels, and antibody-modified nanoparticles. Detection can be achieved through optical, electrochemical, or physical methodologies. | ||
3.2 On-chip integration of optical CTC detection
As briefly mentioned in section 2, optical CTC detection is often utilised within integrated on-chip platforms, perhaps the simplest being direct brightfield or scanning electron microscopy (SEM) of the chip region in which cells were isolated.137,142 Such methods have been performed by simply placing the microfluidic chip under a microscope. However, these basic methods offer little information on the captured CTCs, and no prior works have reported a detection limit.IF staining is by far the most prevalent technique employed thus far for on-chip CTC detection (Table 1), but other available optical techniques include up-conversion luminescence (UCL) imaging and immunoblotting. Researchers frequently default to IF staining, despite the above noted limitations, due to the well-established protocols and literature-supported staining signatures.69 IF within integrated platforms is performed by fixing cells within the channels, flowing staining reagents through, and conducting fluorescence imaging of the chip. For this reason, IF is ubiquitously used to validate CTC detection on platforms where the primary assay was completed by other means (e.g., electrochemical impedance spectroscopy (EIS)112 or deformability cytometry138).
The majority of integrated platforms detect CTCs based on a DAPIpositive, CKpositive, and CD45negative signature56,115,139 with numerous teams including EpCAM positivity in their CTC definition.55,137,140,149 Some reports have enhanced the specificity of their assays towards metastases of distinct origin by including markers beyond the typical selection described above. In one example of this, Lee et al., conducted IF staining of the oestrogen receptor (OR) and progesterone receptor (PR) with an aim to discriminate OR and PR positive CTC subtypes; highly relevant in breast cancer diagnoses (Fig. 5B).122 Similarly, Shi et al. included single cell staining for HER2 on their valve controlled PDMS microfluidic chip. CTCs were shuttled through each region of the device, and subsequently stained, washed and blocked before the entire chip was imaged beneath an inverted microscope, an approach with minimal opportunity for cell loss or damage.121
Of the optical integrated methods, CTC detection by IF has been shown to achieve some of the most impressive detection limits ranging from 1 cell/1.5 mL (ref. 139) to 10 cells/1 mL in whole blood.149 However, gains are being made in lowering this value in an attempt to sample smaller blood volumes using alternative optical techniques such as isothermal nucleic acid detection and immunoblotting.
One unique report of this by Su and team detailed the use of rapid isothermal nucleic detection assay (RIDA) for CTC detection following upstream isolation by antibody-functionalised microbeads and hydrodynamic separation.120 Detection was achieved using biotinylated oligo-DNA conjugated to an anti-EpCAM antibody via streptavidin. This difunctional complex enabled both high specificity capture and sensitive PCR amplified optical detection of the fluorescent signal attribute to the DNA–antibody conjugates. This approach is reported to be effective enough to support a detection limit of 50 cells/1 mL of whole blood.
Another unique report of optical detection in an integrated platform described by Abdulla et al. utilised an immune-affinity technique via immunoblotting to study protein expression within captured CTCs.41 In this work, the team conducted single-cell western blot analysis of captured CTCs by inverting the PDMS chip to transfer the captured cells into microwells in a standard electrophoresis gel (Fig. 7). The cells were lysed in situ, enabling single cell Western blot using primary and secondary antibodies. This method achieved a detection limit of 23 cells/2 mL whole blood, a capture efficiency exceeding 98%, and purity of 90%. Importantly, this work demonstrated an ability to analyse protein expression on the single-cell level, allowing for profiling of CTCs (not just detection) beyond the cell surface markers or physical attributes revealed by IF.
In one example, Wang et al. first captured CTCs using antibody functionalised MNPs, which could be manipulated and detected on chip.118 The cell–particle conjugates proceeded to a silicon nanowire substrate (placed above a permanent magnet) within a PDMS microfluidic channel. The immobilised MNPs/CTC complexes were then quantified using up-conversion luminescence (UCL) by placing the entire chip beneath a 980 nm laser source, allowing for immediate visualisation of CTCs on the chip. This method required only 5 mL of blood from patient samples, demonstrated a detection limit of 10 cells/2 mL of PBS, and a release efficiency of ∼80% with in vitro culture of released cells for 7 days following.
Immunofluorescent staining of on-chip isolated CTCs provides a viable approach for CTC isolation and detection in microfluidic platforms, akin to methods applied in traditional protein immunohistochemistry. This approach has been applied to the label-free isolation of CTCs from ascites and peritoneal lavages using a specially designed continuous flow, size-based, cell trapping array-chip followed by immunofluorescence mapping of EpCAM, YAP-1, HER-2, CD45 to identify CTCs down to a single cell.159 Despite the well-established nature of immunofluorescence-based assays, Cho et al. have, for example, acknowledged a challenge in mapping multiple cell surface markers because of fluorophore spectral overlap and associated difficulty in interpretation.113 Thus, the researchers developed 5 antibody-functionalised gold nanoparticles (Raman-active nanoprobes/RANs), to isolate and tag CTCs from whole blood. The probes were functionalised towards HER2, CD133, EGFR, EpCAM and MUC1, with each RAN having a known spectral fingerprint. By examining the CTC enriched sample with surface enhanced Raman spectroscopy (SERS), the team used the RANs characteristic peaks to identify and quantify circulating cancer stem cells (CCSC), a rare sub-type of CTCs (Fig. 9). SERS mapping imaging was conducted directly on the chip itself using an inverted optical microscope. This allowed for on-chip, multiplexed phenotypic analysis of the captured CTCs based on their spectral identities. This configuration was shown to predict metastasis in a xenograft model with an exact correlation between CCSC concentration and prevalence of liver metastasis. In a recent development, a comparable methodology employed three distinct subtypes of Ab-functionalized Raman-active AuNPs to characterize the heterogeneity of surface protein biomarkers on-chip isolated CTCs. The configuration used on-chip under alternating current-induced mixing of the cells close to chip-laden electrodes, improving CTC capture efficiency, while minimizing non-specific adsorption of loosely bound species within an Ab-functionalized microfluidic chip.160 This configuration was capable of stratifying CTCs and identifying drug-resistant forms by mapping cancer-specific cell-surface biomarkers. Another approach for mapping CTC surface biomarkers through after a semi-automated single step isolation of CTCs on a micropore membrane filter coupled to single cell Raman mapping was proposed.161 In this approach, isolated cells were labelled with SERS-active AuNPs functionalized with specifically engineered aptamers capable of distinguishing isolated CTCs from residual WBCs. While this set-up did not incorporate microfluidic control, it has potential to serve as a readily accessible single platform for such applications.
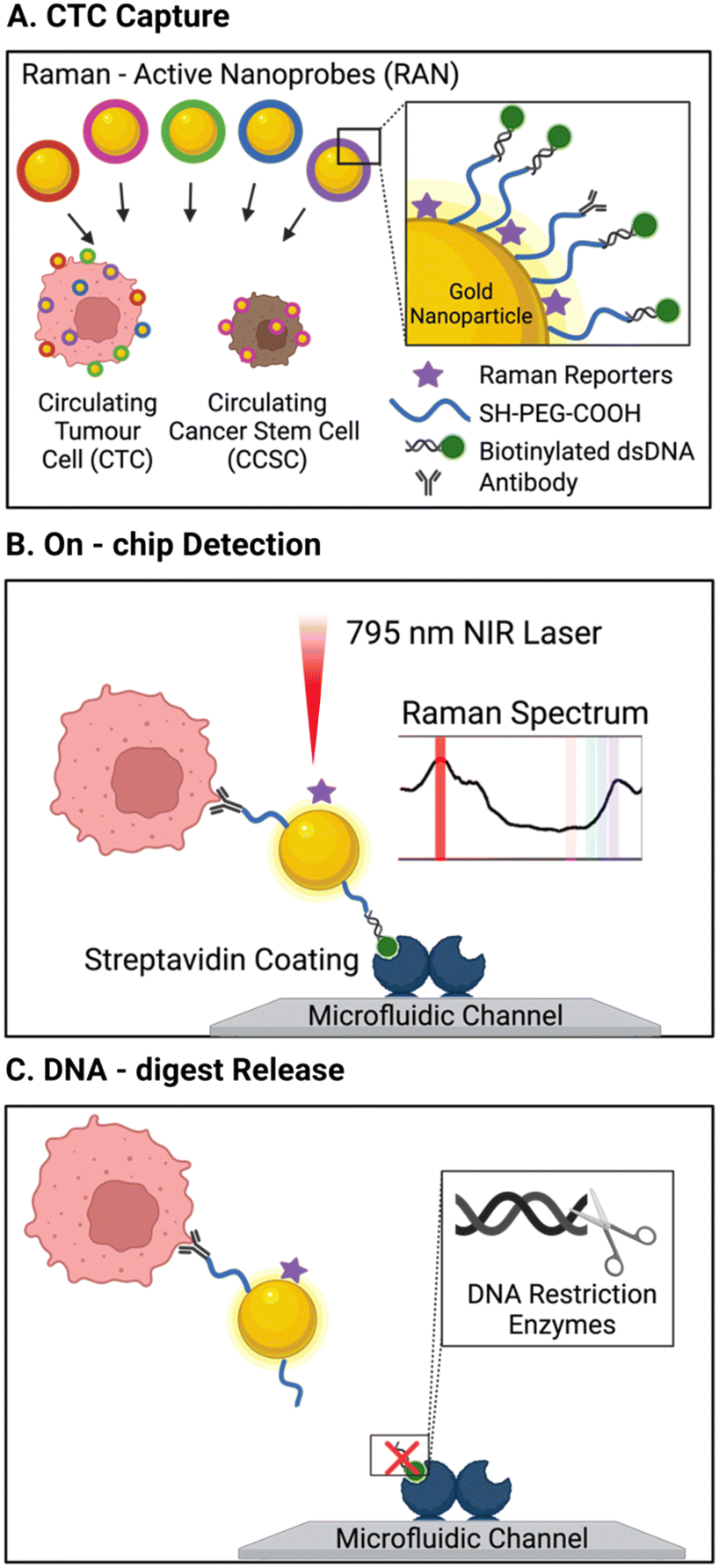 | ||
| Fig. 9 Schematic of a platform with integrated isolation of CTCs via antibody-conjugated Raman-active nanoprobes (Au-NP) wand Raman spectral detection. (A) CTCs were immuno-labelled with 5 types of surface antibody/Raman reporter functionalised AuNP. (B) Biotin units on the capture particles enabled CTC isolation on the streptavidin modified channels, where Raman spectral intensities revealed the degree surface marker expression. (C) CTC release was then accomplished by the addition of a restriction enzyme to cleave dsDNA AuNP linker. Reproduced with permission.113 | ||
As described earlier, the predominant means of CTC isolation prior to on-chip electrochemical assays are Ab-functionalised channels.112,114 An exception to this, is the work of Safaei et al. where CTCs were first enriched in whole blood by anti-EpCAM modified MNPs and subsequently assayed for on-chip using differential pulse voltammetry (DPV), this is an exemplar of an electrochemical ELISA arrangement. In DPV, the current response associated with the voltage pulse is recorded and analysed to examine the faradaic interactions at the electrode surface. The researchers integrated working, counter and reference electrodes to their microfluidic platform by lithographic patterning of 50 nm gold on a glass substrate, and these tracks were subsequently passivated to create isolated electrode apertures. Microfluidic channels were then constructed above the electrodes using photolithography and subsequently capped with a PDMS layer. To obtain a DPV signal, Safaei et al. immobilised and fixed (using 4% paraformaldehyde) their MNP enriched CTCs on ‘X’ shaped geometries within the chip. Prior to this, these conjugates were tagged with biotinylated anti-CK and then exposed to a solution of streptavidin/alkaline phosphatase (ALP) (Fig. 6B).117 Subsequent DPV scans generate substrate dependent amperometric currents that report directly on CTC concentration within the sample. Although this assay involved the numerous incubation steps characteristic of a typical microtiter plate ELISA, the overall protocol complexity was reduced. This is chiefly due to the fabricated microfluidic chip which harboured MNP enriched CTCs for the duration of the ELISA protocol. By simply exchanging the running solution, CTC isolation and detection was completed on-chip in a facile manner.
