
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced copper-catalyzed asymmetric cyanoalkylalkynylation of alkenes, terminal alkynes, and oximes†
Shuang
Xin
,
Jibang
Liao
,
Qi
Tang
,
Xiaoming
Feng
 and
Xiaohua
Liu
*
and
Xiaohua
Liu
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, P. R. China. E-mail: liuxh@scu.edu.cn
First published on 14th October 2024
Abstract
The asymmetric dicarbofunctionalization of alkenes via a radical relay process can provide routes to diverse hydrocarbon derivatives. Three-component carboalkynylation, limited to particular alkyl halides and using readily available cycloketone oxime esters as redox-active precursors, is restricted by the available pool of suitable chiral ligands for broadening the redox potential window of copper complexes and simultaneously creating the enantiocontrol environment. Herein, we report a new hybrid tridentate ligand bearing a guanidine–amide–pyridine unit for photoinduced copper-catalyzed cyanoalkylalkynylation of alkenes. Leveraging the copper catalyst’s redox capability is achieved via merging the electron-rich ligand with a readily organized configuration and enhanced absorption in the visible light range, which also facilitates the enantioselectivity. The generality of the catalyst system is exemplified by the efficacy across a number of alkenes, terminal alkynes and cycloketone oxime esters, working smoothly to give alkyne-bearing nitriles with good yields and excellent enantioselectivity. A mechanistic study reveals that the chiral copper catalyst meets the requirements of possessing sufficient reduction ability, good light absorption properties, and appropriate steric hindrance.
Introduction
Alkene difunctionalization is widely recognized as a pivotal approach in organic synthesis owing to its unique ability to introduce molecular complexity in a single synthetic step.1 Transition-metal-catalyzed three-component alkene dicarbofunctionalization via a radical relay approach has gained significant traction,2 but asymmetric catalytic versions enabling construction of chiral carbon centers are still scarce. Chiral copper-catalyzed alkylcyanation, alkylarylation and alkylalkynylation (Scheme 1a) of styrenes using alkyl halides,3 Togni reagents,4 alkyl peroxides,5 or alkyl N-hydroxyphthalimide esters6 as the alkyl radical sources have been realized, usually with the assistance of oxazoline-type ligands, with or without light assistance. The alkylation of terminal alkynes into internal alkynes via an enantioselective coupling reaction has drawn