
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-component modular synthesis of chiral 1,3-dioxoles via a Rh-catalyzed carbenic olefination cascade†
Shisheng
Huang
a,
Jilong
Luo
a,
Ping
Chen
b,
Jiean
Chen
 *c and
Zhaofeng
Wang
*a
*c and
Zhaofeng
Wang
*a
aState Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha, Hunan 410082, P. R. China. E-mail: zfwangchem@hnu.edu.cn
bInstitute of Chemical Biology, Shenzhen Bay Laboratory, Shenzhen, Guangdong 518118, P. R. China
cPingshan Translational Medicine Center, Shenzhen Bay Laboratory, Shenzhen, Guangdong 518118, P. R. China. E-mail: chenja@szbl.ac.cn
First published on 15th October 2024
Abstract
The advance of organic synthesis and the discovery of novel chemical transformations are often propelled by the rational programming of various bond-forming mechanisms and sequences that involve delicate reactive intermediates. In this study, we present an innovative Rh(II)-catalyzed asymmetric three-component cascade reaction involving IIII/PV-hybrid ylides, aldehydes, and carboxylic acids for the synthesis of 1,3-dioxoles with moderate to good yields and high enantioselectivity. This method utilizes IIII/PV-hybrid ylides as carbene precursors to form α-PV-Rh-carbenes, which initiate the formation of carbonyl ylides, followed by stereoselective cyclization with carboxylate anions and an intramolecular Wittig olefination cascade, ultimately resulting in the modular assembly of chiral 1,3-dioxoles. By employing this strategy, we successfully coupled various aldehydes and carboxylic acids to give chiral non-benzofused 1,3-dioxole scaffolds, highlighting the potential for late-stage functionalization of biologically relevant molecules, versatile synthetic manipulation, and the production of poly-1,3-dioxole macromolecules.
Introduction
Cyclic 1,3-dioxoles are significant structural motifs in organic chemistry,1 being present in natural products,2 designed chiral ligands,3 and synthetic pharmaceutical agents4 (Scheme 1A). Their wide range of applications has driven considerable efforts towards developing new synthesis methods. Most of the research has focused on synthesizing benzo[d][1,3]dioxoles, which possess fused aromatic rings and methylene units.5 Asymmetric acetalization to install an O,O-disubstituted stereogenic carbon center has been widely developed as an efficient pro-drug strategy.6 However, the asymmetric construction of non-benzofused 1,3-dioxoles remains a challenging task.7 Conventional approaches to these non-benzofused 1,3-dioxoles primarily rely on cycloaddition of diazodicarbonilic derivatives with carbonyls such as aldehydes,7b,h ketones,7d,e,j amides7l or esters.7k In these reported cases, the key steps are the nucleophilic attack from oxygen of carbonyls to carbenes with di-carbonyl substitutions. The newly generated carbonyl ylides underwent intramolecular cyclization to close the 1,3-dioxole ring. To date, only one asymmetric attempt, by Lacour, using a combination of [CpRu(CH3CN)3][PF6] and a chiral pymox ligand, has achieved an enantiomeric excess (e.e.) of up to 50%.7j A catalyzed multi-component modular synthesis of chiral 1,3-dioxoles from readily available substrates remains an unknown but highly desirable goal. | ||
| Scheme 1 Importance and synthetic strategies for 1,3-dioxoles. | ||
The Wittig reaction, discovered by Wittig and Geissler in 1953, has been extensively studied for constructing carbon–carbon double bonds in modern synthetic chemistry.8 Typically, this transformation involves treating active carbonyls, such as aldehydes and ketones, with phosphorous ylides, resulting in alkenes with complete chemoselectivity and controlled E/Z selectivity (Scheme 1B).9 Recently, the scope of Wittig olefination has expanded with the design of novel α-cationic PV carbon intermediates, such as α-PV radicals,10 α-PV carbenes,11 and α-PV carbynes,12 which can be used to install multiple bonds on one carbon in a single cascade step. Inspired by these advances and our long-standing interest in developing α-PV carbenes,11b,c we envisioned that the double bond in the 1,3-dioxole structure could be accessed via the Wittig olefination approach between an O-substituted phosphonium ylide and an ester functionality. The former species could be generated by the attack of oxygen nucleophiles on electrophilic α-PV metal carbenes.
Iodonium ylides have emerged as essential carbene precursors, crucial in various transformations.13 In 1984, Moriarty et al. reported the synthesis of bench-stable hybrid ylides (IIII-ylide and PV-ylide) with partial C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P and CI bonding on the central carbon.14 We anticipated that chiral dirhodium(II) carboxylate could decompose IIII/PV-hybrid ylides to generate α-PV Rh(II)-carbenes. According to our recent report, this α-phosphonium metal-carbenoid showed exclusive reactivity toward unsaturated carbonyl groups rather than mediating generally favorable X–H insertions. In this context, the subsequent capture of the Rh(II)-carbenoid by an aldehyde could yield the carbonyl ylide intermediate. Due to the potential stabilization of PV substitutes, this carbonyl ylide tends to exist as a metal-associated form,15 initiating the next stereoselective cycloaddition with a carboxylate ion. Finally, intramolecular Wittig olefination closes the 5-membered ring, affording 1,3-dioxoles in good to high yields with high enantioselectivities (Scheme 1C).
P and CI bonding on the central carbon.14 We anticipated that chiral dirhodium(II) carboxylate could decompose IIII/PV-hybrid ylides to generate α-PV Rh(II)-carbenes. According to our recent report, this α-phosphonium metal-carbenoid showed exclusive reactivity toward unsaturated carbonyl groups rather than mediating generally favorable X–H insertions. In this context, the subsequent capture of the Rh(II)-carbenoid by an aldehyde could yield the carbonyl ylide intermediate. Due to the potential stabilization of PV substitutes, this carbonyl ylide tends to exist as a metal-associated form,15 initiating the next stereoselective cycloaddition with a carboxylate ion. Finally, intramolecular Wittig olefination closes the 5-membered ring, affording 1,3-dioxoles in good to high yields with high enantioselectivities (Scheme 1C).