The assay achieved a detection limit of 2 cells per mL of whole blood, requiring only 1 mL to complete the analysis. Additionally, the researchers reported a capture efficiency of 85% and, due to the MNP separation, low contamination from WBCs at ∼100 cells in 1.5 μL buffer. The electrochemical assay time was not reported; however, each DPV measurement required around 1 minute per sample, a notable contrast to the multiple hours typically required for the IF methods discussed in section 3.1.
In a further example of a fully integrated voltammetric CTC assay, researchers assembled a simple, single channel PDMS microfluidic chip using soft lithography above a screen-printed electrode (SPE). The latter was functionalised with polymer and anti-MC1R antibody to capture cells via immune-affinity interactions.114 The group showed that an increasing CTC (SK-MEL-2) concentration suspended in a redox active running buffer resulted in a log linear decrease (over 10–9000 cells per mL) in voltammetric peak amplitude due to the blocking effects of surface captured cells. In an attempt to reduce non-specific binding from potentially interfering proteins present in real samples, the Ab-modified SPEs were further incubated with bovine serum albumin (BSA). The performance of the sensor was expected to diminish in progressively complex media, and indeed a 20% signal loss was observed over a 3-hour experiment in spiked buffer. In whole blood or other complex media, this degradation is almost certainly substantially higher. The myriad of small cells and proteins present in a human sample are a known issue for electrochemical biosensors as these can readily foul the electrode surface, erroneously reducing the signal from a solution phase redox probe (i.e., poor signal-to-noise ratio).
This can, of course, be remediated by a greater consideration of electrode surface engineering, or integration of this detection method with any of the antigen dependant or physical methods of CTC isolation discussed section 2. A porous 3D channelled PDMS architecture coated with Ab-functionalized conductive polymer was proposed as a novel platform to first, capture EpCAMpositive CTCs, then detecting isolated CTCs leveraging the preferential adsorption of platelets onto the CTCs' surface. The adsorbed platelets activate the catalytic decomposition of H2O2 mediated by specifically designed metalloporphyrin (cobalt hematoporphyrin).163 While this study demonstrates a novel signal generation strategy in identifying patients at risk of hematogenous metastasis (as quantified by the increase in the number of platelet-interacted CTCs), it does not make any reference to the impact of background noise (due to nonspecific adsorption of platelets to the electrode surface) or explore the detection limits.
Chronoamperometric methods, where current is recorded as a function of time, can be employed within microfabricated chips to obtain transient data as CTCs flow through a channel as an alternative to an endpoint assay. This approach has been employed by researchers to quantify CTCs through their association with redox active daunomycin (DM), a potent anti-cancer drug.54 DM was shown to specifically adsorb to a model CTC (HeLa) with low response to a non-cancerous control (HEK-293). A spike in the oxidation current recorded by the electrodes at the channel terminus indicated the presence of a DM labelled CTC eluted from the channel. Due to the upstream physical CTC isolation strategy discussed in section 2.2.1, the time lapsed from the start of sample flow could be used to estimate CTC size, aiding in discrimination between any erroneously labelled healthy cells and, additionally, differentiate between multiple species of CTC. This is an interesting, direct faradaic alternative of the cell tagging immunogenic strategies described earlier. The entire protocol was successfully integrated into a microfluidic chip where the channels and electrodes were formed by screen-printing carbon ink between two glass slides, these then used to probe the oxidation of CTC-tethered DM (Fig. 6A). Importantly, in analysing a significant number of cancer patient samples, with 1 mL volumes incubated off-chip with the drug (∼45 minutes), prior to isolation and detection on-chip, detection limits were reported at 7 cells per mL.
Electrochemical impedance spectroscopy (EIS) involves the application of an AC potential between electrodes and records the resultant current, with respect to frequency or time. This method enables researchers to discriminate between electrode interactions occurring at characteristic frequencies, e.g., simultaneously probing diffusion and charging of the electrochemical double layer and is exquisitely responsive to capture events at a suitably modified electrode. In one report, model CTCs (MCF-7) suspended in PBS at 100 cells per mL were flowed through a simple PDMS channel, captured at the electrode surface via sensor functionalisation with anti-EpCAM and anti-CD36 antibodies, and quantified through the associated change in charge transfer resistance (Rct).112 The researchers reported the quantification of 3 MCF-7 breast cancer cells as CTC analogues in buffer and claim to detect the presence of CTCs in 1 mL of canine cancer patient blood, although no downstream analysis or recovery experiments were reported.
In another example of integrated EIS quantification, CTCs were isolated via pre-concentration onto the electrode surface by application of a dielectrophoretic (DEP) current. This approach offered a rapid TAT, with pre-concentration of CTCs by DEP in 2 minutes, and EIS quantification within 1–2 minutes.146 Similar to the DPV CTC detection method discussed earlier, these sensors are subject to reduced performance in blood due to unwanted electrode fouling. The analyses here were thus completed in lysed blood (requiring pre-processing for RBC lysis) or sucrose buffer, respectively, to present CTCs in a low complexity buffer to the functionalised electrodes.
In summary, electrochemical methods of detection have been shown to be both easy to integrate, and analytically powerful. The typical three electrode configuration has been implemented by directly sealing SPEs beneath a PDMS chip or using tracks of conductive carbon ink as both the electrodes and the walls of microfluidic channels. These rapid assays do not present the notably high costs and TAT presented by IF and have been shown to support an outstanding performance, with detection limits of 2–7 cells per mL in only 1–2 mL of whole blood.54,112,114,117,119,146
In a second example of a purely physical assay, Che et al. utilised deformability cytometry where CTCs were shown to deform to a greater extent than other healthy blood cells when introduced to a microfluidic channel that induced hydrodynamic stretching.138 This was analysed using a high-speed camera, focussed on this deformation channel, synchronised to the release of cells from reservoirs in which they were isolated. The team used additional off-chip IF for validation of detection following CTC elution, using a CKpositive/CD45negative/DAPIpositive signature. Importantly, it was noted that deformability cytometry successfully detected CTCs in 94% of non-small cell lung cancer patients, whereas IF reported only 71% of such samples to be CTC positive.
