
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDriving tert-butyl axial: the surprising cyclopropyl effect†
Anthony R.
Izzotti
and
James L.
Gleason
 *
*
Department of Chemistry, McGill University, 801 SherbrookeW., H3A 0B8, Montreal, QC, Canada. E-mail: jim.gleason@mcgill.ca
First published on 8th October 2024
Abstract
The presence of a small spirocyclic ring at an adjacent position alters the conformational preference for equatorial substitution in six-membered rings. DFT calculations and low-temperature 1H NMR experiments demonstrate that alkyl groups larger than methyl possess negative A-values when geminal to a spirocyclopropane, with larger groups such as isopropyl and tert-butyl being exclusively axial at −78 °C. Similar effects are found for heteroatoms, including halogens, and for a range of other electron-withdrawing substituents. Similar effects are observed for other strained rings (epoxide, cyclobutane, oxetane) and the concepts extend to acyclic models as well as heterocycles such as piperidines and piperazines. The origin of the effect is traced to an increase in torsional strain in combination with hyperconjugative effects in the case of electron-poor groups.
Introduction
The use of small ring structures as isosteres is a prominent technique for molecular design in medicinal chemistry.1 In particular, in recent years the use of bicyclopentanes and oxetanes as replacements for aromatic and carbonyl groups has allowed medicinal chemists to increase the three-dimensionality of drug candidates while maintaining relative dispositions of pendant groups.2,3 Even more common is the use of cyclopropanes (Fig. 1c) which have been widely employed as isosteres for alkenes, isopropyl (e.g. Pitavastatin, Fig. 1c) isobutyl and tert-butyl groups and even aromatic rings.4,5 Notably, spiro-cyclopropanes are featured in many bioactive compounds, including Pim-kinase inhibitors,6 ORL-1 antagonist MK-0911 (ref. 7) and the antitumor natural product illudins.8 While significant attention is paid to the exit vectors for groups attached to small rings,9,10 the conformational control elements adjacent to strained rings are less commonly examined.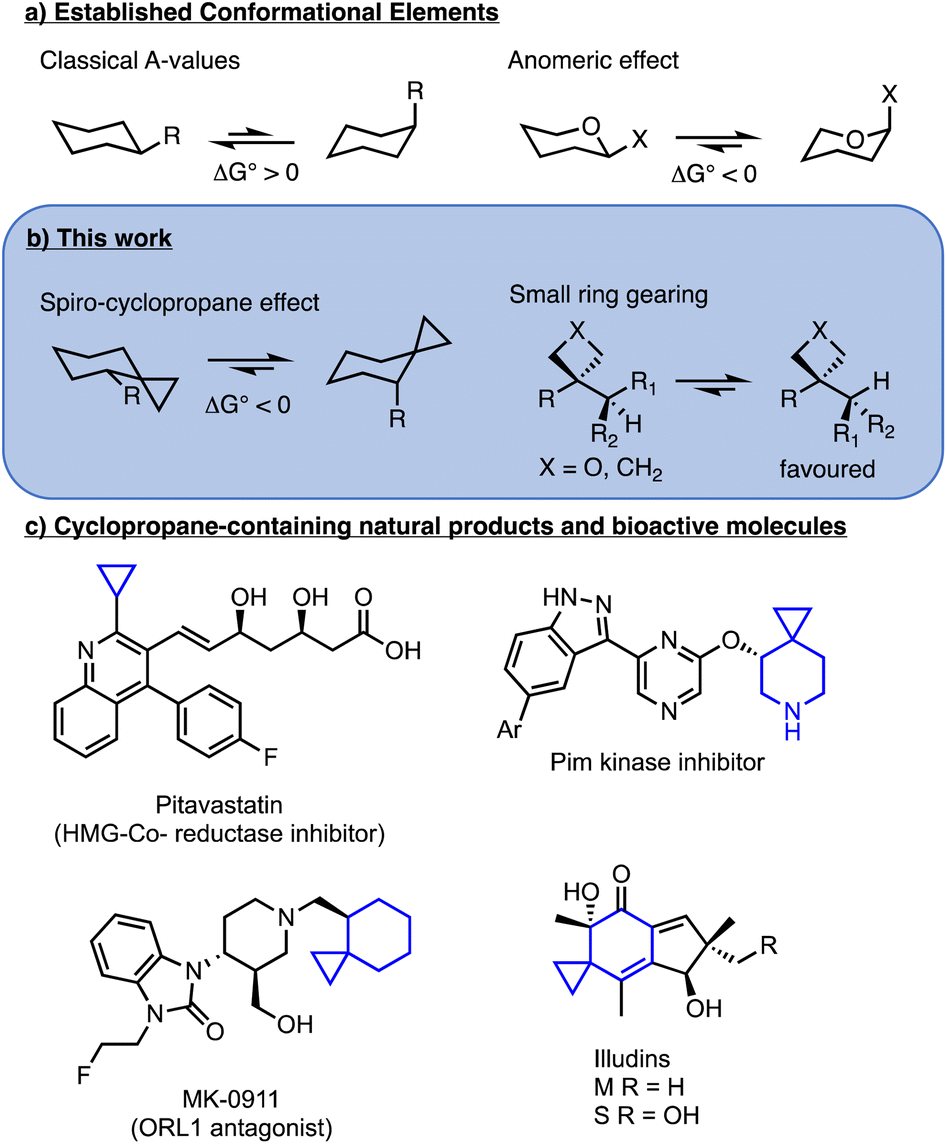 | ||
| Fig. 1 Conformational elements in cyclohexanes and examples of cyclopropane-containing relevant molecules. | ||
The quintessential example of conformational analysis in organic chemistry is the understanding of structure in cyclohexanes (Fig. 1a), where the chair form is preferred over the twist-boat and substituents prefer equatorial positions over axial to minimize gauche interactions with C3 and C5. The equatorial preference is quantified in the form of A-values and in general, larger groups possess larger A-values.11 Known exceptions to the preference for equatorial substitution do exist, with the most common examples resulting from stereoelectronic effects, including anomeric effects in carbohydrates and related acetals, hyperconjugative stabilization observed in 1,3,5-triazinanes,12,13 vinylogous anomeric effects in 2-chlorocyclohexanone oximes,14 and dipole minimization in α-halo-cyclohexanones,15 as well as cases where hydrogen bonding stabilizes axial substitution.16–18
In the context of examining an organocatalytic Cope rearrangement,19 we observed anomalous preference for axial orientation of alkyl groups adjacent to a cyclopropane in chair-like transition states (Fig. 2).20 Modest preference for axial disposition of α-ethers and acetates adjacent to spirocyclopropanes had previously been noted and had been attributed mainly to stereoelectronic effects.6,21 In addition, a spiro-oxetane was previously found to induce an N-alkyl group to adopt an axial conformation in a heterocycle in the solid state.22 As our studies here will show, the effect is in fact broadly applicable to a range of groups and is more significant for large alkyl groups, such that isopropyl and tert-butyl are exclusively axial. In addition, we show that the effect is not limited to cyclopropanes, but is operative with other three- and four-membered rings and the effects can be generalized into heterocycles and acyclic systems. These conformational effects have potential for application in molecular design of catalysts and rational design in medicinal chemistry.