Several challenges are associated with this novel cascade multi-component reaction (MCR): (1) multiple active intermediates, including metal carbene, carbonyl ylides, carboxylate ions, and phosphonium ylides, may lead to predictable, competitive reactions, such as direct O–H insertion of a carboxylic acid into metal-carbene, non-selective carbonyl ylide formation, epoxidation of carbonyl ylide, and undesired Wittig olefination of an aldehyde; (2) poor asymmetric induction due to the rapid dissociation of the metal from the in situ generated metal-bound carbonyl ylide. Additional chiral auxiliaries, such as proton-shuttle catalysis or Lewis acid catalysis, may be required to achieve stereo-control;16 and (3) as a type of cyclic acetal, the 1,3-dioxole ring is highly fragile and could quickly decompose or racemize under harsh conditions.
Results and discussion
To validate the feasibility of the above-mentioned hypothesis, we initiated our investigation with a three-component reaction involving IIII/PV-hybrid ylide (1d), p-methoxybenzoic acid (2), and benzaldehyde (3) under Rh catalysis in basic dichloroethane (DCE) at room temperature (Table 1). We began by employing commercially available chiral Rh(II) catalysts, which were successfully used in various asymmetric MCRs involving carbene transfer processes.17 Using Rh2(S-DOSP)4 as the catalyst, we achieved a moderate yield and 28% e.e. (entry 1). Next, we screened phthalimide-based chiral Rh(II) catalysts with varying electron densities and steric bulk.18 The results showed that enantioselectivities were significantly influenced by different α-substituted alkyl chains on the chiral carboxylate ligand.19 Moderate enantioselectivity was observed with cat. 1m, which has a tethered isoamyl group (entry 2). Further improvements in ee were obtained with cat. 1q, which contains bridged polycyclic N-protecting groups (entry 3). The base additive was critical for this transformation, with nBuMe2N being the optimal choice (entries 4–6). Significant improvements in ee were achieved by conducting the reaction at low temperatures and changing the catalyst to 1v (entries 7–9). Yield enhancement to 43% was achieved by replacing molecular sieves (entry 10). Lowering the reaction temperature to −10 °C further improved enantiocontrol (entry 11). Finally, by finely adjusting the material ratio and using a more soluble IIII/PV-hybrid ylide 1l, we obtained optimal results in both yield (61%) and enantioselectivity (90%) (entry 12, see ESI† for detailed condition screening).|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | 1 |
|
Base | T (°C) | Yieldb (%) | e.e.c (%) |
a Entries 1–16 were carried with 1 (0.05 mmol), 2 (0.05 mmol), 3 (0.06 mmol) and  (2.0 mol%); entry 12 was carried with 1l (0.22 mmol), 2 (0.2 mmol), 3 (0.24 mmol) and (2.0 mol%); entry 12 was carried with 1l (0.22 mmol), 2 (0.2 mmol), 3 (0.24 mmol) and  (2.0 mol%).
b Unless otherwise noted, the yields refer to GC yield with n-dodecane as the internal standard.
c Determined by HPLC on a chiral stationary phase (for details, see ESI).
d 24 h reaction time.
e 3 Å MS was used instead of 4 Å MS.
f 72 h reaction time.
g Isolated yield after flash column chromatography. (2.0 mol%).
b Unless otherwise noted, the yields refer to GC yield with n-dodecane as the internal standard.
c Determined by HPLC on a chiral stationary phase (for details, see ESI).
d 24 h reaction time.
e 3 Å MS was used instead of 4 Å MS.
f 72 h reaction time.
g Isolated yield after flash column chromatography.
|
||||||
| 1 | 1d | Rh2(S-DOSP)4 | Et3N | 25 | 56 | 28 |
| 2 | 1d | Cat. 1m | Et3N | 25 | 24 | 55 |
| 3 | 1d | Cat. 1q | Et3N | 25 | 33 | 59 |
| 4 | 1d | Cat. 1q | n Pr3N | 25 | 32 | 54 |
| 5 | 1d | Cat. 1q | DIPEA | 25 | 26 | 55 |
| 6 | 1d | Cat. 1q | n BuMe2N | 25 | 28 | 68 |
| 7d | 1d | Cat. 1q | n BuMe2N | 0 | 27 | 82 |
| 8d | 1d | Cat. 1w | n BuMe2N | 0 | 41 | 83 |
| 9d | 1d | Cat. 1v | n BuMe2N | 0 | 39 | 85 |
| 10d,e | 1d | Cat. 1v | n BuMe2N | 0 | 43 | 85 |
| 11e,f | 1d | Cat. 1v | n BuMe2N | −10 | 41 | 89 |
| 12e,f | 1l | Cat. 1v | n BuMe2N | −10 | 61g | 90 |


With the optimized conditions in hand, we then explored the aldehyde scope of this Rh-catalyzed carbenic olefination cascade (Table 2). Various substituted aromatic aldehydes reacted well under this asymmetric MCR condition, yielding chiral 1,3-dioxoles with moderate to good yields and good enantioselectivities (4–23). Mono-substituted benzaldehydes with electron-withdrawing (5–11, 17–18) and electron-donating (12–16, 19) groups showed consistent reactivity. This method demonstrated high chemoselectivity for substrates containing common carbenophiles such as ester, ketone, alkyne, and N–H moieties. X-ray crystallographic analysis of product 19 assigned the absolute configuration of the newly generated stereogenic carbon center (see Section 9 in ESI†). Low conversion was generally observed under standard conditions when using aromatic aldehydes with di-substituted electron-withdrawing groups (20–21) or electron-deficient pyridyl substitutes (22–23). The reaction with linear (24) or branched (25–26) aliphatic aldehydes also proceeded smoothly. When a sterically bulky tertiary aldehyde containing a competing ketone motif was used, we isolated the desired 1,3-dioxole product (27) and a 2,3-dihydrofuran derivative (27′) in 21% yield with 90% ee. This result indicated that during the nucleophilic attack by the carboxylate anion, the in situ-generated carbonyl ylide existed as a Rh-bound form. This enantioselective formation of 27′ strongly supports the designed sequence, where Wittig olefination occurs after the asymmetric nucleophilic addition by the carboxylate ion.