great effort,7 for example using the Sonogashira coupling reaction, as shown in work by the groups of Fu and Eckhardt8 and by Hwang and Sagadevan,9 as well as by several other groups, with or without photoinduced copper-catalyzed conditions.10,11 Based on these studies, three-component carboalkynylation of olefins via an atom transfer radical addition process was also made available (Scheme 1a) by the use of alkyl iodides in Zhang’s study3b and electron-withdrawing substituted alkylhalides in Liu’s work.3a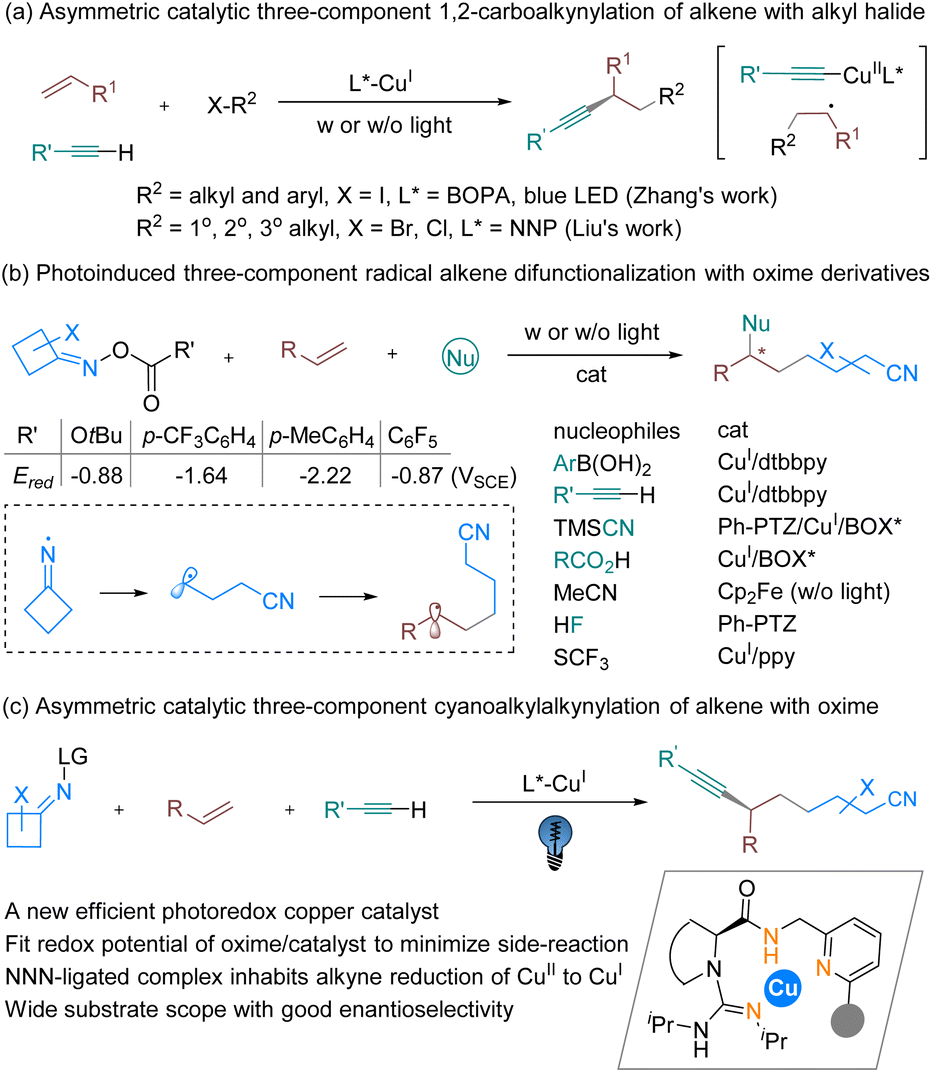 | ||
| Scheme 1 Difunctionalization of alkenes involving cycloketone oxime esters or terminal alkynes. | ||
Cycloketone oxime esters as sources of cyanoalkyl radicals have been extensively explored for synthesizing a broad range of functionalized alkylnitriles,12 pioneered by Zard’s research.13 A variety of difunctionalizations of alkenes using oxime esters as viable coupling partners have been disclosed by Xiao’s group and others (Scheme 1b).14 In comparison with alkyl halides as the radical precursors, the generation of cyanoalkyl radicals provides an improvement via a higher reductive potential and an additional cleavage process from iminyl radicals delaying the radical relay process. Special features of copper complexes under UV irradiation2a,15 provide the ability to accelerate difunctionalization with oxime esters, where in most cases copper photocatalysts bearing particular ligands or combined with an exogenic photocatalyst are able to initiate a single-electron transfer (SET) process to generate a radical species (Scheme 1b). Asymmetric catalytic transformations have been limited to TMSCN14f and carboxylic acid14c as the nucleophilic partners, and aryl boronic acid as the coupling partner has just been reported very recently;16 therefore the process of asymmetric cyanoalkylalkynylation of olefins remains an open subject (Scheme 1c). Although copper acetylides could advance SET by means of a UV-light-absorbing photocatalyst,9,17 the terminal alkyne could reduce the formed Cu(II) species to Cu(I), intruding upon the desired alkylalkynation reaction.18 In addition, side reactions, such as direct coupling between the cyanoalkyl radical and nucleophilic reagents, and dimerization, would accompany the process.19 Therefore, introducing suitable chiral ligands to modulate the reactivity of the excited copper acetylide complex, and to discriminate the sterically hindered aryl/cyanoalkyl subunit of the radical intermediate for enantioselective coupling, is critical.
