
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectron diffraction and solid-state NMR reveal the structure and exciton coupling in a eumelanin precursor†
Kavya
Vinod‡
a,
Renny
Mathew‡
b,
Christian
Jandl
c,
Brijith
Thomas
 *b and
Mahesh
Hariharan
*a
*b and
Mahesh
Hariharan
*a
aSchool of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram (IISER TVM), Maruthamala P.O., Vithura, Thiruvananthapuram 695551, Kerala, India. E-mail: mahesh@iisertvm.ac.in
bScience Division, New York University Abu Dhabi, P.O. Box 129188, Abu Dhabi, United Arab Emirates. E-mail: brijiththomas@nyu.edu
cELDICO Scientific AG, Switzerland Innovation Park Basel Area, Hegenheimermattweg 167A, Allschwil, 4123, Switzerland
First published on 16th September 2024
Abstract
Eumelanin, a versatile biomaterial found throughout the animal kingdom, performs essential functions like photoprotection and radical scavenging. The diverse properties of eumelanin are attributed to its elusive and heterogenous structure with DHI (5,6-dihydroxyindole) and DHICA (5,6-dihydroxyindole-2-carboxylic acid) precursors as the main constituents. Despite DHICA being recognized as the key eumelanin precursor, its crystal structure and functional role in the assembled state remain unknown. Herein, we employ a synthesis-driven, bottom-up approach to elucidate the structure and assembly-specifics of DHICA, a critical building block of eumelanin. We introduce an interdisciplinary methodology to analyse the nanocrystalline assembly of DHICA, employing three-dimensional electron diffraction (3D ED), solid-state NMR and density functional theory (DFT), while correlating the structural aspects with the electronic spectroscopic features. The results underscore charge-transfer exciton delocalization as the predominant energy transfer mechanism within the π–π stacked and hydrogen-bonded crystal network of DHICA. Additionally, extending the investigation to the 13C-labelled DHICA-based polymer improves our understanding of the chemical heterogeneity across the eumelanin pigment, providing crucial insights into the structure of eumelanin.
Introduction
Eumelanin, a natural biomaterial, is instrumental in protecting the skin from harmful radiation and environmental toxins.1–3 Eumelanin is recognized for its broadband UV-visible absorption and efficiency in dissipating excitation energy, thereby preventing cellular damage.4 The structure of eumelanin is highly heterogeneous and ambiguous.3 It is believed to consist of a three-dimensional network of interconnected aromatic units, derived from the oxidative polymerization of 5,6-dihydroxyindole-2-carboxylic acid (DHICA) and 5,6-dihydroxyindole (DHI). Chemical and spectral evidence from eumelanin identified so far points to different levels of chemical, structural and supramolecular disorder within eumelanin.2,3,5–10 Characterizing eumelanin proves challenging due to its disordered, amorphous nature and insolubility in most common solvents, rendering conventional characterization techniques impractical (Table S1†).11,12 The challenges associated with the structure elucidation of eumelanin have evaded the exact mechanism of energy dissipation and photoprotection in the biomaterial. Considering the complexities associated with understanding eumelanin chemistry, a bottom-up approach by elucidating the structure and supramolecular assembly of the monomeric units is crucial (Scheme 1).13–17 Detailed knowledge of the interchromophoric interactions between monomers and their higher-order assemblies can reveal how non-covalent interactions influence the macroscopic properties, providing critical insights into the foundational elements of the bio-pigment.18 Structure elucidation is also a key factor for determining the electronic coupling within the interacting units, allowing further insights into the exciton dynamics of the eumelanin constituents.19 Understanding the inter/intramolecular non-covalent interactions in eumelanin monomers could also reveal hints about the oxidative polymerization mechanism, especially given their susceptibility to excited state reactions.17,20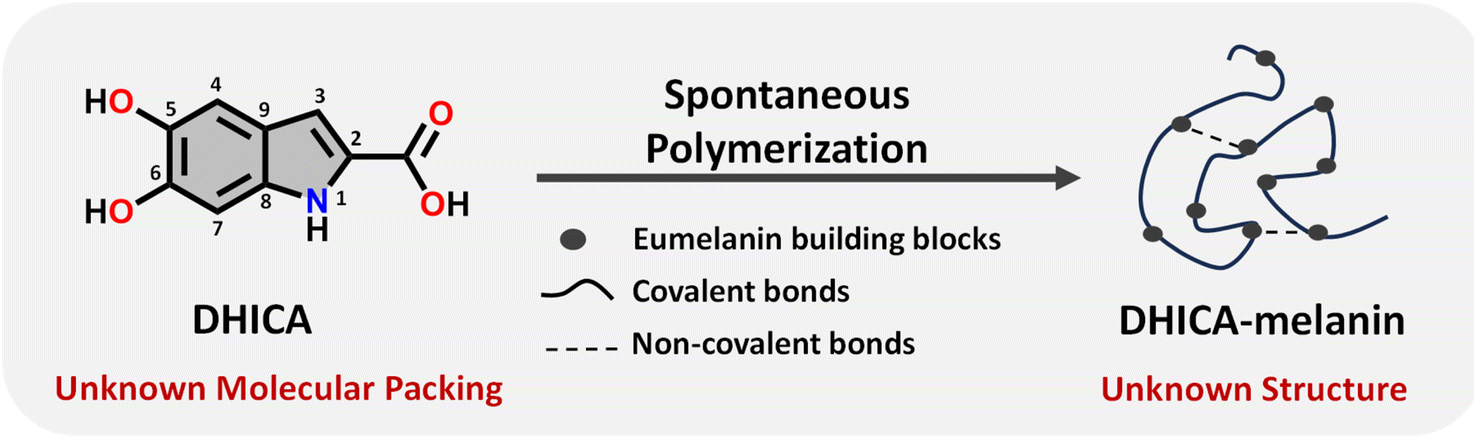 | ||
| Scheme 1 Schematic diagram representing the spontaneous polymerization of DHICA, the monomeric unit of eumelanin into the black polymeric DHICA-melanin. | ||
Among the two eumelanin precursors, previous experimental and theoretical research has focused mostly on DHI while DHICA remains less explored.21–30 Using single-crystal XRD (SC-XRD), the assembly pattern of DHI was previously established where DHI forms hydrogen-bonded enantiomeric helical stacks with Frenkel exciton delocalisation within the helix.22 However, the requirement of suitably large and well-diffracting single crystals is a substantial drawback of structure elucidation with SC-XRD. Unlike DHI, obtaining larger diffracting crystals for DHICA is difficult owing to the superior polymerization tendency of the eumelanin precursor due to which DHICA can only be crystallized as crystals of nanometre size with limited diffraction ability.31 Furthermore, conventional crystallographic techniques encounter challenges in the case of DHICA due to the inherent disorder, warranting the employment of 3D ED and solid-state NMR.
In our prior research, we employed 3D ED and NMR crystallography7,32–34 to elucidate the structure of a disordered microcrystalline nitroperylenediimide system.35 Our continued interest in inspecting the structure–property relationship in eumelanin counterparts motivated us to implement 3D ED,36–39 magnetic resonance40–47 and electronic spectroscopy along with DFT calculations to understand the intricate structure and photophysical properties of DHICA.22,48–53 By utilizing the extensive capabilities of solid-state NMR,54,55 we further extend our investigations to the synthesised 13C-labelled polymer of DHICA, i.e., DHICA-melanin. The applied multi-level approach attempts to unravel the complexities inherent in a disordered material such as eumelanin, opening avenues for advancements in eumelanin chemistry.