| a Unless otherwise specified, all reactions were carried out using IIII/PV-hybrid ylide 1 (0.22 mmol, 1.1 equiv.), carboxylic acid 2 (0.2 mmol, 1.0 equiv.), aldehyde 3 (0.26 mmol, 1.3 equiv.), nBuMe2N (0.40 mmol, 2.0 equiv.), 3 Å MS (40 mg), catalyst 1v (2 mol%) and DCE (2.0 mL), at −10 °C under N2 atmosphere for 72 hours. Isolated yields were calculated. The d. r. value was determined by 1H NMR spectroscopy. a96 hours reaction time. b−20 °C for 120 hours. cCatalyst 1w (2 mol%), at −20 °C for 120 hours. |
|---|

|
Subsequently, we examined the scope of this MCR involving carboxylic acids under standard conditions. We assessed the variation of aromatic acids with different functional groups using p-Cl-benzaldehyde and IIII/PV-hybrid ylide 1l. Benzoic acids with substitutions at para-, meta-, and ortho-positions on the benzene ring, regardless of their electronic properties (electron-deficient, -neutral, or -rich), completed the reaction well, achieving products 28–40 in 52–81% yields with 84–98% ee. To illustrate the synthetic utility, a 5.0 mmol scale reaction was conducted under standard conditions, producing 1,3-dioxole product 34 in 65% yield (0.85 g) with 87% ee. Di-substituted (41) and heteroaromatic analogs (42–45) were also compatible, delivering the corresponding products in satisfactory yields with high ee. The reaction using E-α-methyl cinnamic acid as the starting material afforded the desired product 46 in 61% yield with 89% ee, without isomerization or cyclopropanation of the double bond. Furthermore, aliphatic carboxylic acids with different steric bulky substituents reacted smoothly with the aldehyde and hybrid ylide under optimal conditions, efficiently furnishing the assembled products (47–51). We observed partial racemization of the α-amino chiral center in product 51 when optically pure N-Boc proline was used, likely due to both the strong electron-withdrawing effect of the conjugated ketone and heteroatomic affect. Finally, we investigated the reaction scope of IIII/PV-hybrid ylides. Different lengths of linear alkyl ketyl substituted hybrid ylide reagents performed efficiently in this asymmetric reaction, yielding products 52–54 in moderate yield and good ee. However, using branched alkyl–ketyl IIII/PV hybrid ylide reagents resulted in trace amounts of the desired products with only 16% ee (55), indicating that the metal-carbene formation process is sensitive to the steric hindrance of carbene precursors.
Both carboxylic acids and aldehydes are recurrent functional groups in natural products and bioactive molecules.20 Chiral 1,3-dioxoles were smoothly installed into several natural or drug-related compounds containing carboxylic acids or aldehydes, demonstrating the potential synthetic utility of the presented method (Table 3, A). Notably, the stereochemistry of the chiral catalyst, rather than existing stereocenters on the starting material, predominantly determined the stereochemistry of the assembled 1,3-dioxole products (56 and 58). The excellent efficiency of these late-stage functionalizations is remarkable, as no other reaction products were observed for substrates bearing embedded ketones, esters, ether, nitrile, and alkenes, which could potentially intercept a Rh(II)-carbenoid intermediate. The acid-labile 1,3-dioxoles could also be developed as pH-sensitive linkages between acid-containing fluorescent dyes and bioactive molecules (66 and 67). Additionally, they could be used as pH-sensitive adhesives to bind two bioactive molecules together (68 and 69), potentially applicable in a multi-drug delivery system (Table 3, B).21
| a Unless otherwise specified, all reactions were carried out using IIII/PV-hybrid ylide 1 (0.22 mmol, 1.1 equiv.), carboxylic acid 2 (0.2 mmol, 1.0 equiv.), aldehyde 3 (0.26 mmol, 1.3 equiv.), nBuMe2N (0.40 mmol, 2.0 equiv.), 3 Å MS (80 mg), catalyst 1v (2 mol%) and DCE (2.0 mL), at −10 °C under N2 atmosphere for 72 hours. Isolated yields were calculated. The d.r. value was determined by 1H NMR spectroscopy. aCatalyst 1w (2 mol%), at −20 °C for 120 hours. b96 hours reaction time. cReactions were carried out using IIII/PV-hybrid ylide 1 (0.28 mmol, 1.4 equiv.), carboxylic acid 2 (0.2 mmol, 1.0 equiv.), aldehyde 3 (0.24 mmol, 1.2 equiv.), Et3N (0.44 mmol, 2.2 equiv.), 4 Å MS (40 mg), Rh2(cap)4 (1 mol%) and DCE (2.0 mL), at 25 °C under N2 atmosphere for 12 hours. |
|---|

|
The robust nature of the current method was further highlighted by the synthetic transformations of the double bonds on the 1,3-dioxole ring (Scheme 2A). Despite the significant steric hindrance and poor stability of the acetal structure, Pd-catalyzed hydrogenation of 74 (84% ee) produced saturated 1,3-dioxolane product 75 in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio. Upon treatment with 1 N HCl, the major isomer decomposed to generate optically active cis-diol product 76 with 81% ee. 1,2-Bromo-oxygenation occurred in a highly diastereo- and enantioselective manner, generating chiral 1,3-dioxolanes with two adjacent quaternary stereocenters. To our knowledge, this highly substituted chiral 5-membered ring remains elusive when constructed using established methods. Peroxide-embedded ring systems are widely present in versatile bioactive natural products and pharmaceutical molecules.22 The formation of unsaturated 1,2,4-trioxene remains elusive, with no synthetic methods developed for this scenario. We were delighted to find that using sodium m-chloroperoxybenzoate as a reaction partner, we could obtain a mixture of both 1,3-dioxole (39, 36% yield, 88% ee) and 1,2,4-trioxene product (79, 15% yield, 91% ee). This preliminary result provided a practical synthetic route towards this previously inaccessible ring structure (Scheme 2B).
