
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical reconstruction of metal–organic gels into crystalline oxy-hydroxide heterostructures for efficient oxygen evolution electrocatalysis†
Kang
Liu
 a,
Haikuo
Lan
a,
Yuting
Chen
a,
Weicheng
Tang
a,
Zhenyu
Xiao
a,
Yunmei
Du
b,
Jun
Xing
a,
Zexing
Wu
a and
Lei
Wang
*a
a,
Haikuo
Lan
a,
Yuting
Chen
a,
Weicheng
Tang
a,
Zhenyu
Xiao
a,
Yunmei
Du
b,
Jun
Xing
a,
Zexing
Wu
a and
Lei
Wang
*a
aKey Laboratory of Eco-chemical Engineering, International Science and Technology Cooperation Base of Eco-chemical Engineering and Green Manufacturing, College of Chemistry and Molecular Engineering, School of Materials Science and Engineering, Qingdao University of Science and Technology, China. E-mail: inorchemwl@126.com
bCollege of Environment and Safety Engineering, Qingdao University of Science and Technology, China
First published on 31st October 2024
Abstract
Metal–organic gels (MOGs) are emerging soft materials with distinct metal active centers, multifunctional ligands and hierarchical porous structures, showing promising potential in the field of electrocatalysis. However, the reconfiguration of MOGs during the electrocatalytic process remains underexplored, with current studies in early developmental stages. To deeply investigate the application of MOG materials in electrocatalysis, the compositional transformations and structural changes under an electrochemical activation method were studied in detail, leading to high-performance OER pre-electrocatalysts. XRD and HRTEM results demonstrate the complete reconfiguration of amorphous Fe5Ni5-MOG into crystalline NiOOH/FeOOH heterostructures. The synergistic effect of the bimetallic center and the rich NiOOH–FeOOH interface in the reconstituted Re-Fe5Ni5-MOG exhibit excellent OER activity in alkaline electrolytes, with low overpotentials (205 mV at 10 mA cm−2) and a Tafel slope of 58 mV dec−1. In situ characterization during the electrocatalytic process reveals the gradual transformation of the metal center into metal hydroxyl oxides upon increasing the voltage to 1.55 V. DFT analysis suggests that in the Fe–Ni double site reaction pathway, active substances preferentially adsorb on the Fe site before the Ni sites.
Introduction
The global energy crisis has worsened with the excessive use of traditional fossil fuels, causing severe environmental harm and resource depletion. To address this crisis, the development of new energy sources and improved energy efficiency is crucial. In recent years, emerging energy storage and conversion technologies like electrolysis of water for hydrogen production, fuel cells, metal-air batteries, electrochemical nitrogen reduction for ammonia synthesis, and electrocatalytic carbon dioxide reduction have garnered significant attention for their high efficiency and environmentally friendly characteristics.1–5 Electrochemical energy technologies, particularly hydrogen production through water electrolysis under alkaline conditions, show promise in renewable energy conversion due to their simplicity, efficiency and environmental benefits.6–11 The oxygen evolution reaction (OER) in the overall water-splitting process is the limiting step because of its slow kinetics and need to transfer four electrons/protons. While noble metal Ru-based and Ir-based catalysts are currently the most effective for the OER, their scarcity and cost hinder widespread use.12–15 Therefore, the development of non-noble metal catalysts with superior electrocatalytic performance is essential. Researchers have been actively exploring alternative high-performance OER catalysts and pre-catalysts, such as layered double hydroxides (LDHs), metal oxides, phosphides, and nitrides.16–19In recent years, metal–organic frameworks (MOFs) have shown promise as electrocatalysts due to their tunable structure and high specific surface area. Following the OER in an alkaline environment, MOF materials undergo structural and compositional changes, resulting in the formation of active hydroxyl oxides or hydroxides that serve as the actual catalytic agents.20–26 Mousazade et al. demonstrated that Ni/Co MOF (MOF 1) serves as a precursor and undergoes transformation into Ni/Co oxides, which act as the active catalysts for the OER.27 In a study by Yang et al., the morphology and electronic structure of the MOF-derived bimetal CoFe0.13-MOF were controlled through various reconstruction processes, resulting in a catalyst with exceptional stability.28 Metal–organic gels (MOGs), a subset of MOF materials, are coordination-driven polymers that form gel networks by coordinating metal ions and ligands.29–31 Luo et al. introduced a straightforward and efficient electrochemical reconstruction method for synthesizing NiOOH/FeOOH heterostructures from MOG materials, which displayed remarkable oxygen evolution reactivity.32 Nevertheless, the detailed characterization of the reconstruction process of MOG materials by electrocatalysis and the exploration of how these reconstructed heterostructures catalyze electrochemical processes have not been thoroughly investigated.
In order to develop highly effective MOG-based OER pre-catalysts with superior activity and stability, a series of bimetallic MOG materials were designed and synthesized using a straightforward one-pot method. These MOG pre-catalysts and the in situ electrochemically reconstructed materials were then characterized, and their OER catalytic performance was evaluated and compared. The experimental findings revealed that the fully reconstituted Re-Fe5Ni5-MOG exhibited exceptional OER performance, achieving an overpotential as low as 205 mV at a current density of 10 mA cm−2. Furthermore, a comprehensive analysis of the transformation of MOG during electrocatalysis was conducted using in situ Raman spectroscopy and in situ impedance testing methods. It was observed that the metal ions began to convert into metal hydroxyl oxides when the applied voltage reached 1.55 V. Additionally, DFT results indicated that OH* and O* species had a preference for adsorption on the Fe site before transitioning to the Ni site along the reaction pathway of the Fe–Ni double site. This approach offers a straightforward means of obtaining efficient OER pre-catalysts and broadens the utilization of MOGs as an electrocatalyst in the realm of electrocatalysis.