Results and discussion
Synthesis and optical properties
DHICA was synthesized non-enzymatically using L-DOPA as the starting material, following previous literature with minor modifications to improve the yield (yield = 76%; Scheme S1 and Fig. S1–S5†).5613C-labelled DHICA was synthesized with selectively 13C-labelled L-DOPA (yield = 71%; Scheme S2†). The off-white powder of DHICA upon solubilizing in milliQ water, resulted in a brown-black solution over time. The black powder was collected and centrifuged to obtain DHICA-melanin (Scheme S3 and Fig. S5†). The UV-visible absorption spectrum of DHICA in the solution state ranges in the near UV region peaking at λabsS1 = 315 nm and λabsS2 = 200 nm pertaining to the S0→S1 and S0→S2 electronic absorption bands respectively (Fig. S6a†).57 The solid-state Kubelka–Munk diffuse reflectance transformed absorption spectra of DHICA and DHICA-melanin are presented in Fig. 1. The absorption spectrum of DHICA in the solid state is broader with respect to that of the monomer and ranges from λabs1solid = 200–400 nm, while revealing an additional shoulder band at λabs2solid = 400–550 nm which was absent in the monomeric solution. In extended systems such as molecular crystals, aggregates or polymers, exciton interactions may occur between molecular sites depending on the strength of the electronic coupling, causing differences in the photophysics of the aggregate with respect to the isolated molecule.58 The substantial differences in the solid-state UV-visible spectra of DHICA compared to the monomeric state are suggestive of the intermolecular interactions between the DHICA units in a tightly packed molecular environment. The electronic coupling between the DHICA molecules is intrinsic to the spatial orientation of the chromophores, and to examine the nature of exciton interactions in DHICA, the aggregate architecture has to be considered.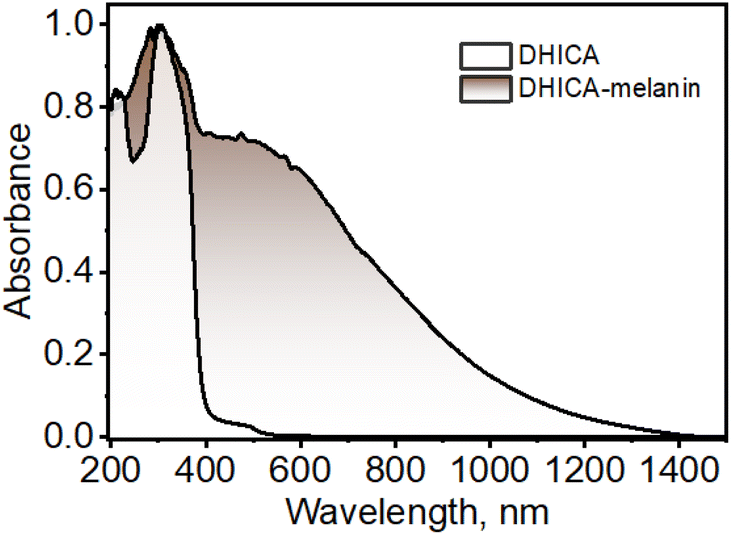 | ||
| Fig. 1 UV-visible-NIR diffuse-reflectance transformed absorption spectra of DHICA and DHICA-melanin in the solid state. | ||
The concentration-dependent UV-visible absorption of DHICA in milliQ water shows visible differences at high (aggregate) concentrations of DHICA compared to low (monomer) concentrations. The emergence of a broad UV-visible absorption band from 400–600 nm is observed with the increase in concentration from 0.01–1.0 mM (Fig. S6b†). The broad red-shifted absorption band for higher concentrated solutions of DHICA resembles the shoulder band in the absorption spectrum in the solid state, pointing to aggregate formation in the high concentrated solutions. The effect of aggregation is also evident in the fluorescence emission where the emission red-shifts from λemimax = 385 nm in the monomer to λemimax = 397 nm in the aggregate (Δλ = 12 nm; Fig. S6c and d†). Temperature-dependent fluorescence measurements show a breakdown of the aggregates at higher temperatures as demonstrated by the blue-shift in the fluorescence emission as temperature increases from 10–90 °C (Fig. S7†). The formation of larger-sized aggregates was also evident from TEM and DLS measurements in higher concentrations of DHICA (Fig. S8 and S9†). The electronic absorption spectrum of the synthesized polymer of DHICA (DHICA-melanin) exhibits differences from previously established synthetic melanins.3,57 The UV-visible-NIR absorption band of DHICA-melanin shows a peak maximum at λabsmax = 310 nm and a broad absorption band covering the entire visible region and tailing towards the NIR region (Fig. 1). The monotonic increase in absorption towards higher energy in DHICA-melanin is reminiscent of the excitonic model proposed for the broadband absorption of natural eumelanin.5 The synthesized DHICA-melanin is distinguished by the absorption in the 1000–1400 nm range while the previously reported DHICA-melanins have an absorption maximum at 320 nm, tailing towards 700 nm. The differences in the absorption spectra between the synthesized DHICA-melanins could be due to the formation of higher-order oligomers that co-exist with the monomer-like units. There could also be extensive π-delocalization within the DHICA substructures, leading to stabilization of energy levels in the synthesized DHICA-melanin, compared to other synthesized eumelanins.59–61 Similar to the non-emissive nature of naturally derived eumelanin, DHICA-melanin did not display an attributable fluorescence emission (Fig. S10†).
Structure elucidation using 3D ED and solid-state NMR
To comprehend the distinct solid-state UV-visible absorption of DHICA, realization of the molecular structure and packing of DHICA is essential. As the polymerization tendency of 5,6-DHICA prohibits traditional crystallization methods based on slowly oversaturating a solution, crystal structure determination needed to be performed with a powder sample. This precluded SC-XRD, but the nanocrystalline powder was still suitable for 3D ED (Fig. 2 and S11, Table S2†). Data were recorded on an ELDICO ED-1 electron diffractometer and analysed as further detailed in the ESI.† DHICA crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (a = 7.49 Å, b = 10.23 Å, c = 10.81 Å, α = 82.27°, β = 87.80°, and γ = 70.10°) with two molecules in the asymmetric unit (Table 1). Each of the independent molecules are interconnected as in chains through extensive hydrogen bonding. Depending on the arrangement of the molecules, two types of H-bonded chains, head-to-tail and head-to-head, are identified (Fig. S12†). For DHICA, based on the electron diffraction data alone, the position of the nitrogen atom is ambiguous due to the low sensitivity of the scattering factors for neighbouring elements. To address this challenge in the 3D ED measurements, we employed solid-state NMR in conjunction with DFT calculations. Three sets of information are obtained from the solid-state NMR: (i) chemical shift information related to the electron distribution around the nuclei, which is sensitive to molecular packing, (ii) orientation related to the spin–spin coupling interactions and (iii) quadrupolar interaction, which is related to the coordination environment around the quadrupolar nuclei 14N. 1H NMR, 13C Cross Polarization (13C CP), 2D CP Heteronuclear Correlation (2D 13C{1H} CP HETCOR), 1H–1H Double Quantum-Single Quantum (DQ-SQ), 2D CP Incredible Natural Abundance Double Quantum Transfer Experiment (INADEQUATE), 1H{14N} Dipolar Heteronuclear Multiple-Quantum Coherence (1H{14N} DHMQC) and 1H{14N} Resonance-Echo Saturation-Pulse Double-Resonance (RESPDOR) along with CASTEP calculations were performed to comprehend