Notably, these examples of physical detection, though underdeveloped, are completely antigen-independent. However, their sensitivity (detection limit of 50 cells per mL in media supplemented with Jurkat cells and 300 cells in 10× diluted blood, respectively) is markedly poorer than that reported to be associated with other methods discussed. Additionally, they both require pre-processing of blood (40 minutes for RBC lysis and removal and 10 times dilution in PBS, respectively). Despite these limits, these are low-cost approaches, simple to operate, and enable rapid CTC analysis on biomarker independent platforms.158
4 Conclusion
CTC analysis can provide crucial insight into cancer metastasis and tumour evolution.47 At present, methods for CTC isolation and detection have been predominantly built to separate isolation (CTC capture) and detection (verification of captured cell identity or quantification of captured cells).47 The multistep nature of the assay increases time, cost, and manual labour required, and introduces cell loss and significant inter-test variability.2–5 The integration of CTC isolation and detection onto a single microfluidic chip promises to decrease time, cost, and variability of both enrichment and analysis. The use of such configurations has thus far supported capture efficiencies as high as 99% (ref. 122) and detection limits as low as 2 cells per mL whole blood (Table 2).117| Target cells | Isolation method | Detection method | Detection limit | Flow rate | Blood pre-processing; duration | Volume | Capture efficiency | Purity | Other notes | Surface antigen targeted | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| μF, microfluidic; MagRC, magnetic ranking cytometry; EpCAM, epithelial cell adhesion molecule; EChem, electrochemical; Ab, antibody; IF, immunofluorescence; UCL, up-conversion luminescence; MNP, magnetic nanoparticle; SiNW, silicon nanowire; EMT, epithelial-to-mesenchymal transition; CD-36, cluster of differentiation 36; EIS, electrochemical impedance spectroscopy; PANI, polyaniline; DPV, differential pulse voltammetry; SPE, screen printed electrode; WBC, white blood cell; HER2, human epidermal growth factor receptor 2; PDMS, polydimethylsiloxane; ODEP, optically induced dielectrophoretic; SMART-chip, system modularity chip for the analysis of rare targets; DEP, dielectrophoresis; EMT, epithelial–mesenchymal transition; SDAM, superwetting droplet-array microchips; MMP9, matrix metalloproteinase 9; FRET, fluorescence resonance energy transfer; IMD, integrated microfluidic device; DFF, dean flow fractionation; DLD, deterministic lateral displacement; CD-45, cluster of differentiation 45; YAP1: yes-associated protein 1; ac-EHD, ac electrohydrodynamic; PBMCs, peripheral blood mononuclear cells; RPMI, Roswell Park Memorial Institute medium; SWV, square wave voltammetry. | |||||||||||
| MDA-MB-231 | Variance in pressure required to initiate cavitation | High speed camera and pressure gauge | 50 cells per mL culture medium suppl. with Jurkat cells | — | >40 minutes for RBC lysis; detection “within minutes” | — | — | — | — | N/A | 158 |
| - Breast cancer | |||||||||||
| H1975 | Dislocation herringbone μF geometry, immobilised anti-EpCAM Ab | IF (CK 18+/CD45−) | 5 cells per mL whole blood | 20 μL min−1 for cells spiked in blood optimized flow rate | No blood pre-processing; 5 hours per sample | — | H1975 87%, A549 78%, H460 72% | 99.6% | — | EpCAM | 116 |
| A549 | |||||||||||
| H460 | |||||||||||
| - Lung cancer | |||||||||||
| A549 | Off-chip incubation with anti-EpCAM MNPs, μF on-chip magnetic capture | UCL imaging | 10 cells/2 mL PBS | 1 mL h−1 optimized flow rate | RBC lysis and 2 h incubation with anti-EpCAM MNPs; not detailed | 0.5 mL spiked whole blood, 5 mL patient blood sample | >80% | — | 80% release efficiency; released cells could be cultured for 7 days | EpCAM | 118 |
| - Lung cancer | |||||||||||
| VcaP | Immuno-modified MNPs, hydrodynamic separation on chip | EChem oxidation of redox-active secondary Ab, IF validation | 2 cells per mL whole blood | — | 20 min incubation with anti-EpCAM MNPs; ∼3 h to pump sample through, signal read out 1 min per sample, ∼1.5 h for IF | — | 85% | Average capture of ∼80 WBCs | — | EpCAM | 117 |
| - Prostate cancer | |||||||||||
| MCF-7 | Hydrodynamic μF cell sorter with size exclusion membrane filter | Single-cell western blotting | 23 cells/2 mL whole blood | 1.4 mL min−1 | 10 min RBC lysis, 15 min washing; not detailed | 7.5 mL blood sample | 87–98.5% | 89.92% | Analysis of EMT in CTCs isolated from patient samples | N/A | 41 |
| - Breast cancer | |||||||||||
| A459 | |||||||||||
| - Lung cancer | |||||||||||
| HeLa | |||||||||||
| -Cervical cancer | |||||||||||
| MCF-7 | Antibody functionalised electrodes | EIS, IF validation | 100 cells per mL PBS | — | RBC lysis; not detailed | — | — | — | — | EpCAM, CD36 | 112 |
| - Breast cancer | |||||||||||
| SK-MEL-2 | Abs on PANI modified carbon SPEs | EChem reduction of solution redox probe | 1 cell/1 mL PBS suppl. with human cells | 1.5 mL min−1 | Not detailed; not detailed | — | — | — | — | MCR1 | 114 |
| - Skin cancer | |||||||||||
| A549 | Off-chip incubation with daunomycin, AC field isolation on μF chip | EChem oxidation of daunomycin | 7 cells per mL whole blood | 5 μL min−1 for 5 minutes followed by 2.5 μL min−1 | 40 min (30 min incubation with daunomycin, 5 min centrifuge); not detailed | 1 mL of whole blood | Average of 95+–1.5% | 92+–0.5% | — | N/A | 54 |
| - Lung cancer | |||||||||||
| MDA-MB-231 | |||||||||||
| - Breast cancer | |||||||||||
| HeLa | |||||||||||
| -Cervical cancer | |||||||||||
| MCF-7 | Off-chip incubation with immuno-modified magnetic beads, on-chip application of magnetic field | IF (CD45−/HER2+/Hoechst 33 342+) | 10 cells/2 mL whole blood | Optimal flow rate 90 μL min−1 for spiking and 1 μL min−1 for patient samples | Dilution of whole blood, incubation with magnetic beads for unknown time; 240 min for entire process | — | 93% | — | On-chip cell lysis for subsequent RNA-seq | EpCAM | 121 |
| - Breast cancer | |||||||||||
| H446 | Size based sorting in trap channel based on flow resistance | IF (CD45−/CK+/DAPI+), additional IF of EpCAM and vimentin | 5 cells per suspension of 100000 WBCS |
Optimized injection pressure of 10 mbar | Ficoll-Paque processing; not detailed | — | 92–99% depending on WBC count | <80–92% depending on WBC concentration | Achieved release efficiency of >97% | N/A | 140 |
| - Small cell lung cancer | |||||||||||
| H1975 | |||||||||||
| - Non-small cell lung cancer | |||||||||||
| MCF-7 | Size-selective vortex trapping with chip geometry | Deformability cytometry (DC), IF for validation (CK+/CD45−/DAPI+) | 300 cells per unknown volume of 10× diluted blood | 8 mL min−1 | 10× dilution of blood in PBS; not detailed | — | 25–35% | 35.1+–7.3% | 98.7+–1.5% of cells transfer from capture to assay region of chip | N/A | 138 |
| - Breast cancer | |||||||||||
| MCF-7 | 1) Off-chip incubation with anti-EpCAM microbeads | Signal amplification using DNA–Ab conjugates in RIDA | 50 cells/1 mL of whole blood | Optimal flow rate of 1–3 mL h−1, 3 mL h−1 used for patient samples | 25-Minute centrifugation for WBC separation; not detailed | — | >85% | 20–40% | Captured cells proliferated normally in culture | EpCAM | 120 |
| - Breast cancer | |||||||||||
| BxPC-3 | |||||||||||
| -Pancreatic cancer | |||||||||||
| HeLa | 2) Inertial micro-flow system | ||||||||||
| - Cervical cancer | |||||||||||
| PANC-1 | |||||||||||
| -Pancreatic cancer | |||||||||||
| PC3 | Dielectrophoresis on-chip | Brightfield microscopy | Not available | Optimal flow rate of 3 μL min−1 | Not detailed; not detailed | — | — | — | DEP buffer can have effects on cells' gene expression (>15 min) | N/A | 142 |
| - Prostate cancer | |||||||||||
| MCF-7 | Off-chip incubation with Ab-conjugated MNPs, magnetic ranking on-chip (MagRC chip) | IF (DAPI+/CK+/CD45−) | 10 cells/1 mL unprocessed blood | 600 μL h−1 | 30 minute incubation with anti-EpCAM nanobeads; not detailed | 1 mL of blood used for patient samples | SKBR3 97+–3%, PC-3 90+–2%, MDA-MB-231 90+–3% | — | 92% recovery, 98% viable | EpCAM, (plus HER2 and N-cadherin for SKBR3 cells) | 115 |
| SKBR3 | |||||||||||
| - Breast cancer | |||||||||||
| PC3 | |||||||||||
| - Prostate cancer | |||||||||||
| MDA-MB-231 | |||||||||||
| - Breast cancer (mesenchymal properties) | |||||||||||
| MDA-MB-231 | Off-chip incubation with anti-HER2 MNPs, magnetic ranking on-chip | IF of estrogen receptor (ER) and progesterone receptor (PR) | — | 3 mL h−1 | No experiments with patient samples conducted | No experiments with patient samples conducted | MDA-MB-231 65.3+–1.6%, BT-474 99.3+–0.4%, SK-BR-3 99.9+–0.1%, MCF-7 92.9+–1.8% | — | — | HER2 | 122 |
| BT-474 | |||||||||||
| SK-BR-3 | |||||||||||
| MCF-7 | |||||||||||
| - Breast cancer | |||||||||||
| MCF-7 | Size based isolation with CellSieve microfilter | IF (CK8/18/19+, EpCAM+, DAPI+), H&E staining | — | 5 mL min−1 for cells spiked in whole blood | No blood pre-processing; not detailed | — | Approx. 90% | >99.9% removal of WBCs from blood sample | Cell lines adhered and grew on the microfilters, 97+–2% release efficiency of MCF-7 cells | N/A | 137 |
| MDA-MB-231 | |||||||||||
| SK-BR-3 | |||||||||||
| - Breast cancer | |||||||||||
| PANC-1 | |||||||||||
| - Pancreatic cancer | |||||||||||
| A549 | Size based isolation with PDMS membrane filter-based microdevice | IF (CK-PE+/CD45-FITC−/DAPI+) | 1 cell/1.5 mL of whole blood | Optimized flow rate of 10 mL h−1 for cells spiked in whole blood | No blood pre-processing; not detailed | — | >92% for A549 and SK-MES-1 cells; 99% for H446 | — | — | N/A | 139 |
| SK-MES-1 | |||||||||||
| H446 | |||||||||||
| - Lung cancer | |||||||||||
| PC-3 | ODEP following IF identification of cells of interest | IF (CD45−/Hoechst+/EpCAM+) | 10 cells per mL whole blood | Optimized flow rate of 2.5 μL min−1 for cells spiked in whole blood | No blood pre-processing; not detailed | 8 mL of blood for spiking experiments | 41.5% | As high as 100% | Detection occurs prior to isolation | N/A | 149 |