:1 diastereomeric ratio. Upon treatment with 1 N HCl, the major isomer decomposed to generate optically active cis-diol product 76 with 81% ee. 1,2-Bromo-oxygenation occurred in a highly diastereo- and enantioselective manner, generating chiral 1,3-dioxolanes with two adjacent quaternary stereocenters. To our knowledge, this highly substituted chiral 5-membered ring remains elusive when constructed using established methods. Peroxide-embedded ring systems are widely present in versatile bioactive natural products and pharmaceutical molecules.22 The formation of unsaturated 1,2,4-trioxene remains elusive, with no synthetic methods developed for this scenario. We were delighted to find that using sodium m-chloroperoxybenzoate as a reaction partner, we could obtain a mixture of both 1,3-dioxole (39, 36% yield, 88% ee) and 1,2,4-trioxene product (79, 15% yield, 91% ee). This preliminary result provided a practical synthetic route towards this previously inaccessible ring structure (Scheme 2B).
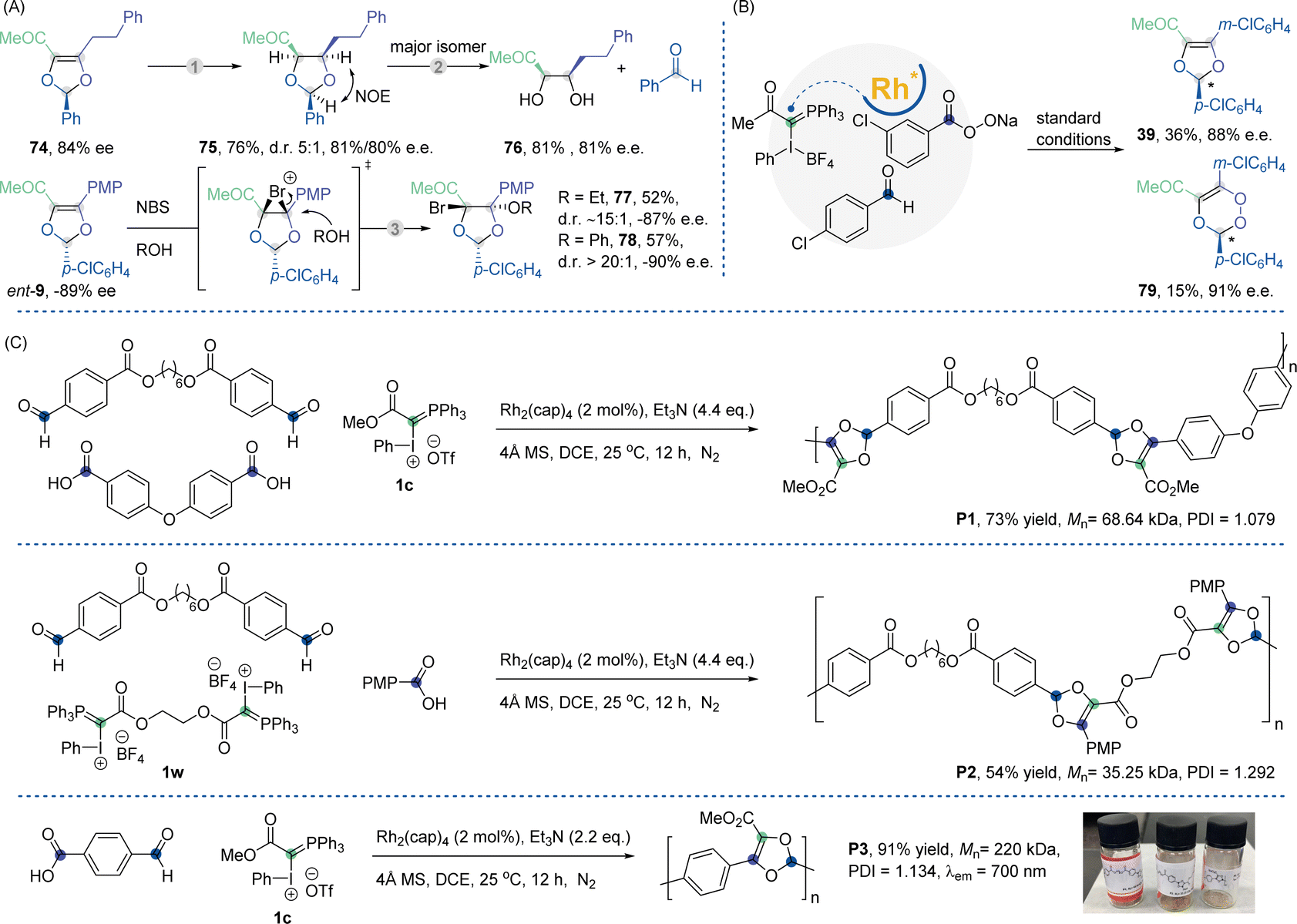 | ||
| Scheme 2 Synthetic transformation, construction of chiral 1,2,4-trioxene and polymerization reaction. (A) Two-step synthesis of chiral 1,2-diols. (1) 5% Pd/C (0.2 equiv.), H2 (1 atm), acetone, rt 16 h. (2) 1 N HCl:acetone (1:1.5), rt, 4 h. (3) Nucleophile (2.0 equiv.), NBS (2.0 equiv.), DCM, 0 °C, 10 min. (B) Synthesis of chiral 1,2,4-trioxene skeleton. (C) Synthesis of polymers with different structures via Rh-catalyzed MCRs using two bifunctional (AA-type and BB-type) and one monofunctional monomer using an AB-type and a monofunctional monomer. | ||
Recently, multi-component reactions have emerged as a new tool in polymer science due to the possibility of creating new libraries of monomers and polymers with various functionalities.23 On the other hand, transition-metal-catalyzed carbenic reactions has been established as a powerful tool for the bonds construction in polymerization processes.24 In this context, our Rh-carbene mediated MCR was also investigated as a step-growth polymerization method. By varying the combination of components, this addition polymerization, which follows a classic step-growth polymerization mechanism, can produce polymers with different substituted 1,3-dioxole structures in the leading chains (Scheme 2C). We polymerized commercially available carboxylic diacids (AA-type monomers) and dialdehydes (BB-type monomers) with ester-substituted IIII/PV-hybrid ylide 1c to yield poly 1,3-dioxoles P1 with a molar mass of up to 68640 g mol−1. The same type of polymerization using the dialdehydes and a newly prepared di-IIII/PV-hybrid ylide 1w with 4-methoxybenzoic acid produced polymer P2 with