
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater splitting over transition metal-doped SrTiO3 photocatalysts with response to visible light up to 660 nm†
Kyohei
Kaiya
a,
Yoshiya
Ueki
a,
Hiromasa
Kawamoto
a,
Kenta
Watanabe
 a,
Shunya
Yoshino
a,
Yuichi
Yamaguchi
ab and
Akihiko
Kudo
*ab
a,
Shunya
Yoshino
a,
Yuichi
Yamaguchi
ab and
Akihiko
Kudo
*ab
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: a-kudo@rs.tus.ac.jp
bCarbon Value Research Center, Research Institute for Science and Technology, Tokyo University of Science, Noda-shi, Chiba-ken 278-8510, Japan
First published on 6th September 2024
Abstract
Highly efficient water splitting under visible light irradiation was achieved using Ir, Sb, and Al-codoped SrTiO3 of a single particulate metal oxide photocatalyst by a solid-state reaction followed by flux treatment using SrCl2 and loading of a RhCrOx cocatalyst. The photocatalytic activity was improved by Al2O3 addition to the flux treatment, and doping of small amounts of Ir and Sb. It is notable that the water splitting over the photocatalyst proceeded with response to visible light up to 660 nm. This response wavelength is the longest compared with previously reported single particulate visible-light-driven photocatalysts for water splitting. The apparent quantum yield at 420 nm of the optimized photocatalyst was 0.73%. This photocatalyst was active for solar water splitting and gave 0.33% solar-to-hydrogen energy conversion efficiency (STH). Notably, water splitting proceeded giving 0.035% STH under visible light (λ > 440 nm) in a solar spectrum. Additionally, Rh, Ru, and Cr-doped SrTiO3 photocatalysts were also successfully developed for highly efficient water splitting under visible light irradiation by application of the strategies of small amounts of doping, flux treatment with SrCl2 with Al2O3 addition, and loading of a RhCrOx cocatalyst.
Introduction
Solar green hydrogen production by photocatalytic water splitting, which is an uphill reaction to convert sunlight energy into storable chemical energy, is a promising method to solve resource, energy, and environmental issues.1–4 For practical use of this technology, a photocatalytic water splitting system employing powdered materials is attractive due to its simplicity and low-cost.2,5,6 It is important to develop a visible light responsive powdered photocatalyst from the viewpoint of efficient utilization of solar light.The photocatalytic water splitting system is classified into single particulate and Z-scheme systems.2,7–10 The Z-scheme system consists of a H2-evolving photocatalyst, an O2-evolving photocatalyst, and an electron mediator. In contrast to it, a single particulate system has the great advantage of being a simple system. To achieve single particulate photocatalytic water splitting, it is necessary to develop a photocatalyst possessing thermodynamically satisfactory valence and conduction bands for the oxidation and reduction of water, respectively. Additionally, a narrow band gap is required for response to visible light. Therefore, development of a single particulate photocatalyst for efficient water splitting under visible light irradiation is a challenging topic.
(Oxy)nitrides and oxysulfides such as GaN–ZnO,11,12 ZnGeN2–ZnO,13,14 Y2Ti2O5S2,15 Ta3N5,16 TaON,17 LaMg1/3Ta2/3O2N,18 BaTaO2N,19,20 and SrTaO2N21 as single particulate photocatalysts have been developed for water splitting under visible light irradiation. The valence bands of these materials consist of hybridized N 2p–O 2p and S 3p–O 2p orbitals, resulting in a narrow band gap. However, these material groups sometimes suffer from self-oxidation by holes generated by light irradiation. In contrast, metal oxides are promising materials because many of them are chemically stable even under light irradiation. However, they often possess a wide band gap because of a deep valence band consisting of O 2p orbitals.
Doping of metal cations to host materials is a useful strategy for sensitization of metal oxides with a wide bandgap to visible light.2,22–27 As an example, a single particulate Rh and Sb-codoped SrTiO3 (SrTiO3:Rh,Sb) photocatalyst split water under visible light irradiation.28 The photocatalyst responded to visible light up to 500 nm for water splitting. Sb is doped as an Sb5+ ion to a Ti4+ site in a SrTiO3 host, leading to the formation of a Rh3+ ion due to charge compensation. Sb codoping is a key issue because SrTiO3:Rh without the Sb codoping showed no activity for water splitting. Recently, we have developed a SrTiO3:Ir photocatalyst responding to the whole range of visible light up to 700 and 800 nm as O2- and H2-evolving photocatalysts in the presence of a sacrificial electron donor, respectively.29,30 Thus, a SrTiO3:Ir photocatalyst is expected to show activity for water splitting under visible light with a long wavelength.
In recent years, Domen and co-workers have reported that an Al-doped SrTiO3 (SrTiO3:Al) photocatalyst31–33 loaded with Rh/Cr2O3 and CoOOH cocatalysts showed high activity for water splitting under UV light irradiation.34 95.9% apparent quantum yield (AQY) at ca. 360 nm was obtained, indicating that the photo-generated carriers could react with water with negligible recombination. The high activity was attributed to the formation of fine particles of ca. 300 nm with the surface facet for water reduction and oxidation by flux treatment and Al doping during the treatment.34 Furthermore, loading of Rh/Cr2O3 and CoOOH cocatalysts which are highly active sites for the reduction and oxidation of water was also essential to achieve efficient water splitting. However, SrTiO3:Al responds to only UV light because of its wide band gap. Therefore, sensitization of the photocatalyst to visible light is necessary.
In the present study, we have developed an Ir-doped SrTiO3:Al photocatalyst prepared by optimization of doping amounts, flux treatment using SrCl2 with/without Al2O3 addition as a dopant of Al, and a cocatalyst aiming at highly efficient water splitting under visible light irradiation. Additionally, we examined various transition metal-doped SrTiO3:Al photocatalysts for water splitting under visible light irradiation.
Experimental
Preparation of photocatalysts
SrTiO3:M(x%),Sb(y%) (M = Ir, Rh, Ru, Cr, Mn, Fe, Co, Ni) photocatalysts were synthesized by a solid-state reaction (SSR). The starting materials of SrCO3 (Kanto Chemical; 99.9%), TiO2 (Kojundo Chemical; 99.99%), IrO2 (Kojundo Chemical), Rh2O3 (Fujifilm Wako Pure Chemical; 99.5%), RuO2 (Fujifilm Wako Pure Chemical), Cr2O3 (Rare Metallic; 99.9%), MnO2 (Fujifilm Wako Pure Chemical; 99.5%), Fe2O3 (Fujifilm Wako Pure Chemical; 99.0%), Co3O4 (Kojundo Chemical; 99.95%), NiO (Kanto Chemical; 99.9%) and Sb2O3 (Nacalai Tesque; 98%) were mixed in an alumina mortar with an atomic ratio of Sr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ti:M:Sb = 1.00:1 − x − y:x:y. The mixture was calcined at 1173 K for 1 h in an alumina crucible, followed by calcination at 1273 K for 10 h in air. The obtained SrTiO3:M(x%),Sb(y%) (M = Ir, Rh, Ru, Cr, Mn, Fe, Co, Ni), SrCl2·6H2O (Fujifilm Wako Pure Chemicals; 99%), and Al2O3 (Kanto Chemical; 99.0%) were mixed in an alumina mortar in a 1:10:z atomic ratio to Ti. The mixture was heated in air at 1373 K for 10 h in an alumina crucible as the flux treatment. It is reported that Al3+ is doped also from an alumina crucible during the flux treatment.34 Therefore, the samples after the flux treatment were described as SrTiO3:M,Sb,Al (M = Ir, Rh, Ru, Cr, Mn, Fe, Co, Ni) even without Al2O3 addition. A flowchart of the preparation of the photocatalysts is provided in the ESI (Fig. S1†). The resulting powder was washed with water to remove the flux reagent.