the spatial arrangement of molecules with respect to each other. The 2D 13C{1H} CP HETCOR spectra provide information about the proton–carbon correlations, relying on the through-space dipolar couplings. To identify the 1H–1H spin pairs in close proximity to each other in DHICA, two dimensional 1H–1H DQ-SQ correlation magic angle spinning (DQ-SQ MAS) NMR experiments were performed (Fig. 3). The 2D CP INADEQUATE spectra rely on J-coupling between adjacent carbon atoms, enabling the mapping of through-bond correlations. The presence of the –NH proton in DHICA enables the utilization of 14N solid-state NMR, offering insights into the coordination environment of the nitrogen species of the molecule.62 The 1H{14N} DHMQC63–66 and 1H{14N} RESPDOR67–69 experiments provide information about the nitrogen environment using proton detection. The assignment of 1H and 13C chemical shifts were done using 2D 13C{1H} CP HETCOR and 2D CP INADEQUATE spectra obtained on labelled and unlabelled samples of DHICA. Additionally, the assignment of solid-state NMR chemical shifts was facilitated by solution state NMR and simulation of chemical shifts using the software package CASTEP.70
(a = 7.49 Å, b = 10.23 Å, c = 10.81 Å, α = 82.27°, β = 87.80°, and γ = 70.10°) with two molecules in the asymmetric unit (Table 1). Each of the independent molecules are interconnected as in chains through extensive hydrogen bonding. Depending on the arrangement of the molecules, two types of H-bonded chains, head-to-tail and head-to-head, are identified (Fig. S12†). For DHICA, based on the electron diffraction data alone, the position of the nitrogen atom is ambiguous due to the low sensitivity of the scattering factors for neighbouring elements. To address this challenge in the 3D ED measurements, we employed solid-state NMR in conjunction with DFT calculations. Three sets of information are obtained from the solid-state NMR: (i) chemical shift information related to the electron distribution around the nuclei, which is sensitive to molecular packing, (ii) orientation related to the spin–spin coupling interactions and (iii) quadrupolar interaction, which is related to the coordination environment around the quadrupolar nuclei 14N. 1H NMR, 13C Cross Polarization (13C CP), 2D CP Heteronuclear Correlation (2D 13C{1H} CP HETCOR), 1H–1H Double Quantum-Single Quantum (DQ-SQ), 2D CP Incredible Natural Abundance Double Quantum Transfer Experiment (INADEQUATE), 1H{14N} Dipolar Heteronuclear Multiple-Quantum Coherence (1H{14N} DHMQC) and 1H{14N} Resonance-Echo Saturation-Pulse Double-Resonance (RESPDOR) along with CASTEP calculations were performed to comprehend the spatial arrangement of molecules with respect to each other. The 2D 13C{1H} CP HETCOR spectra provide information about the proton–carbon correlations, relying on the through-space dipolar couplings. To identify the 1H–1H spin pairs in close proximity to each other in DHICA, two dimensional 1H–1H DQ-SQ correlation magic angle spinning (DQ-SQ MAS) NMR experiments were performed (Fig. 3). The 2D CP INADEQUATE spectra rely on J-coupling between adjacent carbon atoms, enabling the mapping of through-bond correlations. The presence of the –NH proton in DHICA enables the utilization of 14N solid-state NMR, offering insights into the coordination environment of the nitrogen species of the molecule.62 The 1H{14N} DHMQC63–66 and 1H{14N} RESPDOR67–69 experiments provide information about the nitrogen environment using proton detection. The assignment of 1H and 13C chemical shifts were done using 2D 13C{1H} CP HETCOR and 2D CP INADEQUATE spectra obtained on labelled and unlabelled samples of DHICA. Additionally, the assignment of solid-state NMR chemical shifts was facilitated by solution state NMR and simulation of chemical shifts using the software package CASTEP.70
 | ||
| Fig. 2 Selected TEM images and electron diffraction patterns obtained for DHICA. | ||
| Parameters | DHICA |
|---|---|
| a, b, c (Å) | 7.49(7), 10.23(9), 10.81(10) |
| α, β, γ (°) | 82.27(3), 87.79(13), 70.09(13) |
| Space group |
P |
| Formula | C9H7NO4 |
| Independent reflections | 2729 |
| Parameters | 264 |
| Restraints (non-H) | 0 |
| Constraints (non-H) | 0 |
| Resolution (Å) | 0.83 |
| Completeness (%) | 96.9 |
| R int (%) | 14.88 |
| R 1 [l > 2σ(l)] (%) | 13.65 |
| wR 2 [all data] (%) | 36.04 |
| Goodness of fit | 1.09 |
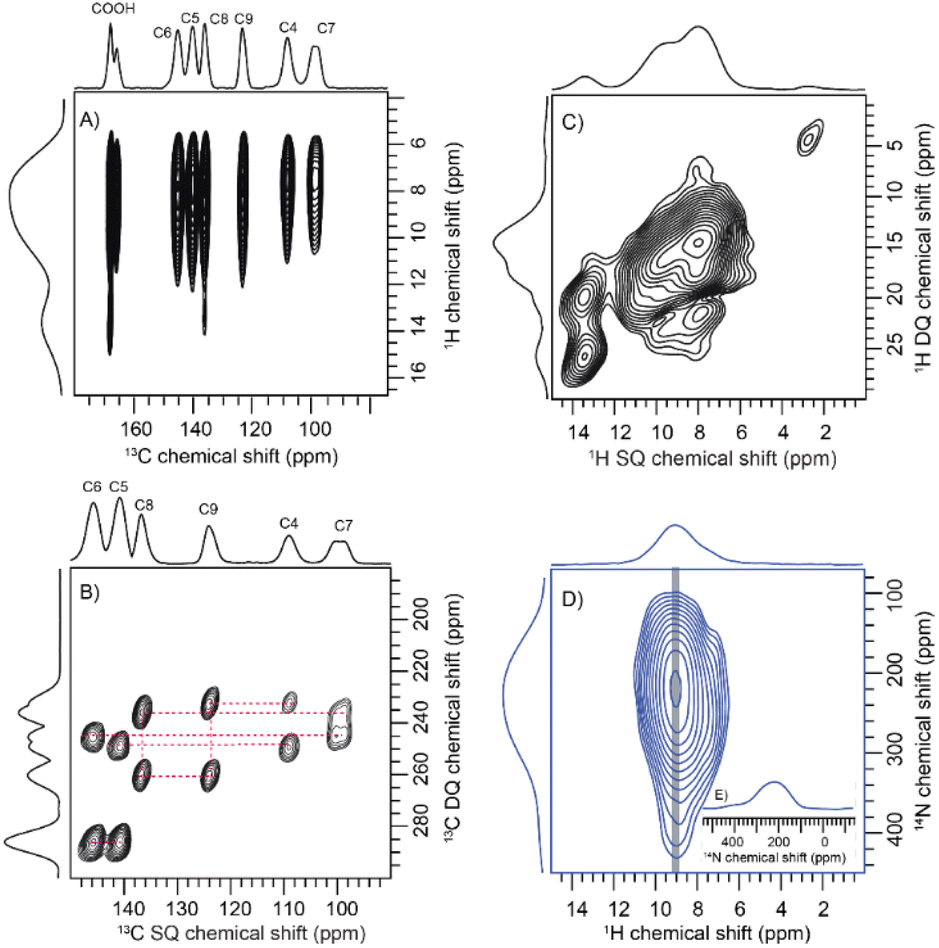 | ||
| Fig. 3 (A) The 2D 13C{1H} cross-polarization heteronuclear correlation (CP HETCOR) NMR spectrum of 13C labelled DHICA obtained at a spinning rate of 60 kHz and a magnetic field of Bo = 14.1 T, utilizing natural isotopic abundance with a contact time of 5 ms; (B) the 2D CP INADEQUATE spectrum of 13C labelled DHICA obtained at 60 kHz spinning speed; (C) the 1H–1H DQ-SQ spectrum of the sample collected at 60 kHz with a recoupling time corresponding to two rotor cycles; (D) the 1H{14N} DHMQC spectrum of the DHICA monomer acquired at a spinning speed of 50 kHz; (E) the one-dimensional slice extracted from the 14N dimension of the 1H{14N} DHMQC spectrum. | ||
The solid-state NMR measurements were performed on two sets of DHICA samples; (i) the unlabelled DHICA and (ii) selectively 13C-labelled DHICA (C2 and C3 carbons are unlabelled). To gather information about the various carbons in DHICA, solid-state 13C NMR is performed (Fig. S13, Table S3–S10†). The 13C{1H} CP HETCOR spectrum (Fig. 3A) of the 13C labelled DHICA monomer at short contact time has mainly two peaks corresponding to C7 and C4 carbons, which appear at 99.25 ppm and 107.70 ppm respectively. Among the aromatic carbons, C3 is challenging to observe in the 13C{1CH} CP HETCOR spectrum of the 13C labelled sample (Fig. S14†). Three types of aromatic C–H cross peaks are observed in the 2D 1H{13C} CP HETCOR correlation spectrum at a short contact time of 0.5 ms on the unlabelled sample. The C–H groups of C3, C4 and C7 appear at 115.39 ppm, 107.70 ppm and 99.25 ppm respectively. In the CP INADEQUATE spectrum, a correlation of C4 carbon with 139.68 ppm and 122.79 ppm is observed. For C7 carbon, a correlation with 144.50 ppm and 135.61 ppm is observed in the CP INADEQUATE spectrum (Fig. 3B). Five sets of new correlation peaks are observed in the 1H{13C} CP HETCOR spectrum collected at a long contact time of 5 ms for the unlabelled DHICA sample. The observed 13C chemical shifts at 122.79 ppm, 135.61 ppm, 139.65 ppm and 144.51 ppm in the long contact times correspond to carbon atoms without directly attached protons. The peak corresponding to 135.61 ppm shows a correlation with C7 carbon and a peak at 122.79 