a molar mass of up to 35250 g mol−1. We also developed polymerization employing AB-type monomers containing an aldehyde and a carboxylic acid moiety. Polymerization of 4-formylbenzoic acid with IIII/PV-hybrid ylide 1c led to polymers P3 with a molar mass of up to 220000 g mol−1 and narrow dispersity of around 1.1, in good yield. Notably, this polymer exhibits fluorescent properties due to its significant hyper-conjugation system.
Control experiments were conducted to gain mechanistic insight into the reaction pathway (Scheme 3A). Firstly, a model reaction involving 1l, 2b, and 3g under standard conditions yielded 1,3-dioxole product 9 in 60% yield and 89% ee. Replacing catalyst 1v with its enantiomer ent-1v caused a reversion of the chiral center in the dioxole product, indicating that the chirality of the α-position of the chiral ligand determines the absolute configuration of the product. Without Rh(II) catalyst 1v, the reaction ceased, and no product 9 was obtained. In the absence of carboxylic acid, 20% of the IIII/PV-hybrid ylide was recovered, indicating that the Rh(II) catalyst is tightly associated with the carbonyl-ylide, preventing catalyst turnover and inhibiting complete consumption of the hybrid ylide substrates. The coupling of IIII/PV-hybrid ylide 1l with 18O-2a and 3g was performed to trace the source of oxygen in the 1,3-dioxole scaffold. This reaction yielded 18O-labeled 1,3-dioxole 80 in 68% yield and 87% ee (56% 18O-inc), along with the generation of PPh3 = 18O (43% 18O-inc). The 18O-labeling experiment supported the hypothesis that two equivalent oxygen atoms from the carboxylate anion nucleophilically attack the Rh(II)-associated carbonyl-ylide (Scheme 3B). The deuterium labeling reaction unambiguously confirmed that the newly generated chiral center originated from the aldehyde component (Scheme 3C).
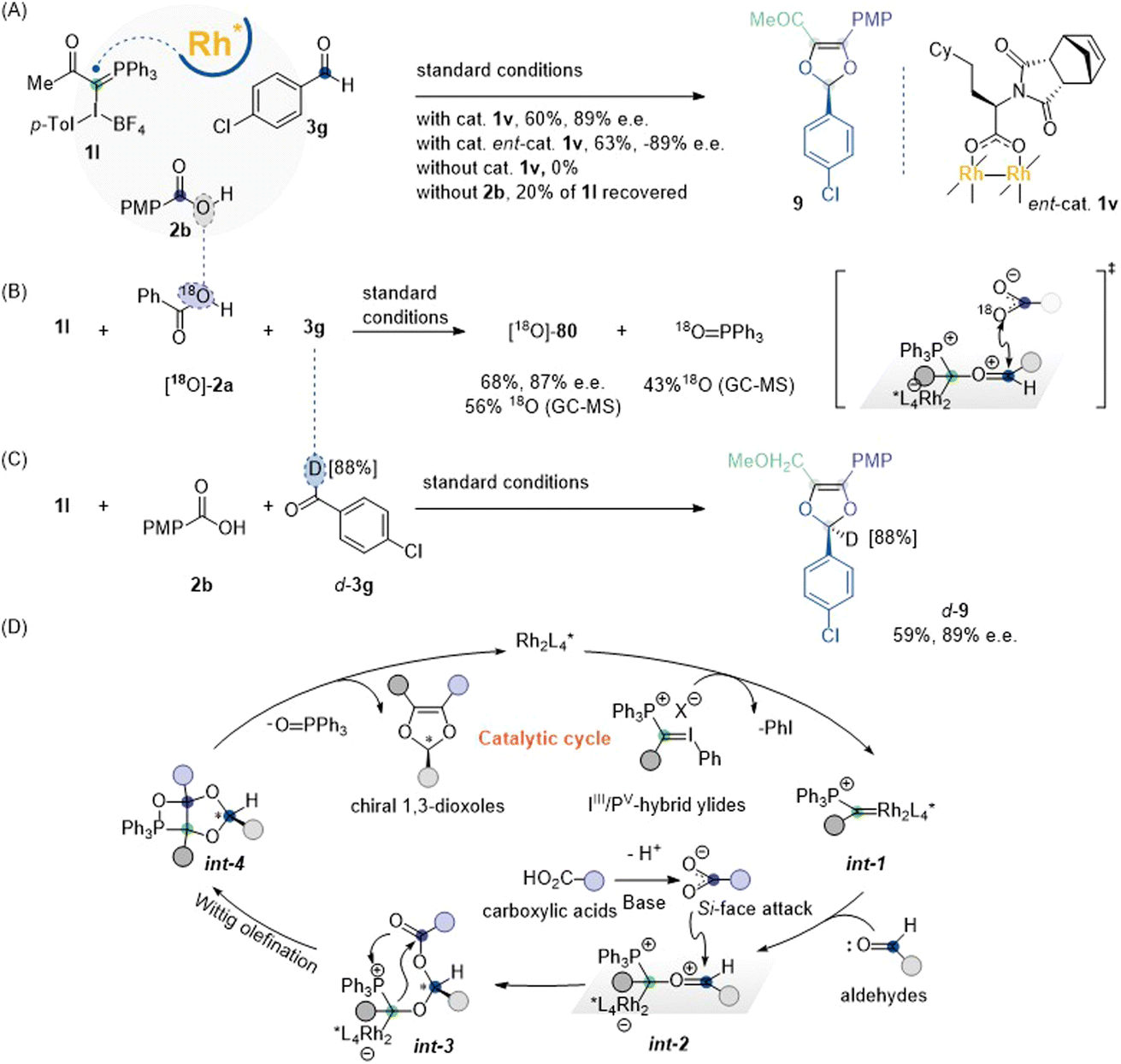 | ||
| Scheme 3 Mechanistic study. (A) Control experiments. (B) 18O-Labeling experiment. (C) Deuterium labeling experiment. (D) Proposed reaction pathway. | ||
Based on these results and previous studies, a plausible mechanism for this cascade MCR is proposed (Scheme 3D). Initially, the Rh catalyst decomposes the IIII/PV-hybrid ylide reagent to generate α-phosphonium Rh-carbenoid int-1, which has been tentatively assigned via real-time mass spectrometric (MS) analysis (see Section 8 in ESI†). This metal-carbene intermediate reacts exclusively with aldehydes through C–O bond formation to furnish Rh-bound carbonyl ylide int-2. The Re-face of this intermediate is shielded by the chiral framework on the Rh center, leaving the Si-face accessible to nucleophilic attack by the carboxylate ion.25 Subsequent intramolecular attack from the Rh-bound carbon atom within int-3 releases the Rh-catalyst and triggers the Wittig olefination step to form oxaphosphetanes fused int-4, which then undergoes cycloreversion to yield chiral 1,3-dioxole products.