| - Prostate cancer | |||||||||||
| SKBR3 | SMART-chip; microchannels with immobilized anti-EpCAM Ab | Impedance counting and viability of unlabelled single cells; IF (DAPI+/CD45−/Pan-CK+) | — | — | No blood pre-processing; 3.5 h | 2 mL of blood processed for patient samples | >80%; 73% for anti-EpCAM PC linker with SKBR3 cells in whole blood | >85% | Quick (2 min) and efficient (>90%) release of affinity-captured CTCs via photo release with visible blue light | EpCAM | 119 |
| - Breast cancer | |||||||||||
| MCF-7 | 3D PDMS scaffold with immobilized anti-EpCAM Ab | IF staining (DAPI+/CK+/CD45−) | 10 cells/1 mL whole blood | Optimized flow rate of 100 μL min−1 for cells spiked in whole blood | No blood pre-processing; ∼4.5 h | 1 mL of whole blood for spiking experiments | Average capture efficiency of 88.4% | — | CTC clusters were also detected and imaged in patient samples | EpCAM | 56 |
| - Breast cancer | |||||||||||
| MCF-7 | Raman active nanoprobes (RANs) conjugated to Ab (anti-CD133, anti-EpCAM, anti-HER2, anti-MUC1) with biotinylated dsDNA for isolation | In situ subtyping by Raman barcoding system | — | 10 μL min−1 | 2× dilution with PBS, Ficoll-Paque processing, centrifugation for WBC exclusion, 30 min incubation with RANs; not detailed | 4 mL blood for spiking experiments | 90% | — | Detects circulating cancer stem cells (CCSCs) as well as CTCs, ability to release captured cells using restriction enzymes | CD133, EpCAM, HER2, MUC1 | 113 |
| MDA-MB-231 | |||||||||||
| SK-BR-3 | |||||||||||
| - Breast cancer | |||||||||||
| A549 | Windmill-like hole array for size-based isolation | IF staining (DAPI+/CD45−/Ep-CAM+ CTC signature) | <10 cells per mL blood | 2 mL min−1 | Unspecified pre-treatment and dilution with 10 mL PBS; not detailed | 1–3 mL whole blood for patient samples | 93% for A549 cells; 90% for HeLa cells | WBC depletion rate of 98.7% | — | N/A | 55 |
| - Lung cancer | |||||||||||
| HeLa | |||||||||||
| - Cervical cancer | |||||||||||
| A549 | DEP | EIS | 3 cells | Flow rate of 1.2 μL min−1 | Not detailed; electrical operation ∼10 min | — | Approx. 80% | — | Detection limit achieved at 50 kHz | N/A | 146 |
| - Lung cancer | |||||||||||
| A549 | SDAM | FRET (peptide probe for MMP9 enzyme activity) | Single cell | — | Diluted 10 mL blood (blood:PBS = 1:1) |
5 mL | — | — | Evaluate EMT and cancer metastasis | MMP6 enzyme activity | 141 |
| - Lung cancer | |||||||||||
| HGC-27 | IMD composed of a spiral chip (using the DFF technique) and a DLD chip | IF staining (EpCAM, YAP1, HER-2, CD45) | 1 cell/10000 cells |
Optimized flow rate of 12 μL min−1 | Ascites and peritoneal lavages | N/A | 63% | 73% | EpCAM | 159 | |
| - Gastric cancer | |||||||||||
| SK-MEL28 | ac-EHD with antibody modified chip | Raman signature for MCSP, MCAM, ErbB3, and LNGFR (melanoma) and PD-L1 and EGFR (lung cancer) | — | Loaded immediately into the chip with ac-EHD applied for 10 min | PBMCs suspended in 2.0 mL RPMI 2 h | — | 60% | N/A | MCSP for melanoma cells | 160 | |
| - Melanoma | |||||||||||
| NSCLC | EGFR for lung cancer cells | ||||||||||
| - Lung cancer | |||||||||||
| A549 | Microporous parylene membrane | Raman intensity | N/A | Dropped onto the micropore membrane for filtration | A549 cells in 100 μL culture medium into 400 μL blood | — | 91% | 90% | Aptamer probe selected for preferential binding to CTCs | Size based filtration | 161 |
| - Lung cancer | |||||||||||
| SK-BR-3 | 3D electrode integrated within a microfluidic flow channel | SWV | — | 40 μL min−1 | Not discussed | — | 71% | N/A | EPCAM | 163 | |
| - Breast cancer | |||||||||||
The majority of CTC isolation methods in integrated platforms rely on immuno-affinity,112,114,115,118,120,122 but others, based on cell deformability,138 inertia force,41 and size variance158 have been documented. Integrated detection methods range from direct imaging of a transparent PDMS chip, often employing some derivative of immunofluorescent staining, to electrochemical detection using electrodes embedded within the platform.54,112,114,117,119,146
Antigen-dependent methods of isolation used in integrated systems have achieved capture efficiencies of 80–97%,56,112–122 and purities of up to 99.9%.56,119,122 Physical methods of isolation completed on a single chip are reportedly capable of obtaining CTCs with capture efficiencies of 25–99% (ref. 41, 54, 55, 120, 137–139, 146) and associated purities spanning 20–92%.54,120,138 While the purity metric is typically lower with physical means of isolation, their utility should not be underestimated as they allow for the recovery of label-free CTCs, delivering an ideal starting point for interference-free downstream analysis. CTC recovery following antigen-dependent isolation has been also reported with release efficiencies ranging from 80–97%,115,118,119 with one report even demonstrating a post-assay proliferation of isolated cells in culture.120 Such downstream release and culture represents an advantage for physical means of isolation, although antigen-dependent isolation methods are also able to achieve release if appropriately designed.
Looking beyond isolation, the platforms discussed in this review have achieved CTC detection from low volumes (down to 1 mL whole blood),54,120 with limits as low as 2 cells/1 mL whole blood.117 Electrochemical detection methods have already demonstrated detection limits ranging from 2–7 cells per mL of whole blood, or 1 cell/1 mL in PBS supplemented with human mononuclear cells.114 Detection limits for optical means typically range from 1–50 cells per mL of whole blood,41,56,115,116,118,120,121,139 with physical methods somewhat poorer.138,158
4.1 Future directions in integrated CTC isolation & detection
While scalable and potentially very cost-effective integrated platforms hold great promise for supporting high capture efficiency and sensitivities high enough to detect as low as 1 cell per mL, such methods are early in development and have yet to translate into clinical practice. Reducing the turnaround time to less than 1 hour per sample (rather than the ∼1/2 day typically associated with a CellSearch analysis) and substantially lowering cost and increasing reliability would enable the application of this currently prohibitively expensive technique to routine clinical practice. While the long-standing optical method of detection via IF imaging is able to achieve low detection limits,116,121 this comes with inherent time challenges, as discussed in section 3.116,121Methods of CTC isolation involving sample incubation with Ab-functionalised MNPs show promise due to their high capture efficiencies. However, reports to date have required off-chip sample incubation with MNPs, increasing processing time and the potential for cell loss with manual transfer.115,117,118,120–122 Thus, there is still need for development of more intimately integrated derivatives to increase throughput. Purely physical methods of CTC analysis/assaying remain rare, and are associated with relatively poor detection limits.138,158 While the long-standing optical method of detection via IF imaging sensitive,116,121 this comes with inherent time and throughput challenges, as discussed in section 3.116,121 Simple forms of optical assay such as brightfield142 or up-conversion luminescence imaging118 can be chip-integrated offer reduced labour and time, but these do not offer any phenotypic or profiling information about the captured CTCs. Reports discussed above which implemented innovative designs such as single-cell immunoblotting,41 rapid isothermal nucleic acid detection assay,120 and in situ Raman barcoding113 represent, then, the most interesting avenues for optical detection. Such methods offer phenotypic information through either multiplexing cell-surface marker analysis or uncovering intracellular protein expression. These approaches enable an on-chip classification of CTC subtype based on their expression of a range of established markers or receptors. This is particularly advantageous, as the clinician is given more than a mere binary i.e., CTC positive/negative result, enabling more informed decisions on diagnosis or treatment avenues.
The integrated platforms in this review represent a significant advance down the road towards a routine low blood volume CTC analysis sufficiently robust to underpin new biological investigations and reliable assays. These analyses can reveal important insight into genomic markers of disease metastasis and ultimately inform clinical decision making.115,122 Future integrated devices could play a major role in conveniently sorting CTCs based on the cell surface expression of specific markers.115,122 Such platforms could facilitate downstream analysis and phenotypic evaluation by genomic profiling of, potentially, individual cells. Ultimately, we have sought to summarise the broad range of scalable and fully integrated toolboxes that are being developed. These may ultimately create opportunities for routine and convenient liquid biopsies and be applied in disease identification and staging, assessing progression risk, and guiding treatment.
Author contributions
Sophia M. Abusamra: writing – original draft preparation, writing – review & editing. Robert Barber: writing – original draft preparation, writing –review & editing. Mohamed Sharafeldin: writing – original draft preparation, writing – review & editing. Claire M. Edwards: writing – review & editing, supervision. Jason J. Davis: writing – original draft preparation, writing – review & editing, supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank CRUK for funding (grant 100029). SMA would also like to thank Dr. Alastair Lamb, MBChB, PhD, Dr. Ian Mills, PhD, and Dr. Todd Morgan, MD for their supervision.References
- D. C. Danila, G. Heller, G. A. Gignac, R. Gonzalez-Espinoza, A. Anand, E. Tanaka, H. Lilja, L. Schwartz, S. Larson, M. Fleisher and H. I. Scher, Clin. Cancer Res., 2007, 13, 7053–7058 CrossRef CAS PubMed.