The variety of chiral ligands that can straightforwardly support copper-catalyzed photoreactions in a stereocontrolled manner is consequently somewhat limited to a handful N- or P-based heteroleptic compounds.20 It is important that the chiral ligand must stabilize the copper complex in specific oxidation states and electronic configurations. Inspired by the entatic-state principle, which has been applied to copper electron-transfer processes21via lowering the reorganization energy during redox processes, thus facilitating a fast electron transfer, we rationalized that tridentate hybrid N-donor ligands containing a guanidine,22,23 amide and pyridine subunit (Scheme 1c), which has been disclosed to accelerate Sonogashira coupling of alkyl bromides,24 are also potential ligands for copper-based photocatalysts. Multiple donors and ready modification of the substituents of the ligand are useful to increase the rigidity for pre-organizing the configuration, and a methylene linker between the amide and pyridine provides some conformational flexibility. Further, taking multiple N-donor characters together, these kinds of ligands have a preference for stabilizing copper in various oxidation states. Additionally, these subunits are thought of as redox non-innocent, which may contribute to changes in the electronic nature of the metal center and contribute to reactivity. Herein, we describe the development of a new chiral photoredox copper catalyst for a general three-component radical 1,2-cyanoalkylalkynylation of alkenes with cycloketone oxime esters and terminal alkynes.
Results and discussion
Our initial survey started with alkylalkynylation of 2-vinylnaphthalene B1 with Cu(I)/chiral guanidine as the catalyst in the presence of Cs2CO3 and under irradiation by 3 W blue LEDs, and cyclobutanone O-(4-methoxybenzoyl) oxime A1 and phenyl-acetylene C1 were used as the cyanoalkyl reagent and alkynyl reagent, respectively. As shown in Table 1, the desired three-component coupling product D1 could be obtained with moderate enantioselectivity and a low yield in tetrahydrofuran (THF) at 20 °C upon irradiation with 440 nm light (entries 1–5). Competitive formation of two-component coupling byproducts between a nitrogen-centered radical or cyanoalkyl radical and the terminal alkyne (BP-1 and BP-2) accounted for the unsatisfactory yield. It was delightful that the addition of a phenyl substituent at the ortho-position of the methylpyridine subunit of the ligand (G-TqPy-3) led to increased enantioselectivity with a slightly higher yield (entry 3). When the reaction was conducted with CuOTf as the metal precursor, the enantioselectivity could be improved to 81% ee (entry 6). In a mixed solvent of p-xylene (PX) and dichloromethane (DCM) at 0 °C, a 91% ee was obtained but the yield remained unsatisfactory (entry 7). Initiation with stronger light (10 W) could increase the yield at 5 °C, but the enantioselectivity slightly dropped (entry 8). If A2 was used as the cyanoalkyl reagent, it participated in the reaction to give a 52% yield and 91% ee (entry 9). Further ligand screening via the exchange of several classic nitrogen-based oxazoline or imidazoline ligands revealed that the reaction was sluggish with neutral nitrogen-based ligands (entries 11–13), while bisoxazoline diphenylaniline (BOPA) served as an exception and could afford the product in 23% yield with 35% ee, accompanied by a lot of two-component byproducts.|
|
|||
|---|---|---|---|
| Entrya | Conditions | Yieldb (%) | eec (%) |
a Unless otherwise noted, all reactions were performed with [Cu]/L* (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2, 10 mol%), A (3.0 equiv.), B1 (0.1 mmol), C1 (3.0 equiv.) and Cs2CO3 (4.0 equiv.) in THF (1.0 mL) at 20 °C for 24 h, with 440 nm LED irradiation.
b NMR yield using 1,1,2,2-tetrabromoethane as an internal standard.
c The ee values were determined via ultraperformance convergence chromatography (UPC2).
d In PX/DCM (v/v = 7/3, 1.0 mL) for 44 h.
e At 0 °C.
f At 5 °C. :1.2, 10 mol%), A (3.0 equiv.), B1 (0.1 mmol), C1 (3.0 equiv.) and Cs2CO3 (4.0 equiv.) in THF (1.0 mL) at 20 °C for 24 h, with 440 nm LED irradiation.
b NMR yield using 1,1,2,2-tetrabromoethane as an internal standard.
c The ee values were determined via ultraperformance convergence chromatography (UPC2).
d In PX/DCM (v/v = 7/3, 1.0 mL) for 44 h.
e At 0 °C.
f At 5 °C.