 | ||
| Fig. 2 Preference for axial orientation in a Cope-like transition state. | ||
Results
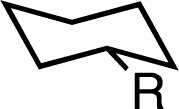
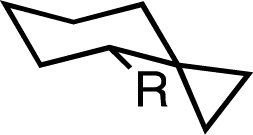
We first examined, computationally and experimentally, the effect of an adjacent spirocyclopropane substitution on the A-values of a variety of simple alkyl groups. Comparisons were made to both simple cyclohexanes and those bearing geminal-dimethyl substitution adjacent to the group of interest to focus on the specific effect of a small ring. Density functional calculations were performed employing the M06-2X functional,23 which has previously been employed in examining A-values,24 with a 6-311++G(2d,2p) basis set. We employed an acetone solvation model (SMD25), as we intended to examine structures experimentally at low temperature by NMR. A-values were calculated from a Boltzmann population analysis of all stable conformers identified by conformational analysis.26,27The effect of a spirocyclopropane on the A-value of a simple methyl group was significant (Table 1). While a geminal dimethyl group adjacent had, as previously observed,28,29 little effect on the A-value, an adjacent cyclopropane resulted in a computed A-value of −0.09 kcal mol−1, a change of −2.04 kcal mol−1vs. the computed value for methylcyclohexane. Even more striking were the effects on groups larger than methyl. While the presence of a geminal dimethyl group generally had minimal effect, the A-values for groups adjacent to a spirocyclopropane were uniformly negative and generally shifted by −3 kcal mol−1 or greater. For instance, an ethyl group was predicted to have an A-value of −0.89 kcal mol−1, indicating a clear preference for axial substitution and a change of −2.95 kcal mol−1 relative to ethylcyclohexane. Larger groups such as isopropyl and tert-butyl were predicted to have even larger preferences for the axial conformation. For tert-butyl the predicted A-value of −2.00 kcal mol−1 represents a change of nearly −8 kcal mol−1 from the normal equatorial preference in tert-butylcyclohexane.30,31
| Calculated A-valuea (kcal mol−1) | Expt. A-valueb (kcal mol−1) | |||
|---|---|---|---|---|
| R |
|
|
|
|
| a ΔG° calculated at 25 °C, M06-2X/6-311++G(2d,2p), SMD = acetone. b Experimental value determined by 1H NMR at −78 °C in d6-acetone. | ||||
| Me | +1.95 | +1.76 | −0.09 (δ = −2.04) | +0.05 |
| Et | +2.06 | +1.90 | −0.89 (δ = −2.95) | −0.46 |
| iPr | +2.33 | +3.27 | −2.10 (δ = −4.43) | <–1.5 |
| CH2OH | +1.98 | +1.71 | −1.01 (δ = −2.99) | −0.72 |
| Bn | +1.85 | +1.78 | −1.10 (δ = −2.95) | −0.75 |
| tBu | +5.83 | +3.44 | −2.00 (δ = −7.83) | <–1.5 |


We examined these species experimentally to corroborate the computational results. We prepared the spirocyclopropane substrates by cyclopropanation32 of the corresponding alkenes, themselves generated by olefination33 of the corresponding ketones (Scheme 1 and ESI†). We measured the equilibrium ratios of axial and equatorial conformers by 1H NMR at −78 °C in d6-acetone. Axial and equatorial isomers were assigned from coupling constants while confirmation that specific protons were interconverting was achieved by saturation transfer in selective irradiation experiments.34 Notably, in the spirocyclopropyl system, the α-equatorial protons are significantly shielded by anisotropy,35 which facilitated the assignment.
