
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-driven synthesis of alkenyl thiocyanates: novel building blocks for assembly of diverse sulfur-containing molecules†
Helena F.
Piedra
 and
Manuel
Plaza
*
and
Manuel
Plaza
*
Departamento de Química Orgánica e Inorgánica, Instituto Universitario de Química Organometálica “Enrique Moles”, Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universidad de Oviedo, Julián Clavería 8, 33006 Oviedo, Spain. E-mail: plazamanuel@uniovi.es
First published on 23rd October 2024
Abstract
The first visible-light-induced protocol for the general preparation of alkenyl thiocyanates from alkenyl bromides is presented. The reaction is simple, proceeds under very mild conditions and demonstrates broad functional group tolerance. Additionally, a flow protocol was developed to enable efficient scale-up of alkenyl thiocyanate synthesis, further enhancing the practicality and value of the method. Importantly, these alkenyl thiocyanates serve as valuable building blocks for the construction of diverse families of sulfur-containing molecules through trifluoromethylations, cycloadditions, oxidations, and C–S or P–S bond forming reactions.
1 Introduction
An important domain of modern organic chemistry research is centred around devising new synthetic approaches for C–S bond formation.1 Over the past few years, several research groups have been dedicated to advancing photochemical methods for this purpose. This focus is motivated by the mild reaction conditions and the intrinsic sustainability of contemporary visible-light-driven processes, which allow for the construction of diverse sulfur-containing molecules in an efficient manner.2 In particular, the recent development of sustainable methodologies for the preparation of organic thiocyanates, an important family of organosulfur compounds, has gained considerable attention.3 These compounds are valued for their inherent biological properties and potential as valuable building blocks in organic synthesis.4 The thiocyanate functional group is highly versatile, with molecules containing this motif being intermediates for the modular construction of other relevant sulfur-containing compounds.5 However, the exploitation of molecules featuring the C(alkenyl)–S–CN motif as building blocks, which would potentially provide a general platform to construct alkenyl-containing organosulfur compounds, remains underexplored. Given the above considerations, an ideal reaction to synthesize alkenyl thiocyanates simply and efficiently would involve the light-driven activation of alkenyl bromides,6 which are easily prepared or commercially available. However, the photochemical excitation of alkenyl bromides is reported to require toxic and dangerous UV light or γ-rays, which promote either homolytic or heterolytic cleavage of the C–Br bond.7 This activation grants access to highly reactive alkenyl radicals or cations, which can engage in radical or polar mechanisms (Scheme 1a).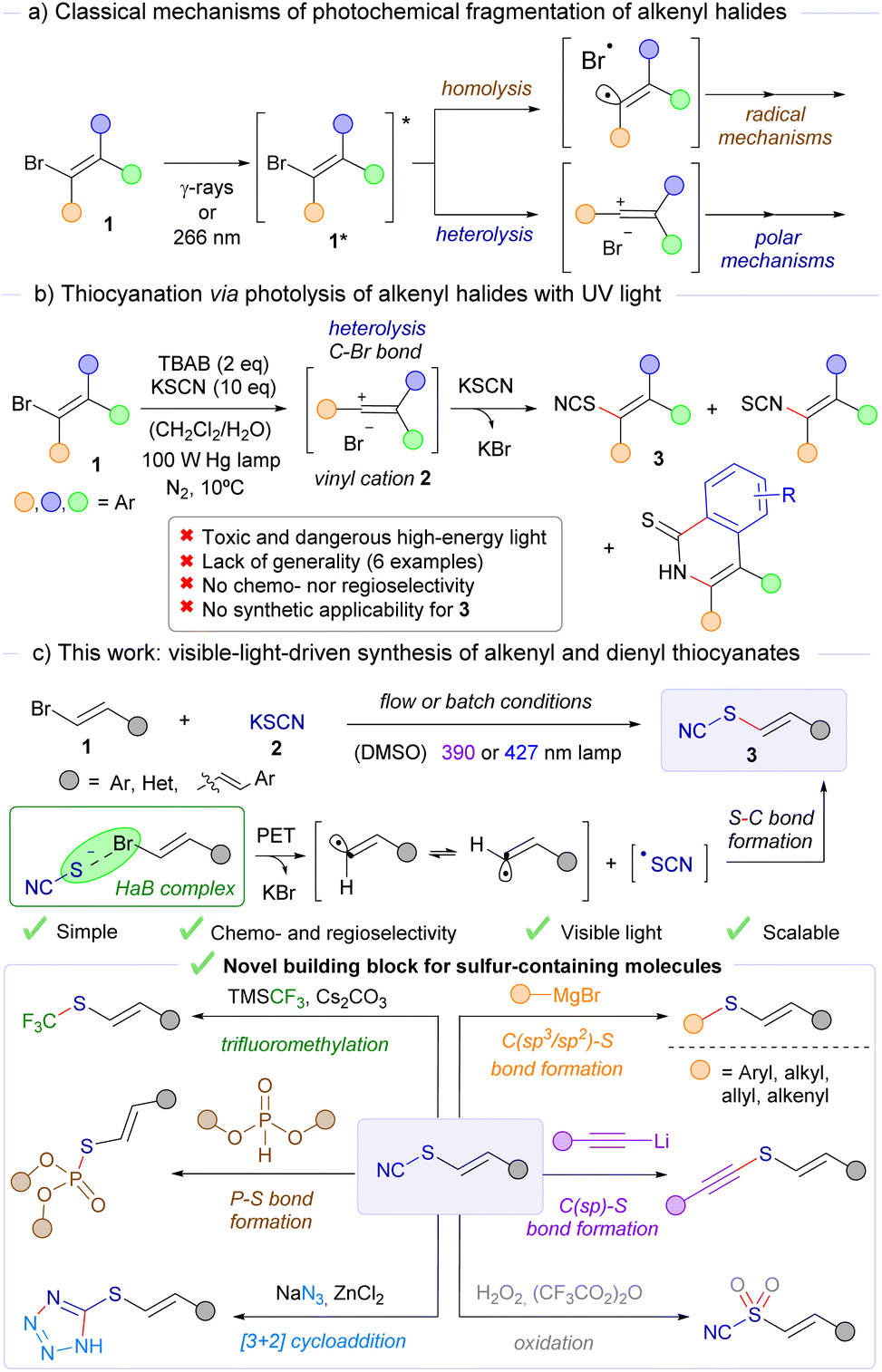 | ||