Conclusions
In summary, we have developed a Rh(II)-catalyzed asymmetric three-component reaction that utilizes a programmed sequence of carbonyl ylide formation, cycloaddition, and Wittig olefination. This method provides efficient access to chiral 1,3-dioxoles with good to high yields and excellent enantioselectivity. The enantioselectivity is controlled by the chiral carboxylate ligands in the Rh catalysts, which form a Rh-associated carbonyl ylide complex, enabling stereoselective cycloaddition with the carboxylate ion during the C–O bonding step. This robust method employs simple and readily accessible materials, exhibits exceptional functional group tolerance, and has a broad substrate scope. It also allows for the facile late-stage introduction of optically pure 1,3-dioxoles into a wide range of natural and bioactive molecules. Further efforts are underway to expand the scope of in situ carbonyl ylide generation with ketones and to construct 1,3-dioxole derivatives with a chiral quaternary center.Data availability
All data presented in this manuscript are available in the ESI.†Author contributions
S. S. H. performed the experiments with contributions from J. L. L.; P. C. conduct real-time HRMS detection experiment; Z. F. W. conceived the study; J. A. C. and Z. F. W. supervised the project; Z. F. W. wrote the manuscript with input from all authors. All authors have read and approved the final manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was funded by the Natural Science Foundation of China (22101079, Z. W.) and the Shenzhen Science and Technology Innovation Commission (KCXFZ20201221173404013, J. C.). Z. Wang thanks the National Program for Thousand Young Talents of China. We thank the Analytical Instrumentation Center of Hunan University for mass spectrometry analysis. This work is dedicated to prof. Yong Huang (The Hong Kong University of Science and Technology) on the occasion of his 50th birthday.Notes and references
- (a) R. A. Aitken and L. A. Power, Adv. Heterocycl. Chem., 2013, 108, 163–193 Search PubMed; (b) R. A. Aitken, in Comprehensive Heterocyclic Chemistry IV, ed. D. S. Black, J. Cossy and C. V. Stevens, Elsevier, 2022, vol. 4, pp. 724–765 Search PubMed.
- (a) J. Lv, Z. Li, A. Deng and H. Qin, Org. Biomol. Chem., 2022, 20, 658–666 Search PubMed; (b) A. Zhang, Y. Zhang, A. Branfman, R. Baldessarini and J. Neumeyer, J. Med. Chem., 2007, 50, 171–181 Search PubMed.