ppm. Similarly, 122.79 ppm shows correlation with C4 carbon and a peak at 135.61 ppm. In the 2D CP INADEQUATE spectrum (Fig. 3B), the peak at 139.65 ppm shows a correlation with both C4 carbon and a peak at 144.51 ppm. Similarly, the peak at 144.51 ppm exhibits a correlation with both C7 carbon and a peak at 139.65 ppm. Thus, it can be concluded that 139.65 ppm corresponds to the C5 carbon and 144.51 ppm corresponds to the C6 carbon. The carbon on the –COOH group appears at 167.28 ppm and 165.32 ppm and correlation with any nearby carbon is not observed in the CP INADEQUATE spectrum due to the absence of nearby labelled carbons. In the proposed packing the –COOH moiety exists in two different environments, in one asymmetric unit it exists as a dimer whereas in the other the –COOH is in proximity to the –OH group. The carboxylic acid group which is closer to the protons will give an intense peak in the CP spectrum relative to the other (Fig. S14 and S15†). To investigate the –NH group, the 1H{14N} DHMQC and 1H{14N} RESPDOR spectra were acquired. The quadrupolar coupling constant (Cq) of the –NH in DHICA is around 4 MHz. The 1H{14N} DHMQC spectrum of the precursor L-DOPA and related compounds are also shown in the ESI for reference.†71 The computed 14N NMR chemical shifts of the NH protons using DFT in CASTEP, are slightly different in the two independent DHICA molecules in the crystal structure. The RESPDOR 1H{14N} curve of DHICA corresponds to the direct N–H bond (Fig. 3B and C), in line with the observations made in a standard sample of histidine hydrochloride monohydrate (Fig. S16†).72
In addition to 13C solid-state NMR, 1H solid-state NMR chemical shifts are utilized to gain insights into the chemical environments surrounding proton atoms (Fig. S17†). Chemical shift data obtained from proton NMR are categorized into four main categories based on the functional groups, –OH, –NH, –CH and –COOH. The 1H NMR peaks observed at 9.2, 7.3, 9.8 and 9.5 ppm are arising from the hydroxyl protons of DHICA. Additionally, the protons in the NH group are observed at 8.7 and 8.9 ppm. In the 1H–1H DQ-SQ spectrum (Fig. 3C), a broad autocorrelation peak appears at around 8.7 ppm and 8.9 ppm corresponding to the NH protons. Due to peak overlap, distinguishing the various protons in the 1H–1H DQ-SQ spectrum is challenging. The aromatic protons appear at around 7.0 ppm in the 1H spectrum and corresponding autocorrelation peaks are observed in the 1H–1H DQ-SQ spectrum. The 1H{13C} CP HETCOR spectrum at short contact time facilitates the assignment of aromatic protons (Table S3†). The protons on carbon C3 were assigned using the 13C{1H} CP HETCOR spectrum of the unlabelled sample, which corresponds to 7.1 ppm. The protons on C4 and C7 correspond to 7.4 and 7.3 ppm respectively. A 1H NMR peak appearing at 14.5 ppm in the 13C{1H} CP HETCOR spectrum at a short contact time aligns with the DFT calculated chemical shift of the carboxylic acid proton (Fig. S14†). In 13C{1H} CP HETCOR at a long contact time of 5 ms (Fig. 3A), a proton chemical shift is observed at 13.4 ppm, correlating with a carbon chemical shift of 167.28 ppm. The root mean square deviation (RMSD) between the 1H chemical shifts observed in solution-state and solid-state NMR is calculated to be 1.4 ppm. The observed longitudinal relaxation time (T1) for carboxylic acid protons is 5 s and that of aromatic protons is 7 s (Fig. S18†). The peak appearing at around 1.0 ppm is possibly due to the trace amounts of hexane residue retained in the sample during purification.
The 13C{1H} CP HETCOR spectrum obtained with long contact time provide details regarding both intermolecular and intramolecular correlations, pointing towards the distance constraints (Fig. 3A and S19, S20†). Though the chemical shifts are distinguishable, it is difficult to differentiate between inter and intramolecular correlations in the case of DHICA. But the chemical shift is sensitive to the packing, and it is a valuable tool to understand the spatial arrangement of the molecules. Four distinct model cases (A, B, C and D) were devised to account for the uncertainties identified in the 3D ED measurements (Fig. S21† and Table 2, S3–S10†). These cases account for the possible orientations related to the flipping around of the long axis of the two independent DHICA molecules. Subsequently, DFT calculations, along with NMR chemical shift predictions using CASTEP, were performed for each model. Among the four cases, case A demonstrated the lowest root mean square deviation (RMSD) of 1.58 ppm in the 13C chemical shift (Table S3–S11†). Similar to the 13C chemical shift, the 1H chemical shift is also sensitive to packing and can be compared with the simulated chemical shift. The obtained 1H RMSD for case A is 0.34 ppm. The calculated energies show a minimum for case A in line with the RMSD of the 13C chemical shift (Table 2). Inspection of the structure reveals that this orientation has the closest interchain N–H⋯O contacts for both the DHICA units (Fig. S22†). Case A was thus used for the final structure refinement which is presented further.
| Label | Relative energy (kcal mol−1) | 13C RMSD (ppm) |
|---|---|---|
| Case A | 0.00 | 1.58 |
| Case B | 6.02 | 4.92 |
| Case C | 4.39 | 5.40 |
| Case D | 7.74 | 7.18 |
Insights into the molecular packing
DHICA crystallized in the P space group (Table 1). Hydrogen bonding and π–π stacking are identified as the dominant interactions in the DHICA crystal architecture (Fig. 4). Among hydrogen bonding, both conventional and unconventional hydrogen bonds are recognized in the crystal packing. Each of the DHICA stacks are linked by C⋯C contacts along the a-axis while inter-stacks are connected by O⋯H and C⋯H contacts. The crystalline assembly is propagated through co-linear stacks (θ = 0.00°) directed by O⋯H contacts as well as near-orthogonal stacks (θ = 83.91°) led by C⋯H contacts (Fig. 4 and S23†). Six dimers (D1–D6, Fig. S24†) are identified within the crystal packing where D1, D2, and D3 are π–π stacked while D4, D5, and D6 are H-bonded. Hirshfeld surface analysis73 enumerates the H⋯H (29.3%), O⋯H (19.5%), C⋯C (10.1%) and C⋯H (7.3%) contacts which majorly contribute to the crystal packing in DHICA (Fig. S25†). The crystal assembly of DHICA exhibits extensive π–π stacking, which is not found in the packing of the other eumelanin monomer, DHI.22 DHI exhibited extended hydrogen bonding between its hydroxyl substituents resulting in a helical assembly. A herringbone packing motif was observed in DHI whereas the π–π stacking influences DHICA to pack in the beta motif.