| Scheme 1 State of the art and our novel approach for the photochemical synthesis and synthetic applications of alkenyl thiocyanates. PET: Photoinduced Electron Transfer. | ||
Notably, only one example of the photochemical synthesis of alkenyl thiocyanates from alkenyl bromides has been reported, using a very powerful light source to excite the organic bromides 1 (Scheme 1b).8 This transformation has additional limitations, including a lack of chemo- and regioselectivity, which restricts the applicability of alkenyl bromides in such reactions to a very limited number of substrates.
Our research group has been a pioneer in developing visible-light-induced transformations for the activation of alkenyl halides, typically proceeding through the transient generation of highly reactive alkenyl radical intermediates.9 These reactions proceed through the visible-light-induced activation of halogen-bonding complexes with visible light,10 a specific type of electron donor–acceptor (EDA) complex.11 In general, the photochemical activation of HaB complexes has recently attracted considerable interest for generating carbon-centred radicals under mild reaction conditions, enabling the development of novel organic transformations.12 The halogen bond, a type of weak interaction classified under σ–hole interactions,13 involves a partial n → σ* charge transfer from a non-bonding orbital of a nucleophilic electron donor (the HaB acceptor) to an antibonding orbital (σ*) of an electron acceptor, the corresponding organic halide (the HaB donor).14 Upon irradiation with visible light, a photochemical fragmentation of the HaB complex through the reduction of the C–halogen bond takes place, where two different radical species can be generated from the synthetic precursors, which can then recombine to form the desired cross-coupling product. Taking all of this into consideration, we envisioned that the visible-light-promoted reaction of alkenyl bromides 1 with a nucleophilic thiocyanate source 2 (such as thiocyanate salt) could produce the desired alkenyl thiocyanates 3 under very simple and mild reaction conditions and in a general manner. Importantly, we found that these molecules further serve as valuable building blocks for the modular synthesis of diverse C(alkenyl)–S-containing compounds, thereby enhancing the significance of this transformation (Scheme 1c).