- (a) N. Arshad and C. Kappe, Adv. Heterocycl. Chem., 2010, 99, 33–59 Search PubMed; (b) T. Saito, T. Yokozawa, T. Ishizaki, T. Moroi, N. Sayo, T. Miura and H. Kumobayashi, Adv. Synth. Catal., 2001, 343, 264–267 Search PubMed; (c) H. Shimizu, T. Ishizaki, T. Fujiwara and T. Saito, Tetrahedron: Asymmetry, 2004, 15, 2169–2172 Search PubMed.
- (a) P. Vanelle, J. Meuche, J. Maldonado, M. Crozet, F. Delmas and P. Timon-David, Eur. J. Med. Chem., 2000, 35, 157–162 Search PubMed; (b) A. Leite, K. da Silva, I. de Souza, J. de Araújo and D. Brondani, Eur. J. Med. Chem., 2004, 39, 1059–1065 Search PubMed; (c) S. Sangeeta, N. Kumari, M. Maruthi, S. Agrawal, A. Sarkar and R. Tomar, J. Mol. Liq., 2024, 398, 124284 Search PubMed; (d) F. Mao, H. Wang, W. Ni, X. Zheng, M. Wang, K. Bao, D. Ling, X. Li, Y. Xu, H. Zhang and J. Li, ACS Chem. Neurosci., 2018, 9, 328–345 Search PubMed; (e) Y.-J. Wu and N. A. Meanwell, J. Med. Chem., 2021, 64, 9786–9874 Search PubMed.
- (a) C. Ramsden, Adv. Heterocycl. Chem., 2010, 100, 1–51 Search PubMed; (b) B. Batanero and F. Barba, Org. Lett., 2005, 7, 2567–2569 Search PubMed; (c) V. Cherkasov, N. Druzhkov, T. Kocherova, G. Fukin and A. Shavyrin, Tetrahedron, 2011, 67, 80–84 Search PubMed; (d) M. Li and R. Hua, J. Org. Chem., 2008, 73, 8658–8660 Search PubMed; (e) A. Giugni, D. Impalà, O. Piccolo, A. Vaccari and A. Corma, Appl. Catal., B, 2010, 98, 72–78 Search PubMed.
- (a) K. M. Huttunen, H. Raunio and J. Rautio, Pharmacol. Rev., 2011, 63, 750–771 Search PubMed; (b) V. J. Stella, W. N. A. Charman and V. H. Naringrekar, Drugs, 1985, 29, 455–473 Search PubMed.
- (a) N. D. Field, J. Am. Chem. Soc., 1961, 83, 3504–3507 Search PubMed; (b) M. E. Alonso and A. Chitty Wladimir, Tetrahedron Lett., 1981, 22, 4181–4184 Search PubMed; (c) M. E. Alonso, M. d. C. Garcia and A. W. Chitty, J. Org. Chem., 1985, 50, 3445–3449 Search PubMed; (d) Y. R. Lee, J. Y. Suk and B. S. Kim, Tetrahedron Lett., 1999, 40, 6603–6607 Search PubMed; (e) C. Batsila, G. Kostakis and L. P. Hadjiarapoglou, Tetrahedron Lett., 2002, 43, 5997–6000 Search PubMed; (f) V. Nikolaev, L. Hennig, H. Heimgartner, B. Schulze and V. Nikolaev, Tetrahedron Lett., 2006, 47, 2643–2647 Search PubMed; (g) V. V. Nikolaev, H. Heimgartner, A. Linden, I. S. Krylov and V. A. Nikolaev, Eur. J. Org Chem., 2006, 2006, 4737–4746 Search PubMed; (h) A. E. Russell, J. Brekan, L. Gronenberg and M. P. Doyle, J. Org. Chem., 2004, 69, 5269–5274 Search PubMed; (i) A. Padwa, J. Boonsombat and P. Rashatasakhon, Tetrahedron Lett., 2007, 48, 5938–5941 Search PubMed; (j) M. Austeri, D. Rix, W. Zeghida and J. Lacour, Org. Lett., 2011, 13, 1394–1397 Search PubMed; (k) C. Tortoreto, T. Achard, L. Egger, L. Guénée and J. Lacour, Org. Lett., 2015, 18, 240–243 Search PubMed; (l) R. Pertschi, E. Brun, A. de Aguirre, L. Guénée, A. I. Poblador-Bahamonde and J. Lacour, Helv. Chim. Acta, 2021, 104, e2100122 Search PubMed.
- (a) G. Wittig and G. Geissler, Liebigs Ann. Chem., 1953, 580, 44–57 Search PubMed; (b) G. Wittig and U. Schöllkopf, Chem. Ber., 1954, 87, 1318–1330 Search PubMed; (c) H. Pommer, Angew. Chem., Int. Ed., 1977, 16, 423–429 Search PubMed; (d) H. J. V. Bestmann and O. Vostrowsky, Top. Curr. Chem., 1983, 109, 85–163 Search PubMed; (e) K. C. Nicolaou, M. W. Härter, J. L. Gunzner and A. Nadin, Liebigs Ann., 1997, 1997, 1283–1301 Search PubMed; (f) M. M. Heravi, V. Zadsirjan, M. Daraie and M. Ghanbarian, ChemistrySelect, 2020, 5, 9654–9690 Search PubMed; (g) D. H. A. Rocha, D. C. G. A. Pinto and A. M. S. Silva, Eur. J. Org Chem., 2018, 2018, 2443–2457 Search PubMed; (h) M. M. Heravi, M. Ghanbarian, V. Zadsirjan and B. Alimadadi Jani, Monatsh. Chem., 2019, 150, 1365–1407 Search PubMed.
- (a) P. A. Byrne and D. G. Gilheany, Chem. Soc. Rev., 2013, 42, 6670–6696 Search PubMed; (b) P. Farfán, S. Gómez and A. Restrepo, J. Org. Chem., 2019, 84, 14644–14658 Search PubMed.