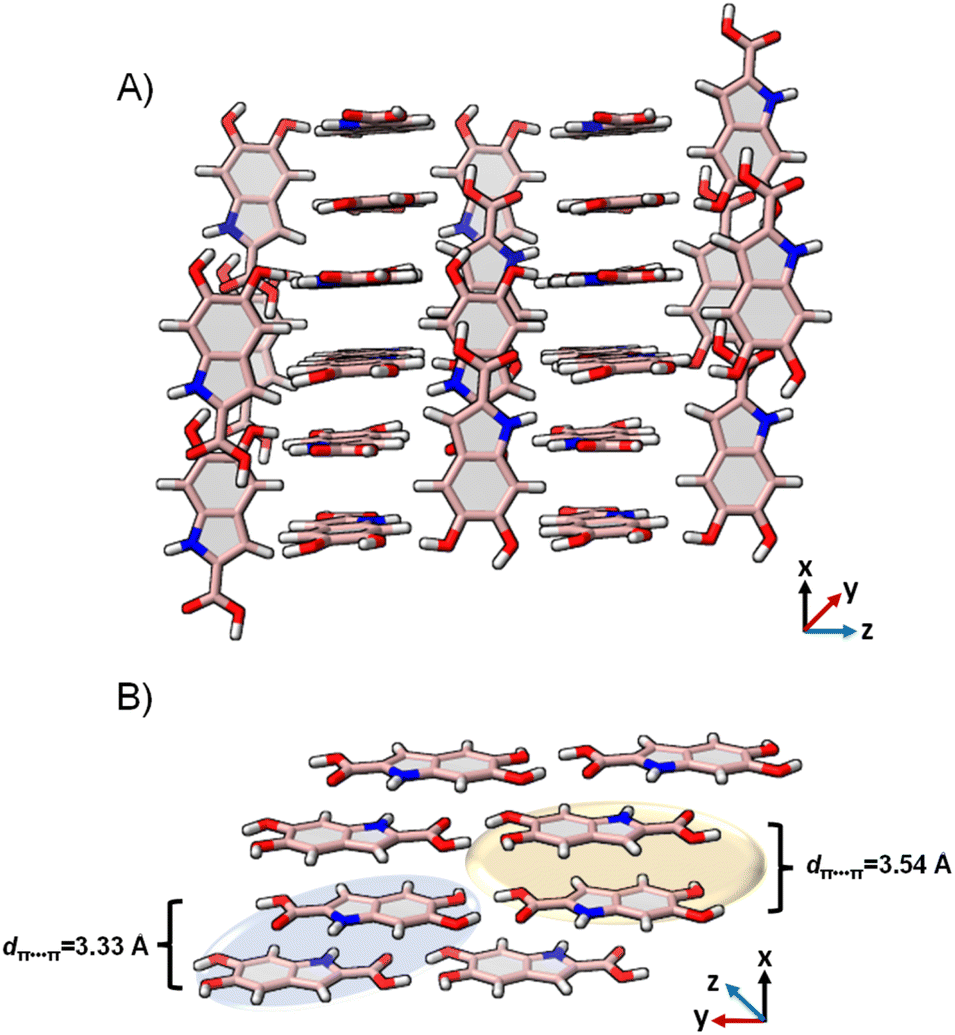 | ||
| Fig. 4 (A) The crystal packing of DHICA showing the near-perpendicular orientation of the π–π stacks; (B) π–π stacked dimers connected to the adjacent stack through hydrogen bonding. | ||
Efficient molecular self-assembly relies on the recognition among intermolecular functionalities, typically resulting in the creation of smaller repeating units or supramolecular synthons. The supramolecular synthons in the DHICA crystals were analysed by using Bader's quantum theory of atoms in molecules (QTAIM) analysis.74 The (3 + 1) ring critical points were identified in each of the hydrogen bonded dimers (Fig. S26 and S27†). The synthons in D1–D3 dimers are facilitated by C⋯C–H, C⋯N–H, C⋯O–H and O⋯C–C contacts while in D4–D6 dimers, synthon formation is orchestrated by the hydrogen bonds between the –COOH and –OH groups. Notably, the D6 dimer demonstrated resonance assisted hydrogen bonding (RAHB) with greater energetic stability (Fig. S28†). Resonance assisted hydrogen bonds (RAHBs) are particularly strong hydrogen bonds present in uncharged molecules, where the conjugated bonds display an equalization of lengths through a pseudo-ring.75 Along with RAHB, bifurcated hydrogen bonding, previously discovered in DHI crystals are also observed in the D6 dimer of DHICA (Fig. S28†).
Truncated symmetry adapted perturbation theory (SAPT(0)) analysis76 of DHICA dimers reveals a notable increase in stabilization for the D6 dimer (ESAPT(0)int = –25.75 kcal mol−1, Table S12†). This enhancement may be attributed to the presence of an eight-membered supramolecular synthon within the dimer, as outlined in Fig. S27.† The overall stabilization of the D6 orientation results from a more substantial contribution of electrostatic (EelcSAPT(0) = –36.46 kcal mol−1) and induction (EindSAPT(0) = –17.83 kcal mol−1) energies to the total SAPT energy. The prominent role of RAHB and bifurcated hydrogen bonding in forming the D6 synthon clarifies the energy stability observed for the D6 dimer in the SAPT(0) analysis with respect to other dimer orientations. The conventionally hydrogen bonded dimers D1 and D2 are mostly stabilized by electrostatic energies while dispersion energies contribute the most to the total stabilization energies of the π–π stacked dimers (D1–D3).
Exciton interactions within the molecular assembly
The molecular architecture of the eumelanin precursor herein presents a valuable framework for understanding the underlying mechanisms of energy dissipation responsible for the characteristic photo-functionality of eumelanin. Previous experimental and theoretical studies have implied the presence of excitation energy transfer phenomena within eumelanin.77–79 The efficiency of energy transfer between assembled chromophores is dictated by the intrinsic electronic coupling over both short and long distances.80 The long-range coulombic coupling stems from the relative orientation of transition dipoles of individual monomers (Eqn S1†). At lower interchromophoric separations, additional contributions due to charge transfer coupling (JCT) are also instrumental to the overall electronic coupling (Eqn S2†).81 The calculated coulombic and charge transfer coupling constants are given in Table 3. While coulombic coupling is prominent in all of the dimers (D1–D6), charge transfer coupling contributes the most in the case of the stacked dimers D1, D2 and D3. Among the π–π stacked dimers, the D1 dimer with an intermolecular distance of dπ–π = 3.33 Å exhibits the highest values of electron (te = 0.146 eV) and hole transfer integrals (th = −0.164 eV), facilitating a higher quotient of CT coupling (JCT = 0.157 eV). Fragment-based excited-state analysis by Plasser was employed to investigate the exciton delocalization within DHICA units in the crystalline structure using the TheoDORE package (Tables S13–S18†).82 The degree of exciton delocalization among the fragment units is assessed through the participation ratio (PR). The level of involvement of fragments in exciton delocalization is represented by the mean position, denoted as the POS value, which signifies the extent of participation of one or more units. The classification of excited states into charge transfer or Frenkel is determined by the CT number, which tends towards one for pure charge transfer states and towards zero for pure Frenkel states. In dimers D1, D2, and D3, exhibiting significant orbital overlap, the higher excited states show high CT character (CTD1 = 0.86, PRD1 = 2.00; CTD2 = 0.91, PRD2 = 2.00; CTD3 = 0.88, PRD3 = 1.99) suggesting the excitation energy transfer of the charge transfer excitons within the π–π stacked units in the crystalline assembly. Conversely, hydrogen-bonded dimers D3, D5, and D6 display minimal CT character in the excited states, indicating their limited contribution to CT exciton generation and subsequent delocalization within the crystalline structure. The strong exciton coupling in the π–π stacks promotes the delocalization of excitons, ultimately leading to efficient charge transfer by channelling the excitation energy within the stack. The charge-density difference plots for the excited states also depict the delocalised excitons within the D1-D3 dimers (Fig. S29†). The remarkable topology and significant charge transfer characteristics of DHICA signify the uniquely efficient energy transfer mechanism adopted by the DHICA monomer architecture compared to that of DHI, reminiscent of the diverse energy funnelling mechanisms in eumelanin/eumelanin-like materials.83–85| Dimer | t e (eV) | t h (eV) | J CT (eV) | J Coul (eV) |
|---|---|---|---|---|
| D1 | 0.146 | −0.164 | 0.157 | −0.123 |
| D2 | 0.181 | −0.096 | 0.081 | −0.488 |
| D3 | 0.043 | 0.149 | −0.017 | −0.009 |
| D4 | 0.025 | −0.022 | 0.000 | −0.097 |
| D5 | 0.024 | 0.010 | 0.000 | 0.066 |
| D6 | 0.017 | 0.081 | −0.002 | 0.115 |
Probing DHICA-melanin with solid-state NMR