- H.-J. Jou, H.-C. Ho, K.-Y. Huang, C.-Y. Chen, S.-W. Chen, P.-H. Lo, P.-W. Huang, C.-E. Huang and M. Chen, Int. J. Mol. Sci., 2022, 23, 15139 CrossRef CAS PubMed.
- H. Gwak, J. Kim, L. Kashefi-Kheyrabadi, B. Kwak, K.-A. Hyun and H.-I. Jung, Micromachines, 2018, 9, 353 CrossRef PubMed.
- H. W. Hou, M. E. Warkiani, B. L. Khoo, Z. R. Li, R. A. Soo, D. S.-W. Tan, W.-T. Lim, J. Han, A. A. S. Bhagat and C. T. Lim, Sci. Rep., 2013, 3, 1259 CrossRef PubMed.
- B. Hong and Y. Zu, Theranostics, 2013, 3, 377–394 CrossRef CAS PubMed.
- C. L. Chaffer and R. A. Weinberg, Science, 2011, 331, 1559–1564 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, Ca-Cancer J. Clin., 2021, 71, 7–33 CrossRef PubMed.
- J. Fares, M. Y. Fares, H. H. Khachfe, H. A. Salhab and Y. Fares, Signal Transduction Targeted Ther., 2020, 5, 28 CrossRef PubMed.
- Z. Deng, S. Wu, Y. Wang and D. Shi, EBioMedicine, 2022, 83, 104237 CrossRef CAS PubMed.
- A. Ring, B. D. Nguyen-Sträuli, A. Wicki and N. Aceto, Nat. Rev. Cancer, 2023, 23, 95–111 CrossRef CAS PubMed.
- D. S. Micalizzi, S. Maheswaran and D. A. Haber, Genes Dev., 2017, 31, 1827–1840 CrossRef CAS PubMed.
- D. Lin, L. Shen, M. Luo, K. Zhang, J. Li, Q. Yang, F. Zhu, D. Zhou, S. Zheng, Y. Chen and J. Zhou, Signal Transduction Targeted Ther., 2021, 6, 404 CrossRef CAS PubMed.
- Z. Feng, J. Wu, Y. Lu, Y.-T. Chan, C. Zhang, D. Wang, D. Luo, Y. Huang, Y. Feng and N. Wang, Int. J. Biol. Sci., 2022, 18, 3251–3265 CrossRef CAS PubMed.
- A. Chauhan, R. Kaur, S. Ghoshal and A. Pal, Indian J. Clin. Biochem., 2021, 36, 131–142 CrossRef PubMed.
- M. Ilié and P. Hofman, Transl. Lung Cancer Res., 2016, 5, 420–423 CrossRef PubMed.
- P. Gilson, J.-L. Merlin and A. Harlé, Cancers, 2022, 14, 1384 CrossRef PubMed.
- I. Martins, I. P. Ribeiro, J. Jorge, A. C. Gonçalves, A. B. Sarmento-Ribeiro, J. B. Melo and I. M. Carreira, Genes, 2021, 12, 349 CrossRef CAS PubMed.
- J. Creaney, D. Yeoman, Y. Demelker, A. Segal, A. W. Musk, S. J. Skates and B. W. S. Robinson, J. Thorac. Oncol., 2008, 3, 851–857 CrossRef PubMed.
- S. Maestranzi, R. Przemioslo, H. Mitchell and R. A. Sherwood, Ann. Clin. Biochem., 1998, 35, 99–103 CrossRef PubMed.
- S. N. Lone, S. Nisar, T. Masoodi, M. Singh, A. Rizwan, S. Hashem, W. El-Rifai, D. Bedognetti, S. K. Batra, M. Haris, A. A. Bhat and M. A. Macha, Mol. Cancer, 2022, 21, 79 CrossRef CAS PubMed.
- H. Zhang, X. Lin, Y. Huang, M. Wang, C. Cen, S. Tang, M. R. Dique, L. Cai, M. A. Luis, J. Smollar, Y. Wan and F. Cai, Front. Oncol., 2021, 11, 652253 CrossRef CAS PubMed.
- G. Galletti, M. S. Sung, L. T. Vahdat, M. A. Shah, S. M. Santana, G. Altavilla, B. J. Kirby and P. Giannakakou, Lab Chip, 2014, 14, 147–156 RSC.
- A. Yadav, A. Kumar and M. H. Siddiqui, World J. Clin. Oncol., 2021, 12, 1169–1181 CrossRef PubMed.
- M. Banys-Paluchowski, T. Fehm, H. Neubauer, P. Paluchowski, N. Krawczyk, F. Meier-Stiegen, C. Wallach, A. Kaczerowsky and G. Gebauer, Arch. Gynecol. Obstet., 2020, 301, 1027–1035 CrossRef CAS PubMed.
- J. Kapeleris, A. Kulasinghe, M. E. Warkiani, I. Vela, L. Kenny, K. O'Byrne and C. Punyadeera, Front. Oncol., 2018, 8, 311 CrossRef PubMed.
- Y. Kiniwa, K. Nakamura, A. Mikoshiba, A. Ashida, Y. Akiyama, A. Morimoto and R. Okuyama, BMC Cancer, 2021, 21, 287 CrossRef CAS PubMed.
- S. Calabuig-Fariñas, E. Jantus-Lewintre, A. Herreros-Pomares and C. Camps, Transl. Lung Cancer Res., 2016, 5, 466–482 CrossRef PubMed.
- M. Nikanjam, S. Kato and R. Kurzrock, J. Hematol. Oncol., 2022, 15, 131 CrossRef CAS PubMed.
- F. Cheng, L. Su and C. Qian, Onco Targets Ther, 2016, 7, 48832–48841 Search PubMed.
- J. Liu, X. Chen, J. Wang, S. Zhou, C. L. Wang, M. Z. Ye, X. Y. Wang, Y. Song, Y. Q. Wang, L. T. Zhang, R. H. Wu, H. M. Yang, S. D. Zhu, M. Z. Zhou, X. C. Zhang, H. M. Zhu and Z. Y. Qian, Ann. Oncol., 2019, 30, 464–470 CrossRef CAS PubMed.
- C. Abbosh, C. Swanton and N. J. Birkbak, Ann. Oncol., 2019, 30, 358–359 CrossRef CAS PubMed.
- R. Okamura, D. E. Piccioni, A. Boichard, S. Lee, R. E. Jimenez, J. K. Sicklick, S. Kato and R. Kurzrock, Int. J. Cancer, 2021, 148, 2839–2847 CrossRef CAS PubMed.
- M. Cristofanilli, G. T. Budd, M. J. Ellis, A. Stopeck, J. Matera, M. C. Miller, J. M. Reuben, G. V. Doyle, W. J. Allard, L. W. M. M. Terstappen and D. F. Hayes, N. Engl. J. Med., 2004, 351, 781–791 CrossRef CAS PubMed.
- B. Rack, C. Schindlbeck, J. Jückstock, U. Andergassen, P. Hepp, T. Zwingers, T. W. Friedl, R. Lorenz, H. Tesch, P. A. Fasching, T. Fehm, A. Schneeweiss, W. Lichtenegger, M. W. Beckmann, K. Friese, K. Pantel and W. Janni, J. Natl. Cancer Inst., 2014, 106, dju066 CrossRef PubMed.
- J.-Y. Pierga, D. Hajage, T. Bachelot, S. Delaloge, E. Brain, M. Campone, V. Diéras, E. Rolland, L. Mignot, C. Mathiot and F.-C. Bidard, Ann. Oncol., 2012, 23, 618–624 CrossRef PubMed.
- D. F. Hayes, M. Cristofanilli, G. T. Budd, M. J. Ellis, A. Stopeck, M. C. Miller, J. Matera, W. J. Allard, G. V. Doyle and L. W. W. M. Terstappen, Clin. Cancer Res., 2006, 12, 4218–4224 CrossRef CAS PubMed.
- E. Yu, A. L. Allan, M. Sanatani, D. Lewis, A. Warner, A. R. Dar, B. P. Yaremko, L. E. Lowes, D. A. Palma, J. Raphael, M. D. Vincent, G. B. Rodrigues, D. Fortin, R. I. Inculet, E. Frechette, J. Bierer, J. Law, J. Younus and R. A. Malthaner, BMC Cancer, 2022, 22, 746 CrossRef CAS PubMed.
- R. Y. Tay, F. Fernández-Gutiérrez, V. Foy, K. Burns, J. Pierce, K. Morris, L. Priest, J. Tugwood, L. Ashcroft, C. R. Lindsay, C. Faivre-Finn, C. Dive and F. Blackhall, Ann. Oncol., 2019, 30, 1114–1120 CrossRef CAS PubMed.
- D.-B. Asante, G. R. K. A. Mohan, E. Acheampong, M. Ziman, L. Calapre, T. M. Meniawy, E. S. Gray and A. B. Beasley, Sci. Rep., 2023, 13, 2552 CrossRef CAS PubMed.
- A. K. Dutta, J.-B. Alberge, E. D. Lightbody, C. J. Boehner, A. Dunford, R. Sklavenitis-Pistofidis, T. H. Mouhieddine, A. N. Cowan, N. K. Su, E. M. Horowitz, H. Barr, L. Hevenor, J. B. Beckwith, J. Perry, A. Cao, Z. Lin, F. K. Kuhr, R. G. Del Mastro, O. Nadeem, P. T. Greipp, C. Stewart, D. Auclair, G. Getz and I. M. Ghobrial, Cancer Discovery, 2023, 13, 348–363 CrossRef CAS PubMed.
- A. Abdulla, T. Zhang, S. Li, W. Guo, A. R. Warden, Y. Xin, N. Maboyi, J. Lou, H. Xie and X. Ding, Microsyst. Nanoeng., 2022, 8, 13 CrossRef CAS PubMed.