Results and discussion
Synthesis and characterization of MOGs
Here we take Fe5Ni5-MOG as an example. The process of sample preparation is shown in Fig. 1a, which involved dissolving the ligand (H5L) in distilled water to create a 20 mM solution, with triethylamine added as a deprotonating agent. FeCl3·6H2O and NiCl2·6H2O were separately dissolved in distilled water to prepare 20 mM solutions of Fe3+ and Ni2+ metal ions. Next, 400 μL of Fe3+, 400 μL of Ni2+ solution and 800 μL of ligand solution were added dropwise into a 2 mL centrifuge tube, and the mixture became condensed after approximately 2 h of standing. The target product (Fe5Ni5-MOG) was obtained after freeze-drying (24 h), washing, and drying steps (Fig. 1a). | ||
| Fig. 1 (a) Schematic illustration of the fabrication process of a FeNi metal–organic gel (FeNi-MOG). (b–d) SEM images at varying magnifications of Fe5Ni5-MOG. (e–g) TEM images at different magnifications of Fe5Ni5-MOG. (h) HAADF-STEM image and corresponding elemental mapping images of Fe5Ni5-MOG. | ||
Three transition metals (Co, Fe and Ni) were combined in pairs to create a variety of MOGs with different metal ratios. Images of the original MOGs and the powders post freeze-drying were captured (Fig. S1 and S2†). Among the MOGs containing various ratios of the three metals, the mono-metallic Co-MOG exhibited the quickest gelation rate and most effective gelation. In addition, the carboxyl and imido groups in the ligands exhibited different coordination patterns and promoted hydrogen bonding in the MOG formation process.33
To investigate the crystallinity of the MOGs, X-ray diffraction (XRD) analyses were performed on Fe-MOG, Ni-MOG and Fe5Ni5-MOG. The XRD spectrum (Fig. S3†) displayed a single wide-angle diffraction peak at 2θ = 26.7°, indicating that the synthesized MOGs exhibit very low crystallinity and are amorphous, consistent with the previously reported gel.34,35 Subsequently, an in situ Raman test was conducted (Fig. S4†), clearly showing M–O vibration (∼800 cm−1) and the C–C stretching pattern (∼1000 cm−1) in the MOG samples. Both XRD and in situ Raman analyses of the three samples suggested similarity in the synthesized MOGs. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) measurements of Fe5Ni5-MOG revealed a Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni mass ratio close to 1:1, aligning with the reaction dosage (Table S1†).
:Ni mass ratio close to 1:1, aligning with the reaction dosage (Table S1†).
During the thermogravimetric analysis (TGA) of Fe5Ni5-MOG (Fig. S5†), two rapid mass decreases were observed. The first stage (25–325 °C) is attributed to the evaporation of solvent water in the sample, while the second stage (325–900 °C) is linked to the decomposition of organic ligands within the sample. The percentage of ligands was estimated to be approximately 65%. Subsequently, the weight stabilizes at around 900 °C, with the remaining weight corresponding to the formation of metal oxides [calculated: 8.8%; actual: 8.4%].
The N2 adsorption curve (Fig. S6†) reveals that the Brunauer–Emmett–Teller (BET) surface area of Fe5Ni5-MOG is as high as 139.11 m2 g−1, surpassing the BET values of many previously reported efficient transition metal-based electrocatalysts (Table S2†). This suggests that Fe5Ni5-MOG facilitates effective contact between the electrolyte and catalytically active sites during the reaction process, ultimately enhancing the catalyst's performance. Notably, N2 adsorption occurs rapidly at P/P0 < 0.01; the isotherm of adsorption–desorption has a lag period at 0.01 < P/P0 < 0.90; and N2 adsorption is sharply increased when P/P0 > 0.90. This phenomenon proves that there are hierarchical pore structures in MOG (Fig. S7†).
The catalysts were morphologically characterized using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). SEM images revealed a nanofibrous structure in freeze-dried gels (Fig. 1b–d and S8†), which not only improved electrical conductivity but also facilitated mass transfer between reactants and catalyst active sites, consistent with the large BET surface area mentioned before. TEM images also confirmed this nanofibrous structure (Fig. 1e–g and S9†), aiding in electric charge transfer and enhancing electrical conductivity. Selected Area Electron Diffraction (SAED) did not exhibit a clear diffraction pattern (Fig. 1e inset), indicating poor crystallinity of Fe5Ni5-MOG as supported by the XRD results. Energy dispersive spectroscopy (EDS) element mapping confirmed a highly uniform distribution of the C, N, O, Fe and Ni elements in the material (Fig. 1h and S10†).
To investigate the self-reconstruction of MOG through electrochemical reactions in an alkaline environment, a series of characterization studies were conducted. Initially, the electrochemical reconstruction process of Re-Fe5Ni5-MOG formation is illustrated in Fig. 2a (200 CV scans in the voltage range of 0–0.6 V). XRD results indicated that Re-Fe5Ni5-MOG consists of FeOOH and NiOOH phases (Fig. 3a), and this means electrochemical reconstruction of Fe5Ni5-MOG into crystalline oxy-hydroxide heterostructures. Subsequently, to elucidate the chemical structure of Re-Fe5Ni5-MOG further, SEM and TEM were employed for detailed morphology and structural characterization (Fig. 2b and c). Compared to the previous MOG structure, the reconstructed material now exhibits a significant shift from a nanofiber network structure to a nanosheet structure. The high-resolution transmission electron microscopy (HRTEM) images of the nanofibers displayed two distinct lattice streaks with lattice spacings of 0.023 and 0.027 nm, corresponding to the (060) crystallographic plane of FeOOH and the (104) crystallographic plane of NiOOH, respectively. Additionally, a distinct heterogeneous interface can be observed between the two lattice stripes (white lines in Fig. 2d). Fourier transform infrared spectroscopy (FT-IR) tests were conducted to investigate the mechanism of MOG formation (Fig. S11†). Absorption bands at 1724, 1251, and 991 cm−1 were associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, and O–H vibrations in the H5L ligand, respectively. The CO vibration peak shifted to 1706 cm−1 upon metal ion coordination with the ligand.36,37 Additionally, the stretching vibration of the C–O bond and the plane bending vibration of the O–H bond disappeared.38 After electrochemical reconstruction, characteristic peaks were not observed in Re-Fe5Ni5-MOG.