 | ||
| Scheme 1 Synthesis of alkyl-susbstituted spirocyclopropanes. | ||
The predicted effect of a spiro-cyclopropane was borne out experimentally. For the substrate bearing a methyl group, a 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :47 ratio of equatorial to axial conformers was observed. Although the equatorial isomer was very slightly preferred, the observed A-value of +0.05 kcal mol−1 was close to the predicted value and, importantly, the ratio was significantly different from the normal ∼95:5 ratio in methylcyclohexane. Moreover, for all other isomers, the axial isomer was clearly preferred. For instance, for an ethyl group, the axial isomer predominated in a 77:23 ratio, indicating an A-value of −0.46 kcal mol−1. Larger groups such as benzyl, isopropyl and tert-butyl more heavily favored the axial conformer – for the latter two substrates the equatorial isomers could not be observed at −78 °C. We estimated our limit of detection of the minor conformer to be roughly 2% of the major conformer, setting an upper limit for measuring the magnitude of A-values at 1.5 kcal mol−1 at −78 °C for these examples. We note that the DFT calculations slightly overestimated the effect of the cyclopropane by 0.1–0.4 kcal mol−1, with the ethyl group having the largest error. Examination of other computational methods for ethyl substitution afforded either similar (B3LYP-D3 (ref. 36–40)) or more significant (WB97XD,41,42 MP2 (refs. 43 and 44)) prediction errors (see ESI†) and thus the M06-2X functional was maintained for subsequent calculations.
:47 ratio of equatorial to axial conformers was observed. Although the equatorial isomer was very slightly preferred, the observed A-value of +0.05 kcal mol−1 was close to the predicted value and, importantly, the ratio was significantly different from the normal ∼95:5 ratio in methylcyclohexane. Moreover, for all other isomers, the axial isomer was clearly preferred. For instance, for an ethyl group, the axial isomer predominated in a 77:23 ratio, indicating an A-value of −0.46 kcal mol−1. Larger groups such as benzyl, isopropyl and tert-butyl more heavily favored the axial conformer – for the latter two substrates the equatorial isomers could not be observed at −78 °C. We estimated our limit of detection of the minor conformer to be roughly 2% of the major conformer, setting an upper limit for measuring the magnitude of A-values at 1.5 kcal mol−1 at −78 °C for these examples. We note that the DFT calculations slightly overestimated the effect of the cyclopropane by 0.1–0.4 kcal mol−1, with the ethyl group having the largest error. Examination of other computational methods for ethyl substitution afforded either similar (B3LYP-D3 (ref. 36–40)) or more significant (WB97XD,41,42 MP2 (refs. 43 and 44)) prediction errors (see ESI†) and thus the M06-2X functional was maintained for subsequent calculations.
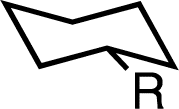
We next examined the effects of spirocyclopropanes on heteroatom substitution (Table 2). As with alkyl substituents, in all cases, we predicted a decrease in the A-value with absolute changes ranging from −1.2 to −4.0 kcal mol−1 and the axial conformation preferred for all substitution patterns. Notably, even small groups such as fluoro were predicted to have significant negative A-values. We prepared all but the chloro and bromo substrates, as the latter were prone to ring-opening of the cyclopropane. The heteroatom substrates were prepared by Simmons–Smith cyclopropanation of 2-methylene-1-cyclohexanol followed by functional group interconversions (see ESI† for details). We found that the calculated A-value reflected well the observed equilibrium concentrations in almost all cases. A slightly larger discrepancy between predicted and observed values was observed with ammonium and acetamide groups, with the latter being the only incorrect prediction of axial preference. We tentatively attribute the difference in the latter case to potential hydrogen bonding (either to itself or to solvent d6-acetone) which was not modeled explicitly by DFT, while the error in the former case may also reflect the difficulty in properly modelling solvation of the cation.
| Calculated A-valuea (kcal mol−1) | Expt. A-valueb (kcal mol−1) | |||
|---|---|---|---|---|
| R |
|
|
|
|
| a ΔG° calculated at 25 °C, M06-2X/6-311++G(2d,2p), SMD = acetone. b Experimental value determined by 1H NMR at −78 °C in d6-acetone, except as note. c Determined in d2-dichloromethane. | ||||
| OH | +0.89 | +0.83 | −0.37 (δ = −1.26) | −0.13 |
| OMe | +0.87 | +0.74 | −0.67 (δ = −1.54) | −0.42 |
| OAc | +0.74 | +0.53 | −0.54 (δ = −1.28) | −0.57 |
| NH2 | +1.41 | +1.36 | −0.15 (δ = −1.56) | −0.24c |
| NH3+ | +2.21 | +0.84 | −1.83 (δ = −4.04) | −1.23c |
| NHAc | +0.91 | +0.84 | −0.57 (δ = −1.48) | +0.19 |
| N3 | +0.63 | +0.86 | −0.92 (δ = −1.55) | −0.72 |
| F | +0.31 | +0.29 | −1.05 (δ = −1.36) | −0.82 |
| Cl | +0.63 | +1.16 | −1.81 (δ = −2.44) | nd |
| Br | +0.61 | +1.21 | −2.23 (δ = −2.84) | nd |



Finally, we examined a range of π- and electron withdrawing groups (Table 3). As with alkyl and heteroatom groups, all substituents also displayed a shift towards a more negative A-value. For phenyl the shift was not sufficient to prefer the axial conformation, although the absolute change is still significant (−2.04 kcal mol−1) and for vinyl an equal population of axial and equatorial was predicted. In contrast, alkynes and all electron withdrawing groups were predicted to have a negative A-value, with the CF3 group having the most significant change relative to a simple cyclohexane among this group and the largest predicted negative A-value among all substituents examined. We prepared four examples from this series; the phenyl substrate was indeed equatorial (92:8 ratio) as predicted, for ester and acid substrates the axial conformer was favored by roughly 70:30 ratios at −78 °C and for a cyano group the ratio was 80:20, again favoring axial.
| Calculated A-valuea (kcal mol−1) | Expt. A-valueb (kcal mol−1) | |||
|---|---|---|---|---|
| R |
|
|
|
|
| a ΔG° calculated at 25 °C, M06-2X/6-311++G(2d,2p), SMD = acetone. b Experimental value determined by 1H NMR at −78 °C in d6-acetone. | ||||
| Ph | 3.03 | 3.76 | 0.99 (δ = −2.04) | +0.95 |
CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
1.80 | 1.74 | −0.02 (δ = −1.82) | nd |
C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH CH |
0.46 | 0.96 | −0.23 (δ = −0.69) | nd |
| CO2H | 1.16 | 1.52 | −0.75 (δ = −1.31) | −0.38 |
| CO2Me | 1.34 | 1.59 | −0.59 (δ = −1.93) | −0.32 |
| CN | 0.13 | 0.62 | −0.69 (δ = −0.82) | −0.54 |
| NO2 | 1.02 | 1.31 | −2.02 (δ = −3,04) | nd |
| CF3 | 2.50 | 1.66 | −3.02 (δ = −5.52) | nd |



An important question was whether this effect could be extended to other spiro ring systems. We probed this computationally with small (Me, Et), medium (iPr) and large (tBu) substituents with a variety of different ring sizes and rings incorporating oxygen (Table 4). Calculations showed that the effect was generally maintained with cyclobutane, albeit with a partial loss of expected axial preference. However, when the ring expands to a cyclopentane, the equatorial isomer is now preferred for all group sizes, though still less compared to a simple dimethyl substituted system. Examining other three-membered rings, we found that spiro-epoxides were predicted to have a decrease in A-values, although the magnitude of the effect was reduced and dependent on the relative stereochemistry. For small substituents, the equatorial isomers are often still preferred, though by a much smaller margin than for a regular cyclohexane. However, for larger groups such as isopropyl or tert-butyl, the axial conformers are again predicted to be dominant. We examined cyclopropene substitution and noted that while A-values were again diminished, it was not sufficient to favor the axial isomer for groups other than for isopropyl. We also examined cyclobutanone and oxetane units which have been extensively employed as isosteres. Oxetane had a significant effect on medium and large groups, though its effects on smaller groups was not sufficient to prefer the axial conformer and the effect of cyclobutanone was also muted even with large groups (Table 4).
|
|
||||
|---|---|---|---|---|
| X | Me | Et | iPr | tBu |
| a ΔG° values calculated at 25 °C at M06-2X/6-311++(2g,2p), SMD = acetone and reported in kcal mol−1. Experimental values determined at −78 °C in d6-acetone in parentheses. b Determined at −98 °C. | ||||
| CH2 | +1.95 | +2.06 | +2.33 | +5.83 |
| CMe2 | +1.76 | +1.90 | +3.27 | +3.44 |

|
−0.09 (+0.3) | −0.89 (−0.46) | −2.10 (<−1.5) | −2.00 (<−1.5) |

|
+0.26 | −0.25 | −0.52 | −0.72 (<0) |

|
+0.67 | +0.66 | +1.46 | +0.11 |

|
+0.69 | 0.00 | −1.31 | −1.64 (<−1.5) |

|
+0.24 | −0.42 | −0.98 | −0.57 (−0.20)b |

|
+1.47 | +1.07 | +0.42 | +1.14 |

|
+0.17 | +0.15 | −1.80 | −1.91 |

|
+0.16 | +0.18 | −0.28 | −0.27 (−0.23) |

We examined experimentally several different ring systems bearing tert-butyl groups (see ESI† for synthesis) and all were found to be generally in line with the calculations (see values in parentheses, Table 4). Due to overlapping signals, the ratio in the cyclobutane substrate could not be quantified. However, using computational NMR shift prediction with the GIAO method45 and cross-peak intensity in low-temperature HSQC, the axial conformer was assigned as the major isomer. The anti-epoxide46 heavily favored the axial conformer, as predicted, with no equatorial isomer observed while the corresponding syn-epoxide had a lower preference for axial, again as predicted. The tert-butyl substituted spirocyclobutanone could be quantified and was found to have a 64:36 ratio. Although we were unable to assign the conformers with NOEs alone, again NMR chemical shift prediction allowed us to assign the major conformer as axial (see ESI†), indicating an A-value of −0.23 kcal mol−1 which was consistent with the computations.
Finally, the trends observed in cyclic frameworks were expected to have parallels in acyclic structures. Specifically, there should be an energetic penalty to place an alkyl group above a small ring, similar to the equatorial placement in the spiro systems above. We examined the orientation of an isopropyl group situated on a 1-methylcyclopropane, -cyclobutane and -oxetane. In all cases, the preferred conformation where the hydrogen of the isopropyl group was situated above the ring, was favored by 1.2–1.6 kcal mol−1 (Fig. 3). It would be expected that other similar substitutions, such as secondary stereocenters, would adopt a similar conformation. The acyclic directing effect of oxetanes has been previously noted22 and is highly relevant in their use as bioisosteres, as it alters the normal preference for syn-periplanar orientation of alpha groups in ketones.47
 | ||
| Fig. 3 Conformations in acyclic systems. | ||
Application
The surprising cyclopropane effect has potential as a simple but elegant way to influence conformation, particularly in cyclic systems. In medicinal chemistry, the ability of molecules to occupy specific chemical space is critical to ligand/target interactions. Heterocycles including piperidines and piperazines are common features of approved drugs, often adding an element of three-dimensionality to aromatic-rich structures. We considered whether the use of cyclopropanes could influence conformation about a heterocycle in similar fashion to the cyclohexanes above. We first examined a series of N-alkylpiperidines which, like cyclohexanes, display a preference for equatorial orientation of the N-alkyl group.48N-Methyl, N-benzyl and N-tert-butyl spiropiperidines 1–3 (Fig. 4) were easily prepared by Kulinkovich reaction on the corresponding lactams.49 DFT calculations on all three substrates suggested the alkyl groups on nitrogen should assume an axial disposition, with predicted A-values of −1.16 kcal mol−1, −1.83 kcal mol−1 and −2.37 kcal mol−1 for 1–3, respectively. Notably, unlike in cyclohexanes, even the small methyl group is predicted to be mainly axial. Experimentally, all three exhibited only a single conformer at −78 °C.50 For 2 and 3, NOE studies clearly identified these as the axial conformer while for 1 the NOE data was inconclusive but DFT chemical shift analysis suggested the axial conformer (see ESI†). In the case of 3, it was possible to crystallize the amine as its HCl salt (calculated A-value for 3·HCl: −2.61 kcal mol−1). X-Ray diffraction analysis confirmed the remarkable axial orientation of the tert-butyl group (Fig. 2b). | ||
| Fig. 4 Conformational control in 2-spirocyclopropyl piperidines. (A) Calculated piperidine A-values (B) X-ray crystal structure of 3·HCl. | ||
Piperazines are among the most common heterocycles in active pharmaceutical ingredients, being found in almost 10% of the top selling small-molecule drugs.51 We prepared mono- and bis-cyclopropyl piperazines by Kulinkovich reaction of N,N′-dibenzyldiketopiperazine to afford a mixture of 4 and 5 (Scheme 2A). Lactam 5 could be further reduced with lithium aluminum hydride to afford mono-cyclopropyl piperazine 6. All three piperazines (4–6) were found to display a single conformation at low temperature. While it was not possible to establish the conformations by NOE, the expected preference for diaxial conformation in 4 was observed in an X-ray crystal structure (Scheme 2b). Similarly, mono-cyclopropane 6 when crystallized as a hydrated bis-p-toluenesulfonic acid salt displayed an axial/equatorial conformation with the benzyl group adjacent to the cyclopropane selectively forced into the axial position (Scheme 2c). These studies establish the ability to modulate the conformation of 6-membered ring heterocycles by straightforward incorporation of spirocyclic cyclopropanes. Notably, comparing 4, 5, and 6, it is possible to maintain one benzyl group axial while displaying the other axial, pseudo-equatorial, or equatorial, respectively, depending upon the identity of the adjacent group.
 | ||
| Scheme 2 (A) Synthesis and conformation of spirocyclopropylpiperazines (B) SCXRD of 4 (C) SCXRD of 6·(TsOH)2·H2O (tosylates and water have been removed for clarity – see ESI† for full structure). | ||
Discussion
The ability of small rings to act as isosteres, mimicking functionality such as alkenes, arenes, carbonyl and other groups is well recognized in medicinal chemistry. A key factor in the implementation of small rings is their ability to project groups into three-dimensional space in a way that reproduces the replaced functionality. Cyclopropanes are excellent mimics for multiple groups, including alkenes given their frozen torsion angles at 0° and ∼145°. Similarly, the oxetane has seen significant use as a carbonyl mimic, with the slightly widened exocyclic bond angle mimicking that of the sp2 carbon.3Our study has demonstrated that small rings commonly used as isosteres can also be used for elements of conformational control in cyclohexanes, six-membered ring heterocycles and in acyclic systems. Compared to simple di-alkyl substitution, small constrained rings shift the equilibria of adjacent groups towards axial conformers for most substituents.
The main origin of the effect for alkyl groups is a change in steric strain. In a simple geminal dimethyl system, the torsion angle between an equatorial substituent (e.g. methyl) and the two methyl groups is 60° ± 5° (Fig. 5A). The constraint of the cyclopropane ring significantly reduces this angle (34° and 38°), resulting in increased steric interactions. In addition, in the axial form, the torsion angle in the cyclopropane is opened significantly from 47° to 77°, suggesting a potential reduction of steric interactions. An NBO-STERIC calculation52,53 indicated the relative difference between axial and equatorial conformers is reduced by 2.08 kcal mol−1 when comparing the dimethyl (2.72 kcal mol−1) to cyclopropyl (0.64 kcal mol−1) substituents. This 2 kcal mol−1 reduction in steric interactions is consistent with the change in A-value observed experimentally. To examine this effect in more depth, butane was used to model the steric interactions of individual torsion components. Freezing butane torsion angles at the equivalent angles from the cyclohexane system revealed that the smaller torsional angles in the equatorial form for the cyclopropane add 1.96 kcal mol−1 in steric interactions relative to the torsional angles in a dimethyl group (Fig. 5b). Conversely, in the axial form, the steric interactions are lower by −0.81 kcal mol−1 for the cyclopropane.54 While only an approximation, these results are suggestive of a combination of destabilization of the equatorial form and a net stabilization (relative to dimethyl) in the axial form for spirocyclopropanes. These steric strain effects are presumably increased in N-heterocycles (e.g.1–6) due to reduced bond length along the torsion axis (1.45 vs.1.52 Å) as well as with larger groups (e.g. tert-butyl).