- (a) M. Das, M. Vu, Q. Zhang and X. Liu, Chem. Sci., 2019, 10, 1687–1691 Search PubMed; (b) M. Vu, W. Leng, H. Hsu and X. Liu, Asian J. Org. Chem., 2019, 8, 93–96 Search PubMed; (c) D. Filippini and M. Silvi, Nat. Chem., 2021, 14, 66–70 Search PubMed.
- (a) E. D. Matveeva, T. A. Podrugina, A. S. Pavlova, A. V. Mironov, R. Gleiter and N. S. Zefirov, Eur. J. Org Chem., 2009, 2009, 2323–2327 Search PubMed; (b) Z. Wang and X. Ye, Synlett, 2023, 35, 1153–1159 Search PubMed; (c) X. Ye, H. Pan, Y. Huang, J. Chen and Z. Wang, Chem. Sci., 2024, 15, 6515–6521 Search PubMed.
- (a) T. Koike, J.-K. Yu and M. M. Hansmann, Science, 2024, 385, 305–311 Search PubMed; (b) R. J. Ward, M. Jörges, H. Remm, E. Kiliani, F. Krischer, Q. Le Dé and V. H. Gessner, J. Am. Chem. Soc., 2024, 146, 24602–24608 Search PubMed.
- (a) V. V. Zhdankin, M. S. Yusubov, A. Yoshimura and V. V. Zhdankin, Arkivoc, 2016, 2016, 342–374 Search PubMed; (b) X. Mi, C. Pi, W. Feng and X. Cui, Org. Chem. Front., 2022, 9, 6999–7015 Search PubMed; (c) P. Müller, Acc. Chem. Res., 2004, 37, 243–251 Search PubMed.
- (a) R. M. Moriarty, I. Prakash, O. Prakash and W. A. Freeman, J. Am. Chem. Soc., 1984, 106, 6082–6084 Search PubMed; (b) Z. Huang, X. Yu and X. Huang, J. Org. Chem., 2002, 67, 8261–8264 Search PubMed.
- C.-Y. Zhou, J.-C. Wang, J. Wei, Z.-J. Xu, Z. Guo, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2012, 51, 11376–11380 Search PubMed.
- D. Zhang and W. Hu, Chem. Rec., 2017, 17, 739–753 Search PubMed.
- (a) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911–936 Search PubMed; (b) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 Search PubMed.
- F. G. Adly, M. G. Gardiner and A. Ghanem, Chem.–Eur. J., 2016, 22, 3447–3461 Search PubMed.
- (a) K. Liao, W. Liu, Z. L. Niemeyer, Z. Ren, J. Bacsa, D. G. Musaev, M. S. Sigman and H. M. L. Davies, ACS Catal., 2017, 8, 678–682 Search PubMed; (b) Z. J. Garlets, Y. T. Boni, J. C. Sharland, R. P. Kirby, J. Fu, J. Bacsa and H. M. L. Davies, ACS Catal., 2022, 12, 10841–10848 Search PubMed.
- A. S. Kalgutkar and J. S. Daniels, in Metabolism, Pharmacokinetics and Toxicity of Functional Groups: Impact of Chemical Building Blocks on ADMET; RSC Drug Discovery Series, ed. D. A. Smith, Royal Society of Chemistry, Cambridge, 2010, pp. 99–167 Search PubMed.
- (a) M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Biomaterials, 2016, 85, 152–167 Search PubMed; (b) Y. Zhai, X. Zhou, L. Jia, C. Ma, R. Song, Y. Deng, X. Hu and W. Sun, Polymers, 2017, 9, 698 Search PubMed; (c) J. Li, X. Zhang, M. Zhao, L. Wu, K. Luo, Y. Pu and B. He, Biomacromolecules, 2018, 19, 3140–3148 Search PubMed; (d) N. Deirram, C. Zhang, S. S. Kermaniyan, A. P. R. Johnston and G. K. Such, Macromol. Rapid Commun., 2019, 40, 1800917 Search PubMed; (e) E. R. Gillies, A. P. Goodwin and J. M. J. Fréchet, Bioconjugate Chem., 2004, 15, 1254–1263 Search PubMed.
- (a) J. Wang, C. Xu, Y. K. Wong, Y. Li, F. Liao, T. Jiang and Y. Tu, Engineering, 2019, 5, 32–39 Search PubMed; (b) T. Efferth, Drug Resistance Updates, 2005, 8, 85–97 Search PubMed; (c) J. N. Cumming, D. Wang, S. B. Park, T. A. Shapiro and G. H. Posner, J. Med. Chem., 1998, 41, 952–964 Search PubMed.
- (a) O. Kreye, T. Tóth and M. A. Meier, J. Am. Chem. Soc., 2011, 133, 1790–1792 Search PubMed; (b) R. Kakuchi, Polym. J., 2019, 51, 945–953 Search PubMed; (c) R. Kakuchi, Angew. Chem., Int. Ed., 2014, 53, 46–48 Search PubMed; (d) P. Theato, Multi-Component and Sequential Reactions in Polymer Synthesis, Springer International Publishing, Heidelberg, 2015 Search PubMed; (e) E. Blasco, M. Sims, A. Goldmann, B. Sumerlin and C. Barner-Kowollik, Macromolecules, 2017, 50, 5215–5252 Search PubMed.
- L. Yu, X. Yao and J. Wang, Acta Chim. Sin., 2023, 81, 1015–1029 Search PubMed.
- Based on the catalysts' conformation and guiding principles proposed by Prof. Huw Davies, we proposed a possible asymmetric induction model. See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2294355. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06166g |
| This journal is © The Royal Society of Chemistry 2024 |