|
|||
| 1 | A1, G-TqPy-1, CuI, THF, (3 W) | 22 | 45 |
| 2 | A1, G-TqPy-2, CuI, THF, (3 W) | 24 | 49 |
| 3 | A1, G-TqPy-3, CuI, THF, (3 W) | 38 | 74 |
| 4 | A1, G-TqPy-4, CuI, THF, (3 W) | 18 | 45 |
| 5 | A1, G-TqPy-5, CuI, THF, (3 W) | 20 | 45 |
| 6 | A1, G-TqPy-3, CuOTf, THF, (3 W) | 38 | 81 |
| 7d,e | A1, G-TqPy-3, CuOTf, (3 W) | 34 | 91 |
| 8d,f | A1, G-TqPy-3, CuOTf, (10 W) | 51 | 87 |
| 9d,f | A2, G-TqPy-3, CuOTf, (10 W) | 52 | 91 |
| 10d,f | A2, BOPA, CuOTf, (10 W) | 23 | 35 |
| 11d,f | A2, Box, CuOTf, (10 W) | Trace | — |
| 12d,f | A2, PyImz, CuOTf, (10 W) | Trace | — |
| 13d,f | A2, PyBox, CuOTf, (10 W) | Trace | — |

With the optimized reaction conditions established, the substrate scope of the terminal alkynes was explored in the presence of CuOTf/G-TqPy-3 catalyst under 440 nm light irradiation (Scheme 2). Various substituted ethynylbenzenes could undergo the coupling reaction with moderate yields (48–58%) and excellent ee values (85–91%), regardless of the positions and electronic nature (D2-D12, and D17). Terminal alkynes bearing a substituent such as an alkyl, cyclopropyl or TMS were also tolerable with slightly higher yields (55–60%) and good enantioselectivities (79–85% ee; D13–D16). In addition, 2-ethynylnaphthalene and 3-ethynylthiophene could also be installed into the desired products (D18 and D19) with comparable outcomes.
 | ||
| Scheme 2 Substrate scope of terminal alkynes. Unless otherwise noted, all reactions were performed with CuOTf/G-TqPy-3 (1:1.2, 10 mol%), A2 (3.0 equiv.), B1 (0.1 mmol), C (3.0 equiv.), and Cs2CO3 (4.0 equiv.) in PX/DCM (v/v = 7/3, 1.0 mL) under 10 W LED (440 nm) irradiation at 5 °C for 44 h. Isolated yields. The ee values were determined via high-performance liquid chromatography (HPLC) on a chiral stationary phase. | ||
Next, we examined the scope of the alkenes and cyanoalkyl reagents (Scheme 3). The investigation of the generality of the alkenes when reacting with A2 and phenylacetylene C1 revealed that an array of alkenes was well-tolerated (D20–D42). The 6-substituted 2-vinylnaphthalenes bearing electron-donating or -withdrawing groups all worked well (D20–D22). The aryl groups could also be hetero-containing ones, such as 5-benzo[b]thiophene, 5-benzo[d][1,3]dioxole, N-tosyl-indole or 5-benzofuran, and the corresponding products (D23–D26) could be isolated in 45–55% yield with 82–85% ee. Next, styrene derivatives with electron-donating and -withdrawing substituents at different positions were tested (D27–D40), which showed less effect on both the yield (47–64%) and enantioselectivity (83–91% ee), with para-OtBu-substituted D41 as an exception, which was isolated in reduced yield but with a good ee value. The coupling with 3-vinylthiophene led to the formation of product D42 in 58% yield and 87% ee. Interestingly, the alkynylated alkene B25 can provide the corresponding product D43 in 63% yield with 92% ee. This provides a new method for constructing chiral dialkynyl compounds.
 | ||
| Scheme 3 Substrate scope of alkenes and cycloketone oxime esters. Unless otherwise noted, all reactions were performed with CuOTf/G-TqPy-oPh (1:1.2, 10 mol%), A (3.0 equiv.), B (0.1 mmol), C1 (3.0 equiv.), and Cs2CO3 (4.0 equiv.) in PX (0.7 mL) and DCM (0.3 mL) under 10 W LED (440 nm) irradiation for 44 h. Yields are of the isolated products. The ee values were determined via HPLC on a chiral stationary phase. | ||
In addition, we continued to examine the cyanoalkyl radical precursors. Mono-substituted cyclobutanone oxime ester A3, 4,4-pyridine-substituted A5 and 3,3-azetidine-substituted A6 could be delivered into the products (D44–D46) bearing substitution at the alkyl-chain, with the results slightly diminished (80–85% ee). Unfortunately, while the bicyclo[4.2.0]octa-1(6),2,4-trien-7-one oxime ester A4 and 2,2,4,4-tetramethylcyclobutane-1,3-dione oxime ester A7 could transfer the related cyanoalkyl group into the products, both the yield and ee value dropped significantly (D47 and D48), which might be due to steric hindrance.
In order to evaluate the application potential of this catalytic system, we conducted a scaled-up experiment using A2, styrene (B10) and phenylacetylene (C1) as the substrates (Scheme 4a). Product D28 could be obtained with a 60% yield (0.92 g) and 90% ee value, indicating the suitability of this catalytic system for gram-scale synthesis. Subsequently, the alkyne group was reduced under Pd/C and H2 conditions to yield 6,8-diphenyloctanenitrile E with 82% yield, maintaining the enantioselectivity. The cyano group was further converted to a carbonyl group, affording 1,6,8-triphenyloctan-1-one F, whose absolute configuration was determined to be (R)-F based on X-ray single-crystal diffraction analysis (Scheme 4a).25 Thus, the absolute configuration of the product D28 was rationalized as the (S)-isomer.
 | ||
| Scheme 4 Synthetic application and mechanistic investigation. | ||
To detect the radical intermediates generated in the system, EPR experiments and control experiments were conducted (Scheme 4b–d). A solution of oxime A2 with one equivalent of 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) and chiral copper catalyst in a mixture of p-xylene and dichloromethane produced obvious signals under visible-light irradiation. The experimental signals matched the simulated spectra of the mixture of intermediates H and I (Scheme 4b), indicating the generation of an iminyl radical and the related alkyl radical through a photoinduced copper(I)-catalyzed SET process. When (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) was introduced into the reaction system, the cyanoalkylalkynylation reaction was inhibited and only the radical cross-coupling byproduct G was detected via ESI-MS and 1H NMR analysis (Scheme 4c). Furthermore, under the standard conditions, when a radical clock experiment was conducted using alkene B26, the cyclopropyl group was opened to give product D49 in a yield of 44% (Scheme 4d), giving evidence of a radical relay process via addition to the alkene.
The photophysical and electrochemical properties of the chiral copper complexes prepared in situ were investigated. UV-visible absorption spectra of the free ligand and the copper complexes in a dichloromethane solution were recorded (Scheme 4e). This revealed that the absorption above 400 nm increased gradually upon coordination with CuOTf, and the chiral copper acetylide complexes featured a further intense absorption band. The irradiation with 440 nm light enables the formation of excited copper catalytic species. In comparison, the ortho-phenyl decorated ligand G-TQPy-3 showed a slight enhancement over ligand G-TQPy-1. This enhancement is characterized by a less intense metal-to-ligand charge-transfer (MLCT) band, implying a charge transfer in the coordination structure where Cu(I) is oxidized to a relatively high valence state. The phosphorescence emission spectra of the chiral copper complexes in solution (Scheme 4f) revealed a small blue-shift for the complex of G-TQPy-3 in comparison with G-TQPy-1 (508 nm vs. 513 nm), but the corresponding chiral copper acetylide complexes exhibited a more intense signal, showing that the anionic alkyne is a strong ligand for forming the photocatalytic species. In addition, the ligated guanidine reduces the lifetime of excited copper(I) acetylide (from τ = 15.95 μs to 5.25 μs),26 but it remains longer than that of species in the singlet excited state to support the redox process (see ESI for details†). It is rationalized that the excited chiral copper complexes undergo intersystem crossing to become triplet excited-state metal complexes, acting as a photo-reductant to undergo single-electron transfer with the oxime.
Cyclic voltammetry was performed on an MeCN solution of each complex in order to probe their electronic properties in the ground state. The voltammograms are given in the ESI† and the relevant potentials are listed in Scheme 4g. The measurements showed that oxime A2 has a low Ered (−2.44 VSCE), implying that a strongly reductive catalyst is required to initiate iminyl radical generation. Hwang and coworkers have evaluated the redox potential of excited copper(I) acetylide to be −2.048 VSCE in CH3CN, with a long lifetime.26 A pseudo-reversible oxidation wave of CuOTf was observed above −1.0 V versus SCE, which can be assigned to the Cu(II)/Cu(I) redox couple.
The guanidine-ligated copper complexes (G-TQPy-1 and G-TQPy-3) showed similar Eap/Ecp, as did the related chiral copper acetylide complexes. The chiral copper acetylide species have lower oxidation potential than the corresponding chiral precursors. The difference between the redox waves (ΔE) is much smaller for the chiral copper acetylide species than for the precursors. This implies that a flattening of the coordination sphere in the guanidine–ligand–copper catalysts occurs slightly when the distorted Cu(I) acetylide is oxidized to square planar Cu(II), owing to the similar coordination cage. In addition, a rather prominent reduction wave (below −2.1 VSCE) is displayed for the guanidine–copper complexes, corresponding to one-electron ligand-centred reduction, where ligand G-TQPy-3 itself exhibits multiple reduction waves (see the ESI for details†). The oxidative potential of the excited copper complexes was calculated (Scheme 4g) based on their excitation and emission profiles. This redox potential is sufficiently higher than that of oxime A2 (−2.44 VSCE). Therefore, in connection with the photophysical properties, SET from a photoexcited triplet chiral copper(I) phenylacetylide to the oxime is exothermic and can occur smoothly.
Based on the above mechanistic data, a plausible mechanism is proposed in Scheme 5. In the presence of a base and the basic tridentate ligand G-TQPy-3, a chiral anionic guanidine–amide copper(I) acetylide complex forms. Upon light irradiation, it reaches the triplet state *Int1, acting as a strong reductant to perform a SET process with oxime ester A2. Thus, square planar copper(II) acetylide Int2 and an iminyl radical via fragmentation are generated, releasing the cesium carboxylate. The iminyl radical Int3 transforms into cyanoalkyl radical Int3′via carbon–carbon cleavage, then is trapped by the alkene to give rise to Int4. Otherwise, the cross coupling with copper species Int2 will generate two-component byproducts. The enantioselective coupling between Int4 and Int2 may proceed via elimination of copper(III) species Int5 or via radical substitution pathways, which is hard to clarify at present. This affords the cyanoalkylalkynylation product with release of the copper(I) species.
 | ||
| Scheme 5 Proposed catalytic cycle. | ||
Conclusions
In summary, we disclose that a properly designed chiral copper complex with a hybrid tridentate ligand enables photocatalytic asymmetric cyanoalkylalkynylation of olefins using cycloketone oxime esters as the precursors. The ligand, based on a guanidine–amide embellished with a (6-phenylpyridin-2-yl)methanyl group, allows ready electron transfer with lower reorganization energy during the redox processes of the copper acetylide in the excited state. The stereoinduction in the C(sp3)–C(sp) bond formation is also well-controlled with the readily accessible new ligand. The system permits a broad substrate scope with good functional group tolerance, allowing construction of a series of enantiomerically enriched alkyne–nitrile derivatives. We believe that this ligand will bring more opportunities to the development of asymmetric cross-coupling chemistry with detailed studies to understand the spectroscopic origins and the structure–property relationships.Data availability
Further details of experimental procedure, 1H, 13C{1H} and 19F{1H} NMR, HPLC and UPC2 spectra, and X-ray crystallographic data for F, are available in the ESI.†Author contributions
S. X. performed the experiments. J. B. L. repeated some experiments. Q. T. participated in the article discussions. X. M. F. and X. H. L. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22188101), and Sichuan University (2020SCUNL204) for financial support. We are grateful to Dr Yuqiao Zhou (Sichuan University) for the X-ray single-crystal diffraction analysis and Dr Hanjiao Chen (Sichuan University) for the EPR measurement and simulation.Notes and references
- For selected reviews of alkene difunctionalization:
(a) L. M. Wickham and R. Giri, Acc. Chem. Res., 2021, 54, 3415–3437 Search PubMed
; (b) M. Patel, B. Desai, A. Sheth, B. Z. Dholakiya and T. Naveen, Asian J. Org. Chem., 2021, 10, 3201–3232 Search PubMed
-
(a) L. Song, L. Cai, L. Gong and E. V. Van der Eycken, Chem. Soc. Rev., 2023, 52, 2358–2376 Search PubMed
-
(a) X. Y. Dong, J. T. Cheng, Y. F. Zhang, Z. L. Li, T. Y. Zhan, J. J. Chen, F. L. Wang, N. Y. Yang, L. Ye, Q. S. Gu and X. Y. Liu, J. Am. Chem. Soc., 2020, 142, 9501–9509 Search PubMed
- For selected examples of Togni reagents:
(a) F. Wang, D. Wang, X. Wan, L. Wu, P. Chen and G. Liu, J. Am. Chem. Soc., 2016, 138, 15547–15550 Search PubMed
- S. Sakurai, A. Matsumoto, T. Kano and K. Maruoka, J. Am. Chem. Soc., 2020, 142, 19017–19022 Search PubMed
- W. Sha, L. Deng, S. Ni, H. Mei, J. Han and Y. Pan, ACS Catal., 2018, 8, 7489–7494 Search PubMed
- Z. H. Zhang, H. Wei, Z. L. Li and X. Y. Liu, Synlett, 2021, 32, 362–369 Search PubMed
- M. Eckhardt and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 13642–13643 Search PubMed
- A. Sagadevan and K. C. Hwang, Adv. Synth. Catal., 2012, 354, 3421–3427 Search PubMed
-
(a) X. Y. Dong, Z. L. Li, Q. S. Gu and X. Y. Liu, J. Am. Chem. Soc., 2022, 144, 17319–17329 Search PubMed
- X. Y. Dong, Y. F. Zhang, C. L. Ma, Q. S. Gu, F. L. Wang, Z. L. Li, S. P. Jiang and X. Y. Liu, Nat. Chem., 2019, 11, 1158–1166 Search PubMed
-
(a) W. Yin and X. Wang, New J. Chem., 2019, 43, 3254–3264 Search PubMed
- J. Boivin, E. Fouquet and S. Z. Zard, J. Am. Chem. Soc., 1991, 113, 1055–1057 Search PubMed
- For selected examples of cycloketone oxime esters:
(a) X. Y. Yu, Q. Q. Zhao, J. Chen, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 15505–15509 Search PubMed
-
(a) C. Sandoval-Pauker, G. Molina-Aguirre and B. Pinter, Polyhedron, 2021, 199, 115105 Search PubMed
- P. Z. Wang, Z. Zhang, M. Jiang, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2024, 63, e202411469 Search PubMed
- J. K. McCusker, Science, 2019, 363, 484–488 Search PubMed
- G. Zhang, H. Yi, G. Zhang, Y. Deng, R. Bai, H. Zhang, J. T. Miller, A. J. Kropf, E. E. Bunel and A. Lei, J. Am. Chem. Soc., 2014, 136, 924–926 Search PubMed
- H. D. Zuo, S. S. Zhu, W. J. Hao, S. C. Wang, S. J. Tu and B. Jiang, ACS Catal., 2021, 11, 6010–6019 Search PubMed
-
(a) A. Hossain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, eaav9713 Search PubMed
- B. Dicke, A. Hoffmann, J. Stanek, M. S. Rampp, B. Grimm-Lebsanft, F. Biebl, D. Rukser, B. Maerz, D. Göries, M. Naumova, M. Biednov, G. Neuber, A. Wetzel, S. M. Hofmann, P. Roedig, A. Meents, J. Bielecki, J. Andreasson, K. R. Beyerlein, H. N. Chapman, C. Bressler, W. Zinth, M. Rübhausen and S. Herres-Pawlis, Nat. Chem., 2018, 10, 355–362 Search PubMed
- For reviews on guanidine catalysts:
(a) W. D. Cao, X. H. Liu and X. M. Feng, Chin. Chem. Lett., 2018, 29, 1201–1208 Search PubMed
- For selected examples of chiral guanidine–metal complex catalysis:
(a) Y. Tang, Q. G. Chen, X. H. Liu, G. Wang, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2015, 54, 9512–9516 Search PubMed
- J. Z. Li, L. C. Ning, Q. F. Tan, X. M. Feng and X. H. Liu, Org. Chem. Front., 2022, 9, 6312–6318 Search PubMed
- CCDC 2331715 for F contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- A. Sagadevan, A. Ragupathi and K. C. Hwang, Angew. Chem., Int. Ed., 2015, 54, 13896–13901 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR, HPLC and UPC2 spectra. X-ray crystallographic data for F. CCDC 2331715. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05642f |
| This journal is © The Royal Society of Chemistry 2024 |