Having established the molecular assembly and spectroscopic features of the key eumelanin monomer, DHICA, we analyse the structure of the polymer of DHICA, that is, DHICA-melanin. DHICA-melanin exhibits an amorphous nature, and is insoluble in most common solvents, posing challenges for conventional characterization techniques. The amorphous nature of DHICA-melanin is evident from the broad peak observed in the PXRD pattern (Fig. S30†) as well as the absence of diffraction patterns in 3D ED experiments. However, by extending the solid-state NMR experiments previously discussed for DHICA, we can gain insights into the molecular structure of DHICA-melanin (Table S19 and S20†). The 13C{1H} CP HETCOR experiments along with CP INADEQUATE (Fig. 5A) experiments were performed on 13C labelled samples to fully assign the chemical shifts in DHICA-melanin (Fig. S31–S34†).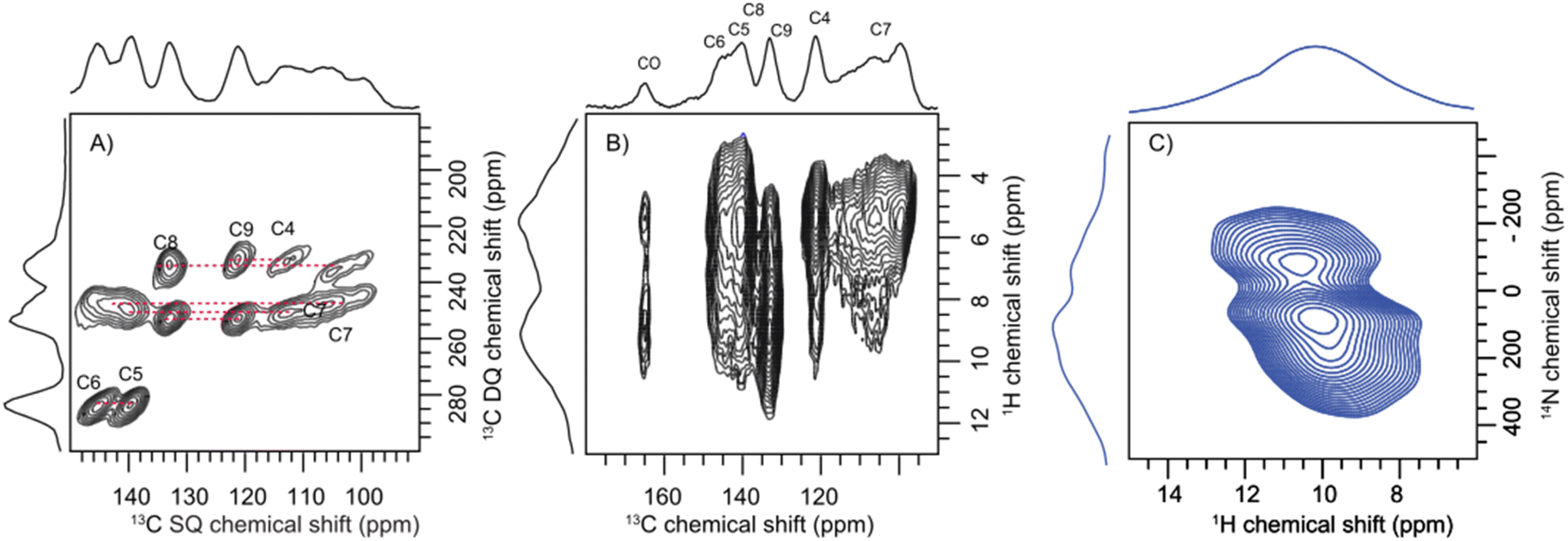 | ||
| Fig. 5 (A) The 2D CP INADEQUATE spectrum of 13C selectively labelled DHICA-melanin showing the correlation between the adjacent carbon atoms. (B) The 2D 13C{1H} CP HETCOR NMR spectrum of DHICA-melanin at a spinning speed of 60 kHz and a magnetic field strength of Bo = 14.1 T; (C) the 1H{14N} DHMQC spectrum of DHICA-melanin collected at 50 kHz spinning speed. | ||
A comparison of the solid-state 1H NMR spectra of DHICA and DHICA-melanin is shown in Fig. S17.† The two dimensional 13C{1H} CP HETCOR spectrum of DHICA-melanin at short contact time shows the CH carbons (Fig. 5). As expected, the 13C{1H} CP HETCOR spectrum of DHICA-melanin exhibits broader features relative to the monomer, reflecting the amorphous nature of DHICA-melanin. The 13C{1H} CP HETCOR spectrum acquired with long contact time, assists in identifying all the individual carbon atoms within DHICA-melanin. To resolve the individual carbon peaks, CP INADEQUATE spectrum was collected for 13C labelled DHICA-melanin. While most correlations observed in DHICA are also present in DHICA-melanin, there is a noticeable distribution of chemical shifts for C7 in DHICA-melanin. The localized broadening observed at around C7 may reflect variations in the packing of melanin units or the presence of multiple conformational states. A similar broadening is seen for C4, which could also be attributed to disorder. To further elucidate the potential heterogeneities in the structure and packing of DHICA-melanin, advanced theoretical calculations are necessary.
The carboxylic acid proton which appeared at around 13.4 ppm in DHICA is not present in DHICA-melanin. The 13C{1H} CP HETCOR correlation peak at 160 ppm indicates that the carbonyl group is in proximity to protons on the hydroxyl group. The peak observed at 145.2 ppm in the HETCOR correlation spectrum of DHICA-melanin is in line with the observation of a peak at 145.60 ppm in DHICA, which belongs to C6 carbon. In the case of C8 and C9 carbons in the monomer, peaks appear at 135.61 ppm and 122.79 ppm, respectively, whereas in DHICA-melanin, these peaks shift to 132.92 ppm and 121.65 ppm. To understand about the nitrogen environments, 1H{14N} DHMQC spectrum were acquired at various recoupling times. The broad 1H{14N} DHMQC spectrum suggests the presence of more than one nitrogen species in the system, primarily attached to protons, as indicated by the experiments conducted with a short recoupling time of 80 μs (Fig. 5C). The NH peaks shift from 9.0 ppm to 10.0 ppm and 10.7 ppm clearly showing the formation of new nitrogen species in DHICA-melanin. Compared to the 1H{14N} DHMQC spectrum of histidine hydrochloride monohydrate and L-DOPA, the broad peak is possibly a combination of different nitrogen environments (Fig. S35 and S36†).
The FT-IR spectrum of DHICA has a prominent N–H stretching band at 3435 cm−1, a broad O–H and COO–H stretching band at 3265 cm−1 and aromatic sp2-hybridized C–H stretching peaks.86 In the case of DHICA-melanin, in addition to the N–H and O–H stretching bands, a sp3-hybridized C–H stretching peak is observed at 2980 cm−1. The sp3-hybridized C–H bending, which is not found in DHICA is another prominent vibrational mode in the FT-IR spectrum of DHICA-melanin (Fig. S5†), corroborating the presence of saturated indoline products along with indole moieties.87,88 Additionally, the scanning electron microscope (SEM) images of DHICA-melanin exhibited a lamellar morphology with superficial attachment of globular particles on the surface (Fig. S37†). Comprehensive insights into the structure of DHICA-melanin will necessitate the application of advanced NMR and theoretical methods, which could be a potential direction for future research.
Conclusions
To conclude, the detailed structure and excitonic properties of DHICA, a critical yet underexplored constituent of eumelanin are elucidated in this work. DHICA crystallizes in an antiparallel stacking arrangement within a hydrogen-bonded network and exhibits the P space group with unit cell parameters of a = 7.49 Å, b = 10.23 Å, c = 10.81 Å, α = 82.27°, β = 87.80°, and γ = 70.10 Å. One of the challenges in correlating the photophysics of DHICA with its structure lies in the difficulty in crystallizing DHICA, which is hindered by its inherent disorder and nanocrystalline nature. Hence, we employed an interdisciplinary approach integrating 3D ED, solid-state NMR and DFT calculations to solve the crystal structure of DHICA. The ambiguity in the orientation of the N–H group of DHICA in 3D ED experiments was addressed by generating a library of structures with solid-state NMR and DFT calculations. The broad absorption spectrum of DHICA in the solid state is due to the significant charge transfer and coulombic coupling within the crystal architecture of DHICA. Charge transfer exciton delocalization within the π–π stacks of DHICA serves as the major energy dissipation channel within the crystalline assembly of the eumelanin precursor, as suggested by fragment based excited state analysis. A series of solid-state NMR experiments was performed to get an insight into the structure of the insoluble polymer of 13C labelled DHICA, i.e., DHICA-melanin. Distribution of chemical shifts at the C7 carbon and multiple nitrogen species has been recognized from the solid-state NMR spectra of 13C-labelled DHICA-melanin. Advanced solid-state NMR experiments, coupled with theoretical modelling, will be required to enhance our understanding of the intricate structure of eumelanin. Extending our methodology to other members of the melanin family could be intriguing and holds promise for advancing melanin-centred biomedical research.