 | ||
| Fig. 5 Increased steric strain adjacent to cyclopropane. (A) NBO-STERIC calculations suggest a reduced difference between axial and equatorial epimers in the presence of a cyclopropane. (B) Modeling the effect of specific torsion angles using butane. NBO-STERIC energies were calculated fixing butane at angles found in the dimethyl and spirocyclopropyl systems. The sum of steric interactions are approximately 2 kcal mol−1 higher in the equatorial isomer with cyclopropyl substitution compared to dimethyl whereas in the axial isomer the cyclopropane interactions are 0.81 kcal mol−1 less. | ||
While simple torsional strain arguments can be used to explain the preference in alkyl-substituted cyclohexanes, the trends with halides and other electron poor groups suggest that stereoelectronic factors also contribute. For instance, both fluoro and chloro groups have smaller van der Waals radius than methyl55,56 but both clearly prefer the axial conformation whereas for methyl the axial and equatorial conformations are roughly equal in energy. One potential stabilizing factor may be hyperconjugative donation from the cyclopropyl group into the C–X antibonding orbitals.21 While the C–C bond axis is not well aligned for donation, the electron density of the cyclopropane lies outside of the bond axis and is positioned appropriately to donate to σ* (Fig. 6). Notably, an NBO analysis revealed stronger donation into the C–X σ* from the cyclopropane vs. an axial methyl, on the order of 0.8–1 kcal mol−1 for both F and Cl. This hyperconjugative stabilization presumably stabilizes the axial form and, in combination with increased torsional strain in the equatorial isomer, results in a more significant preference for axial orientation. Notably, the hyperconjugation is significantly reduced in the corresponding cyclobutyl fluoride (predicted A-value: +0.15 kcal mol−1, σ–σ* donation only 0.2 kcal mol−1 greater than methyl).
 | ||
| Fig. 6 NBO orbital overlap of cyclopropane C–C σ-bond with axial C–F σ*, calculated at the M06-2X/6-311++G(2d,2p) level. | ||
Conclusions
In summary, we have found that the presence of small spirocyclic rings on cyclohexane has a profound effect on the axial/equatorial orientation of adjacent groups. The effect of increased torsional strain and, in certain instances, hyperconjugation, results in a significant shift towards greater relative stability of the axial conformation. Effects are significant for a range of groups, with larger groups experiencing a more significant shift towards axial preference. The effect is observed most acutely with cyclopropane, but also extends to other three and four-membered rings as well as acyclic systems, and can be incorporated within heterocycles to control nitrogen stereochemistry. Importantly, these effects have significant potential for use as a design element in a wide range of applications. In medicinal chemistry, installation of a cyclopropane into a structure can allow the exploration of different chemical space. Similarly, small rings could be incorporated in cyclic catalysts to project large groups into specific areas to alter catalyst selectivity.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Both authors conceptualized the project, conducted computational studies and wrote the manuscript. ARI conducted all experimental work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the Natural Sciences and Engineering Research Council of Canada is gratefully acknowledged.Notes and references
- M. R. Bauer, P. Di Fruscia, S. C. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, Put a Ring on It: Application of Small Aliphatic Rings in Medicinal Chemistry, RSC Med. Chem., 2021, 12, 448–471 RSC.
- Y. P. Auberson, C. Brocklehurst, M. Furegati, T. C. Fessard, G. Koch, A. Decker, L. La Vecchia and E. Briard, Improving Nonspecific Binding and Solubility: Bicycloalkyl Groups and Cubanes as para-Phenyl Bioisosteres, ChemMedChem, 2017, 12, 590–598 CrossRef PubMed.
- J. J. Rojas and J. A. Bull, Oxetanes in Drug Discovery Campaigns, J. Med. Chem., 2023, 66, 12697–12709 CrossRef.
- T. Talele, The “cyclopropyl fragment” is a versatile player that frequently appears in preclinical/clinical drug molecules, J. Med. Chem., 2016, 59(19), 8712–8756 CrossRef.
- M. Suzuki, H. Iwasaki, Y. Fujikawa, M. Kitahara, M. Sakashita and R. Sakoda, Synthesis and biological evaluations of quinoline-based HMG-CoA reductase inhibitors, Bioorg. Med. Chem., 2001, 9(10), 2727–2743 CrossRef PubMed.
- H.-L. Wang, V. J. Cee, F. Chavez Jr, B. A. Lanman, A. B. Reed, B. Wu, N. Guerrero, J. R. Lipford and C. Sastri, Winston, The discovery of novel 3-(pyrazin-2-yl)-1H-indazoles as potent pan-Pim kinase inhibitors, Bioorg. Med. Chem. Lett., 2015, 25(4), 834–840 CrossRef PubMed.
- T. McMorris and M. Anchel, Fungal metabolites. The structures of the novel sesquiterpenoids illudin-S and-M, J. Am. Chem. Soc., 1965, 87(7), 1594–1600 CrossRef PubMed.