:Ti:M:Sb = 1.00:1 − x − y:x:y. The mixture was calcined at 1173 K for 1 h in an alumina crucible, followed by calcination at 1273 K for 10 h in air. The obtained SrTiO3:M(x%),Sb(y%) (M = Ir, Rh, Ru, Cr, Mn, Fe, Co, Ni), SrCl2·6H2O (Fujifilm Wako Pure Chemicals; 99%), and Al2O3 (Kanto Chemical; 99.0%) were mixed in an alumina mortar in a 1:10:z atomic ratio to Ti. The mixture was heated in air at 1373 K for 10 h in an alumina crucible as the flux treatment. It is reported that Al3+ is doped also from an alumina crucible during the flux treatment.34 Therefore, the samples after the flux treatment were described as SrTiO3:M,Sb,Al (M = Ir, Rh, Ru, Cr, Mn, Fe, Co, Ni) even without Al2O3 addition. A flowchart of the preparation of the photocatalysts is provided in the ESI (Fig. S1†). The resulting powder was washed with water to remove the flux reagent.
RhCrOx,12,35,36 Rh2O3, Cr2O3, IrO2, RuO2, and CoOx cocatalysts were loaded on the samples by an impregnation method. The obtained sample was dispersed in aqueous solutions dissolving Rh(NO3)3, Cr(NO3)3, (NH4)2IrCl6, RuCl4, and Co(NO3)2 in a porcelain crucible. The dispersion was heated in air to evaporate water following calcination in air at 623 K for 1 h for loading of RhCrOx, Rh2O3, and Cr2O3 and at 673 K for 2 h for loading of IrO2, RuO2, and CoOx. Rh and CrOx cocatalysts37,38 were also loaded in situ by a photodeposition method using aqueous solutions of Rh(NO3)3 and Cr(NO3)3.
Characterization
The crystal phase of the obtained sample was confirmed by X-ray diffraction (XRD; Rigaku; MiniFlex600) using Cu Kα radiation. Diffuse reflectance spectra were measured using a UV-Vis-NIR spectrometer (JASCO; UbeatV-570) equipped with an integrating sphere and were converted to a K–M function by the Kubelka–Munk method. The chemical composition of the sample was analyzed using an X-ray fluorescence spectrometer (XRF; Rigaku; NEXDE). The morphology and particle size of the sample were observed using a scanning electron microscope (SEM; JEOL; JSM-6700F).Photocatalytic activity
Photocatalytic water splitting was carried out using a top-irradiation reaction cell with a Pyrex window connected to a gas-tight circulation system. A 300 W Xe-arc lamp (PerkinElmer; CERMAX PF300BF) attached to a long-pass filter (HOYA; Y44) and a solar simulator (ASAHI SPECTRA, HAL-320, 0.1 W cm−2) were used as light sources. Irradiated areas using a 300 W Xe-arc lamp and a solar simulator were 33 and 16 cm2, respectively. The evolved H2 and O2 were measured using an online gas chromatograph (Shimadzu; GC-8A, MS-5A column, TCD, Ar carrier). The apparent quantum yield (AQY) was calculated using the following equation.| AQY (%) = 100 × [the number of reacted holes]/[the number of incident photons] = 100 × [the number of evolved O2 molecules] × 4/[the number of incident photons] |
The solar-to-hydrogen energy conversion efficiencies (STH) under full arc and visible light (λ > 440 nm) measured using a solar simulator were calculated using the following equations. Powers of a solar simulator were 0.1 W cm−2.
| STH (%) = (100 × ([ΔG°(H2O)/J mol−1] × [rate of H2 evolution/mol h−1]))/([3600/s h−1] × [solar energy/W cm−2] × [irradiation area/cm2]) = (100 × 237000 × [rate of H2 evolution/mol h−1])/(3600 × 0.1 × 16) |
Results and discussion
Characterization and water splitting over SrTiO3:Ir,Sb,Al photocatalysts prepared by SSR followed by flux treatment in an alumina crucible
X-ray diffraction patterns of SSR-prepared SrTiO3 and SrTiO3:Ir(x%),Sb(y%) (x = 0.001–0.3, y = 0.002–0.6) with and without the flux treatment in an alumina crucible were measured. A single phase of SrTiO3 was obtained for all samples before and after the flux treatment without any impurities of Ir and Sb species. As previously reported, it was considered that Ir and Sb ions were substituted for Ti sites in SrTiO3, judging from the ionic radii; Ti4+: 0.605 Å, Ir3+: 0.68 Å, Ir4+: 0.625 Å, Sb5+: 0.60 Å for 6 coordination. Peak shifts between non-doped SrTiO3 and SrTiO3:Ir(x%),Sb(y%) were not observed, due to the doping amounts of Ir and Sb being very small and/or the ionic radii of Ir and Sb ions being larger and smaller than that of a Ti ion, respectively. The Raman spectra of SSR-prepared SrTiO3:Ir(x%),Sb(2x%),Al (x = 0–0.3) with the flux treatment were also measured (Fig. S2†). New Raman bands around 800 cm−1 were clearly observed with an increase in doping amounts of Ir and Sb although they were not observed for the non-doped sample. It has been reported that the Raman band at around 800 cm−1 appears when different elements are substituted for the B-site of ABO3 with the perovskite structure due to the formation of an ordered distribution at the B-site.39 These XRD patterns and Raman spectra indicated that Ir and Sb ions were successfully doped at the Ti sites of SrTiO3. Next, XRF analysis of SrTiO3:Ir(x%),Sb(2x%) (x = 0.025–0.3) was carried out to confirm the amounts of Ir and Sb doped in the photocatalyst (Table S1†). The Ir and Sb ions of x = 0.025 could not be detected due to the doping amounts being very small. The amounts of Ir and Sb in the samples with x = 0.05–0.3 before the flux treatment were almost the same as those after the flux treatment. It is suggested that dopants of Ir and Sb were not lost to a flux reagent.Fig. 1 shows the diffuse reflectance spectra of SrTiO3:Ir(x%),Sb(2x%) (x = 0–0.3) before and after the flux treatment. The absorption in the visible light region increased upon an increase in the doping amount of Ir. The absorption in the range of visible light negligibly changed on the sample with x = 0.001–0.01% after the flux treatment. In contrast to it, the absorption of the samples with x = 0.05–0.3% greatly increased after the flux treatment. The increase in the visible light absorption region would mainly be due to the increase in the particle size as shown in Fig. 2. It was confirmed by previous reports that absorptions at around 500–800 nm and 400–450 nm were assigned to transitions from Ir3+ ions to a conduction band, and a valence band to Ir4+ ions, respectively.29,40
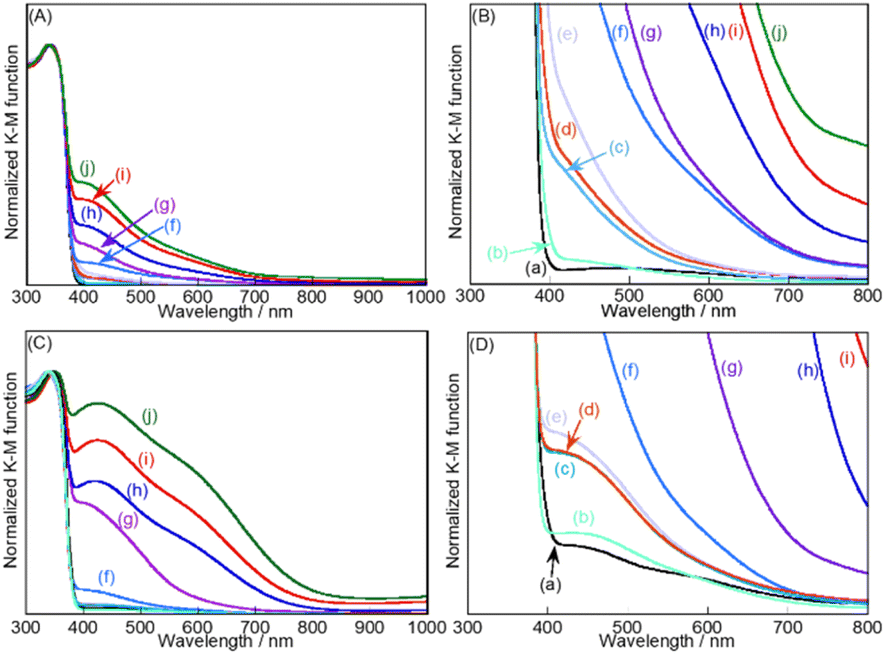 | ||
| Fig. 1 Diffuse reflectance spectra of SSR-prepared SrTiO3:Ir(x%),Sb(2x%) (A and B) without and (C and D) with the flux treatment without Al2O3 addition. Samples were doped with (a) x = 0, (b) x = 0.001, (c) x = 0.008, (d) x = 0.009, (e) x = 0.01, (f) x = 0.025, (g) x = 0.05, (h) x = 0.1, (i) x = 0.2, and (j) x = 0.3. (A and C) Full and (B and D) enlarged scales. | ||
 | ||
| Fig. 2 SEM images of SSR-prepared SrTiO3:Ir(x%),Sb(2x%) (a–j) without and (k–t) with the flux treatment in an alumina crucible. Samples were doped with (a and k) x = 0, (b and l) x = 0.001, (c and m) x = 0.008, (d and n) x = 0.009, (e and o) x = 0.01, (f and p) x = 0.025, (g and q) x = 0.05, (h and r) x = 0.1, (i and s) x = 0.2, and (j and t) x = 0.3. | ||
The morphologies of SrTiO3:Ir(x%),Sb(2x%) particles before and after the flux treatment in an alumina crucible were observed by scanning electron microscopy measurements as shown in Fig. 2. The flux-treated samples had a cubic-like shape with the specific facets whereas non-flux-treated samples were sphere-like particles with no specific facet. It was previously reported that the flux treatment contributed to the formation of truncated particles.31,34,41 The flux-treated particles of x = 0.001–0.025 were mainly 0.1–0.5 μm whereas large particles of submicrometer order were partially observed. In contrast to them, those of x = 0, 0.05–0.3% were mainly 0.3–10 μm. It was found that small amounts of Ir and Sb doping suppressed crystal growth.
Photocatalytic overall water splitting over RhCrOx-loaded SrTiO3:Ir(x%),Sb(2x%) under visible light irradiation was carried out as shown in Table 1. Efficient water splitting proceeded over Ir,Sb-codoped samples with the flux treatment compared with a non-doped sample. However, the Ir,Sb-codoped sample without the flux treatment showed negligible activity, indicating that the flux treatment was essential for efficient water splitting. It has been reported by Ham and Domen that the photocatalytic activity for water splitting was greatly improved by usage of an alumina crucible but not usage of an Y2O3 crucible for the flux treatment.31 Al was doped into a SrTiO3 photocatalyst from the alumina crucible during the flux treatment. Therefore, Al is also doped into the Ir,Sb co-doped SrTiO3 photocatalyst in the present study even without intentional Al2O3 addition. The Al doping by the flux treatment using an alumina crucible actually affects water splitting in the present Ir,Sb co-doped SrTiO3 photocatalyst. The flux treatment accompanied by Al doping to the sample doped with the small amounts of Ir and Sb contributed to suppression of particle growth and formation of the specific facet which worked as the active sites for water splitting and efficient charge separation of photo-generated electrons and holes.34 The activity increased with an increase in the doping amount up to x = 0.009% (Table 1, entries 1–4 and 7), which was due to an increase in visible light absorption by formation of an impurity level of an Ir dopant. In contrast to it, the activity of the samples decreased with a further increase in the doping amounts of Ir and Sb (Table 1, entries 7 and 9–14), which was due to an increase in recombination centers by dopants and formation of large particles as shown in Fig. 2.
| Entry | Dopant (mol% to Ti) | SrCl2-flux treatment | Water splitting activity/μmol h−1 | |
|---|---|---|---|---|
| H2 | O2 | |||
| a Photocatalyst: 0.2 g, cocat.: RhCrOx (Rh 0.1 mol%, Cr 0.1 mol%) impregnation, reactant solution: pure water (120 mL), light source: a 300 W Xe lamp (λ > 440 nm), cell: a top-irradiation cell with a Pyrex window, system: a gas-tight circulation system. | ||||
| 1 | None | Yes | 0.14 | 0.05 |
| 2 | Sb(0.018%) | Yes | 0.17 | 0.07 |
| 3 | Ir(0.001%),Sb(0.002%) | Yes | 4.2 | 1.9 |
| 4 | Ir(0.008%),Sb(0.016%) | Yes | 33 | 16 |
| 5 | Ir(0.009%) | Yes | 20 | 9.4 |
| 6 | Ir(0.009%),Sb(0.009%) | Yes | 29 | 14 |
| 7 | Ir(0.009%),Sb(0.018%) | Yes | 41 | 21 |
| 8 | Ir(0.009%),Sb(0.027%) | Yes | 16 | 7.8 |
| 9 | Ir(0.01%),Sb(0.02%) | Yes | 36 | 15 |
| 10 | Ir(0.025%),Sb(0.05%) | Yes | 21 | 11 |
| 11 | Ir(0.05%),Sb(0.1%) | Yes | 8.2 | 4.0 |
| 12 | Ir(0.1%),Sb(0.2%) | Yes | 3.1 | 1.4 |
| 13 | Ir(0.2%),Sb(0.4%) | Yes | 1.1 | 0.5 |
| 14 | Ir(0.3%),Sb(0.6%) | Yes | 0.15 | 0.08 |
| 15 | None | No | 0.01 | 0.005 |
| 16 | Ir(0.009%),Sb(0.018%) | No | 0.02 | 0.01 |
Next, the effect of the codoping amount of Sb on photocatalytic water splitting over SrTiO3:Ir(0.009%),Sb(y%),Al under visible light irradiation was examined. The activity was increased by Sb codoping up to 0.018% (Table 1, entries 5–7) because of charge compensation by Sb doping in the oxidation number of Ir. In contrast, the activity of the sample doped with a large amount of Sb decreased (Table 1, entry 8). Many large particles of the sample were observed by SEM (Fig. S3(g)†), which is the main factor in the low activity. It was clarified that 0.009% of Ir and 0.018% of Sb were the optimum doping amounts. We have reported that stoichiometric codoping of Sb5+ ions was not enough for charge compensation to doped transition metal ions such as Rh and Cr.28,42–44 Electron spin resonance (ESR) measurements of TiO2:Rh,Sb and SrTiO3:Rh,Sb revealed that the ESR signals related to Rh4+ ions were considerably suppressed by Sb codoped with more than the stoichiometric ratio, 1:1.43,44 Therefore, an excess amount of Sb doping (Ir:Sb = 1:2) was necessary for a high enough charge compensation that had a positive effect on water splitting over the SrTiO3:Ir,Sb,Al photocatalyst.
Effect of intentional Al2O3 addition for the flux treatment on photocatalytic water splitting
Doping of a cation with a lower oxidation number than the parent cation into a host material is sometimes effective for the improvement of photocatalytic activity.45,46 It has been reported that doping of an Al3+ ion to Ti4+ sites in the SrTiO3 host plays an important role in suppressing Ti3+ ions which work as recombination centers for photogenerated carriers, and decreasing the n-type semiconductor character of SrTiO3.47,48 In addition, Al2O3 addition to the flux treatment produces fine particles of ca. 300 nm. Therefore, the effect of intentional Al2O3 addition in the flux reagent to the SrTiO3:Ir(0.009%),Sb(0.018%),Al photocatalyst on water splitting under visible light irradiation was investigated. No impurities of Ir, Sb, and Al species, and no peak shifts were observed in the X-ray diffraction patterns of the samples prepared by the flux treatment with various amounts of Al2O3 (0–3%), suggesting that Al ions were successfully doped to Ti4+ sites in SrTiO3 more or less.The effect of an amount of Al2O3 addition in the flux for the preparation of SrTiO3:Ir(0.009%),Sb(0.018%),Al on photocatalytic water splitting under visible light irradiation was investigated (Table S2†). All samples prepared with Al2O3 addition showed higher activity than that without Al2O3 addition. The optimum amount of added Al3+ was 1–2 mol%.
Effect of loading of cocatalysts on photocatalytic water splitting
The activity for photocatalytic water splitting greatly depends on cocatalysts as active sites. Therefore, the effect of loading with various cocatalysts on water splitting over SrTiO3:Ir(0.009%),Sb(0.018%),Al prepared with Al2O3 (1%) addition in the flux was examined. Photocatalytic water splitting proceeded with a reasonable activity when RhCrOx (H2: 62 μmol h−1, O2: 30 μmol h−1) and Rh2O3 (H2: 12 μmol h−1, O2: 5.6 μmol h−1) cocatalysts were loaded (Table S3†). The RhCrOx cocatalyst was more effective among these cocatalysts. It was previously reported that RhCrOx promoted the H2 evolution reaction and suppressed a backward reaction from evolved H2 and O2 to form water and photoreduction of O2.49 We carried out photodeposition of CrOx and Rh cocatalysts on a SrTiO3:Ir,Sb,Al photocatalyst for water splitting under visible light irradiation. The photodeposition method did not give a higher activity (H2: 41 μmol h−1, O2: 18 μmol h−1) than an impregnation method (H2: 62 μmol h−1, O2: 30 μmol h−1) with the same loading amount. The impregnated cocatalysts would dissolve and re-photodeposit during irradiation, resulting in the impregnated cocatalyst becoming similar to the photodeposited one. The optimum loading amount of the RhCrOx cocatalyst was 0.1 mol% (Table S3, entries 2–5†). The photocatalyst optimized for doping amounts, Al2O3 addition in a flux treatment, and the cocatalyst split water into H2 and O2 in a stoichiometric amount under visible light irradiation (Fig. S4†).For an Al-doped SrTiO3 photocatalyst, the facets of (100) as a reduction site and (110) as an oxidation site were exposed on a particle, promoting H2 and O2 evolution from water separately.34 Therefore, we observed reductively and oxidatively photodeposited Ag and PbO2, respectively, under visible light irradiation (λ > 440 nm) on SrTiO3:Ir(0.009%),Sb(0.018%),Al particles prepared by Al2O3 addition in the flux by SEM as shown in Fig. 3. The SEM images showed that Ag and PbO2 particles were deposited on a reductive facet of (100) and an oxidative facet of (110), respectively. This indicated that photo-generated carriers separated to the reductive and oxidative facets on the photocatalyst surface.
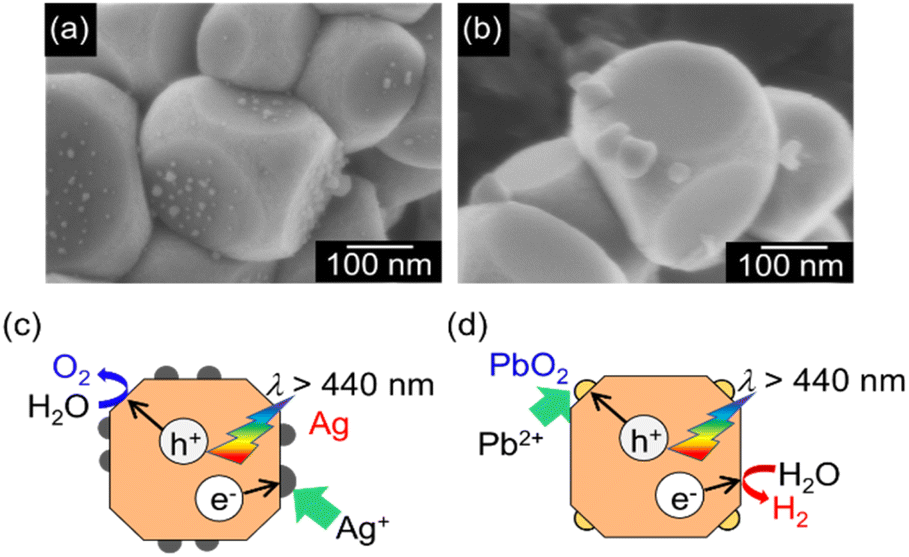 | ||
| Fig. 3 SEM images of (a) Ag (3 wt%)- and (b) PbO2 (5 wt%)-deposited SrTiO3:Ir(0.009%),Sb(0.018%),Al prepared by the flux treatment with Al2O3 (1%) addition after SSR. Ag and PbO2 were loaded by a photo-deposition method under visible light irradiation (λ > 440 nm). The mechanism of photo-deposition of (c) Ag and (d) PbO2 on the photocatalyst. | ||
Mechanism of water splitting under visible light irradiation over a RhCrOx-loaded SrTiO3:Ir,Sb,Al photocatalyst
The action spectra of RhCrOx (0.1 mol%)-loaded SrTiO3:Ir(0.009%),Sb(0.018%),Al prepared with Al2O3 (1%) addition in the flux for photocatalytic water splitting are shown in Fig. 4. The photocatalyst responded up to 660 nm which is the longest wavelength among the previously reported visible-light-driven single particulate photocatalysts being active for water splitting. Stoichiometric O2 evolution was also observed up to 660 nm. The profiles of action spectra and the onset were similar to the action spectra of Ir-doped SrTiO3 for the photoanodic properties for oxidation of water.40,50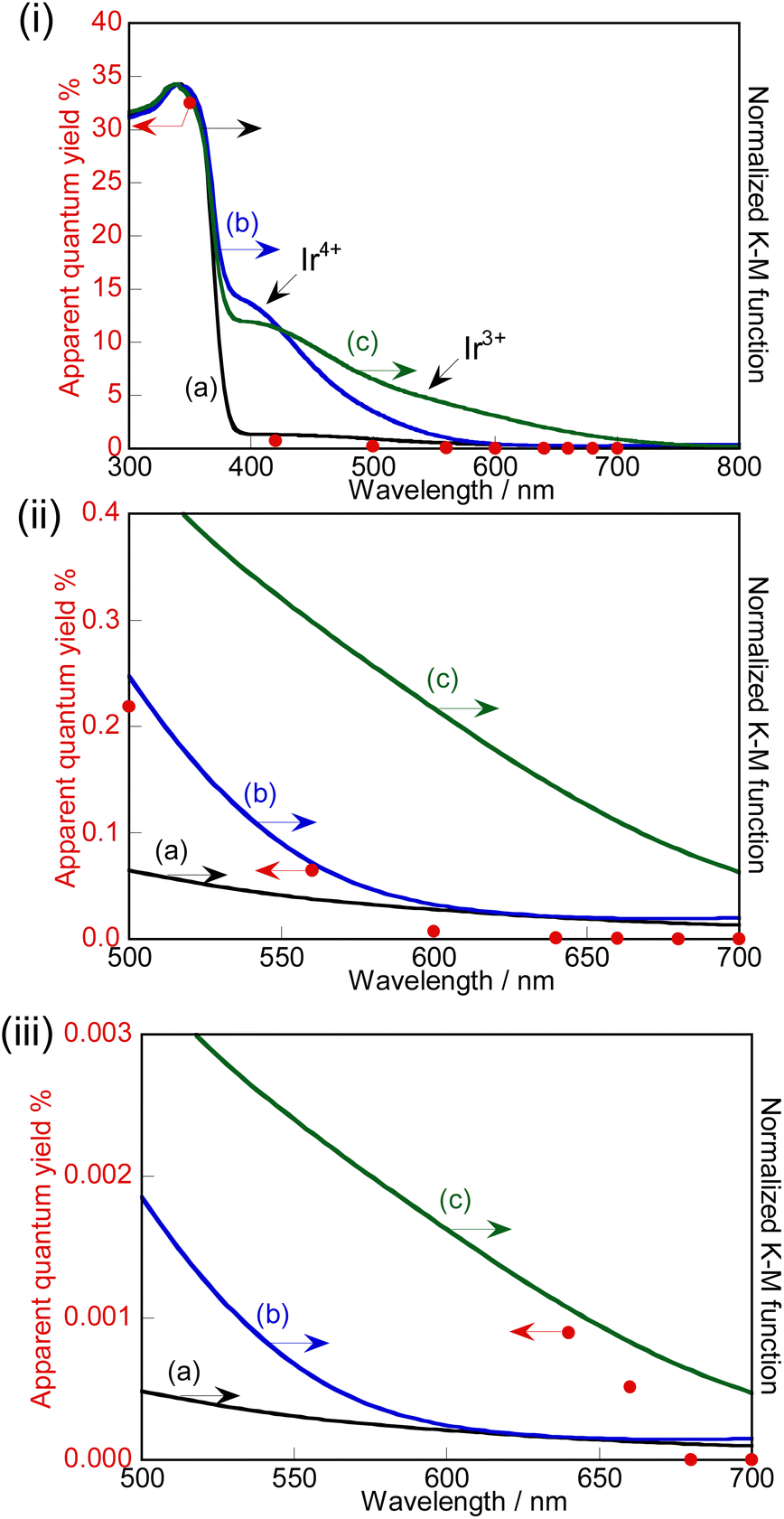 | ||
| Fig. 4 Action spectrum of photocatalytic water splitting over RhCrOx(0.1 mol%)-loaded SrTiO3:Ir(0.009%),Sb(0.018%),Al prepared by the flux treatment with Al2O3 (1%) addition after SSR and diffuse reflectance spectra of (a) SrTiO3:Ir(0.009%),Sb(0.018%) prepared by the flux treatment with Al2O3 (1%) addition after SSR, (b) SrTiO3:Ir(0.2%), and (c) H2-reduced (673 K, 1 h) SrTiO3:Ir(0.2%). (ii) An enlarged scale of (i); (iii) an enlarged scale of (ii). Photocatalyst: 0.5 g, cocat.: RhCrOx (impregnation), reactant solution: pure water (120 mL), light source: a 300 W Xe lamp with band-pass filters, cell: a top-irradiation cell with a Pyrex window, system: a gas-tight circulation system. | ||
We discuss the photoexcitation mechanism for water splitting on a SrTiO3:Ir(0.009%),Sb(0.018%),Al photocatalyst based on an action spectrum and diffuse reflectance spectra. The absorption edge of a non-H2-treated SrTiO3:Ir(0.2%) was around 580 nm, indicating that the absorption was due to photoexcitation from a valence band to the impurity level consisting of Ir4+ ions as shown in Fig. 4(i). The absorption edge was red-shifted to around 750 nm by H2 reduction, indicating that the photoexcitation was due to transition from an impurity level consisting of Ir3+ ions to a conduction band.29,40
The photocatalyst activity was observed by utilizing absorptions related to Ir4+ (λ ≤ ca. 560 nm) and Ir3+ (λ ≤ ca. 660 nm) ions. In the one-step photoexcitation process as shown in Fig. 5(i), the conduction band level and holes transiently photogenerated in the impurity level consisting of Ir3+ ions thermodynamically satisfy the reduction and oxidation of water, with water splitting being able to proceed. However, the activity by photoexcitation at wavelengths longer than 600 nm was low due to poor migration of holes in the low concentration of an impurity level and small potential difference between the impurity level consisting of Ir3+ and water oxidation as shown in Fig. 4(iii). In contrast to it, AQY greatly increased with simultaneous photoexcitation (λ ≤ ca. 560 nm) of both absorption bands related to Ir3+ and Ir4+ ions as shown in Fig. 4(ii). In this scheme, Ir4+ ions are reduced to Ir3+ ions by photoexcitation from a valence band to Ir4+ ions as shown in Fig. 5(ii), which is a similar trend to the case of a Rh-doped SrTiO3 photocatalyst,23 and subsequently the electrons at the formed Ir3+ ions are excited to a conduction band. Then, Ir3+ ions return to Ir4+ ions after photoexcitation from Ir3+ ions. This results in e− and h+ being photogenerated in the conduction and valence bands of SrTiO3 through this two-step photoexcitation, respectively. Therefore, migration of photogenerated carriers in the conduction and valence bands becomes smooth compared with that via the one-step photoexcitation process, resulting in the high activity. Here, the redox in Ir4+/Ir3+ occurs in one Ir cation, but not Z-scheme electron flow. The maximum AQY was estimated to be 0.73% at 420 nm.
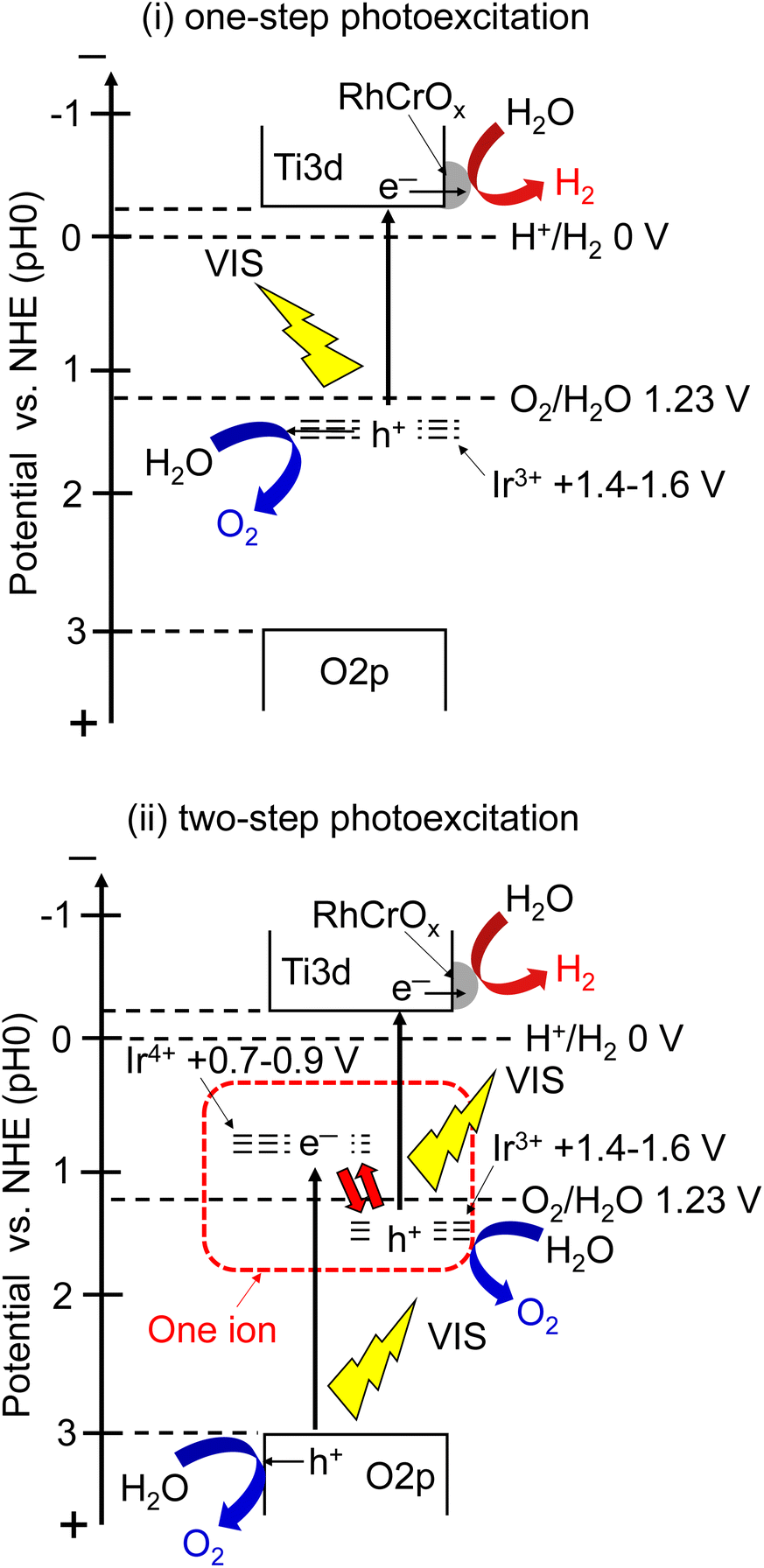 | ||
| Fig. 5 Proposed mechanism of water splitting over a SrTiO3:Ir,Sb,Al photocatalyst under visible light irradiation. (i) One-step photoexcitation, (ii) two-step photoexcitation. | ||
Photocatalytic solar water splitting over RhCrOx-loaded SrTiO3:Ir(0.009%),Sb(0.018%),Al prepared with Al2O3 (1%) addition in the flux proceeded as shown in Fig. 6, giving 0.33% STH. H2 and O2 stoichiometrically evolved for 20 h. The amounts of H2 and O2 did not decrease under dark conditions, suggesting that the backward reaction of the formation of H2O hardly occurred. In addition, 0.030% STH was also obtained by using only visible light (λ > 440 nm) from a solar simulator. This is currently the highest value for water splitting with a single particulate photocatalyst compared with previously reported visible-light-driven photocatalysts. The turnover numbers estimated from electrons used for evolved H2 (65.6 μmol) for 10 h under visible light (λ > 440 nm) from a solar simulator to the mole number of an Ir dopant (0.245 μmol) of SrTiO3:Ir,Sb,Al and a RhCrOx cocatalyst (2.73 μmol) were about 536 and 48.1, respectively, indicating that this reaction proceeded photocatalytically.
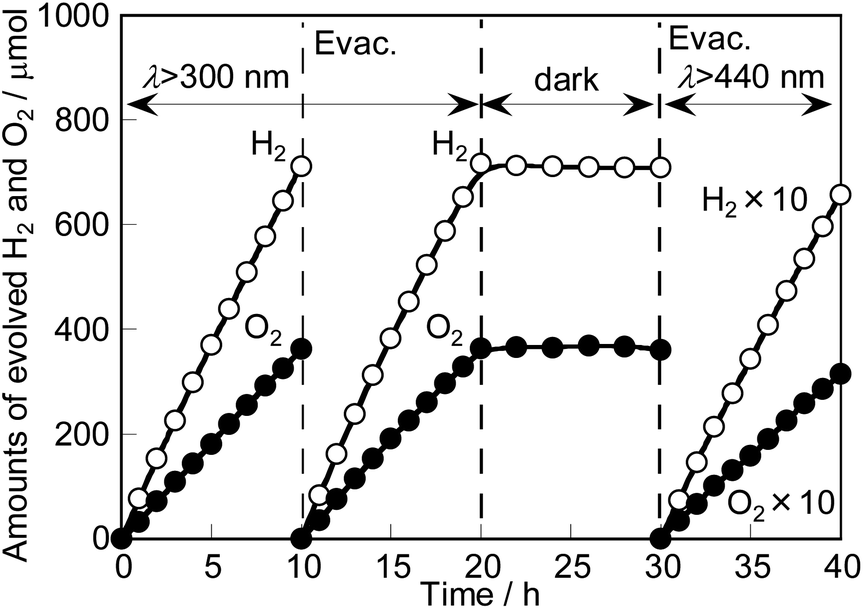 | ||
| Fig. 6 Photocatalytic water splitting over RhCrOx-loaded SrTiO3:Ir(0.009%),Sb(0.018%),Al prepared by the flux treatment with Al2O3 (1%) addition after SSR under simulated sunlight irradiation. Photocatalyst: 0.5 g, cocat.: RhCrOx (Rh 0.1 mol%, Cr 0.1 mol%) impregnation, reactant solution: pure water (120 mL), light source: a solar simulator (AM 1.5G), irradiation area: 16 cm2, system: a gas-tight circulation system. | ||
Water splitting over RhCrOx-loaded various transition metal-doped SrTiO3:Al photocatalysts
To expand the doping species of the transition metal, water splitting over various transition metal-doped SrTiO3:Al photocatalysts prepared with Al2O3 (1%) addition for the flux treatment was investigated as shown in Table 2. We would have investigated the XPS and XRF of SrTiO3:Ir,Sb,Al after flux treatment with Al2O3 addition. However, the Al signal was not clearly detected due to a small amount of doped Al ions in the photocatalyst. Therefore, we could not determine the quantity of doped Al here. Rh, Ru, and Cr-doped samples showed high activity for water splitting under visible light as well as with an Ir dopant. It was notable that solar water splitting proceeded under visible light (λ > 440 nm) over all samples (Table S4†). We have preliminarily reported that the flux-treated SrTiO3:Al doped with small amounts of Rh, Ru and Cr was active for photocatalytic water splitting.51–53 Recently, single particulate KTaO3:Ir,A (A = Ca, Sr, Ba, La), SrTiO3:Cr,La,Al, and KTaO3:Ru,La oxide photocatalysts for water splitting under visible light irradiation have been developed by doping small amounts of transition metal dopants, and loading with Rh–Cr-based cocatalysts,54–56 this being similar to our strategy.51–53 Our SrTiO3:Ir,Sb,Al and SrTiO3:Ru,Sb,Al showed much higher activity than the KTaO3:Ir,A and KTaO3:Ru,La. In our present study, the activities of SrTiO3:Cr,Al and SrTiO3:Ru,Sb,Al were improved by optimization of the doping amount and the preparation conditions. The Rh and Sb-codoped, Ru and Sb-codoped, and Cr-doped photocatalysts responded to 570, 600 and 660 nm, respectively. Among the samples, SrTiO3:Ir(0.009%),Sb(0.018%),Al showed the highest activity. Thus, the development of various transition metal-doped photocatalysts as single particulate photocatalysts for highly efficient water splitting was successful with small amounts of dopants, the flux treatment, and RhCrOx cocatalyst loading.| Dopant (mol% to Ti) | Responsive wavelength/nm | Water splitting activity/μmol h−1 | |
|---|---|---|---|
| H2 | O2 | ||
| a Photocatalyst: 0.2 g, cocatalyst: RhCrOx (Rh 0.1 mol%, Cr 0.1 mol%), reactant solution: pure water (120 mL), light source: a 300 W Xe lamp (λ > 440 nm), cell: a top-irradiation cell with a Pyrex window, system: a gas-tight circulation system. | |||
| Ir(0.009%),Sb(0.018%),Al | 660 | 62 | 30 |
| Rh(0.02%),Sb(0.04%),Al | 570 | 56 | 28 |
| Ru(0.03%),Sb(0.06%),Al | 600 | 54 | 27 |
| Cr(0.01%),Al | 660 | 21 | 12 |
| Mn(0.01%),Al | — | 0.81 | 0.43 |
| Fe(0.01%),Al | — | 0.29 | 0.15 |
| Co(0.01%),Al | — | 1.5 | 0.72 |
| Ni(0.01%),Al | — | 0.77 | 0.38 |
Conclusions
Strategies of a small doping amount of transition metals to suppress recombination between photogenerated electrons and holes, flux treatment with Al2O3 addition for formation of homogeneous fine particles with exposed reductive and oxidative facets, and loading of a RhCrOx cocatalyst as an active site were effective for the development of a highly efficient doping photocatalyst for water splitting under visible light. Efficient photocatalytic water splitting under visible light irradiation over RhCrOx-loaded SrTiO3:Ir,Sb,Al as a single particulate metal oxide photocatalyst was achieved by the strategy. The high activity was obtained by the flux treatment with Al2O3 addition to the SSR-prepared samples, doping of small amounts of Ir and Sb, and loading of a RhCrOx cocatalyst. The optimized photocatalyst responded to 660 nm that is currently the highest value for water splitting with a single particulate photocatalyst compared with previously reported visible-light-driven photocatalysts. The maximum AQY reached 0.73% at 420 nm. Solar water splitting under full arc and visible light (λ > 440 nm) proceeded, giving 0.33% and 0.030% STHs (λ > 440 nm), respectively. It is proposed that photocatalytic water splitting under visible light irradiation over RhCrOx-loaded SrTiO3:Ir,Sb,Al proceeded via two-step photoexcitation utilizing subsequent absorptions from a valence band to Ir4+ ions and from Ir3+ ions to a conduction band in one Ir cation based on the results of diffuse reflectance spectra and an action spectrum. Additionally, various transition metal doped-SrTiO3:Al photocatalysts were developed for water splitting as a single particulate photocatalyst in the same manner as the Ir,Sb-codoped sample. In particular, Ru(0.03%) and Sb(0.06%)-codoped and Cr(0.01%)-doped samples showed activity for water splitting under visible light up to 600 and 660 nm, respectively. These strategies are expected to be applied for doping other photocatalysts for water splitting.Data availability
All data are available in the main manuscript and the ESI.†Author contributions
K. K.: methodology, investigation, visualization, and writing – original draft. Y. U., H. K.: methodology, investigation, and visualization. K. W., S. Y., Y. Y.: methodology and writing – review and editing. A. K.: conceptualization, supervision and writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI, Grant Numbers 17H06433 and 23H00248 in Scientific Research on “Innovative Areas for Light-Energy Conversion (I4LEC)”, 17H01217 and 22K14770, and the Tokyo University of Science Grant for President's Research Promotion.References
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- S. Nandy, T. Hisatomi, T. Takata, T. Setoyama and K. Domen, J. Mater. Chem. A, 2023, 11, 20470–20479 RSC.
- A. Kudo, H. Kato and I. Tsuji, Chem. Lett., 2004, 33, 1534–1539 CrossRef CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- Y. Ma, L. Lin, T. Takata, T. Hisatomi and K. Domen, Phys. Chem. Chem. Phys., 2023, 25, 6586–6601 RSC.
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286–8287 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- Y. Lee, H. Terashima, Y. Shimodaira, K. Teramura, M. Hara, H. Kobayashi, K. Domen and M. Yashima, J. Phys. Chem. C, 2007, 111, 1042–1048 CrossRef CAS.
- X. Wang, K. Maeda, Y. Lee and K. Domen, Chem. Phys. Lett., 2008, 457, 134–136 CrossRef CAS.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Nat. Mater., 2019, 18, 827–832 CrossRef CAS PubMed.
- Z. Wang, Y. Inoue, T. Hisatomi, R. Ishikawa, Q. Wang, T. Takata, S. Chen, N. Shibata, Y. Ikuhara and K. Domen, Nat. Catal., 2018, 1, 756–763 CrossRef CAS.
- K. Maeda, D. Lu and K. Domen, Chem.–Eur. J., 2013, 19, 4986–4991 CrossRef CAS PubMed.
- C. Pan, T. Takata and K. Domen, Chem.–Eur. J., 2016, 22, 1854–1862 CrossRef CAS PubMed.
- H. Li, J. Xiao, J. J. M. Vequizo, T. Hisatomi, M. Nakabayashi, Z. Pan, N. Shibata, A. Yamakata, T. Takata and K. Domen, ACS Catal., 2022, 12, 10179–10185 CrossRef CAS.
- S. Nishimae, J. J. M. Vequizo, Y. Inoue, A. Yamakata, M. Nakabayashi, T. Higashi and K. Domen, J. Mater. Chem. A, 2023, 11, 6299–6310 RSC.
- K. Chen, J. Xiao, J. J. M. Vequizo, T. Hisatomi, Y. Ma, M. Nakabayashi, T. Takata, A. Yamakata, N. Shibata and K. Domen, J. Am. Chem. Soc., 2023, 145, 3839–3843 CrossRef CAS PubMed.
- Y. Sakata, T. Yamamoto, T. Okazaki, H. Imamura and S. Tsuchiya, Chem. Lett., 1998, 27, 1253–1254 CrossRef.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992–8995 CrossRef CAS.
- D. W. Hwang, H. G. Kim, J. S. Jang, S. W. Bae, S. M. Ji and J. S. Lee, Catal. Today, 2004, 93–95, 845–850 CrossRef CAS.
- Y. Shimodaira, H. Kato, H. Kobayashi and A. Kudo, Bull. Chem. Soc. Jpn., 2007, 80, 885–893 CrossRef CAS.
- Y. Yamaguchi and A. Kudo, Front. Energy, 2021, 15, 568–576 CrossRef.
- S. Sun, T. Hisatomi, Q. Wang, S. Chen, G. Ma, J. Liu, S. Nandy, T. Minegishi, M. Katayama and K. Domen, ACS Catal., 2018, 8, 1690–1696 CrossRef CAS.
- R. Asai, H. Nemoto, Q. Jia, K. Saito, A. Iwase and A. Kudo, Chem. Commun., 2014, 50, 2543–2546 RSC.
- S. Suzuki, H. Matsumoto, A. Iwase and A. Kudo, Chem. Commun., 2018, 54, 10606–10609 RSC.
- A. Kudo, S. Yoshino, T. Tsuchiya, Y. Udagawa, Y. Takahashi, M. Yamaguchi, I. Ogasawara, H. Matsumoto and A. Iwase, Faraday Discuss., 2019, 215, 313–328 RSC.
- Y. Ham, T. Hisatomi, Y. Goto, Y. Moriya, Y. Sakata, A. Yamakata, J. Kubota and K. Domen, J. Mater. Chem. A, 2016, 4, 3027–3033 RSC.
- T. H. Chiang, H. Lyu, T. Hisatomi, Y. Goto, T. Takata, M. Katayama, T. Minegishi and K. Domen, ACS Catal., 2018, 8, 2782–2788 CrossRef CAS.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2018, 2, 509–520 CrossRef CAS.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13753–13758 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, N. Saito, Y. Inoue and K. Domen, J. Catal., 2006, 243, 303–308 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806–7809 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. C, 2007, 111, 7554–7560 CrossRef CAS.
- H. Zheng, I. M. Reaney, G. D. C. Csete de Györgyfalva, R. Ubic, J. Yarwood, M. P. Seabra and V. M. Ferreira, J. Mater. Res., 2004, 19, 488–495 CrossRef CAS.
- S. Kawasaki, R. Takahashi, K. Akagi, J. Yoshinobu, F. Komori, K. Horiba, H. Kumigashira, K. Iwashina, A. Kudo and M. Lippmaa, J. Phys. Chem. C, 2014, 118, 20222–20228 CrossRef CAS.
- H. Kato, M. Kobayashi, M. Hara and M. Kakihana, Catal. Sci. Technol., 2013, 3, 1733–1738 RSC.
- H. Kato and A. Kudo, J. Phys. Chem. B, 2002, 106, 5029–5034 CrossRef CAS.
- R. Niishiro, R. Konta, H. Kato, W.-J. Chun, K. Asakura and A. Kudo, J. Phys. Chem. C, 2007, 111, 17420–17426 CrossRef CAS.
- R. Niishiro, S. Tanaka and A. Kudo, Appl. Catal., B, 2014, 150–151, 187–196 CrossRef CAS.
- T. Ishihara, H. Nishiguchi, K. Fukamachi and Y. Takita, J. Phys. Chem. B, 1999, 103, 1–3 CrossRef CAS.
- T. Takata and K. Domen, J. Phys. Chem. C, 2009, 113, 19386–19388 CrossRef CAS.
- D. H. K. Murthy, V. Nandal, A. Furube, K. Seki, R. Katoh, H. Lyu, T. Hisatomi, K. Domen and H. Matsuzaki, Adv. Energy Mater., 2023, 13, 2302064 CrossRef CAS.
- R. Li, T. Takata, B. Zhang, C. Feng, Q. Wu, C. Cui, Z. Zhang, K. Domen and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202313537 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, H. Masuda, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13107–13112 CrossRef CAS PubMed.
- S. Kawasaki, R. Takahashi, T. Yamamoto, M. Kobayashi, H. Kumigashira, J. Yoshinobu, F. Komori, A. Kudo and M. Lippmaa, Nat. Commun., 2016, 7, 11818 CrossRef CAS PubMed.
- K. Kaiya, K. Watanabe, S. Yoshino, Y. Yamaguchi and A. Kudo, Abstracts of Papers (in Japanese), 126th CATSJ Meeting, Online, Japan, 2020, p. P058 Search PubMed.
- K. Kaiya, Master thesis (in Japanese), Tokyo University of Science, Tokyo, Japan, 2021.
- H. Kawamoto, Master thesis (in Japanese), Tokyo University of Science, Tokyo, Japan, 2022.
- A. Iwase and H. Misono, Chem. Commun., 2021, 57, 10331–10334 RSC.
- A. Iwase and H. Misono, Energy Technol., 2023, 11, 2200739 CrossRef CAS.
- Y. Qin, R. Xu, L. Ding, T. Wang, D. Guo, F. Fang and K. Chang, J. Catal., 2024, 430, 115317 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Flowchart, details of characterisation (XRF, SEM), and the activities for water splitting of the materials. See DOI: https://doi.org/10.1039/d4sc03978e |
| This journal is © The Royal Society of Chemistry 2024 |