Data availability
All experimental procedures and characterization data are available in the ESI.†Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
M. H. acknowledges the Science and Engineering Research Board, Department of Science and Technology, Govt. of India (CRG/2019/002119). B. T. and R. M. acknowledges the Tamkeen New York University Abu Dhabi (NYUAD, UAE) for the financial support. The research was partially carried out using the Core Technology Platforms at New York University Abu Dhabi. K. V. acknowledges CSIR for financial support. We acknowledge the early assistance provided by Ms Medha Gangopadhyay and Ms Diana Thomas. The authors greatly acknowledge the support involving high-performance computing time at the high-performance computing resources at New York University Abu Dhabi (NYUAD, UAE) and the Padmanabha cluster at IISER TVM, India.References
- M. A. Pathak, F. J. Riley, T. B. Fitzpatrick and W. L. Curwen, Nature, 1962, 193, 148–150 CrossRef CAS PubMed.
- M. D'Ischia, A. Napolitano, V. Ball, C. T. Chen and M. J. Buehler, Acc. Chem. Res., 2014, 47, 3541–3550 CrossRef PubMed.
- P. Meredith and T. Sarna, Pigm. Cell Res., 2006, 19, 572–594 CrossRef CAS PubMed.
- J. E. Mcginness, Science, 1972, 177, 896–897 CrossRef CAS PubMed.
- C. T. Chen, C. Chuang, J. Cao, V. Ball, D. Ruch and M. J. Buehler, Nat. Commun., 2014, 5, 1–10 Search PubMed.
- M. D'Ischia, A. Napolitano, A. Pezzella, P. Meredith and T. Sarna, Angew. Chem., Int. Ed., 2009, 48, 3914–3921 CrossRef PubMed.
- C. Guzmán-Afonso, Y. Hong, H. Colaux, H. Iijima, A. Saitow, T. Fukumura, Y. Aoyama, S. Motoki, T. Oikawa, T. Yamazaki, K. Yonekura and Y. Nishiyama, Nat. Commun., 2019, 10, 3537 CrossRef.
- S. Chatterjee, R. Prados-Rosales, S. Tan, B. Itin, A. Casadevall and R. E. Stark, Org. Biomol. Chem., 2014, 12, 6730–6736 RSC.
- Q. Z. Ni, B. N. Sierra, J. J. La Clair and M. D. Burkart, Chem. Sci., 2020, 11, 7836–7841 RSC.
- A. Ilina, K. E. Thorn, P. A. Hume, I. Wagner, R. R. Tamming, J. J. Sutton, K. C. Gordon, S. K. Andreassend, K. Chen and J. M. Hodgkiss, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2212343119 CrossRef CAS PubMed.
- M. Chergui, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2216527119 CrossRef CAS PubMed.
- W. Cao, X. Zhou, N. C. McCallum, Z. Hu, Q. Z. Ni, U. Kapoor, C. M. Heil, K. S. Cay, T. Zand, A. J. Mantanona, A. Jayaraman, A. Dhinojwala, D. D. Deheyn, M. D. Shawkey, M. D. Burkart, J. D. Rinehart and N. C. Gianneschi, J. Am. Chem. Soc., 2021, 143, 2622–2637 CrossRef CAS PubMed.
- S. P. Nighswander-Rempel, I. B. Mahadevan, P. V. Bernhardt, J. Butcher and P. Meredith, Photochem. Photobiol., 2008, 84, 620–626 CrossRef CAS PubMed.
- A. Corani, A. Huijser, A. Iadonisi, A. Pezzella, V. Sundström and M. D'Ischia, J. Phys. Chem. B, 2012, 116, 13151–13158 CrossRef CAS PubMed.
- C. Grieco, F. R. Kohl, A. T. Hanes and B. Kohler, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- M. Gauden, A. Pezzella, L. Panzella, M. T. Neves-Petersen, E. Skovsen, S. B. Petersen, K. M. Mullen, A. Napolitano, M. D'Ischia and V. Sundström, J. Am. Chem. Soc., 2008, 130, 17038–17043 CrossRef CAS PubMed.
- S. Olsen, J. Riesz, I. Mahadevan, A. Coutts, J. P. Bothma, B. J. Powell, R. H. McKenzie, S. C. Smith and P. Meredith, J. Am. Chem. Soc., 2007, 129, 6672–6673 CrossRef CAS PubMed.
- C. M. R. Clancy, J. B. Nofsinger, R. K. Hanks and J. D. Simon, J. Phys. Chem. B, 2000, 104, 7871–7873 CrossRef CAS.
- A. Choudhury, D. Ghosh and R. Li, Chem. Commun., 2020, 56, 10481–10484 RSC.
- A. Corani, A. Pezzella, T. Pascher, T. Gustavsson, D. Markovitsi, A. Huijser, M. D'Ischia and V. Sundström, J. Phys. Chem. Lett., 2013, 4, 1383–1388 CrossRef CAS PubMed.
- M. D'Ischia, A. Napolitano and A. Pezzella, Eur. J. Org Chem., 2011, 5501–5516 CrossRef.
- D. Sasikumar, K. Vinod, J. Sunny and M. Hariharan, Chem. Sci., 2022, 13, 2331–2338 RSC.
- S. W. Crane, O. Ghafur, T. Y. Cowie, A. G. Lindsay, J. O. F. Thompson, J. B. Greenwood, M. W. P. Bebbington and D. Townsend, Phys. Chem. Chem. Phys., 2019, 21, 8152–8160 RSC.
- J. Wang and L. Blancafort, Angew. Chem., Int. Ed., 2021, 60, 18800–18809 CrossRef CAS PubMed.
- M. Gauden, A. Pezzella, L. Panzella, A. Napolitano, M. D'Ischia and V. Sundstro, J. Phys. Chem. B, 2009, 113, 12575–12580 CrossRef CAS PubMed.
- J. J. Nogueira, A. Corani, A. El Nahhas, A. Pezzella, M. D'Ischia, L. González and V. Sundström, J. Phys. Chem. Lett., 2017, 8, 1004–1008 CrossRef CAS PubMed.
- A. Pezzella, L. Panzella, O. Crescenzi, A. Napolitano, S. Navaratman, R. Edge, E. J. Land, V. Barone and M. D'Ischia, J. Am. Chem. Soc., 2006, 128, 15490–15498 CrossRef CAS.
- A. Pezzella, A. Iadonisi, S. Valerio, L. Panzella, A. Napolitano, M. Adinolfi and M. D'Ischia, J. Am. Chem. Soc., 2009, 131, 15270–15275 CrossRef CAS PubMed.
- R. Micillo, L. Panzella, M. Iacomino, G. Prampolini, I. Cacelli, A. Ferretti, O. Crescenzi, K. Koike, A. Napolitano and M. D'Ischia, Sci. Rep., 2017, 7, 1–12 CrossRef PubMed.
- P. Ghosh and D. Ghosh, Phys. Chem. Chem. Phys., 2019, 21, 6635–6642 RSC.
- A. Pezzella, A. Napolitano, M. D'Ischia and G. Prota, Tetrahedron, 1996, 52, 7913–7920 CrossRef CAS.
- B. Thomas, J. Rombouts, K. B. S. S. Gupta, R. V. A. Orru, K. Lammertsma and H. J. M. de Groot, Chem.–Eur. J., 2017, 23, 9346 CrossRef CAS PubMed.
- B. Thomas, J. Rombouts, G. T. Oostergetel, K. B. S. S. Gupta, F. Buda, K. Lammertsma, R. Orru and H. J. M. de Groot, Chem.–Eur. J., 2017, 23, 3280–3284 CrossRef CAS PubMed.
- B. Thomas, R. K. Dubey, M. T. B. Clabbers, K. B. S. S. Gupta, E. van Genderen, W. F. Jager, J. P. Abrahams, E. J. R. Sudholter and H. J. M. de Groot, Chem.–Eur. J., 2018, 24, 14989 CrossRef CAS PubMed.
- R. Mathew, A. Mazumder, P. Kumar, J. Matula, S. Mohamed, P. Brazda, M. Hariharan and B. Thomas, Chem. Sci., 2024, 15, 490–499 RSC.
- M. Gemmi, E. Mugnaioli, T. E. Gorelik, U. Kolb, L. Palatinus, P. Boullay, S. Hovmöller and J. P. Abrahams, ACS Cent. Sci., 2019, 5, 1315–1329 CrossRef CAS PubMed.
- P. Brázda, M. Klementová, Y. Krysiak and L. Palatinus, IUCrJ, 2022, 9, 735–755 CrossRef PubMed.