O, C–O, and O–H vibrations in the H5L ligand, respectively. The CO vibration peak shifted to 1706 cm−1 upon metal ion coordination with the ligand.36,37 Additionally, the stretching vibration of the C–O bond and the plane bending vibration of the O–H bond disappeared.38 After electrochemical reconstruction, characteristic peaks were not observed in Re-Fe5Ni5-MOG.
 | ||
| Fig. 2 (a) Sketch of the electrochemical in situ reconstruction of Fe5Ni5-MOG. (b) SEM image, (c) TEM images, and (d) HRTEM images of Re-Fe5Ni5-MOG. | ||
 | ||
| Fig. 3 (a) XRD patterns of Re-Fe5Ni5-MOG. (b) In situ Raman spectroscopy and (c) corresponding contour of the Fe5Ni5-MOG pre-electrocatalyst at various potentials. (d) The XPS full survey spectra of Fe5Ni5-MOG and Re-Fe5Ni5-MOG. The high-resolution XPS spectra of (e) Fe 2p and (f) Ni 2p of Fe5Ni5-MOG and Re-Fe5Ni5-MOG. Nyquist plots for (g) Fe5Ni5-MOG, (h) Ni-MOG and (i) Fe-MOG at different applied potentials. | ||
In order to investigate the surface atomic structure and elemental composition of the prepared pre-catalysts, and compare the changes before and after reconstruction, X-ray photoelectron spectroscopy (XPS) tests were conducted, with all XPS spectra calibrated using C 1s at 284.8 eV. The XPS results revealed the presence of C, N, O, Fe, and Ni elements in the Fe5Ni5-MOG spectra. However, the N element in Re-Fe5Ni5-MOG disappeared after reconstruction, in line with expectations (Fig. 3d). The C 1s spectra of Fe5Ni5-MOG displayed four peaks at 284.6, 285.9, 288.3, and 290.5 eV, corresponding to C–C, C–N/CN, C–O, and CO groups, respectively (Fig. S12a†).39,40 The N 1s spectra of Fe5Ni5-MOG exhibited peaks at 398.6, 400, 402 and 406.1 eV (Fig. S12b†), representing pyridine nitrogen, pyrrole nitrogen, graphitic nitrogen and oxidized nitrogen, respectively.41,42 The contents of pyridine nitrogen and pyrrole nitrogen were 35.80% and 51.97%, respectively, which were much higher than those of the other two kinds of nitrogen. The O 1s spectra of Fe5Ni5-MOG can be deconvoluted into two characteristic peaks at 831.5 and 532.6 eV, attributed to Fe/Ni–O and CO (Fig. S12c†).43,44 In the high-resolution XPS spectrum of Fe (Fig. 3e), Fe5Ni5-MOG exhibits two main peaks at 711.6 and 724.9 eV corresponding to Fe 2p1/2 and Fe 2p3/2, along with two satellite peaks at 724.9 and 731.5 eV, indicating the presence of Fe3+ in the compound. The Fe3+ peak in Re-Fe5Ni5-MOG shows a slight negative shift (0.6 eV) compared to Fe5Ni5-MOG, suggesting a strong electronic interaction between Fe and Ni.45 The XPS spectrum of Ni from Fe5Ni5-MOG shows main peaks at 856.0 and 873.7 eV and satellite peaks at 861.0 and 879.4 eV, indicating the valence state of Ni2+.46 In Re-Fe5Ni5-MOG, the Ni peak has a positive shift (1.56 eV) compared to Fe5Ni5-MOG, indicating that Ni2+ is oxidized to form NiOOH (Fig. 3f).47
In order to further clarify the structural evolution of the pre-catalyst Fe5Ni5-MOG, we performed in situ Raman spectroscopy and obtained the Raman spectra while varying the applied potential at 0.05 V intervals (Fig. 3b and c). The results show that Fe5Ni5-MOG exhibits no significant Raman signals when the applied potential is below 1.50 V. Upon increasing the potential to 1.55 V, distinctive NiOOH characteristic peaks appeared at 472 cm−1 and 543 cm−1, indicating the oxidation of Ni2+ to Ni3+.48,49 Subsequent potential increments did not alter the peak position. It is commonly accepted that highly oxidized nickel species are present in NiOOH, exhibiting a more disordered structure that can enhance OER activity.50,51
Electrochemical impedance spectroscopy (EIS) is a valuable method for examining interface properties of the electrode/electrolyte and adsorption kinetics of reactants on the electrode surface. To delve deeper into electrochemical reaction kinetics, operando EIS tests were conducted. Fig. 3g–i display Nyquist plots of Fe5Ni5-MOG, Fe-MOG and Ni-MOG in 1 M KOH with applied voltages ranging from 0.18 V to 0.53 V. The total resistance of Fe5Ni5-MOG, Fe-MOG and Ni-MOG at various applied potentials was quantified based on Nyquist plots. Fe5Ni5-MOG showed a low charge transfer resistance in the tested potential range due to the synergistic effect of multiple metal oxides, enhancing interfacial charge transfer and promoting surface activation of electrocatalysts.52–54 At 1.55 V, Fe5Ni5-MOG exhibited significantly lower resistance compared to Fe-MOG and Ni-MOG, indicating faster OH* adsorption kinetics. Nyquist plots of Fe-MOG and Ni-MOG displayed nearly vertical lines at 1.25–1.40 V, suggesting infinite resistance (R). However, a distinct semicircle emerged when the applied voltage exceeded 1.55 V, indicating the occurrence of the electrocatalytic OER. Overall, Fe5Ni5-MOG demonstrated faster surface and inner layer charge transfer rates, leading to superior electrochemical performance compared to Fe-MOG and Ni-MOG. This improvement can be attributed to the synergistic effect between Fe3+ and Ni2+.55
Electrochemical performance
The electrochemical performance of MOG with varying Fe–Ni–Co ratios was evaluated in this study. Among the CoFe bimetallic gels, Co5Fe5-MOG exhibited the highest performance (336 mV (j10)); within the CoNi bimetallic gels, Co1Ni9-MOG showed the best performance (330 mV (j10)), while among the FeNi bimetallic gels, Fe5Ni5-MOG demonstrated the best OER performance (320 mV (j10)) (Fig. S19†); Consequently, Fe5Ni5-MOG was chosen as the primary sample for further investigation. The OER performance of RuO2, Ni-MOG, Fe-MOG, Fe5Ni5-MOG and Re-Fe5Ni5-MOG as electrocatalysts in a 1.0 M KOH alkaline electrolyte was investigated. All electrochemical tests were performed in a 1.0 M KOH solution using a standard three-electrode system, with potential values calibrated against a typical reversible hydrogen electrode (RHE).