- E. D. Hostetler, S. Sanabria-Bohórquez, W. Eng, A. D. Joshi, S. Patel, R. E. Gibson, S. O'Malley, S. M. Krause, C. Ryan and K. Riffel, Evaluation of [18F] MK-0911, a positron emission tomography (PET) tracer for opioid receptor-like 1 (ORL1), in rhesus monkey and human, Neuroimage, 2013, 68, 1 CrossRef PubMed.
- O. O. Grygorenko, P. Babenko, D. M. Volochnyuk, O. Raievskyi and I. Komarov, Following Ramachandran: exit vector plots (EVP) as a tool to navigate chemical space covered by 3D bifunctional scaffolds. The case of cycloalkanes, RSC Adv., 2016, 6(21), 17595–17605 RSC.
- M. A. Subbaiah and N. A. Meanwell, Bioisosteres of the phenyl ring: recent strategic applications in lead optimization and drug design, J. Med. Chem., 2021, 64(19), 14046–14128 CrossRef.
- K. B. Wiberg, J. D. Hammer, H. Castejon, W. F. Bailey, E. L. DeLeon and R. M. Jarret, Conformational Studies in the Cyclohexane Series. 1. Experimental and Computational Investigation of Methyl, Ethyl, Isopropyl, and Tert -Butylcyclohexanes, J. Org. Chem., 1999, 64(6), 2085–2095 CrossRef PubMed.
- F. P. dos Santos and C. F. Tormena, Orbital interactions and their effects on the conformational stability in six-membered rings containing nitrogen atoms, J. Mol. Struct.: THEOCHEM, 2006, 763(1–3), 145–148 CrossRef.
- E. Juaristi and Y. Bandala, Anomeric Effect in Saturated Heterocyclic Ring Systems, Adv. Heterocycl. Chem., 2012, 105, 189–222 CrossRef.
- S. E. Denmark, M. S. Dappen, N. L. Sear and R. T. Jacobs, The Vinylogous Anomeric Effect in 3-Alkyl-2-Chlorocyclohexanone Oximes and Oxime Ethers, J. Am. Chem. Soc., 1990, 112(9), 3466–3474 CrossRef.
- E. A. Basso, C. Kaiser, R. Rittner and J. B. Lambert, Axial Equatorial Proportions for 2-Substituted Cyclohexanones, J. Org. Chem., 1993, 58(27), 7865–7869 CrossRef.
- A. Sun, D. C. Lankin, K. Hardcastle and J. Snyder, 3-Fluoropiperidines and N-methyl-3-fluoropiperidinium salts: The persistence of axial fluorine, Adv. Heterocycl. Chem., 2005, 11(5), 1579–1591 Search PubMed.
- Q. Shen, The molecular structure and conformation of 2-chlorocyclohexanone as determined by gas-phase electron diffraction, J. Mol. Struct., 1983, 96(1–2), 133–139 CrossRef.
- C.-Y. Huang, L. A. Cabell and E. V. Anslyn, Molecular recognition of cyclitols by neutral polyaza-hydrogen-bonding receptors: The strength and influence of intramolecular hydrogen bonds between vicinal alcohols, J. Am. Chem. Soc., 1994, 116(7), 2778–2792 CrossRef.
- D. Kaldre and J. L. Gleason, An Organocatalytic Cope Rearrangement, Angew. Chem., Int. Ed., 2016, 55(38), 11557–11561 CrossRef PubMed.
- The rearrangements depicted in Fig. 2 eventually produce novel ring-expansion products. A full report will be detailed elsewhere.
- N. S. Zefirov, E. G. Chalenko, A. V. Aripovsky, I. G. Mursakulov, M. M. Guseinov and E. A. Ramazanov, New conformational effect: predominance of the axial conformation in spiro[5.2]octan-4-ol derivatives, J. Chem. Soc., Chem. Commun., 1978,(3), 147–148 RSC.
- G. Wuitschik, E. M. Carreira, B. r. Wagner, H. Fischer, I. Parrilla, F. Schuler, M. Rogers-Evans and K. Müller, Oxetanes in drug discovery: structural and synthetic insights, J. Med. Chem., 2010, 53(8), 3227–3246 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. F. He, B. A. Piscelli, R. A. Cormanich and D. O'Hagan, Conformational Analysis Explores the Role of Electrostatic Nonclassical CF···HC Hydrogen Bonding Interactions in Selectively Halogenated Cyclohexanes, J. Org. Chem., 2024, 89(6), 4009–4018 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef.
- Boltzmann population analysis was computed using Good Vibes: G. Luchini, J. V. Alegre-Requena, I. Funes-Ardoiz and R. S. Paton, GoodVibes: Automated Thermochemistry for Heterogenous Computational Chemistry Data, F1000Research, 2020, 9, 291 Search PubMed.
- Calculated values are for standard Boltzmann populations at 25 °C. Repeating calculations at −78 °C, to match the conditions of experimental measurements, resulted in insignificant differences in the predicted A-values. For example, the computed A-value for ethyl in the presence of the spirocyclopropane changes from 0.89 kcal mol−1 at 25 °C to 0.85 kcal mol−1 at −78 °C.
- E. L. Eliel and S. Chandrasekaran, Conformational Analysis. 44. 1,1,2-Trimethylcyclohexane, J. Org. Chem., 1982, 47(24), 4783–4786 CrossRef.
- E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, Wiley, 1994, p. 705 Search PubMed.
- Notably, unlike other simple systems such as cis-1,4-di-tert-butylcyclohexane, the twist boat was predicted to be significantly higher in energy (3.4 kcal mol−1 above the chair conformation bearing the axial group) and thus would not be expected to be present to any significant extent.
- G. Gill, D. M. Pawar and E. A. Noe, Conformational study of cis-1,4-di-tert-butylcyclohexane by dynamic NMR spectroscopy and computational methods. Observation of chair and twist-boat conformations, J. Org. Chem., 2005, 70(26), 10726–10731 CrossRef PubMed.
- K. Maruoka, Y. Fukutani and H. Yamamoto, Trialkylaluminum-Alkylidene Iodide. A Powerful Cyclopropanation Agent with Unique Selectivity, J. Org. Chem., 1985, 50(22), 4412–4414 CrossRef.
- T. H. Yan, C. C. Tsai, C. T. Chien, C. C. Cho and P. C. Huang, Dichloromethane activation. Direct methylenation of ketones and aldehydes with CH2Cl2 promoted by Mg/TiCl4/THF, Org. Lett., 2004, 6(26), 4961–4963 CrossRef.
- N. Lokesh, A. Seegerer, J. Hioe and R. M. Gschwind, Chemical exchange saturation transfer in chemical reactions: A mechanistic tool for NMR detection and characterization of transient intermediates, J. Am. Chem. Soc., 2018, 140(5), 1855–1862 CrossRef PubMed.
- E. Kleinpeter, S. Krüger and A. Koch, Anisotropy effect of three-membered rings in 1H NMR spectra: Quantification by TSNMRS and assignment of the stereochemistry, J. Phys. Chem. A, 2015, 119(18), 4268–4276 CrossRef.
- A. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648 CrossRef.
- S. H. Vosko, L. Wilk and M. Nusair, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis, Can. J. Phys., 1980, 58(8), 1200–1211 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B, 1988, 37(2), 785 CrossRef PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98(45), 11623–11627 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10(44), 6615–6620 RSC.
- J.-D. Chai and M. Head-Gordon, Systematic optimization of long-range corrected hybrid density functionals, J. Chem. Phys., 2008, 128(8), 084106 CrossRef PubMed.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, A direct MP2 gradient method, Chem. Phys. Lett., 1990, 166(3), 275–280 CrossRef CAS.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, Semi-direct algorithms for the MP2 energy and gradient, Chem. Phys. Lett., 1990, 166(3), 281–289 CrossRef CAS.
- K. Wolinski, R. Haacke, J. F. Hinton and P. Pulay, Methods for parallel computation of SCF NMR chemical shifts by GIAO method: Efficient integral calculation, multi-Fock algorithm, and pseudodiagonalization, J. Comput. Chem., 1997, 18(6), 816–825 CrossRef CAS.
- The syn-O epoxide was also prepared but was challenging to assess, as significant peak broadening was observed at −78 °C, suggesting that ring flip was still occurring. This was corroborated computationally; the syn-epoxide was predicted to have an 8.4 kcal mol−1 barrier to ring flip vs. 11.3 kcal mol−1 for the corresponding anti-epoxide. This difference is attributed to reduced eclipsing interactions between the epoxide oxygen and the tert-butyl in the transition state for the syn isomer.
- T. Sakurai, M. Ishiyama, H. Takeuchi, K. Takeshita, K. Fukushi and S. Konaka, Molecular structure and conformation of 3-methyl-2-butanone: A gas electron diffraction investigation combined with ab initio calculation and vibrational spectroscopy, J. Mol. Struct., 1989, 213(C), 245–261 CrossRef CAS.
- J. B. Lambert and S. I. Featherman, Conformational analysis of pentamethylene heterocycles, Chem. Rev., 1975, 75(5), 611–626 CrossRef CAS.
- A. de Meijere and S. I. Kozhushkov, Facile syntheses of aminocyclopropanes: N,N-dibenzyl-N-(2-ethenylcyclopropyl)amine [benzenemethanamine, N-(2-ethenylcyclopropyl)-N-(phenylmethyl)], Org. Synth., 2018, 95, 289–309 CrossRef CAS.
- The NMR spectrum of 1 was also acquired at −98 °C and continued to show a single set of signals. The possibility of coalesced peaks due to ring-inversion at this temperature is viewed as unlikely given the higher barrier to ring inversion of piperidines relative to cyclohexane J. B. Lambert and R. G. Keske, J. Am. Chem. Soc., 1966, 88, 620–622 CrossRef CAS.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, A graphical journey of innovative organic architectures that have improved our lives, J. Chem. Ed. , 2010, 87(12), 1348–1349 CrossRef CAS . Based on the “top 200 small molecule drugs by sales in 2023 poster”..
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsincity - Madison, 2018 Search PubMed.
- J. Badenhoop and F. Weinhold, Natural bond orbital analysis of steric interactions, J. Chem. Phys., 1997, 107(14), 5406–5421 CrossRef CAS.
- It is important to note that the torsion angles between axial methyl groups and the ring carbons do not change significantly between the spirocyclopropyl and dimethyl systems and thus are expected to be roughly equivalent.
- A. Bondi, van der Waals volumes and radii, J. Phys. Chem., 1964, 68(3), 441–451 CrossRef CAS.
- E. Kutter and C. Hansch, Steric parameters in drug design. Monoamine oxidase inhibitors and antihistamines, J. Med. Chem., 1969, 12(4), 647–652 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2375096–2375098. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05470a |
| This journal is © The Royal Society of Chemistry 2024 |