2 Results and discussion
We initiated our studies by selecting β-bromostyrene 1a as the starting alkenyl bromide, employing 5 equivalents of potassium thiocyanate (2a) in DMSO as solvent, and illuminating the reaction vessel with a 390 nm lamp for three hours. Much to our delight, under these reaction conditions, the desired alkenyl thiocyanate 3a was forged in an 80% isolated yield (d.r. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 trans/cis). A control experiment revealed that light was fundamental for the reaction to take place (Table 1, entry 2). Running the reaction without KSCN led to a complete recovery of 1a, which shows that the thiocyanate salt is essential to promote the photochemical activation of β-bromostyrene (Table 1, entry 3). The implementation of an argon atmosphere did not lead to any improvement in the reaction efficiency (Table 1, entry 4). Either decreasing the equivalents of potassium thiocyanate or changing it for a different salt (ammonium thiocyanate) did not enhance the reaction yield (Table 1, entries 5 and 6). However, as the work progressed, we encountered specific cases where increasing the amount of KSCN to 5.0 equivalents led to a slight improvement in reaction yields. To ensure consistent and optimal results across a broader range of substrates, and given the low cost of KSCN, the reaction conditions were https://www.google.com/search?sca_esv=dcbcef83952848e4%26rlz=1C1GCEB_enIN1086IN1086%26q=standardized%26spell=1%26sa=X%26ved=2ahUKEwikj-LG_aSJAxUGzzgGHXpUIPcQkeECKAB6BAgTEAE using 5.0 equivalents of this salt. Decreasing the reaction time to one hour led to an incomplete conversion (Table 1, entry 7). On the other hand, prolonged reaction times (12 hours) were found to be unsuitable for the reaction, leading to photochemical decomposition of the compound 3a (Table 1, entry 8). Therefore, the selection of an adequate reaction time is crucial to ensure full conversion and avoid photochemical decomposition of the alkenyl thiocyanate at the same time (UV-vis profile of 3a included in the ESI†). Changing the irradiation wavelength to 427 nm led, as could be easily anticipated, to a diminished conversion in 3 hours (Table 1, entry 9) due to inefficient excitation of the corresponding HaB complex. Other polar solvents such as DMF or acetonitrile were also compatible with this transformation, although compound 3a was obtained with less isolated yield (Table 1, entries 10 and 11). Eventually, the employment of β-iodostyrene and β-chlorostyrene as alkenyl halides led to a very diminished reaction efficiency (Table 1, entries 12 and 13, respectively). In the first case, this is mainly attributed to the high photochemical instability of the alkenyl iodide at 390 nm, preventing the formation of the halogen-bonding complex, whereas in the case of the alkenyl chloride, the reaction is less efficient due to the unfavoured formation of the corresponding HaB complex, essential for the reaction to occur. These observations are in line with other previous reports on different photochemical HaB-assisted transformations.9
:1 trans/cis). A control experiment revealed that light was fundamental for the reaction to take place (Table 1, entry 2). Running the reaction without KSCN led to a complete recovery of 1a, which shows that the thiocyanate salt is essential to promote the photochemical activation of β-bromostyrene (Table 1, entry 3). The implementation of an argon atmosphere did not lead to any improvement in the reaction efficiency (Table 1, entry 4). Either decreasing the equivalents of potassium thiocyanate or changing it for a different salt (ammonium thiocyanate) did not enhance the reaction yield (Table 1, entries 5 and 6). However, as the work progressed, we encountered specific cases where increasing the amount of KSCN to 5.0 equivalents led to a slight improvement in reaction yields. To ensure consistent and optimal results across a broader range of substrates, and given the low cost of KSCN, the reaction conditions were https://www.google.com/search?sca_esv=dcbcef83952848e4%26rlz=1C1GCEB_enIN1086IN1086%26q=standardized%26spell=1%26sa=X%26ved=2ahUKEwikj-LG_aSJAxUGzzgGHXpUIPcQkeECKAB6BAgTEAE using 5.0 equivalents of this salt. Decreasing the reaction time to one hour led to an incomplete conversion (Table 1, entry 7). On the other hand, prolonged reaction times (12 hours) were found to be unsuitable for the reaction, leading to photochemical decomposition of the compound 3a (Table 1, entry 8). Therefore, the selection of an adequate reaction time is crucial to ensure full conversion and avoid photochemical decomposition of the alkenyl thiocyanate at the same time (UV-vis profile of 3a included in the ESI†). Changing the irradiation wavelength to 427 nm led, as could be easily anticipated, to a diminished conversion in 3 hours (Table 1, entry 9) due to inefficient excitation of the corresponding HaB complex. Other polar solvents such as DMF or acetonitrile were also compatible with this transformation, although compound 3a was obtained with less isolated yield (Table 1, entries 10 and 11). Eventually, the employment of β-iodostyrene and β-chlorostyrene as alkenyl halides led to a very diminished reaction efficiency (Table 1, entries 12 and 13, respectively). In the first case, this is mainly attributed to the high photochemical instability of the alkenyl iodide at 390 nm, preventing the formation of the halogen-bonding complex, whereas in the case of the alkenyl chloride, the reaction is less efficient due to the unfavoured formation of the corresponding HaB complex, essential for the reaction to occur. These observations are in line with other previous reports on different photochemical HaB-assisted transformations.9
|
|
|||
|---|---|---|---|
| Entry | Deviation from standard conditions | Yield 3a | d.r. trans/cis |
| a Recovery of 1a (98%). | |||
| 1 | None | 80% | 10.0:1 |
| 2 | Dark, 72 h | — | — |
| 3 | Without KSCN | a0% | 10.0:1 |
| 4 | Ar, 2 h | 79% | 10.0:1 |
| 5 | 3 eq. of KSCN | 80% | 10.0:1 |
| 6 | NH4SCN instead of KSCN | 66% | 10.0:1 |
| 7 | 1 h | 65% | 10.0:1 |
| 8 | 12 h | 4% | — |
| 9 | 427 nm lamp, 3 h | 45% | 10.0:1 |
| 10 | DMF instead of DMSO | 78% | 10.0:1 |
| 11 | MeCN instead of DMSO | 65% | 10.0:1 |
| 12 | X = I | 12% | — |
| 13 | X = Cl | 6% | — |
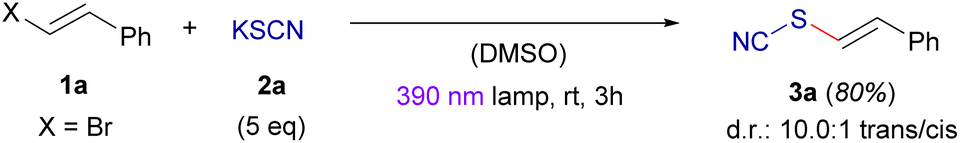
Having set the optimal conditions under batch conditions for the photochemical cross-coupling between alkenyl bromides and thiocyanate salts, we started to evaluate the scope of the reaction (Scheme 2a). Gratifyingly, the reaction was found to be compatible with a wide range of functional groups at the aryl fragment. In this regard, it was possible to synthesize with moderate to very good yields alkenyl thiocyanates with neutral groups (3b, 3g, 3j), halogens (3d, 3e, 3f, 3k), esters (3c), ethers (3h, 3l, 3m) and thioethers (3i). The transformation was also compatible with heteroaromatic groups such as benzofuran (3o) and thiophene (3p).
 | ||
| Scheme 2 Scope of the photochemical thiocyanation reaction under batch (light purple) and continuous flow (orange) conditions. (a) General reaction conditions for the batch protocol: alkenyl bromide 1 (0.2 mmol) and potassium thiocyanate 2a (1.0 mmol) in 2 mL of DMSO, irradiation with either a 390 nm lamp (GP A) or a 427 nm lamp (GP B) for 3 hours. (b) General reaction conditions for the continuous flow protocol: a solution of alkenyl bromide 1 (0.40 mmol) and potassium thiocyanate 2a (1.60 mmol) in dry DMSO (4 mL) was prepared and loaded into a syringe. This syringe was connected to a high-pressure pump and a photoreactor. The solution was continuously pumped through at a rate of 4 mL h−1 while illuminated with two 390 nm lamps (GP C). Isolated yields after flash chromatography are presented. | ||
Interestingly, the reaction proceeded well when employing a β-bromostyrene derived from the natural product (−)-citronellol (3r) and (−)-menthol (3s). Moreover, the reaction could be extended to construct 1,3-dienyl thiocyanates such as compound 3q, where the optimal irradiation wavelength was 427 nm. It is essential to note that the presence of the β-styrene fragment is crucial for the absorption of visible light by the corresponding HaB complexes. Consequently, the reaction is incompatible with alkyl-substituted alkenyl or 1,3-dienyl bromides, as evidenced by the failure to produce compounds 3t and 3u.
As will be discussed later in the manuscript, the alkenyl thiocyanates 3 serve as novel building blocks for the construction of a variety of alkenyl-containing organosulfur compounds. Therefore, to enhance the utility of the photochemical transformation, efforts were made to develop a continuous flow protocol, which would allow for an efficient scale up. Additionally, the implementation of synthetic procedures on continuous flow presents advantages like enhanced safety, reproducibility and reaction control.15 As explained before, in this transformation the control of the illumination period is crucial to promote the reaction completion and avoid light-driven decomposition of compounds 3. Due to the increased reaction efficiency of the flow protocol, an exhaustive optimization study was carried out in order to enable scale up of the transformation in our flow reactor (see the ESI† for a detailed description). Under the continuous flow conditions, the scale up of the photochemical reaction for compounds 3a, 3b, 3f and 3p could be achieved; however, slightly lower yields and diastereoselectivity when compared to the corresponding batch procedures were obtained (Scheme 2b). Nevertheless, it is worth noticing that in some specific examples, the stereoselectivity (compound 3f) could be improved through this protocol. Overall, our method allows for the efficient scale-up of these transformations, where compound 3a could also be obtained in a 77% isolated yield in a 5 mmol scale.
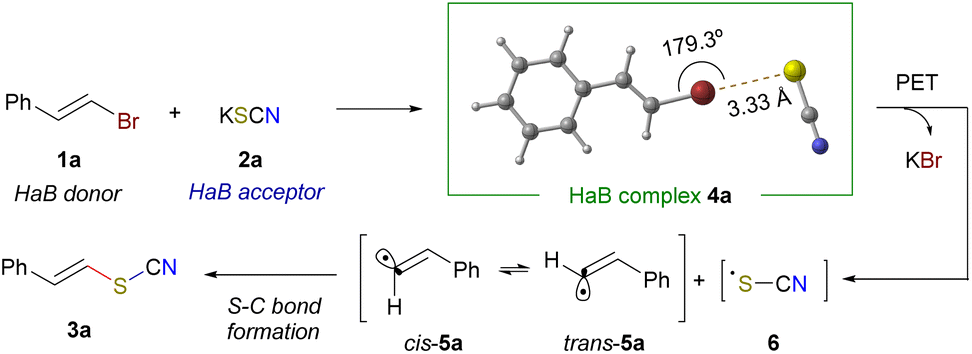
Mechanistically, the reaction is proposed to proceed through the photochemical fragmentation of a halogen-bonding complex between the alkenyl bromide 1 and thiocyanate anion, which generates the corresponding alkenyl radical 5 (which can undergo trans/cis isomerization) and the sulfur-centered radical 6. A radical recombination would forge the final compounds 3 (Scheme 3). In order to detect the formation of a halogen-bonding complex between the alkenyl bromide 1a and the thiocyanate salt, a set of mechanistic experiments was carried out. First, a UV-vis analysis of independent solutions of 1a and the thiocyanate salts was conducted. Notably, when mixing both starting materials, no change in the UV-vis profile when compared to the absorbance of 1a alone was observed (see the ESI† for details). However, it has been documented that halogen-bonding complexes in which a charge-transfer band is not detected in the UV-vis spectra can also undergo photochemical fragmentation with visible light.12a In order to evidence the formation of a halogen-bonding complex between 1a and the thiocyanate salt, 13C-NMR titration experiments were run. Importantly, when preparing solutions of increasing molar concentrations of the thiocyanate salt, maintaining the same amount of alkenyl bromide 1a, a progressive downfield shift was tracked through 13C-NMR of both alkenyl hydrogens of 1a, nearing 0.3 ppm (see the ESI† for a detailed analysis). This fact supports the formation of a HaB complex between 1a and the thiocyanate salt,13,16 where the thermodynamic binding constant was calculated (Ka = 3 × 10−2 M).17 Additionally, through DFT calculations, a supramolecular aggregate between the alkenyl bromide 1a and the thiocyanate salt was identified as a minimum at the potential energy surface. It was observed that the dihedral angle between the C(sp2)–Br–S bonds is close to 180°, which accounts for the high directionality of the halogen bond and sets a nearly linear structure for the HaB complex 4a.18 It was also observed that the calculated Br–S distance is shorter than the sum of the individual van der Waals radii of both atoms. Next, different mechanistic studies were run to evaluate the radical nature of this transformation and its intermediates involved. A complete shutdown of the reaction was observed upon addition of a radical scavenger like TEMPO. Additionally, a combination of light ON/OFF experiments and the quantum yield determination of the reaction (ϕ = 0.004, see the ESI† for details) pointed at no radical chain propagation events taking place.19
 | ||
| Scheme 3 Mechanistic proposal for the photochemical reaction. | ||
The final section of this work presents the findings on the potential applications of alkenyl thiocyanates 3 as building blocks for the construction of high-added-value, sulfur-containing compounds. In particular, we were pleased to discover that a variety of relevant alkenyl-containing organosulfur scaffolds could be synthesized using molecules 3 as reaction precursors (Scheme 4). First, recognizing the importance of the SCF3 group in the pharmaceutical and agrochemical industries,20 we developed a method to convert compounds 3 into the corresponding alkenyl sulfanes 7 through trifluoromethylthiolation. Importantly, there is a scarcity of methodologies for the preparation of alkenyl-containing thiotrifluoromethylated compounds which proceed under operationally simple and mild conditions.21 In this regard, our procedure relies on the employment of TMSCF3 as the trifluoromethylation reagent using cesium carbonate as base, allowing for the formation at room temperature of diverse trifluoromethyl compounds 7a–7d from the corresponding alkenyl thiocyanates 3 in good isolated yields (Scheme 4a). Next, we developed a series of transformations based on the nucleophilic substitution of the cyano group in alkenyl thiocyanates 3 with various reagents, facilitating the construction of a range of important organosulfur compounds. In this context, we initially devised synthetic procedures utilizing different Grignard reagents for C(sp3)–S and C(sp2)–S bond-forming reactions. This approach enabled the creation of diverse alkenyl thioethers 8 (Scheme 4b). Additionally, the corresponding alkynylation of the alkenyl thiocyanate 3a with lithium phenyl acetylide allowed for the preparation of the alkynyl vinyl thioether 9a through a C(sp)–S bond-forming transformation (Scheme 4c). It should be noted that the development of synthetic procedures for the construction of alkenyl thioethers has garnered significant attention from various research groups in recent years.22 Furthermore, the carbothiophosphorylation of 3a by a nucleophilic phosphite source led to the formation of compound 10a (Scheme 4d). Noticeably, the preparation of this class of organophosphorus compounds remained challenging until very recently through alternative methodologies.9b,23 Next, a synthetic procedure based on a [3 + 2] cycloaddition reaction between compound 3a and sodium azide mediated by ZnCl2 was explored (Scheme 4e). This transformation granted access to the tetrazole-containing alkenyl thioether 11a, expanding in this way the synthetic utility of the alkenyl thioethers to the construction of relevant heterocycles in medicinal chemistry.24 Eventually, the oxidation of 3a with an aqueous solution of hydrogen peroxide mediated by trifluoroacetic anhydride allowed for the preparation of the S-cyanosulfone 12a, a class of compounds that have been reported to be useful as nitrile-transfer reagents.25 All in all, it was demonstrated that alkenyl thiocyanates 3 can serve as highly versatile building blocks for synthesizing a variety of important organosulfur compounds. This fact underscores the significance of the synthetic protocol presented here, which allows for the scalable preparation of compounds 3 under very operationally simple, mild conditions.
 | ||
| Scheme 4 Set of synthetic transformations employing the alkenyl thiocyanates 3 for the construction of the organosulfur compounds 7, 8, 9, 10, 11 and 12 (see the ESI† for a detailed description of the synthetic protocols). | ||
3 Conclusions
In this work, we have developed the first photochemical general method for synthesizing alkenyl thiocyanates from alkenyl bromides and thiocyanide salts, which makes use of visible light as the sole energy source. Our methodology is exceptionally straightforward, requiring only both coupling partners and solvent, with no need for catalysts. The reaction proceeds under mild conditions (room temperature and short reaction times), being also scalable through continuous flow. The substrate scope is extensive, with very good functional group tolerance, including applicability to the structural modification of biologically relevant molecules. Mechanistic studies revealed that radical intermediates are generated through photochemical excitation of halogen-bonding complexes. Eventually, we demonstrate the utility of alkenyl thiocyanates through new multiple synthetic applications, including trifluoromethylations, cycloadditions, oxidations, and C–S or P–S bond forming reactions, showcasing their potential in constructing diverse organosulfur architectures.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
H. F. Piedra conducted all the reactions, DFT calculations, experimental mechanistic studies and full characterization of the compounds. M. Plaza conceptualized and directed the project and wrote the manuscript. Both authors wrote the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
A “Severo Ochoa” predoctoral fellowship (BP21/050) to H. F. P. by FICYT (Principality of Asturias) and a “Ramón y Cajal” postdoctoral grant to M. P. (MCINN-24-RYC2022-035485-I) by Ministerio de Ciencia e Innovación of Spain (Agencia Estatal de Investigación) are gratefully acknowledged. Financial support of this work by Ministerio de Ciencia e Innovación of Spain (Agencia Estatal de Investigación: MCINN-23-PID2022-140635NB-I00/MCIN/AEI/10.13039/501100011033/FEDER, UE) and the University of Oviedo (PAPI-23-GR-COM-04) is also acknowledged. We wish to thank Prof. Carlos Valdés for the insightful discussions.Notes and references
- Selected reviews on C–S bond formation: (a) C.-F. Lee, R. S. Basha and S. S. Badsara, Top. Curr. Chem., 2018, 376, 25 CrossRef; (b) P. Annamalai, K. Liu, S. Singh Badsara and C. Lee, Chem. Rec., 2021, 21, 3674–3688 CrossRef CAS PubMed; (c) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2022, 122, 16110–16293 CrossRef CAS PubMed.
- Recent reviews on photochemical creation of C–S bonds: (a) Z. Wu and D. A. Pratt, Nat. Rev. Chem, 2023, 7, 573–589 Search PubMed; (b) J. Feng, Y. Zhang, X. Wang, J. Liu, V. Benazzi, K. Lu, X. Zhao and S. Protti, Adv. Synth. Catal., 2023, 65, 3413–3431 Search PubMed.
- Selected recent reviews: (a) P. G. Karmaker, M. A. Alam and F. Huo, RSC Adv., 2022, 12, 6214–6233 RSC; (b) H. Chen, X. Shi, X. Liu and L. Zhao, Org. Biomol. Chem., 2022, 20, 6508–6527 RSC.
- Selected examples: (a) E. Elhalem, B. N. Bailey, R. Docampo, I. Ujváry, S. H. Szajnman and J. B. Rodriguez, J. Med. Chem., 2002, 45, 3984–3999 CrossRef CAS; (b) R. A. E. Yasman, V. Wray and P. Proksch, J. Nat. Prod., 2003, 66, 1512–1514 Search PubMed; (c) V. A. Kokorekin, A. O. Terent’ev, G. V. Ramenskaya, N. É. Grammatikova, G. M. Rodionova and A. I. Ilovaiskii, Pharm. Chem. J., 2013, 47, 422–425 CrossRef CAS.
- (a) T. Castanheiro, J. Suffert, M. Donnard and M. Gulea, Chem. Soc. Rev., 2016, 45, 494–505 Search PubMed; (b) X. Qing, Z. Lianyang, F. Gaofeng and J. Chengan, Chin. J. Org. Chem., 2019, 39, 287–300 CrossRef; (c) B. An, L. Zhou, S. Liu, Y. Zheng, C. Li, F. Cui, C. Yue, H. Liu, Y. Sui, C. Ji, J. Yan and Y. Li, Angew. Chem., Int. Ed., 2024, 63, e202402511 CrossRef CAS PubMed.
- H. F. Piedra and M. Plaza, Photochem. Photobiol. Sci., 2024, 23, 1217–1228 CrossRef CAS PubMed.
- R. Gronheid, H. Zuilhof, M. G. Hellings, J. Cornelisse and G. Lodder, J. Org. Chem., 2003, 68, 3205–3215 Search PubMed.
- T. Kitamura, S. Kobayashi and H. Taniguchi, J. Org. Chem., 1990, 55, 1801–1805 CrossRef CAS.
- (a) H. F. Piedra and M. Plaza, Chem. Sci., 2023, 14, 650–657 RSC; (b) H. F. Piedra, V. Gebler, C. Valdés and M. Plaza, Chem. Sci., 2023, 14, 12767–12773 RSC; (c) H. F. Piedra, C. Valdés and M. Plaza, Adv. Synth. Catal., 2024, 366, 1422–1429 CrossRef CAS.
- For a recent review, see: H. F. Piedra, C. Valdés and M. Plaza, Chem. Sci., 2023, 14, 5545–5568 RSC.
- For some recent reviews covering the photochemistry of EDA-complexes, see: (a) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed; (b) Y. Yuan, S. Majumder, M. Yang and S. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS; (c) Y. Sempere, M. Morgenstern, T. Bach and M. Plaza, Photochem. Photobiol. Sci., 2022, 21, 719–737 CrossRef CAS PubMed.
- Selected recent examples: (a) T. Li, K. Liang, J. Tang, Y. Ding, X. Tong and C. Xia, Chem. Sci., 2021, 12, 15655 RSC; (b) Y. Shen, N. Lei, C. Lu, D. Xi, X. Geng, P. Tao, Z. Su and K. Zheng, Chem. Sci., 2021, 12, 15399–15406 RSC; (c) S. Cuadros, C. Rosso, G. Barison, P. Costa, M. Kurbasic, M. Bonchio, M. Prato, G. Filippini and L. Dell'Amico, Org. Lett., 2022, 24, 2961 CrossRef CAS PubMed; (d) N. Kato, T. Nanjo and Y. Takemoto, ACS Catal., 2022, 12, 7843 CrossRef CAS; (e) C. Zhang, H. Zuo, G. Y. Lee, Y. Zou, Q.-D. Dang, K. N. Houk and D. Niu, Nat. Chem., 2022, 14, 686 CrossRef CAS PubMed; (f) A. Bourboula, O. G. Mountanea, G. Krasakis, C. Mantzourani, M. G. Kokotou, C. G. Kokotos and G. Kokotos, Eur. J. Org Chem., 2023, 26, e202300008 CrossRef CAS; (g) Z. Jiang, K. You, H. Wu, M. Xu, T. Wang and J. Luo, Org. Lett., 2024, 26, 636–641 Search PubMed.
- Reviews and selected publications on the importance of halogen-bonding interactions: (a) T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC; (b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed; (c) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (d) R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS; (e) M. Breugst and J. J. Koenig, Eur. J. Org Chem., 2020, 2020, 5473–5487 CrossRef CAS; (f) A. C. Keuper, K. Fengler, F. Ostler, T. Danelzik, D. G. Piekarski and O. García Mancheño, Angew. Chem., Int. Ed., 2023, 62, e202304781 CrossRef CAS PubMed.
- A. Karpfen, J. Phys. Chem. A, 2000, 104, 6871–6879 CrossRef CAS.
- Recent reviews on continuous flow photochemistry: (a) L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed; (b) L. Capaldo, Z. Wen and T. Noël, Chem. Sci., 2023, 14, 4230–4247 RSC; (c) A. I. Alfano, J. García-Lacuna, O. M. Griffiths, S. V. Ley and M. Baumann, Chem. Sci., 2024, 15, 4618–4630 RSC.
- (a) P. Metrangolo, W. Panzeri, F. Recupero, G. Resnati and J. Fluor, Chem, 2002, 114, 27–33 CAS; (b) M. Erdélyi, Chem. Soc. Rev., 2012, 41, 3547 RSC.
- Determination of association constants from titration experiments in supramolecular chemistry: (a) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC; (b) N. Schulz, S. Schindler, S. M. Huber and M. Erdelyi, J. Org. Chem., 2018, 83, 10881–10886 CrossRef CAS PubMed; (c) C. Rosso, J. D. Williams, G. Filippini, M. Prato and C. O. Kappe, Org. Lett., 2019, 21, 5341–5345 CrossRef CAS PubMed.
- (a) M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646–1653 CrossRef CAS; (b) L. Maugeri, E. M. G. Jamieson, D. B. Cordes, A. M. Z. Slawin and D. Philp, Chem. Sci., 2017, 8, 938–945 RSC.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- (a) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed; (b) S. Rossi, A. Puglisi, L. Raimondi and M. Benaglia, ChemCatChem, 2018, 10, 2717–2733 CrossRef CAS; (c) A. Modak, E. N. Pinter and S. P. Cook, J. Am. Chem. Soc., 2019, 141, 18405–18410 CrossRef CAS PubMed; (d) F. Toulgoat, F. Liger and T. Billard, in Organofluorine Chemistry, ed. K. Szabó and N. Selander, Wiley, 2021, pp. 49–97 Search PubMed; (e) D. Cahard, N. Marie, J.-A. Ma and V. Tognetti, Angew. Chem., Int. Ed., 2024, 63, e202407689 CrossRef.
- Y. Ren, Q. Yan, Y. Li, Y. Gao, J. Zhao, L. Li, Z.-Q. Liu and Z. Li, J. Org. Chem., 2022, 87, 8773–8781 CrossRef CAS.
- Representative recent examples: (a) J. V. Burykina, N. S. Shlapakov, E. G. Gordeev, B. König and V. P. Ananikov, Chem. Sci., 2020, 11, 10061–10070 RSC; (b) J. V. Burykina, A. D. Kobelev, N. S. Shlapakov, A. Yu. Kostyukovich, A. N. Fakhrutdinov, B. König and V. P. Ananikov, Angew. Chem., Int. Ed., 2022, 61, e202116888 CrossRef CAS; (c) T. Jiang, L. Chen, S. Wen, L. Zhang, T. Wang and F. Xiong, J. Org. Chem., 2024, 89, 1296–1300 CrossRef CAS PubMed.
- S. M. Urvashi and N. T. Patil, Chem. Sci., 2023, 14, 13134–13139 RSC.
- Y. Zou, L. Liu, J. Liu and G. Liu, Future Med. Chem., 2020, 12, 91–93 CrossRef CAS.
- (a) D. Wang, J. Zhou, Z. Hu and T. Xu, J. Am. Chem. Soc., 2022, 144, 22870–22876 CrossRef CAS; (b) F. Yue, H. Ma, H. Song, Y. Liu, J. Dong and Q. Wang, Chem. Sci., 2022, 13, 13466–13474 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06550f |
| This journal is © The Royal Society of Chemistry 2024 |