Fig. 4a displays the linear scanning voltammetry (LSV) curves of Re-Fe5Ni5-MOG, Fe5Ni5-MOG, Fe-MOG, Ni-MOG and RuO2. The overpotential of Re-Fe5Ni5-MOG is notably lower at 205 mV compared to Fe5Ni5-MOG (320 mV), Fe-MOG (506 mV) and Ni-MOG (490 mV), and even the commercial noble metal RuO2 (450 mV) when operating at a current density of 10 mA cm−2. Furthermore, Re-Fe5Ni5-MOG exhibits superior OER activity compared to most reported transition metal-based catalysts (Fig. S20†). The lower Tafel slope of Re-Fe5Ni5-MOG (58 mV dec−1) indicates faster surface kinetics among all samples (Fig. 4b). EIS was used to further explore the resistance (R) used to assess the charge transfer capability of interfacial properties, and the tested data were fitted using the Randles equivalent circuit model, which consists of the solution resistance (Rs), constant phase element (CPE) and charge transfer resistance (Rct) (Fig. 4c inset). The Nyquist plots indicate that the Rct value of Re-Fe5Ni5-MOG is only 31.1 Ω, which is much lower than that of other samples (Fig. 4c), suggesting that Re-Fe5Ni5-MOG has a faster electron transfer rate and better conductivity, which is conducive to interfacial reactions. Additionally, cyclic voltammetry (CV) curves at different scanning rates (2–10 mV s−1) were obtained for all samples (Fig. S21†), and the corresponding double layer capacitance (Cdl) was calculated to evaluate the intrinsic electrochemically active surface area (ECSA) of the catalysts (Fig. 4d). Among them, the Cdl value of the Re-Fe5Ni5-MOG sample was 7.5 mF cm−2, which was higher than that of Fe-MOG (3.7 mF cm−2), Ni-MOG (3.3 mF cm−2), and Fe5Ni5-MOG (5.3 mF cm−2), highlighting the superior electron storage capacity of Re-Fe5Ni5-MOG as an electrode material. Moreover, the specific activity (ECSA normalized current density) analysis revealed that the intrinsic activity of the Re-Fe5Ni5-MOG catalyst outperformed other samples under similar conditions (Fig. 4e). Electrochemical stability was assessed using the chrono current method, showing only a 7.8% decrease in current density after 240000 s OER at 320 mV overpotential (j10) (Fig. 4f), indicating good stability of Re-Fe5Ni5-MOG possibly attributed to its unique porous nanofiber network structure and synergistic effects of Fe and Ni. The slight decline in current could be attributed to the improper drip method leading to sample detachment from the substrate, especially under high current conditions generating bubbles. It was postulated that Fe and Ni are active sites for the OER. The turnover frequency (TOF) value for Fe5Ni5-MOG in the OER was determined to be 3.84 s−1 at 300 mV overpotential based on ICP data, showcasing the superior performance of this pre-electrocatalyst compared to most of the reported catalysts (Table S3†).
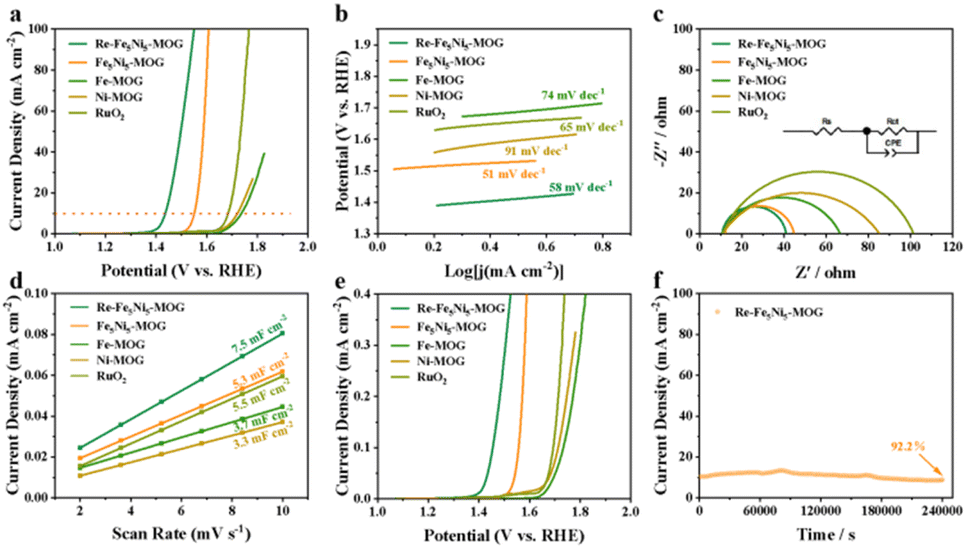 | ||
| Fig. 4 (a) LSV curves, (b) Tafel plots, (c) EIS Nyquist plots, (d) capacitive current densities plotted against scan rates and (e) LSV curves normalized by ECSA for Re-Fe5Ni5-MOG, Fe5Ni5-MOG, Fe-MOG, Ni-MOG and RuO2. (f) The chronoamperometry test of Re-Fe5Ni5-MOG. | ||
The electrochemical properties of Re-Fe5Ni5-MOG, FeNi-MOG, Fe-MOG, Ni-MOG, and RuO2 were comprehensively compared. From the radar plots, it is evident that Re-Fe5Ni5-MOG exhibits significant advantages in terms of potential and ECSA when compared to Fe5Ni5-MOG (Fig. 5a). To further facilitate a comparative analysis of the overpotential and Tafel slope, a bar chart was created, revealing that Re-Fe5Ni5-MOG demonstrates the lowest potential and a lower Tafel slope. These results suggest that Re-Fe5Ni5-MOG exhibits superior electrocatalytic performance (Fig. 5b). An electrolytic cell was constructed to assess the water splitting performance. Three sets of pairings were tested, and Re-Fe5Ni5-MOG ‖ Pt/C required only 1.49 V to achieve a current density of 10 mA cm−2, outperforming RuO2 ‖ Pt/C (1.60 V) and NF ‖ NF (1.79 V) (Fig. 5c). The nickel foam in the NF ‖ NF cell seemed to serve solely as a self-supporting substrate to increase specific surface area, with no significant impact on catalyst performance. In addition, we evaluated the stability of overall water splitting by the chrono current method, which showed only a slight decay (12.5%) after 240000 s (Fig. 5d). Faraday efficiency was evaluated by comparing actual H2 and O2 gas production during water splitting with theoretical values based on charge passage (Fig. S22†). The volume ratio of collected H2 to O2 was 2.02:1, close to the theoretical ratio of 2:1, and the calculated Faraday efficiency approached 100% (Fig. S23†).
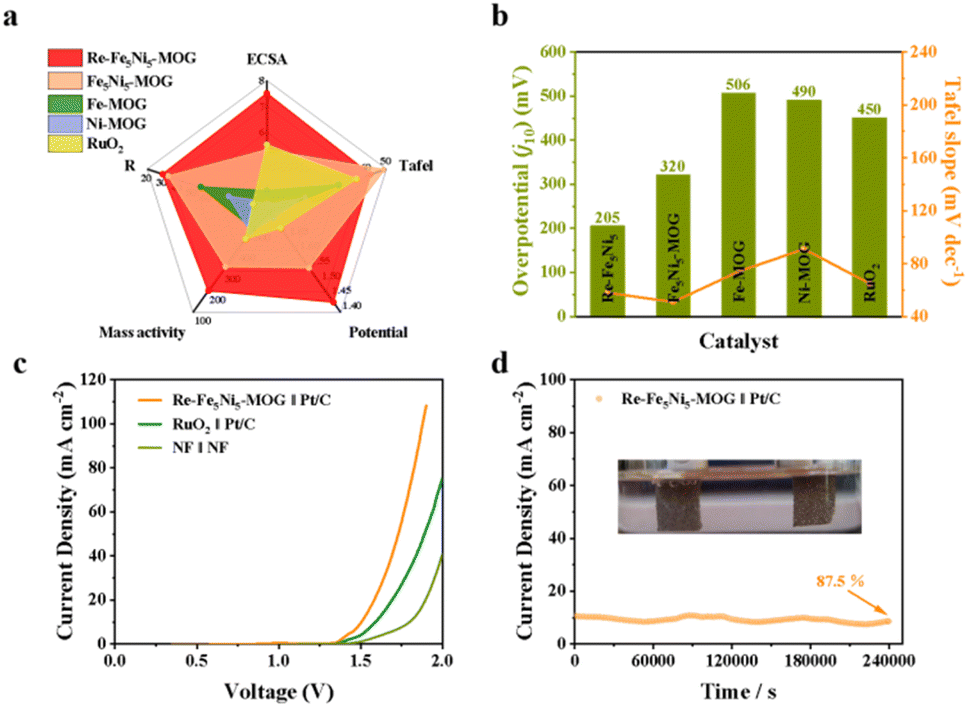 | ||
| Fig. 5 (a) Comparison of the catalytic performances of catalysts. (b) Comparison of the overpotential and Tafel slope of electrocatalysts. (c) Overall water splitting electrocatalysis by NF ‖ NF, Re-Fe5Ni5-MOG ‖ Pt/C and the benchmark RuO2 ‖ Pt/C. (d) The chronoamperometry test of overall water splitting electrocatalysis (Re-Fe5Ni5-MOG ‖ Pt/C). | ||
Theoretical calculation
To investigate the rationale behind the increased OER activity and identify potential active sites of the NiOOH/FeOOH heterogeneous structure during the OER process, density functional theory (DFT) calculations were performed at the interface. Models of NiOOH and NiOOH/FeOOH were developed to determine the Gibbs free energy at each step of the OER process (Fig. S24† and 6a). | ||
| Fig. 6 (a) Atomic model of NiOOH/FeOOH. (b) The calculated DOS of NiOOH and NiOOH/FeOOH. (c) OER steps on the Fe–Ni dual site 1 of NiOOH/FeOOH (brown balls: Fe atoms; silvery balls: Ni atoms; red balls: O atoms; white balls: H atoms). (d) The calculated Gibbs free energy diagrams. | ||
The total density of states (TDOS) of NiOOH/FeOOH, positioned closer to the Fermi energy level (Ef), is slightly higher than that of NiOOH. This suggests that the inclusion of Fe can modulate the distribution of electron density of states on the surface of materials, thereby enhancing its intrinsic conductivity (Fig. 6b).56 Meanwhile, the energy levels of the Ni-3d orbitals increase towards Ef, as seen in the partial density of states (PDOS) (Fig. S25†). The results of TDOS and PDOS indicate that the incorporation of FeOOH can greatly optimize the electronic structure of NiOOH, resulting in higher conductivity in the heterogeneous NiOOH/FeOOH system.
The study focused on the Ni site of NiOOH and the Fe–Ni dual site of NiOOH/FeOOH in the OER process. It is important to highlight that the reaction mechanisms differ between the single site and the double site. In the case of single sites, the Gibbs free energy is influenced by the adsorption energies of OH*, O*, and OOH* intermediates (Fig. S26†), with the Ni site in NiOOH representing the single site (Fig. S27†).
For the Fe–Ni dual site, the intermediates in the second step are different. We have categorized the OER process of the Fe–Ni dual site into four pathways, as illustrated in Fig. S28.† Pathways 2 and 4 are not feasible due to the instability of the Fe–OH* + Ni–O* structure, leading us to focus solely on pathways 1 and 3 in our calculation for the dual site. These pathways are denoted as Fe–Ni dual site 1 and Fe–Ni dual site 2, respectively. Taking Fe–Ni dual site 1 as an example, the adsorbed intermediates during the OER step are detailed in Fig. 6c. Initially, OH* is adsorbed on the Fe site at the interface, followed by the adsorption of another OH* on the Ni site to form Fe–OH* + Ni–OH*. Subsequently, the proton H is removed from Ni–OH* to yield O2. On the other hand, for Fe–Ni dual site 2, the OH* intermediates first adsorb on the Ni site, as depicted in Fig. S29.† The change in maximum free energy is considered the rate-determining step (RDS) in the OER process, prompting us to calculate the free energy of these three reaction pathways, as shown in Fig. 6d and Table S4.† Our analysis reveals that △G3 serves as the RDS for both the Ni site of NiOOH and the Fe–Ni dual site of NiOOH/FeOOH. Furthermore, the theoretical overpotential of the Ni site of NiOOH is calculated to be 0.58 V, while both pathways of the NiOOH/FeOOH dual site exhibit a theoretical overpotential of 0.31 V. Notably, during the OER, FeOOH/NiOOH displays a preference for pathway 1, with OH* and O* species showing a tendency to adsorb initially on the Fe site before the Ni site.
The enhanced OER activity of the NiOOH/FeOOH heterostructure can be attributed to two main factors: (1) the nanofibrous structure of the freeze-dried gel, which promotes mass transfer and improves the interaction between the electrolyte and electrocatalyst; and (2) the strong electronic coupling effect between NiOOH and FeOOH, which facilitates electron and charge transport.
Conclusions
In summary, an electrochemical in situ strategy is proposed for synthesizing NiOOH/FeOOH heterostructure materials using Fe5Ni5-MOG pre-catalysts. During the OER process, irreversible deep self-reconstruction of Fe5Ni5-MOG leads to the formation of NiOOH/FeOOH. The nanosheet structures and the synergic coupling between Fe and Ni hydroxyl oxide particles, generated by in situ electrochemical reconstruction, contribute to the superior OER catalytic performance of Re-Fe5Ni5-MOG (205 mV (j10)), which surpasses that of commercial Ru-based catalysts. Detailed characterization of the transformation of MOGs during the electrocatalytic process was conducted using in situ Raman spectroscopy and in situ impedance testing. DFT results indicate that in the Fe–Ni dual site reaction pathway, OH* and O* species preferentially adsorb on Fe sites before Ni sites. This strategy is believed to enhance the design of high-performance OER electrocatalysts, offering a promising pathway for developing durable electrochemical catalysts. It also expands the research scope of MOG-derived materials, contributing to the exploration of more efficient electrocatalysts.Experimental section
The synthesis of the MOGs was borrowed from previously reported literature. Herein, we take Fe5Ni5-MOG as an example. FeCl3·6H2O and NiCl2·6H2O were dissolved in distilled water to prepare 20 mM solutions of Fe3+ and Ni2+ metal ion solutions, respectively. The synthesized ligands were also prepared as 20 mM concentrated solutions, in which 20 μL of triethylamine (triethylamine acts as a deprotonating hydrolyser and coagulant) was added to each milliliter of solution. Then, 400 μL of Fe3+, 400 μL of Ni2+, and 800 μL of the solution of the ligand are added dropwise to a small 5 mL centrifuge tube, and the mixture takes approximately 2 h to coagulate in air. Then, the target product (Fe5Ni5-MOG) was obtained after three steps of freeze-drying (24 h), washing with distilled water and drying.Data availability
All the data supporting the key findings in the paper have been included as part of the ESI.†Author contributions
Kang Liu: writing-review & editing, data curation, formal analysis, validation, supervision, funding acquisition. Haikuo Lan: conceptualization, data curation, formal analysis, validation, writing-original draft. Yuting Chen: formal analysis, validation, discussion. Weicheng Tang: validation, discussion. Yunmei Du: writing-review & editing. Zhenyu Xiao: writing-review & editing. Jun Xing: writing-review & editing. Zexing Wu: writing-review & editing. Lei Wang: writing-review & editing, funding acquisition, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Shandong Province, China (No. ZR2022MB022), the National Natural Science Foundation of China (52072197 and 52272222), the Youth Innovation and Technology Foundation of Shandong Higher Education Institutions, China (2019KJC004), the Outstanding Youth Foundation of Shandong Province, China (ZR2019JQ14), the Taishan Scholar Young Talent Program (tsqn201909114), the Major Scientific and Technological Innovation Project (2019JZZY020405), and the Major Basic Research Program of the Natural Science Foundation of Shandong Province (ZR2020ZD09).References
- Y. X. Wang, X. Z. Cui, J. Q. Zhang, J. L. Qiao, H. T. Huang, J. L. Shi and G. X. Guo, Prog. Mater. Sci., 2022, 128, 100964 CrossRef CAS.
- J. Wang, N. Zang, C. J. Xuan, B. H. Jia, W. Jin and T. Y. Ma, Adv. Funct. Mater., 2021, 31, 2104620 CrossRef CAS.
- X. B. Liao, R. H. Lu, L. X. Xia, Q. Liu, H. Wang, K. Zhao, Z. Y. Wang and Y. Zhao, Energy Environ. Mater., 2022, 5, 157–185 CrossRef CAS.
- X. Cui, M. J. Wu, X. Q. Liu, B. He, Y. H. Zhu, Y. L. Jiang and Y. K. Yang, Chem. Soc. Rev., 2024, 53, 1447–1494 RSC.
- H. Zhang, Z. H. Zhou, Y. N. Yin, H. Xu, Y. M. Wang, K. Yang, Z. J. Zhang, J. L. Wang and X. M. He, EcoEnergy, 2023, 1, 217–247 CrossRef.
- S. B. Chen, Y. L. Zhuo, X. Wang, S. P. Li, J. X. Lu, D. Liu, H. Pan and Z. B. Wang, Coord. Chem. Rev., 2024, 510, 215832 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- M. A. Khan, T. Al-Attas, S. Roy, M. M. Rahman, N. Ghaffour, V. Thangadurai, S. Larter, J. Hu, P. M. Ajayan and M. G. Kibria, Energy Environ. Sci., 2021, 14, 4831–4839 RSC.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef PubMed.
- Q. He, H. Sun, W. T. Bi, X. Y. Wang, B. Li, F. Li, Z. G. Guo, J. Ding and J. B. He, Chem. Eng. J., 2024, 495, 153461 CrossRef CAS.
- L. N. Chong, J. G. Wen, E. H. Song, Z. Z. Yang, I. D. Bloom and W. J. Ding, Adv. Energy Mater., 2023, 13, 2302306 CrossRef CAS.
- J. He, X. Zhou, P. Xu and J. M. Sun, Adv. Energy Mater., 2021, 11, 2102883 CrossRef CAS.
- M. S. A. S. Shah, G. Y. Jang, K. Zhang and J. H. Park, EcoEnergy, 2023, 1, 344–374 CrossRef.
- Y. L. Zhu, W. Zhou, Y. J. Zhong, Y. F. Bu, X. Y. Chen, Q. Zhong, M. L. Liu and Z. P. Shao, Adv. Energy Mater., 2017, 7, 1602122 CrossRef.
- A. Hameed, M. Batool, Z. Y. Liu, M. A. Nadeem and R. C. Jin, ACS Energy Lett., 2022, 7, 3311–3328 CrossRef CAS.
- R. P. Luo, Z. Y. Qian, L. X. Xing, C. Y. Du, G. P. Yin, S. L. Zhao and L. Du, Adv. Funct. Mater., 2021, 31, 2102918 CrossRef CAS.
- G. R. Chang, Y. T. Zhou, J. H. Wang, H. Zhang, P. Yan and H. B. Wu, Small, 2023, 19, 2206768 CrossRef CAS PubMed.
- B. Singh, A. Yadav and A. Indra, J. Mater. Chem. A, 2022, 10, 3843–3868 RSC.
- Q. Z. Qian, Y. P. Li, Y. Liu, L. Yu and G. Q. Zhang, Adv. Mater., 2019, 31, 1901139 CrossRef PubMed.
- S. S. Li, Y. Q. Gao, N. Li, L. G, X. H. Bu and P. Y. Feng, Energy Environ. Sci., 2021, 14, 1897–1927 RSC.
- K. Ge, S. J. Sun, Y. Zhao, K. Yang, S. Wang, Z. H. Zhang, J. Y. Cao, Y. F. Yang, Y. Zhang, M. W. Pan and L. Zhu, Angew. Chem., Int. Ed., 2021, 60, 12097–12102 CrossRef CAS PubMed.
- Y. Liu, S. J. Wang, Z. Z. Li, H. Q. Chu and W. Zhou, Coord. Chem. Rev., 2023, 484, 215117 CrossRef CAS.
- Z. H. Zou, T. T. Wang, X. H. Zhao, W. J. Jiang, H. R. Pan, D. Q. Gao and C. L. Xu, ACS Catal., 2019, 8, 7356–7364 CrossRef.
- W. B. Chen, C. S. Wang, S. B. Su, H. Wang and D. D. Cai, Chem. Eng. J., 2021, 414, 128784 CrossRef CAS.
- M. Kuang, J. M. Zhang, D. B. Liu, H. T. Tan, K. N. Dinh, L. Yang, H. Ren, W. J. Huang, W. Feng, J. D. Yao, X. D. Hao, J. W. Xu, C. T. Liu, L. Song, B. Liu and Q. Y. Yan, Adv. Energy Mater., 2020, 10, 2002215 CrossRef CAS.
- Y. Mousazade, M. R. Mohammadi, P. Chernev, R. Bagheri, Z. L. Song, H. Dau and M. M. Najafpour, Inorg. Chem., 2020, 59, 15335–15342 CrossRef CAS PubMed.
- K. X. Yang, Z. Q. Jin, Q. C. Zhang, Q. M. Chen, W. C. Peng, Y. Li, F. B. Zhang, Q. Xia and X. B. Fan, Chem. Commun., 2022, 58, 1115–1118 RSC.
- J. W. Hou, A. F. Sapnik and T. D. Bennett, Chem. Sci., 2020, 11, 310–323 RSC.
- H. Wang, B. H. Chen and D. J. Liu, Adv. Mater., 2021, 33, 2008023 CrossRef CAS PubMed.
- S. J. Liu, M. H. Liu, X. W. Li, S. Yang, Q. Y. Miao, Q. Xu and G. F. Zeng, Carbon Energy, 2023, 5, e303 CrossRef CAS.
- R. P. Luo, Z. Y. Qian, L. X. Xing, C. Y. Du, G. P. Yin, S. L. Zhao and L. Du, Adv. Funct. Mater., 2021, 31, 2102918 CrossRef CAS.
- H. Zhao and Z. Y. Yuan, J. Energy Chem., 2021, 54, 89–104 CrossRef CAS.
- F. Zhao, W. X. Yang, Y. Han, X. L. Luo, W. Z. Tang, T. L. Yue and Z. H. Li, Chem. Eng. J., 2021, 407, 126744 CrossRef CAS.
- H. M. Khater and A. M. El-Nagar, Adv. Compos. Hybrid Mater., 2020, 3, 375–389 CrossRef CAS.
- J. Y. Zhang and C. Y. Su, Coord. Chem. Rev., 2013, 257, 1373–1408 CrossRef CAS.
- S. Hirata, R. Kusaka, S. Meiji, S. Tamekuni, K. Okudera, S. Hamada, C. Sakamoto, T. Honda, K. Matsushita, S. Muramatsu, T. Ebata, D. Kajiya, K. Saitow, T. Ikeda, T. Hirao, T. Haino, M. Watanabe and Y. Inokuchi, Inorg. Chem., 2023, 62, 474–486 CrossRef CAS PubMed.
- Z. W. Jiang, T. T. Zhao, Y. F. Li and C. Z. Huang, Nanoscale Horiz., 2020, 5, 119–123 RSC.
- J. H. Shi, F. Qiu, W. B. Yuan, M. M. Guo and Z. H. Lu, Chem. Eng. J., 2021, 403, 126312 CrossRef CAS.
- Q. Wang, H. F. Qi, Y. J. Ren, K. Junge, R. V. Jagadeesh and M. Beller, Chem, 2024, 10, 1897–1909 CAS.
- T. T. Jiang, Y. Wang, K. Wang, Y. R. Liang, D. C. Wu, P. Tsiakaras and S. Q. Song, Appl. Catal., B, 2016, 189, 1–11 CrossRef CAS.
- X. Chen, W. D. Oh, Z. T. Hu, Y. M. Sun, R. D. Webster, S. Z. Li and T. T. Lim, Appl. Catal., B, 2018, 225, 243–257 CrossRef CAS.
- C. Q. Dong, T. Y. Kou, H. Gao, Z. Q. Peng and Z. H. Zhang, Adv. Energy Mater., 2018, 8, 1701347 CrossRef.
- C. F. Li, L. J. Xie, J. W. Zhao, L. F. Gu, H. B. Tang, L. R. Zheng and G. R. Li, Angew. Chem., Int. Ed., 2022, 61, e202116934 CrossRef CAS PubMed.
- J. Meng, Z. Xu, H. X. Li, D. J. Young, C. J. Hu and Y. G. Yang, ChemCatChem, 2021, 13, 1396–1402 CrossRef CAS.
- S. Q. Niu, X. P. Kong, S. W. Li, Y. Y. Zhang, J. Wu, W. W. Zhao and P. Xu, Appl. Catal., B, 2021, 297, 120442 CrossRef CAS.
- X. Cao, P. Wen, R. Ma, Y. Liu, S. C. Sun, Q. Ma, P. X. Zhang and Y. J. Qiu, Chem. Eng. J., 2022, 449, 137792 CrossRef CAS.
- J. W. Huang, Y. Y. Li, Y. D. Zhang, G. F. Rao, C. Y. Wu, Y. Hu, X. F. Wang, R. F. Lu, Y. R. Li and J. Xiong, Angew. Chem., Int. Ed., 2019, 58, 17458–17464 CrossRef CAS PubMed.
- X. Ding, R. Jiang, J. L. Wu, M. H. Xing, Z. L. Qiao, X. F. Zeng, S. T. Wang and D. P. Cao, Adv. Funct. Mater., 2023, 33, 2306787 Search PubMed.
- H. Y. Zhong, X. P. Wang, G. X. Sun, Y. X. Tang, S. D. Tan, Q. He, J. Zhang, T. Xiong, C. Z. Diao, Z. G. Yu, S. B. Xi, W. S. V. Lee and J. M. Xue, Energy Environ. Sci., 2023, 16, 641–652 RSC.
- Y. J. Zhu, C. Liu, S. W. Cui, Z. R. Lu, J. Y. Ye, Y. Z. Wen, W. J. Shi, X. X. Huang, L. Y. Xue, J. J. Bian, Y. Y. Li, Y. F. Xu and B. Zhang, Adv. Mater., 2023, 35, 2301549 CrossRef CAS PubMed.
- X. R. Zheng, X. P. Han, Y. H. Cao, Y. Zhang, D. Nordlund, J. H. Wang, S. L. Chou, H. Liu, L. L. Li, C. Zhong, Y. D. Deng and W. B. Hu, Adv. Mater., 2020, 32, 2000607 CrossRef CAS PubMed.
- B. You, Y. D. Zhang, Y. Jiao, K. Davey and S. Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 11796–11800 CrossRef CAS PubMed.
- C. G. Liao, B. P. Yang, N. Zhang, M. Liu, G. X. Chen, X. M. Jiang, G. Chen, J. L. Yang, X. H. Liu, T. S. Chan, Y. J. Lu, R. Z. Ma and W. Zhou, Adv. Funct. Mater., 2019, 29, 1904020 CrossRef.
- P. Babar, K. Patil, V. Karade, K. Gour, A. Lokhande, S. Pawar and J. H. Kim, ACS Appl. Mater. Interfaces, 2021, 13, 52620–52628 CrossRef CAS PubMed.
- M. H. Du, J. Q. Yan, V. R. Cooper and M. Eisenbach, Adv. Funct. Mater., 2021, 31, 2006515 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05799f |
| This journal is © The Royal Society of Chemistry 2024 |