- L. Carter, D. G. Rothwell, B. Mesquita, C. Smowton, H. S. Leong, F. Fernandez-Gutierrez, Y. Li, D. J. Burt, J. Antonello, C. J. Morrow, C. L. Hodgkinson, K. Morris, L. Priest, M. Carter, C. Miller, A. Hughes, F. Blackhall, C. Dive and G. Brady, Nat. Med., 2017, 23, 114–119 CrossRef CAS PubMed.
- J. P. Winer-Jones, B. Vahidi, N. Arquilevich, C. Fang, S. Ferguson, D. Harkins, C. Hill, E. Klem, P. C. Pagano, C. Peasley, J. Romero, R. Shartle, R. C. Vasko, W. M. Strauss and P. W. Dempsey, PLoS One, 2014, 9, e86717 CrossRef PubMed.
- D. Sefrioui, F. Blanchard, E. Toure, P. Basile, L. Beaussire, C. Dolfus, A. Perdrix, M. Paresy, M. Antonietti, I. Iwanicki-Caron, R. Alhameedi, S. Lecleire, A. Gangloff, L. Schwarz, F. Clatot, J.-J. Tuech, T. Frébourg, F. Jardin, J.-C. Sabourin, N. Sarafan-Vasseur, P. Michel and F. Di Fiore, Br. J. Cancer, 2017, 117, 1017–1025 CrossRef CAS PubMed.
- J. S. de Bono, H. I. Scher, R. B. Montgomery, C. Parker, M. C. Miller, H. Tissing, G. V. Doyle, L. W. W. M. Terstappen, K. J. Pienta and D. Raghavan, Clin. Cancer Res., 2008, 14, 6302–6309 CrossRef CAS PubMed.
- Z. R. Reichert, T. Kasputis, S. Nallandhighal, S. M. Abusamra, A. Kasputis, S. Haruray, Y. Wang, S. Williams, U. Singhal, A. Alva, F. C. Cackowski, M. E. V. Caram, P. L. Palmbos, S. E. Yentz, D. C. Smith, J. J. Alumkal and T. M. Morgan, Int. J. Mol. Sci., 2021, 23, 4 CrossRef PubMed.
- Z. Deng, S. Wu, Y. Wang and D. Shi, EBioMedicine, 2022, 83, 104237 CrossRef CAS PubMed.
- S. Park, D. J. Wong, C. C. Ooi, D. M. Kurtz, O. Vermesh, A. Aalipour, S. Suh, K. L. Pian, J. J. Chabon, S. H. Lee, M. Jamali, C. Say, J. N. Carter, L. P. Lee, W. G. Kuschner, E. J. Schwartz, J. B. Shrager, J. W. Neal, H. A. Wakelee, M. Diehn, V. S. Nair, S. X. Wang and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E8379–E8386 CAS.
- M. J. M. Magbanua, E. V. Sosa, R. Roy, L. E. Eisenbud, J. H. Scott, A. Olshen, D. Pinkel, H. S. Rugo and J. W. Park, Cancer Res., 2013, 73, 30–40 CrossRef CAS PubMed.
- R. Negishi, H. Yamakawa, T. Kobayashi, M. Horikawa, T. Shimoyama, F. Koizumi, T. Sawada, K. Oboki, Y. Omuro, C. Funasaka, A. Kageyama, Y. Kanemasa, T. Tanaka, T. Matsunaga and T. Yoshino, Commun. Biol., 2022, 5, 20 CrossRef CAS PubMed.
- P. Neužil, S. Giselbrecht, K. Länge, T. J. Huang and A. Manz, Nat. Rev. Drug Discovery, 2012, 11, 620–632 CrossRef PubMed.
- S. Regmi, C. Poudel, R. Adhikari and K. Q. Luo, Biosensors, 2022, 12, 459 CrossRef CAS PubMed.
- L. Convert, V. Chabot, P.-J. Zermatten, R. Hamel, J.-P. Cloarec, R. Lecomte, V. Aimez and P. G. Charette, Sens. Actuators, B, 2012, 173, 447–454 CrossRef CAS.
- N. G. Gurudatt, S. Chung, J.-M. Kim, M.-H. Kim, D.-K. Jung, J.-Y. Han and Y.-B. Shim, Biosens. Bioelectron., 2019, 146, 111746 CrossRef CAS PubMed.
- H. Li, J. Li, Z. Zhang, Z. Guo, C. Zhang, Z. Wang, Q. Guo, C. Li, C. Li, J. Yao, A. Zheng, J. Xu, Q. Gao, W. Zhang and L. Zhou, Microsyst. Nanoeng., 2022, 8, 23 CrossRef CAS PubMed.
- S.-B. Cheng, M. Xie, J.-Q. Xu, J. Wang, S.-W. Lv, S. Guo, Y. Shu, M. Wang, W.-G. Dong and W.-H. Huang, Anal. Chem., 2016, 88, 6773–6780 CrossRef CAS PubMed.
- D. Yin, A. Shi, B. Zhou, M. Wang, G. Xu, M. Shen, X. Zhu and X. Shi, Langmuir, 2022, 38, 11080–11086 CrossRef CAS PubMed.
- K. Shirai, G. Guan, T. Meihui, P. Xiaoling, Y. Oka, Y. Takahashi, A. A. S. Bhagat, M. Yanagida, S. Iwanaga, N. Matsubara, T. Mukohara and T. Yoshida, Lab Chip, 2022, 22, 4418–4429 RSC.
- A. A. Kajani, L. Rafiee, M. Samandari, M. A. Mehrgardi, B. Zarrin and S. H. Javanmard, RSC Adv., 2022, 12, 32834–32843 RSC.
- A. A. Pore, S. S. Bithi, M. Zeinali, H. Bin Navaid, S. Nagrath, R. Layeequr Rahman and S. A. Vanapalli, Biomicrofluidics, 2022, 16, 64107 CrossRef CAS PubMed.
- V. Murlidhar, M. Zeinali, S. Grabauskiene, M. Ghannad-Rezaie, M. S. Wicha, D. M. Simeone, N. Ramnath, R. M. Reddy and S. Nagrath, Small, 2014, 10, 4895–4904 CrossRef CAS PubMed.
- C. Launiere, M. Gaskill, G. Czaplewski, J. H. Myung, S. Hong and D. T. Eddington, Anal. Chem., 2012, 84, 4022–4028 CrossRef CAS PubMed.
- X. Jiang, K. H. K. Wong, A. H. Khankhel, M. Zeinali, E. Reategui, M. J. Phillips, X. Luo, N. Aceto, F. Fachin, A. N. Hoang, W. Kim, A. E. Jensen, L. V. Sequist, S. Maheswaran, D. A. Haber, S. L. Stott and M. Toner, Lab Chip, 2017, 17, 3498–3503 RSC.
- Y. Kang, T. Hadlock, T. Lo, E. Purcell, A. Mutukuri, S. Fouladdel, M. D. S. Raguera, H. Fairbairn, V. Murlidhar, A. Durham, S. A. McLean and S. Nagrath, Adv. Sci., 2020, 7, 2001581 CrossRef CAS PubMed.
- S. Dey, R. Vaidyanathan, L. G. Carrascosa, M. J. A. Shiddiky and M. Trau, ACS Sens., 2016, 1, 399–405 CrossRef CAS.
- T. Gerber, S. Taschner-Mandl, L. Saloberger-Sindhöringer, N. Popitsch, E. Heitzer, V. Witt, R. Geyeregger, C. Hutter, R. Schwentner, I. M. Ambros and P. F. Ambros, J. Mol. Diagn., 2020, 22, 1070–1086 CrossRef CAS PubMed.
- D. Irimia and M. Toner, Lab Chip, 2006, 6, 345 RSC.
- P. P. Lin, O. Gires, D. D. Wang, L. Li and H. Wang, Sci. Rep., 2017, 7, 9789 CrossRef PubMed.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- J. Jiang, H. Zhao, W. Shu, J. Tian, Y. Huang, Y. Song, R. Wang, E. Li, D. Slamon, D. Hou, X. Du, L. Zhang, Y. Chen and Q. Wang, Sci. Rep., 2017, 7, 42612 CrossRef CAS PubMed.
- N. M. Karabacak, P. S. Spuhler, F. Fachin, E. J. Lim, V. Pai, E. Ozkumur, J. M. Martel, N. Kojic, K. Smith, P. Chen, J. Yang, H. Hwang, B. Morgan, J. Trautwein, T. A. Barber, S. L. Stott, S. Maheswaran, R. Kapur, D. A. Haber and M. Toner, Nat. Protoc., 2014, 9, 694–710 CrossRef CAS PubMed.
- Z. Dan and C. Daxiang, Cancer Biol. Med., 2018, 15, 335 CrossRef PubMed.
- W. Qian, Y. Zhang and W. Chen, Small, 2015, 11, 3850–3872 CrossRef CAS PubMed.
- R. T. Krivacic, A. Ladanyi, D. N. Curry, H. B. Hsieh, P. Kuhn, D. E. Bergsrud, J. F. Kepros, T. Barbera, M. Y. Ho, L. B. Chen, R. A. Lerner and R. H. Bruce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10501–10504 CrossRef CAS PubMed.
- J.-S. Chung, Y. Wang, J. Henderson, U. Singhal, Y. Qiao, A. B. Zaslavsky, D. H. Hovelson, D. E. Spratt, Z. Reichert, G. S. Palapattu, R. S. Taichman, S. A. Tomlins and T. M. Morgan, Neoplasia, 2019, 21, 802–809 CrossRef CAS PubMed.
- L. Descamps, D. Le Roy and A. L. Deman, Int. J. Mol. Sci., 2022, 23, 1981 CrossRef CAS PubMed.
- C. Macaraniag, Q. Luan, J. Zhou and I. Papautsky, APL Bioeng., 2022, 6, 31501 CrossRef CAS PubMed.
- M. M. Ferreira, V. C. Ramani and S. S. Jeffrey, Mol. Oncol., 2016, 10, 374–394 CrossRef CAS PubMed.
- M. Labib and S. O. Kelley, Mol. Oncol., 2021, 15, 1622–1646 CrossRef CAS PubMed.
- W. Lv, B. Fu, W. Liu, W. Huang, M. Li, Y. Liu, Y. Kang, J. Wang, S. Bai, C. Lu and X. Dai, Talanta, 2024, 267, 125220 CrossRef CAS PubMed.
- C. Cruz, A. Miranda and T. Santos, in Aptamers Engineered Nanocarriers for Cancer Therapy, Elsevier, 2023, pp. 403–432 Search PubMed.
- E. Racila, D. Euhus, A. J. Weiss, C. Rao, J. McConnell, L. W. M. M. Terstappen and J. W. Uhr, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4589–4594 CrossRef CAS PubMed.
- T. Fehm, A. Sagalowsky, E. J. Clifford, P. Beitsch, H. Saboorian, D. Euhus, S. Meng, L. Morrison, T. Tucker, N. Lane, B. M. Ghadimi, K. Heselmeyer-Haddad, T. Ried, C. Rao and J. Uhr, Clin. Cancer Res., 2002, 8, 2073–2084 CAS.
- L. Hinck and I. Näthke, Curr. Opin. Cell Biol., 2014, 26, 87–95 CrossRef CAS PubMed.
- K. Xiong, W. Wei, Y. Jin, S. Wang, D. Zhao, S. Wang, X. Gao, C. Qiao, H. Yue, G. Ma and H. Xie, Adv. Mater., 2016, 28, 7929–7935 CrossRef CAS PubMed.
- L. Wang, P. Balasubramanian, A. P. Chen, S. Kummar, Y. A. Evrard and R. J. Kinders, Semin. Oncol., 2016, 43, 464–475 CrossRef CAS PubMed.
- J. F. Swennenhuis, G. van Dalum, L. L. Zeune and L. W. M. M. Terstappen, Expert Rev. Mol. Diagn., 2016, 16, 1291–1305 CrossRef CAS PubMed.
- K. Obayashi, J. Akatsuka, Y. Endo, H. Takeda, T. Hayashi, Y. Toyama, Y. Suzuki, T. Hamasaki, G. Kimura, T. Ohnaga and Y. Kondo, Prostate Int., 2019, 7, 131–138 CrossRef PubMed.
- Z. Shen, A. Wu and X. Chen, Chem. Soc. Rev., 2017, 46, 2038–2056 RSC.
- L. Wang, P. Balasubramanian, A. P. Chen, S. Kummar, Y. A. Evrard and R. J. Kinders, Semin. Oncol., 2016, 43, 464–475 CrossRef CAS PubMed.
- C. Rao, D. Chianese, G. Doyle, M. Miller, T. Russell, R. Sanders and L. Terstappen, Int. J. Oncol., 2005, 27, 49–57 CAS.
- T. M. Gorges, I. Tinhofer, M. Drosch, L. Röse, T. M. Zollner, T. Krahn and O. von Ahsen, BMC Cancer, 2012, 12, 178 CrossRef CAS PubMed.
- S. D. Mikolajczyk, L. S. Millar, P. Tsinberg, S. M. Coutts, M. Zomorrodi, T. Pham, F. Z. Bischoff and T. J. Pircher, J. Oncol., 2011, 2011, 1–10 CrossRef PubMed.
- S. Park, R. R. Ang, S. P. Duffy, J. Bazov, K. N. Chi, P. C. Black and H. Ma, PLoS One, 2014, 9, e85264 CrossRef PubMed.
- T. Han, J. Zhang, D. Xiao, B. Yang, L. Chen, C. Zhai, F. Ding, Y. Xu, X. Zhao and J. Zhao, J. Oncol., 2022, 2022, 1–11 Search PubMed.
- R. Sulaiman, P. De, J. C. Aske, X. Lin, A. Dale, E. Vaselaar, C. Ageton, K. Gaster, L. R. Espaillat, D. Starks and N. Dey, Cancers, 2022, 14, 4577 CrossRef CAS PubMed.
- J. P. Gleghorn, E. D. Pratt, D. Denning, H. Liu, N. H. Bander, S. T. Tagawa, D. M. Nanus, P. A. Giannakakou and B. J. Kirby, Lab Chip, 2010, 10, 27–29 RSC.
- P. Bankó, S. Y. Lee, V. Nagygyörgy, M. Zrínyi, C. H. Chae, D. H. Cho and A. Telekes, J. Hematol. Oncol., 2019, 12, 48 CrossRef PubMed.
- M. Lustberg, K. R. Jatana, M. Zborowski and J. J. Chalmers, in Recent Results Cancer Res, 2012, vol. 195, pp. 97–110 Search PubMed.
- K. Cushing, E. Undvall, Y. Ceder, H. Lilja and T. Laurell, Anal. Chim. Acta, 2018, 1000, 256–264 CrossRef CAS PubMed.
- C.-H. Chu, R. Liu, T. Ozkaya-Ahmadov, B. E. Swain, M. Boya, B. El-Rayes, M. Akce, M. A. Bilen, O. Kucuk and A. F. Sarioglu, Sci. Rep., 2021, 11, 20583 CrossRef CAS PubMed.
- Y. Maertens, V. Humberg, F. Erlmeier, S. Steffens, J. Steinestel, M. Bögemann, A. J. Schrader and C. Bernemann, Onco Targets Ther, 2017, 8, 87710–87717 Search PubMed.
- P. Balasubramanian, L. Yang, J. C. Lang, K. R. Jatana, D. Schuller, A. Agrawal, M. Zborowski and J. J. Chalmers, Mol. Pharmaceutics, 2009, 6, 1402–1408 CrossRef CAS PubMed.
- S.-B. Huang, M.-H. Wu, Y.-H. Lin, C.-H. Hsieh, C.-L. Yang, H.-C. Lin, C.-P. Tseng and G.-B. Lee, Lab Chip, 2013, 13, 1371 RSC.
- C.-J. Liao, C.-H. Hsieh, H.-M. Wang, W.-P. Chou, T.-K. Chiu, J.-H. Chang, A.-C. Chao and M.-H. Wu, RSC Adv., 2017, 7, 29339–29349 RSC.
- L. Yang, J. C. Lang, P. Balasubramanian, K. R. Jatana, D. Schuller, A. Agrawal, M. Zborowski and J. J. Chalmers, Biotechnol. Bioeng., 2009, 102, 521–534 CrossRef CAS PubMed.
- O. Lara, X. Tong, M. Zborowski and J. J. Chalmers, Exp. Hematol., 2004, 32, 891–904 CrossRef PubMed.
- M. M. Ferreira, V. C. Ramani and S. S. Jeffrey, Mol. Oncol., 2016, 10, 374–394 CrossRef CAS PubMed.
- Z. Liu, A. Fusi, E. Klopocki, A. Schmittel, I. Tinhofer, A. Nonnenmacher and U. Keilholz, J. Transl. Med., 2011, 9, 70 CrossRef PubMed.
- L. Diéguez, M. A. Winter, K. J. Pocock, K. E. Bremmell and B. Thierry, Analyst, 2015, 140, 3565–3572 RSC.
- C.-H. Chu, R. Liu, T. Ozkaya-Ahmadov, M. Boya, B. E. Swain, J. M. Owens, E. Burentugs, M. A. Bilen, J. F. McDonald and A. F. Sarioglu, Lab Chip, 2019, 19, 3427–3437 RSC.
- T. A. Burinaru, B. Adiaconiţă, M. Avram, P. Preda, A.-M. Enciu, E. Chiriac, C. Mărculescu, T. Constantin and M. Militaru, Mater. Today Commun., 2022, 32, 104016 CrossRef CAS.
- H.-Y. Cho, Md. K. Hossain, J.-H. Lee, J. Han, H. J. Lee, K.-J. Kim, J.-H. Kim, K.-B. Lee and J.-W. Choi, Biosens. Bioelectron., 2018, 102, 372–382 CrossRef CAS PubMed.
- M. U. Anu Prathap, E. Castro-Pérez, J. A. Jiménez-Torres, V. Setaluri and S. Gunasekaran, Biosens. Bioelectron., 2019, 142, 111522 CrossRef CAS PubMed.
- M. Poudineh, P. M. Aldridge, S. Ahmed, B. J. Green, L. Kermanshah, V. Nguyen, C. Tu, R. M. Mohamadi, R. K. Nam, A. Hansen, S. S. Sridhar, A. Finelli, N. E. Fleshner, A. M. Joshua, E. H. Sargent and S. O. Kelley, Nat. Nanotechnol., 2017, 12, 274–281 CrossRef CAS PubMed.
- X. Huang, J. Tang, L. Hu, R. Bian, M. Liu, W. Cao and H. Zhang, Anal. Biochem., 2019, 564–565, 64–71 CrossRef CAS PubMed.
- T. S. Safaei, R. M. Mohamadi, E. H. Sargent and S. O. Kelley, ACS Appl. Mater. Interfaces, 2015, 7, 14165–14169 CrossRef CAS PubMed.
- C. Wang, M. Ye, L. Cheng, R. Li, W. Zhu, Z. Shi, C. Fan, J. He, J. Liu and Z. Liu, Biomaterials, 2015, 54, 55–62 CrossRef CAS PubMed.
- T. N. Pahattuge, I. M. Freed, M. L. Hupert, S. Vaidyanathan, K. Childers, M. A. Witek, K. Weerakoon-Ratnayake, D. Park, A. Kasi, M. F. Al-Kasspooles, M. C. Murphy and S. A. Soper, ACS Sens., 2021, 6, 1831–1839 CrossRef CAS PubMed.
- Y. Su, Q. Tian, D. Pan, L. Hui, Y. Chen, Q. Zhang, W. Tian, J. Yu, S. Hu, Y. Gao, D. Qian, T. Xie and B. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 29569–29578 CrossRef CAS PubMed.
- F. Shi, F. Jia, Z. Wei, Y. Ma, Z. Fang, W. Zhang and Z. Hu, Proteomics, 2021, 21, e2000060 CrossRef PubMed.
- J. Lee and B. Kwak, Biosens. Bioelectron., 2020, 168, 112564 CrossRef CAS PubMed.
- S. J. Shepherd, D. Issadore and M. J. Mitchell, Biomaterials, 2021, 274, 120826 CrossRef CAS PubMed.
- R. A. Harouaka, M. Nisic and S.-Y. Zheng, SLAS Technol., 2013, 18, 455–468 CrossRef CAS PubMed.
- S. Sharma, R. Zhuang, M. Long, M. Pavlovic, Y. Kang, A. Ilyas and W. Asghar, Biotechnol. Adv., 2018, 36, 1063–1078 CrossRef CAS PubMed.
- S. C. Dolfi, L. L.-Y. Chan, J. Qiu, P. M. Tedeschi, J. R. Bertino, K. M. Hirshfield, Z. N. Oltvai and A. Vazquez, Cancer Metab., 2013, 1, 20 CrossRef PubMed.
- M. Diez-Silva, M. Dao, J. Han, C.-T. Lim and S. Suresh, MRS Bull., 2010, 35, 382–388 CrossRef CAS PubMed.
- P. R. Wheater, H. G. Burkitt and V. G. Daniels, Wheater's functional histology: A text and colour atlas, Churchill Livingstone, London, England, 3rd edn, 1993 Search PubMed.
- J. Chen, H. Ho, J. Lichterman, Y. Lu, Y. Zhang, M. A. Garcia, S. Chen, A. Liang, E. Hodara, H. E. Zhau, S. Hou, R. S. Ahmed, D. J. Luthringer, J. Huang, K. Li, L. W. K. Chung, Z. Ke, H. Tseng and E. M. Posadas, Cancer, 2015, 121, 3240–3251 CrossRef PubMed.
- C. Magnusson, P. Augustsson, A. Lenshof, Y. Ceder, T. Laurell and H. Lilja, Anal. Chem., 2017, 89, 11954–11961 CrossRef CAS PubMed.
- S. Zhang, Y. Wang, C. Yang, J. Zhu, X. Ye and W. Wang, Nanotechnol. Precis. Eng., 2022, 5, 013003 CrossRef CAS.
- M. E. Warkiani, B. L. Khoo, D. S.-W. Tan, A. A. S. Bhagat, W.-T. Lim, Y. S. Yap, S. C. Lee, R. A. Soo, J. Han and C. T. Lim, Analyst, 2014, 139, 3245–3255 RSC.
- G. Vona, A. Sabile, M. Louha, V. Sitruk, S. Romana, K. Schütze, F. Capron, D. Franco, M. Pazzagli, M. Vekemans, B. Lacour, C. Bréchot and P. Paterlini-Bréchot, Am. J. Pathol., 2000, 156, 57–63 CrossRef CAS PubMed.
- V. Murlidhar, L. Rivera-Báez and S. Nagrath, Small, 2016, 12, 4450–4463 CrossRef CAS PubMed.
- Q. Huang, F.-B. Wang, C.-H. Yuan, Z. He, L. Rao, B. Cai, B. Chen, S. Jiang, Z. Li, J. Chen, W. Liu, F. Guo, Z. Ao, S. Chen and X.-Z. Zhao, Theranostics, 2018, 8, 1624–1635 CrossRef CAS PubMed.
- F. Chen, S. Wang, Y. Fang, L. Zheng, X. Zhi, B. Cheng, Y. Chen, C. Zhang, D. Shi, H. Song, C. Cai, P. Zhou and B. Xiong, Onco Targets Ther, 2017, 8, 3029–3041 Search PubMed.
- D. L. Adams, R. K. Alpaugh, S. S. Martin, M. Charpentier, S. Chumsri, M. Cristofanilli, D. K. Adams, O. V. Makarova, P. Zhu, S. Li, C.-M. Tang and S. Stefansson, RSC Adv., 2016, 6, 6405–6414 RSC.
- J. Che, V. Yu, E. B. Garon, J. W. Goldman and D. Di Carlo, Lab Chip, 2017, 17, 1452–1461 RSC.
- X. Fan, C. Jia, J. Yang, G. Li, H. Mao, Q. Jin and J. Zhao, Biosens. Bioelectron., 2015, 71, 380–386 CrossRef CAS PubMed.
- K. Wang, L. Zhou, S. Zhao, Z. Cheng, S. Qiu, Y. Lu, Z. Wu, A. H. A. Abdel Wahab, H. Mao and J. Zhao, Talanta, 2019, 200, 169–176 CrossRef CAS PubMed.
- X. Xiao, X. Miao, S. Duan, S. Liu, Q. Cao, R. Wu, C. Tao, J. Zhao, Q. Qu, A. Markiewicz, R. Peng, Y. Chen, A. Żaczek and J. Liu, Small Methods, 2023, 7, 2300096 CrossRef CAS PubMed.
- M. Farasat, S. M. Chavoshi, A. Bakhshi, A. Valipour and M. Badieirostami, J. Micromech. Microeng., 2022, 32, 015008 CrossRef CAS.
- E. Khosrowabadi, A. Rivinoja, M. Risteli, A. Tuomisto, T. Salo, M. J. Mäkinen and S. Kellokumpu, Cell. Mol. Life Sci., 2021, 78, 6283–6304 CrossRef CAS PubMed.
- S. Hwang, D. M. Shin and J. H. Hong, Molecules, 2019, 24, 3409 CrossRef CAS PubMed.
- A. Mishra, T. D. Dubash, J. F. Edd, M. K. Jewett, S. G. Garre, N. M. Karabacak, D. C. Rabe, B. R. Mutlu, J. R. Walsh, R. Kapur, S. L. Stott, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16839–16847 CrossRef CAS PubMed.
- N.-V. Nguyen and C.-P. Jen, Biosens. Bioelectron., 2018, 121, 10–18 CrossRef CAS PubMed.
- Y. Su, Q. Tian, D. Pan, L. Hui, Y. Chen, Q. Zhang, W. Tian, J. Yu, S. Hu, Y. Gao, D. Qian, T. Xie and B. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 29569–29578 CrossRef CAS PubMed.
- H.-Y. Cho, Md. K. Hossain, J.-H. Lee, J. Han, H. J. Lee, K.-J. Kim, J.-H. Kim, K.-B. Lee and J.-W. Choi, Biosens. Bioelectron., 2018, 102, 372–382 CrossRef CAS PubMed.
- T.-K. Chiu, W.-P. Chou, S.-B. Huang, H.-M. Wang, Y.-C. Lin, C.-H. Hsieh and M.-H. Wu, Sci. Rep., 2016, 6, 32851 CrossRef CAS PubMed.
- C. Alix-Panabières and K. Pantel, Nat. Rev. Cancer, 2014, 14, 623–631 CrossRef PubMed.
- C. E. Nwankire, A. Venkatanarayanan, T. Glennon, T. E. Keyes, R. J. Forster and J. Ducrée, Biosens. Bioelectron., 2015, 68, 382–389 CrossRef CAS PubMed.
- B. Dou, L. Xu, B. Jiang, R. Yuan and Y. Xiang, Anal. Chem., 2019, 91, 10792–10799 CrossRef CAS PubMed.
- Y. Chen, J. Peng, Y. Lai, B. Wu, L. Sun and J. Weng, Biosens. Bioelectron., 2019, 142, 111520 CrossRef CAS PubMed.
- B. Bénéteau-Burnat, M. C. Bocque, A. Lorin, C. Martin and M. Vaubourdolle, Clin. Chem. Lab. Med., 2004, 42, 96–101 Search PubMed.
- B. J. Dascombe, P. R. J. Reaburn, A. C. Sirotic and A. J. Coutts, J. Sci. Med. Sport, 2007, 10, 135–140 CrossRef CAS PubMed.
- V. Vásquez, M.-C. Navas, J. A. Jaimes and J. Orozco, Anal. Chim. Acta, 2022, 1205, 339718 CrossRef PubMed.
- S. P. Duffy and H. Ma, in Circulating Tumor Cells, Wiley, Hoboken, NJ, USA, 2016, pp. 147–172 Search PubMed.
- I. Namli, S. Seyedmirzaei Sarraf, A. Sheibani Aghdam, G. Celebi Torabfam, O. Kutlu, S. Cetinel, M. Ghorbani and A. Koşar, ACS Appl. Mater. Interfaces, 2022, 14, 40688–40697 CrossRef CAS PubMed.
- J. Zhao, Z. Han, C. Xu, L. Li, H. Pei, Y. Song, Z. Wang and B. Tang, EBioMedicine, 2023, 90, 104522 CrossRef CAS PubMed.
- E. Ahmed, K. B. Shanmugasundaram, J. Li, A. Behren, R. Lobb, A. Möller, A. Wuethrich, S. Dey, A. A. I. Sina and M. Trau, Adv. Sens. Res., 2023, 2, 2300059 CrossRef.
- W. Lv, B. Fu, W. Liu, W. Huang, M. Li, Y. Liu, Y. Kang, J. Wang, S. Bai, C. Lu and X. Dai, Talanta, 2024, 267, 125220 CrossRef CAS PubMed.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical Methods: Fundamentals and Applications, Wiley, 3rd edn, 2022 Search PubMed.
- X. Wang, T. Gao, J. Zhu, S. Long, S. Zhao, L. Yuan and Z. Wang, Anal. Chem., 2023, 95, 2496–2503 CrossRef CAS PubMed.
Footnote |
| † Denotes equal contribution to the work. |
| This journal is © The Royal Society of Chemistry 2024 |