- D. P. Karothu, Z. Alhaddad, C. R. Göb, C. J. Schürmann, R. Bücker and P. Naumov, Angew. Chem., Int. Ed., 2023, 62, e202303761 CrossRef CAS PubMed.
- P. B. Klar, Y. Krysiak, H. Xu, G. Steciuk, J. Cho, X. Zou and L. Palatinus, Nat. Chem., 2023, 15, 848–855 CrossRef CAS PubMed.
- B. Reif, S. E. Ashbrook, L. Emsley and M. Hong, Nat. Rev. Methods Primers, 2021, 1, 2 CrossRef CAS PubMed.
- P. Hodgkinson, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 118–119, 10–53 CrossRef CAS PubMed.
- D. L. Bryce, IUCrJ, 2017, 4, 350–359 CrossRef CAS PubMed.
- F. Taulelle, Solid State Sci., 2004, 6, 1053–1057 CrossRef CAS.
- C. J. H. Smalley, H. E. Hoskyns, C. E. Hughes, D. N. Johnstone, T. Willhammar, M. T. Young, C. J. Pickard, A. J. Logsdail, P. A. Midgley and K. D. M. Harris, Chem. Sci., 2022, 13, 5277–5288 RSC.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azaïs, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- R. Mathew, B. Stevensson and M. Edén, J. Phys. Chem. B, 2021, 125, 10985–11004 CrossRef CAS PubMed.
- G. N. M. Reddy, M. Malon, A. Marsh, Y. Nishiyama and S. P. Brown, Anal. Chem., 2016, 88, 11412–11419 CrossRef CAS PubMed.
- K. Vinod, S. D. Jadhav and M. Hariharan, Chem.–Eur. J., 2024, e202400499 CrossRef CAS PubMed.
- M. Balodis, M. Cordova, A. Hofstetter, G. M. Day and L. Emsley, J. Am. Chem. Soc., 2022, 144, 7215–7223 CrossRef CAS PubMed.
- M. Baias, J. N. Dumez, P. H. Svensson, S. Schantz, G. M. Day and L. Emsley, J. Am. Chem. Soc., 2013, 135, 17501–17507 CrossRef CAS PubMed.
- J. Cui, D. L. Olmsted, A. K. Mehta, M. Asta and S. E. Hayes, Angew. Chem., Int. Ed., 2019, 58, 4210–4216 CrossRef CAS PubMed.
- D. V Dudenko, J. R. Yates, K. D. M. Harris and S. P. Brown, CrystEngComm, 2013, 15, 8797–8807 RSC.
- B. G. Caulkins, R. P. Young, R. A. Kudla, C. Yang, T. J. Bittbauer, B. Bastin, E. Hilario, L. Fan, M. J. Marsella, M. F. Dunn and L. J. Mueller, J. Am. Chem. Soc., 2016, 138, 15214–15226 CrossRef CAS PubMed.
- Z. Gan, Chem. Commun., 2006, 4712–4714 RSC.
- L. A. O'Dell and C. I. Ratcliffe, Chem. Commun., 2010, 46, 6774–6776 RSC.
- K. Wakamatsu and S. Ito, Anal. Biochem., 1988, 170, 335–340 CrossRef CAS PubMed.
- A. Corani, A. Huijser, T. Gustavsson, D. Markovitsi, P. Å. Malmqvist, A. Pezzella, M. D'Ischia and V. Sundström, J. Am. Chem. Soc., 2014, 136, 11626–11635 CrossRef CAS PubMed.
- Z. Fernández, L. Sánchez, S. S. Babu and G. Fernández, Angew. Chem., Int. Ed., 2024, 63, e202402259 CrossRef PubMed.
- T. Ligonzo, M. Ambrico, V. Augelli, G. Perna, L. Schiavulli, M. A. Tamma, P. F. Biagi, A. Minafra and V. Capozzi, J. Non-Cryst. Solids, 2009, 355, 1221–1226 CrossRef CAS.
- L. Panzella, G. Gentile, G. D'Errico, N. F. Della Vecchia, M. E. Errico, A. Napolitano, C. Carfagna and M. D'Ischia, Angew. Chem., Int. Ed., 2013, 52, 12684–12687 CrossRef CAS PubMed.
- M. L. Tran, B. J. Powell and P. Meredith, Biophys. J., 2006, 90, 743–752 CrossRef CAS PubMed.
- J. A. Jarvis, M. Concistre, I. M. Haies, R. W. Bounds, I. Kuprov, M. Carravetta and P. T. F. Williamson, Phys. Chem. Chem. Phys., 2019, 21, 5941–5949 RSC.
- A. V Wijesekara, A. Venkatesh, B. J. Lampkin, B. VanVeller, J. W. Lubach, K. Nagapudi, I. Hung, P. L. Gor’kov, Z. Gan and A. J. Rossini, Chem.–Eur. J., 2020, 26, 7881–7888 CrossRef PubMed.
- A. Bax, R. H. Griffey and B. L. Hawkins, J. Am. Chem. Soc., 1983, 105, 7188–7190 CrossRef CAS.
- Z. Gan, J. P. Amoureux and J. Trébosc, Chem. Phys. Lett., 2007, 435, 163–169 CrossRef CAS.
- N. T. Duong and Y. Nishiyama, Phys. Chem. Chem. Phys., 2022, 24, 10717–10726 RSC.
- N. T. Duong, F. Rossi, M. Makrinich, A. Goldbourt, M. R. Chierotti, R. Gobetto and Y. Nishiyama, J. Magn. Reson., 2019, 308, 106559 CrossRef CAS PubMed.
- X. Lu, O. Lafon, J. Trébosc and J.-P. Amoureux, J. Magn. Reson., 2012, 215, 34–49 CrossRef CAS PubMed.
- F. A. Perras, A. Arroyave, S. A. Southern, J. V Lamb, Y. Li, A. LaPointe and M. Delferro, Chem. Commun., 2023, 59, 4604–4607 RSC.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr. - Cryst. Mater., 2005, 220, 567–570 CrossRef CAS.
- J. DONOHUE, Nature, 1953, 171, 258 CrossRef CAS PubMed.
- T. Gullion and A. J. Vega, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 47, 123–136 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- A. T. John, A. Narayanasamy, K. P. Sudhakaran and M. Hariharan, Cryst. Growth Des., 2022, 22, 5686–5693 CrossRef CAS.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- V. Petropoulos, A. Mavridi-Printezi, A. Menichetti, D. Mordini, P. Kabacinski, N. C. Gianneschi, M. Montalti, M. Maiuri and G. Cerullo, J. Phys. Chem. Lett., 2024, 15, 3639–3645 CrossRef CAS PubMed.
- F. R. Kohl, C. Grieco and B. Kohler, Chem. Sci., 2020, 11, 1248–1259 RSC.
- J. M. Menter and I. Willis, Pigm. Cell Res., 1997, 10, 214–217 CrossRef CAS PubMed.
- A. Benny, R. Ramakrishnan and M. Hariharan, Chem. Sci., 2021, 12, 5064–5072 RSC.
- N. J. Hestand and F. C. Spano, Acc. Chem. Res., 2017, 50, 341–350 CrossRef CAS PubMed.
- F. Plasser and H. Lischka, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef CAS PubMed.
- C. Grieco, F. R. Kohl, A. T. Hanes and B. Kohler, Nat. Commun., 2020, 11, 1–9 CrossRef.
- M. Matta, A. Pezzella and A. Troisi, J. Phys. Chem. Lett., 2020, 11, 1045–1051 CrossRef CAS PubMed.
- V. Petropoulos, A. Mavridi-Printezi, A. Menichetti, D. Mordini, P. Kabacinski, N. C. Gianneschi, M. Montalti, M. Maiuri and G. Cerullo, J. Phys. Chem. Lett., 2024, 15, 3639–3645 CrossRef CAS PubMed.
- H. Okuda, A. Nakamura, K. Wakamatsu, S. Ito and T. Sota, Chem. Phys. Lett., 2007, 433, 355–359 CrossRef CAS.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428–6435 CrossRef CAS PubMed.
- Q. Z. Ni, B. N. Sierra, J. J. La Clair and M. D. Burkart, Chem. Sci., 2020, 11, 7836–7841 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2347665. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05453a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |