
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of highly soluble zirconium organic cages by iodine substitution toward a CO2/N2 separation membrane†
Junchao
Dong
a,
Dongxu
Gai
a,
Guocai
Cha
b,
Qinhe
Pan
 c,
Jia
Liu
b,
Xiaoqin
Zou
*a and
Guangshan
Zhu
*a
c,
Jia
Liu
b,
Xiaoqin
Zou
*a and
Guangshan
Zhu
*a
aFaculty of Chemistry, Northeast Normal University, Changchun 130024, China. E-mail: zouxq100@nenu.edu.cn; zhugs@nenu.edu.cn
bCollege of Chemistry, Jilin University, Changchun 130012, China
cKey Laboratory of Advanced Materials of Tropical Island Resources of Ministry of Education, School of Chemistry and Chemical Engineering, Hainan University, Haikou 570228, China
First published on 28th October 2024
Abstract
Metal organic cages (MOCs) show promise as fillers in mixed-matrix membranes (MMMs) for gas separation; highly soluble MOCs are desirable for fabrication of high-compatibility membranes. Herein, we report an iodine substitution strategy to substantially increase the MOC solubility. The synthesized MOC of ZrT-NH2-I possesses over 10-fold higher solubility than the parent ZrT-NH2 in organic solvents whilst retaining the original molecular structure and permanent porosity. Such enhanced solubility allows for the effective integration of ZrT-NH2-I with an amidoxime polymer of intrinsic microporosity (PIM-PAO), resulting in a compatible MMM with a uniform distribution of MOC. The ZrT-NH2-I@PIM-PAO MMM demonstrates a CO2 permeability of 1377 barrer and a CO2/N2 gas selectivity of 45 which is 45 times that of the membrane made from ZrT-NH2. The permeability-selectivity performance not only surpasses the 2008 upper bound, but also exceeds those of currently available MMMs.
Introduction
The processability of solid fillers in solutions is crucial for fabrication of mixed-matrix membranes (MMMs).1 Insoluble solid fillers, such as zeolites,2 covalent organic frameworks (COFs)3 and metal organic frameworks (MOFs),4 are typically synthesized for fabricating MMMs.5,6 Due to the insolubility in solvents, the above solid fillers are difficult to be dispersed homogeneously in the casting solution, usually resulting in poor compatibility between the filler and the polymer. The interfacial defects caused by poor compatibility decline the membrane performance. If the fillers are replaced by soluble materials,7 the filler–polymer interfaces are expected to be significantly amended.Metal organic cages (MOCs),8 a new category of porous solid materials, offer a unique combination of structural variability9 and high porosity,10 similar to metal organic frameworks (MOFs),11,12 with the added advantage of good dispersibility.13 A MOC is constituted by the assembly of discrete cage molecules,14 and relatively weak intermolecular forces allow its solubility in some solvents with porosity retention. In this regard, MOCs are candidate materials for fabrication of defect-free MMMs.15,16 Current attention is focused on Zr-MOC owing to its high stability;17 however, Zr-MOC commonly has insufficient solubility in required solvents,18 limiting its application in separation membranes.
In general, two methods have been reported for improving Zr-MOC solubility.13 The first one is the post functionalization of the organic ligand. A typical strategy involves grafting of a large alkyl group to the ligand through a reaction between an amino group and an acyl chloride.19–21 This addition of a bulky chain reduces the interactions between cages, leading to the separation of adjacent cages into distinct entities within the solvent.10,22,23 However, this process also introduces steric hindrance due to the alkyl chain, which can interfere with the cage’s ability to mix with the solvent, resulting in only a marginal increase in solubility. Another approach to enhance solubility is the ion exchange of the counter anion.24 An exchange of Cl− with CF3SO3− has been exemplified to increase the solubility of Me2/Zr-MOC (Cl4{[CpZrμ3-O(μ2-OH)]3[Me2-pBDC]6}).25 The ion of CF3SO3− breaks the hydrogen bond which occurs in bridging the adjacent cages in the Cl− equilibrated cage. However, the coordination tendency of CF3SO3− with Zr cation decreases the net positive charge of the cages leading to reduction of repulsion between cages, thereby limiting the solubility increment. Achieving a reduction in cage-to-cage interactions while simultaneously enhancing cage–solvent solvation presents a paradox, thus the balancing of interaction and solvation effects is a grand challenge in the quest for soluble MOCs.
Herein, we present an iodine substitution strategy to improve Zr-MOC solubility (Fig. 1a) for fabrication of a gas separation MMM. ZrT-NH2 from Cp3Zr3 and 2-aminoterephthalic acid,26 is selected as the platform owing to high stability,27 permanent porosity28 and gas recognition.21 ZrT-NH2 is reacted with methyl iodide to form an I− cage of ZrT-NH2-I with the same charge of I− but better leaving ability than Cl− guarantees the balance of cage dissociation and solvent solvation. The discrete ZrT-NH2-I is processed with a polymer to form the MMM. Such membrane is applied for CO2/N2 separation with superior performance.
 | ||
| Fig. 1 (a) The synthetic route to ZrT-NH2-I. (b) 1H NMR spectra and (c) ESI-MS spectra of ZrT-NH2 (green) and ZrT-NH2-I (blue). | ||
Results and discussion
ZrT-NH2-I is synthesized by iodine substitution of ZrT-NH2 with CH3I (Fig. 1a). ZrT-NH2 was first synthesized according to the previous report.26 The similarity of the measured powder X-ray diffraction (PXRD) pattern with the simulated one proves the successful formation of crystalline ZrT-NH2 (Fig. S1†). ZrT-NH2 loses its crystallinity after activation by high-temperature degassing,29 probably because the solvent removal disrupts the arrangement of molecular cages. ZrT-NH2-I is also amorphous, translating to the loss of long-range order, which can be ascribed to the bulky iodine effect. However, ZrT-NH2-I retains a molecular structure like ZrT-NH2 throughout the reaction, and its structure is analyzed by infrared (IR), mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy.30–32 The hydrogen chemical shifts of amino, phenyl and cyclopentadiene groups are located at 7.2, 7.8 and 6.6 ppm, respectively, in 1H NMR spectra (Fig. 1b). ZrT-NH2 and ZrT-NH2-I have m/z values around 804, 1072 and 1607 in high-resolution mass spectra (Fig. 1c), corresponding to [M]4+, [M − H]3+ and [M − 2H]2+ ions, respectively (Fig. S2–S4†). The contents of Zr, C, H and N in ZrT-NH2-I were determined by elemental analysis. The molar ratios of Zr/C, C/N and C/H are 0.11, 18.1 and 1.07, respectively (Tables S1 and S2†). This consistency with the theoretical molecular composition (0.11, 18 and 1.06 for Zr/C, C/N and C/H ratios) indicates that ZrT-NH2-I contains complete zirconium tetrahedral cages as ZrT-NH2 does (Zr/C, C/N and C/H ratios of 0.11, 17.8 and 1.0). The NH2 and Zr–O associated bands are observed at 1335 and 616 cm−1 in IR spectra (Fig. S5†), confirming the presence of functional groups. MS, IR and NMR characterizations reveal that iodine substitution of ZrT-NH2 gives ZrT-NH2-I with the identical molecular structure.To further prove the intactness of the ZrT-NH2 cage after iodine substitution, the synthesized ZrT-NH2-I was hydrolyzed in an alkaline solution (1.0 mol L−1 NaOH). The solution after centrifugation was then acidified with 1.0 mol L−1 HCl to obtain the dicarboxylic acid ligand. The analysis of the 1H NMR spectrum (Fig. 2a) shows that the ligand contains 96% 2-aminoterephthalic acid and 4% 2-aminomethylterephthalic acid, and this portion is further quantified by NMR yields using 1,3,5-trimethoxybenzene as an internal standard (Fig. S6†). This indicates that the iodide compound is formed in priority and the yield of alkylated product is suppressed. The possible reason is that ZrT-NH2 was synthesized under acidic conditions (pH = 4.0) where the protonated –NH2 was difficult to react with CH3I.33 This protonation effect is supplemented by an increase in zeta potential (19.6, 26.2, 31.8 mV at pH = 6.76, 4.26, 2.17). The yield of alkylated product is almost completely suppressed after intentionally decreasing the pH to 3 (1% 2-aminomethylterephthalic acid in the ligand). Moreover, 2-aminoterephthalic acid, 5-aminoisophthalic acid, 4-aminobenzoic acid and bis(cyclopentadienyl)zirconium dichloride were used as the model molecules to investigate the reaction possibility of CH3I with organic groups on the ligand or Zr cluster. No other chemical shifts are observed in the 1H NMR spectra after 2 h reaction (Fig. S7†), confirming the above conclusion that the iodide compound is the product rather than the alkylated compound.
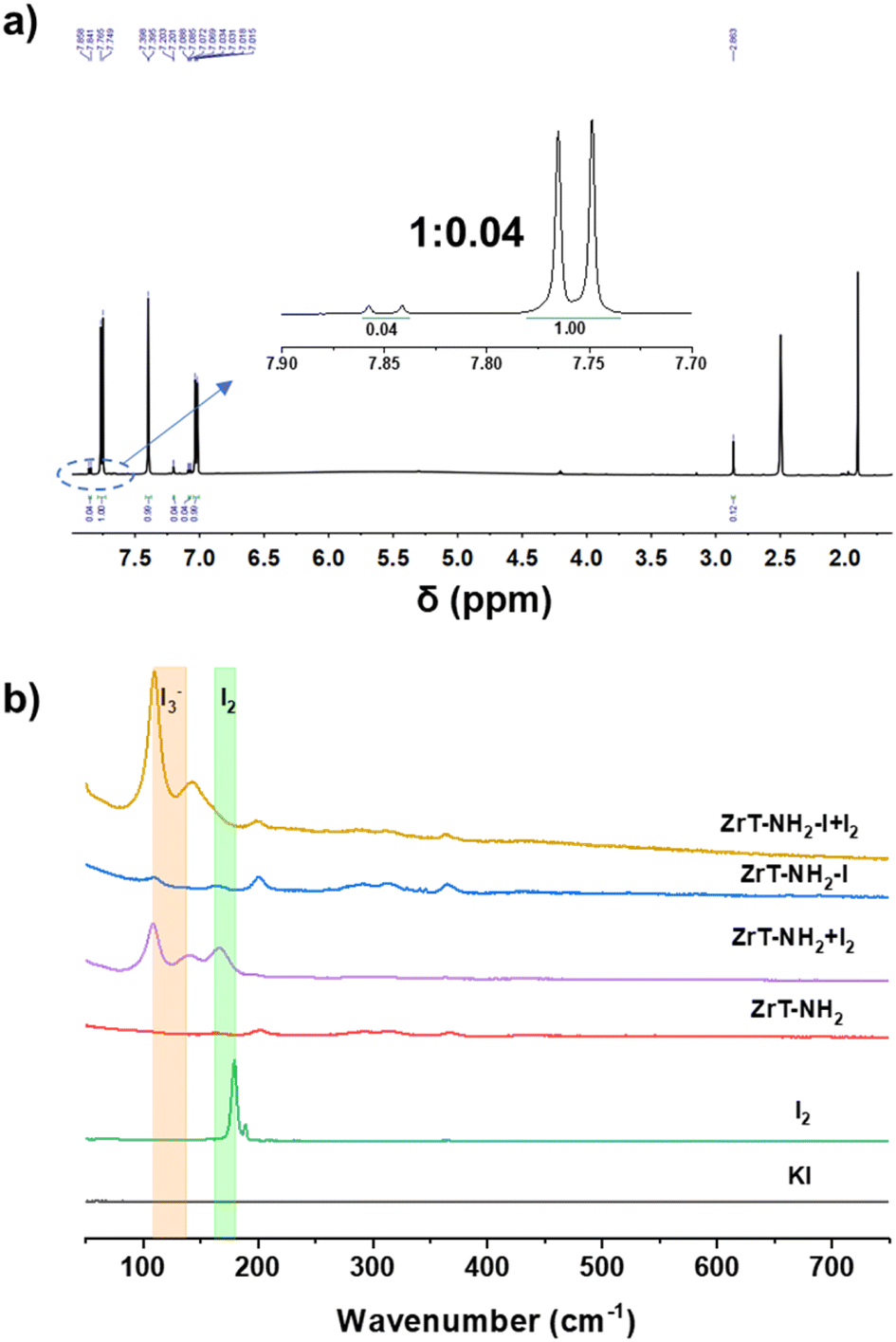 | ||
| Fig. 2 (a) 1H NMR spectrum of ZrT-NH2-I solution after first alkaline hydrolysis and then acidic neutralization. (b) Raman spectra of KI and I2 solids, parent ZrT-NH2 and ZrT-NH2-I, and ZrT-NH2 and ZrT-NH2-I after iodine adsorption (labeled as ZrT-NH2 + I2 and ZrT-NH2-I + I2). | ||
Since I− is silent in Raman spectra, ZrT-NH2-I was first exposed to I2 adsorption and then this product was investigated by Raman spectroscopy to identify the presence of I− ions in ZrT-NH2-I. A strong band associated with I3− appears at 109 cm−1 in ZrT-NH2-I and negligible signals for I2 are detected (Fig. 2b). I3− is mainly formed by transforming I− with I2. In contrast, ZrT-NH2 has both I2 (166 cm−1) and I3− (109 cm−1) signals which are coming from disproportionation of physically adsorbed I2.34 The slight band shift of I2 species in ZrT-NH2 from I2 solid is possibly due to the adsorption effect. The comparison of Raman spectra for ZrT-NH2-I and ZrT-NH2 shows that Zr-MOC is equilibrated with I− ions.
The substitution extent was determined from iodine content using X-ray photoelectron spectroscopy (XPS) and inductively coupled plasma optical emission spectroscopy (ICP-OES). ZrT-NH2 is composed of C, N, O, Zr and Cl. Instead, ZrT-NH2-I not only consists of C, N, O and Zr, but also contains I (Fig. S8 and S9†). Chlorine associated binding energies of 197 and 199 eV vanish, while I 3d5/2 and I 3d3/2 at 618.5 and 630.1 eV appear,35–37 indicating that Cl− ions are almost completely replaced by I− ions. The substitution extent of 96% is confirmed by the ICP analysis (Table S1†). Almost no chlorine element is observed in ZrT-NH2-I from the SEM mapping (Fig. S10 and S11†). The above results indicate that the substitution reaction gives an entirely iodinated cage of ZrT-NH2-I with an approximate chemical formula of Zr12C108H102O40N6I4.
Zeta potentials show that both ZrT-NH2 and ZrT-NH2-I are positively charged (Fig. 3b), in agreement with the cationic nature of Zr–O cages. At 10 mg mL−1, ZrT-NH2 has a lower zeta potential than ZrT-NH2-I, due to the fact that the majority of dispersion is neutral particles. The sizes of ZrT-NH2-I at low and high concentrations are all measured around 1.8 nm by dynamic light scattering (Fig. 3c), consistent with the molecular size of the discrete cage (1.5 nm). However, the size of ZrT-NH2 is increased to 300 nm, due to the cage aggregation in concentrated methanol solution. This aggregation leads to the insolubility, visualized by the turbid liquid (Fig. 3a). This result suggests that the substitution by bulky iodine weakens the cage-to-cage interactions, thereby increasing the solubility of the zirconium cage by 10 times (Fig. 3a and Table S3†).
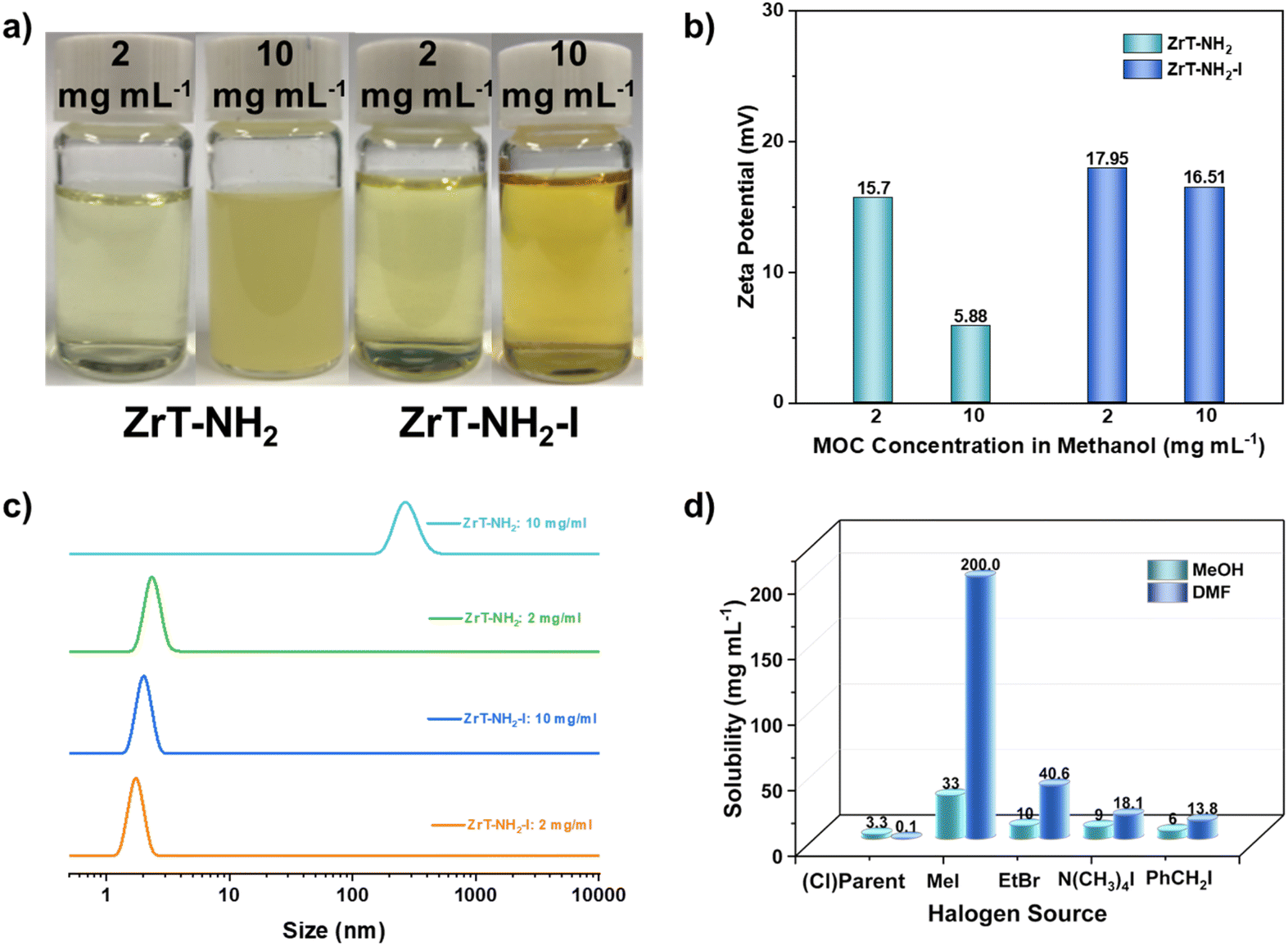 | ||
| Fig. 3 (a) ZrT-NH2 and ZrT-NH2-I solutions or suspensions with different concentrations. (b and c) Zeta potentials (b) and DLS (c) of ZrT-NH2 and ZrT-NH2-I with different concentrations. (d) The solubility of ZrT-NH2 (parent material) and its derived materials after reaction with different halogen sources in CH3OH (MeOH) and DMF. | ||
The solubilities of ZrT-NH2-I in methanol and DMF are plotted in Fig. 3d. Referencing to ZrT-NH2 (3.3 and 0.1 mg mL−1), ZrT-NH2-I can be respectively dissolved as 33 and 200 mg mL−1 in methanol and DMF. In the ZrT-NH2-I structure, tetrahedral cages are loosely packed and bulky iodine ions are weakly attracted by electrostatic forces, thus improving solvent–solute interactions. This enhanced solvation is responsible for the significant increase in solubility. When benzyl iodine (PhCH2I) is used instead of CH3I, the solubility is increased to 6 mg mL−1 in methanol. The minor improvement is due to steric hindrance which makes the substitution incomplete (Tables S1 and S3†). When the substitution reagent is changed to ethyl bromide (EtBr), Cl− is completely converted into Br−, the solubility is increased by 3 times in methanol. The solubility of Zr-MOCs follows the trend of I− > Br− > Cl−. The anion-induced discrepancy is due to the varied leaving abilities of the anions. The halogen substitution is advantageous over ion exchange in solubility improvement. When ZrT-NH2 is ion exchanged with tetramethylammonium iodide (N(CH3)4I), the solubility is slightly increased even after long-time (1 day) exchanging, which is due to low reaction degree with iodine (Table S1†). The high solubility of zirconium cages is expected to expand their application in MMMs.
Rapid N2 adsorptions at low pressures (P/P0 ≤ 0.03) indicate that ZrT-NH2 and ZrT-NH2-I are microporous materials (Fig. 4a). Compared with the isotherm plateau of ZrT-NH2, continuous adsorption of N2 is observed with pressure increase in the ZrT-NH2-I isotherm, ascribed to extra pores generated by insufficient cage packing in ZrT-NH2-I. Surface areas of 474 and 490 m2 g−1 are calculated using the Brunauer–Emmett–Teller (BET) model for ZrT-NH2 and ZrT-NH2-I, respectively. The pore size distribution curves based on the quenching solid density functional theory (QSDFT) show that the pore sizes of ZrT-NH2 and ZrT-NH2-I are both dominantly around 6.0 Å (Fig. 4b). The values close to the crystallographic diameter (6.0 Å) indicate that the tetrahedral cages remain intact.
 | ||
| Fig. 4 (a) Nitrogen adsorption (solid symbols) and desorption (hollow symbols) isotherms at 77 K. (b) Pore size distributions calculated from the QSDFT model. (c) CO2 and N2 adsorption isotherms at 273 K and 298 K. (d) Isosteric heats of adsorption (Qst) calculated from the temperature-dependent CO2 and N2 isotherms for the materials of ZrT-NH2 and ZrT-NH2-I. | ||
The gas affinity is analyzed using CO2 and N2 adsorptions (Fig. 4c). ZrT-NH2-I possesses favorable adsorption for CO2 over N2, evidenced by 6 times higher uptake (22.33 and 3.45 cm3 g−1 for CO2 and N2, respectively, at 298 K and 100 kPa). The high preference for CO2 adsorption is due to the presence of amino groups which more strongly interact with CO2 over N2. The isotherms at 273 K and 298 K derive the adsorption heats (Qst) of 27.3 and 10.4 kJ mol−1 for CO2 and N2, respectively (Fig. 4d). The Qst difference supports the adsorption preference for CO2. The similarity of CO2 and N2 adsorptions for ZrT-NH2 and ZrT-NH2-I in terms of uptake and Qst is originated from the identical surface chemistry of the two molecular cages.
ZrT-NH2-I was mixed with an amidoximated polymer of intrinsic microporosity (PIM-PAO) which is soluble in different solvents such as DMF and DMSO.38 After DMF evaporation under heat (60 °C), a self-standing membrane was formed (this MMM is defined as ZrT-NH2-I@PIM-PAO and its photo is shown in the inset of Fig. 5a). Scanning electron microscopy (SEM) images show that ZrT-NH2-I@PIM-PAO has no crack (Fig. 5a) and ZrT-NH2-I is uniformly dispersed in PIM-PAO without aggregation (Fig. S17†). The homogeneous distribution is attributed to the good solubility of ZrT-NH2-I in DMF. The EDX mapping of zirconium element confirms the uniform distribution of ZrT-NH2-I (Fig. S18†). The thickness of ZrT-NH2-I@PIM-PAO is around 20 μm (Fig. 5a). In contrast, particle aggregation occurs and interface defects form in ZrT-NH2@PIM-PAO (Fig. S19†), caused by the poor solubility of ZrT-NH2 in DMF. This phenomenon is also reflected by the membrane opacity (Fig. S20†). ZrT-NH2-I@PIM-PAO and ZrT-NH2@PIM-PAO were imaged using transmission electron microscopy (TEM). Large particles of ZrT-NH2 appear in ZrT-NH2@PIM-PAO (Fig. 5d); however, no aggregates of ZrT-NH2-I are observed in ZrT-NH2-I@PIM-PAO (Fig. 5c). The TEM result verifies the membrane uniformity and implies the interface compatibility between MOC and PIM. The uniform MMMs were prepared with different loadings, sizes and thicknesses (Fig. S20†). The membrane structure of ZrT-NH2-I@PIM-PAO was further probed by CO2 adsorption at 195 K (Fig. S21†). PIM-PAO has bimodal pores distributed at 0.7 and 1.0 nm. ZrT-NH2-I@PIM-PAO is encoded with 0.7 nm unimodal pores (Fig. 5b), presumably coming from included cages, consistent with dominant pores at 0.65 nm for ZrT-NH2-I. To be noted, large pores of 1.0 nm from PIM-PAO disappear; manifesting that molecular-sized ZrT-NH2-I cages (1.5 nm) occupy these pores. The decreased surface area of ZrT-NH2-I@PIM-PAO in comparison to that of PIM-PAO indicates the phase compatibility between ZrT-NH2-I and PIM-PAO.
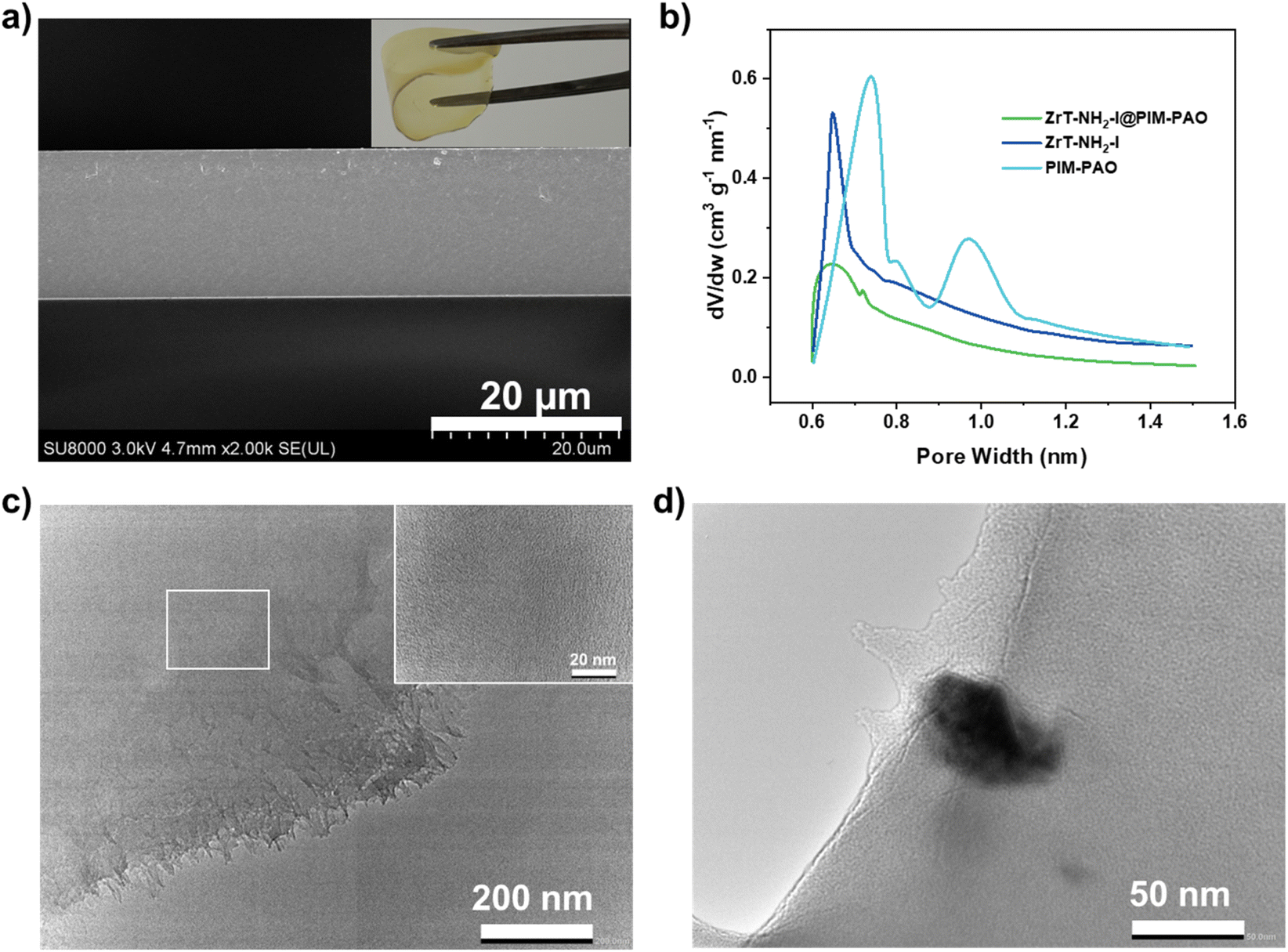 | ||
| Fig. 5 (a) SEM cross-sectional image and photo (inset) of the membrane of ZrT-NH2-I@PIM-PAO (8% ZrT-NH2-I). (b) Pore size distribution curves of PIM-PAO membrane, ZrT-NH2-I powder and ZrT-NH2-I@PIM-PAO membrane (8% ZrT-NH2-I) derived from CO2 adsorption isotherms at 195 K. (c and d) TEM images of ZrT-NH2-I@PIM-PAO (8% ZrT-NH2-I) (c) and ZrT-NH2@PIM-PAO (8% ZrT-NH2) (d). | ||
MMMs fabricated using ZrT-NH2 and ZrT-NH2-I have been tested for CO2/N2 separation. CO2 permeability of 2844.2 barrer, higher than most polymers (e.g. PEO ≤ 200 barrer, PVAm ≤ 200 barrer, PI < 1000 barrer),39–41 is recorded for PIM-PAO in Fig. 6a, showing that PIM-PAO is a suitable membrane matrix.42 With incorporation of 5 wt% ZrT-NH2 or ZrT-NH2-I in PIM-PAO, gas permeability decreases while CO2/N2 selectivity increases. In particular, ZrT-NH2-I derived MMM has CO2 permeability of 1425 barrer and CO2/N2 selectivity of 41. Such observation can be interpreted as the following: ZrT-NH2-I cages in the molecular state fill up large pores of PIM-PAO, reducing the pore volume and thus decreasing the permeability. CO2 can be preferentially adsorbed on amino groups in ZrT-NH2-I, contributing to the increased selectivity. The lower gas selectivity of ZrT-NH2 than ZrT-NH2-I is due to the fact that most of the ZrT-NH2 is present as aggregates. In more detail, the permeability was decoupled into the diffusion coefficient (D) and solubility coefficient (S) (Table S4†). After ZrT-NH2-I is incorporated into PIM-PAO, the solubility coefficient slightly changes (≤20%) but the diffusion coefficient significantly decreases (>260%), affirming the occupancy of PIM-PAO pores by ZrT-NH2-I cages. The diffusion selectivity of CO2/N2 increased by 63% confirming that ZrT-NH2-I eliminates non-selective gas transport pathways. As shown in Fig. 6b, the CO2/N2 selectivity for binary gases (45) is 134% of that for single gases (33.7), owing to the effect of competitive diffusion. The results from membrane structure and gas permeation suggest that ZrT-NH2-I regulates CO2 transport via a selective manner within the membrane (Fig. 6c). A serial pathway is built by connecting ZrT-NH2-I with PIM-PAO. The small pore in PIM-PAO facilitates CO2 diffusion and the functional group in ZrT-NH2-I enables CO2 adsorption. The synergic effects of size and functionality dominate the selective gas permeation of CO2 over N2. The molecular distribution of ZrT-NH2-I in PIM-PAO allows the MMM with an upper-bound-beyond performance (Fig. S23†) which is superior to all MOCs based MMMs and is also approaching to the best MMMs of MOFs and COFs (Table S6†).
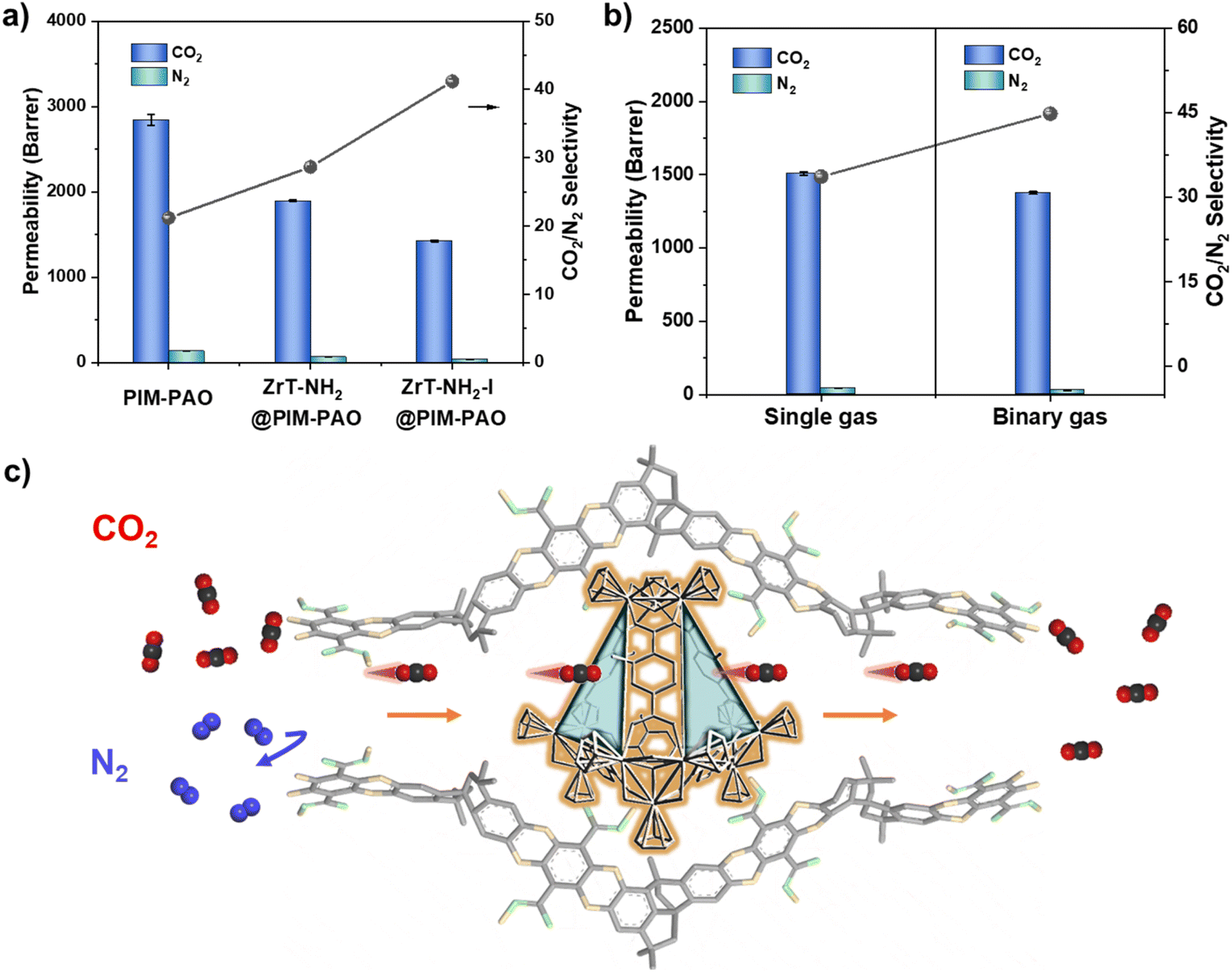 | ||
| Fig. 6 (a) Binary CO2/N2 gas permeation through pure PIM-PAO membrane and MMMs with ZrT-NH2 (5 wt%) and ZrT-NH2-I (5 wt%) (T = 25 °C, P = 2 bar, v/v = 50/50 in volume) (the data are shown here for MMMs with 5 wt% fillers because the membrane with high ZrT-NH2 loading (e.g. 8 wt%) cracks). (b) Single and binary CO2/N2 gas permeation through ZrT-NH2-I@PIM-PAO (8 wt% ZrT-NH2-I). (c) Schematic illustration of CO2 and N2 selective transport through a polymer–MOC serial pathway in the membrane (the MOC is located within a PIM-PAO large pore which is formed by insufficient packing of contorted chains). | ||
CO2/N2 permeations of ZrT-NH2-I@PIM-PAO were tested at various conditions. The decreasing trend of CO2 permeability as a function of loading supplements the inclusion of discrete ZrT-NH2-I cages within large pores of PIM-PAO (Fig. 7a). The curve of CO2/N2 selectivity looks like a parabola and the selectivity reaches a maximum at ZrT-NH2-I loading of 8 wt%. More –NH2 groups present in the membrane promote selective CO2 transport. After the loading exceeds 8 wt%, the selectivity of CO2/N2 gradually decreases, which may be due to polymer densification surrounding ZrT-NH2-I. In Fig. 7b, as the feed pressure increases, CO2 permeability decreases due to the Langmuir adsorption behavior. CO2/N2 selectivity decreases due to the fact that CO2 adsorption preference is reduced at high pressure.43,44 Gas permeability continuously increases from 298 K to 338 K (Fig. 7c), because the movement of gas molecules is accelerated with temperature. The CO2/N2 selectivity decreases with temperature because the interaction of –NH2 groups with CO2 is weakened more than with N2. During a consecutive test for 72 h (Fig. 7d), the CO2 permeability fluctuates marginally around 1377 barrer and the CO2/N2 selectivity averages at 45, translating to high stability of the ZrT-NH2-I MMM. MMMs with other thicknesses also exhibit similar performance (Table S5†).
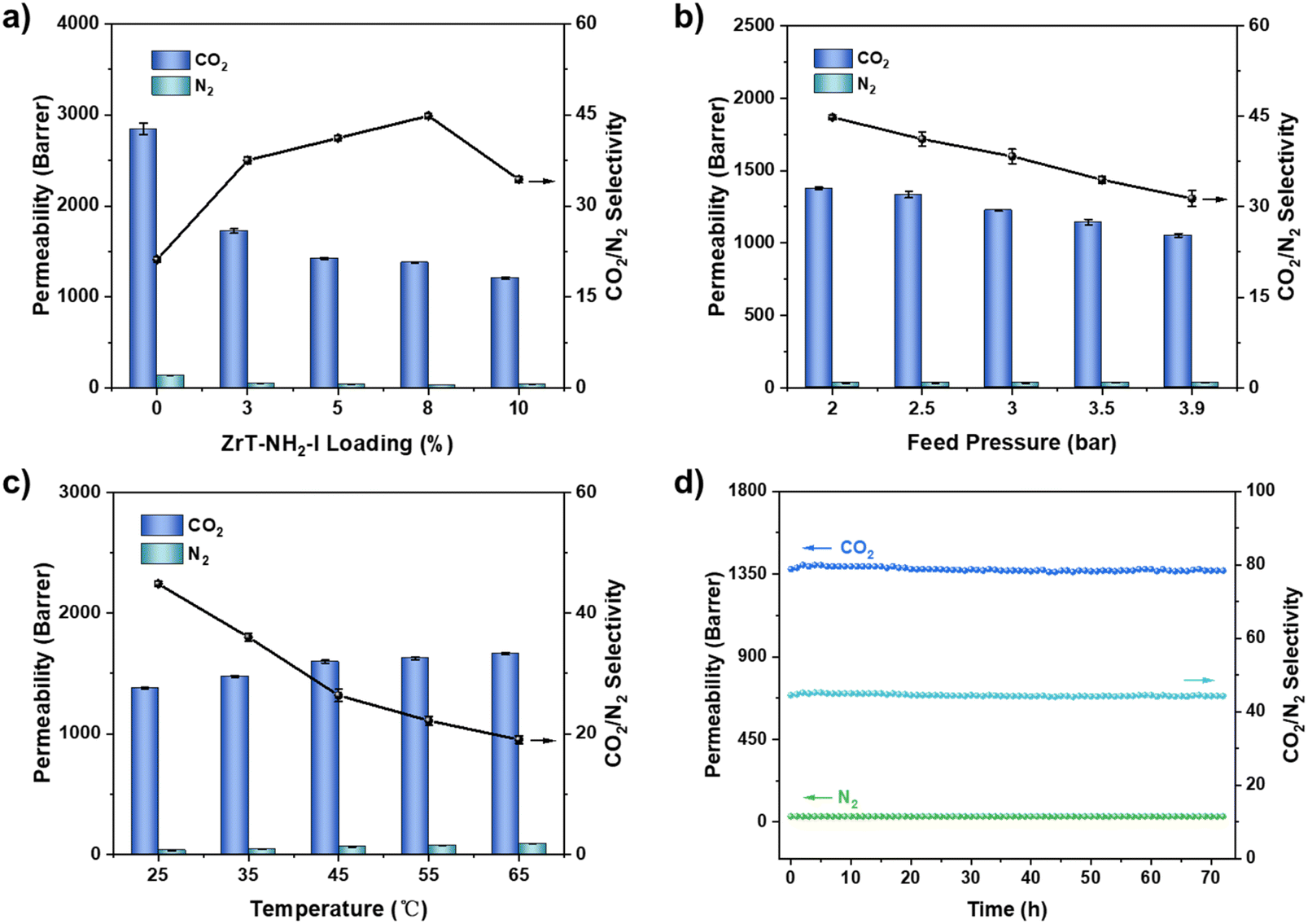 | ||
| Fig. 7 The effects of (a) ZrT-NH2-I loading, (b) feed pressure, (c) test temperature and (d) test time on CO2 and N2 permeability and CO2/N2 selectivity. | ||
Conclusions
In summary, iodine substitution was developed for increasing the solubility of zirconium-based MOCs. This reaction enabled the solubility of the synthesized MOC (ZrT-NH2-I) to be 10 times higher than the parent MOC. Moreover, ZrT-NH2-I preserved intact both tetrahedral molecular structure and permanent porosity. Highly soluble ZrT-NH2-I was processed with PIM-PAO to form uniform compatible MMMs. The membrane performance of ZrT-NH2-I@PIM-PAO was significantly improved after ZrT-NH2-I inclusion owing to the molecular-scaled distribution of ZrT-NH2-I and the compatibility of the MOC and polymer. Gas permeation revealed that ZrT-NH2-I@PIM-PAO possessed CO2 permeability of over 1300 barrer and CO2/N2 selectivity of more than 40 in CO2/N2 separation. Such performance was superior to those of other MMMs and the permeability-selectivity product broke the 2008 upper bound of polymers, promising MOCs based membranes applicable in carbon capture and separation.Experimental methods
Chemicals and solvents were purchased and subsequently used directly from commercial sources. The detailed procedure is described in the ESI.†ZrT-NH2-I synthesis
The parent material of ZrT-NH2 was prepared according to the previous protocol.26 100 mg of ZrT-NH2 was dispersed in 1 mL of DMF in a 5 mL vial, followed by an addition of 20 μL of CH3I. After 2 h reaction, the powder of ZrT-NH2-I was collected and then purified by washing with fresh CHCl3.PIM-PAO synthesis
PIM-1 and PIM-PAO were prepared according to previous protocols.45,46 0.5 g PIM-1 was dissolved in 30 mL THF and the solution was heated to reflux under N2. Then, 5.0 mL NH2OH was added dropwise into the solution above; this mixture was further refluxed for 20 h. The resultant polymer was precipitated by ethanol, filtered and then washed thoroughly with ethanol and water, and finally dried at 110 °C for 10 h.Fabrication of ZrT-NH2-I@PIM-PAO membranes
To fabricate the mixed-matrix membrane, a clear solution containing 3 wt% ZrT-NH2-I and PIM-PAO in DMF was first configured. This solution was subsequently cast on a silicon disc. After slow evaporation of the solvent at 60 °C, a dense ZrT-NH2-I@PIM-PAO membrane was obtained. The loadings of ZrT-NH2-I in the membranes were varied in the range of 0–10 wt%.Gas permeation
The CO2/N2 separation was performed on a self-made membrane permeation unit using a standard Wicke–Kallenbach mode. The feed flow rate for each gas of pure CO2 or N2 and binary mixture of CO2 and N2 was 25 mL min−1. The permeate gas was collected using Ar (15 mL min−1) swept from the membrane back. The feed and permeate gases were detected using on-line gas chromatography (GC-7890B, Agilent). The separation parameters of permeability (P, barrer) and selectivity (α) for CO2 over N2 were reported.Data availability
The data supporting this study are available in the ESI of this article.†Author contributions
X. Z. and G. Z. conceived and designed this work. J. D. conducted the synthesis and characterization of the materials and wrote this paper. D. G. and G. C. helped the characterization of the materials. Q. P. and J. L. helped the data analysis and discussion. All authors reviewed and edited the manuscript.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC grant nos. 22375031, 22131004, U21A20330), National Key R&D Program of China (2022YFB3805902), “111” Program (B18012), the Jilin Natural Science Fund for Excellent Young Scholars (20230508116RC), and the Fundamental Research Funds for the Central Universities (2412023YQ001). We would like to thank Prof. Dan Zhao (National University of Singapore) and Prof. Daqiang Yuan (Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences) for their constructive suggestions on this paper.References
- A. Knebel, A. Bavykina, S. J. Datta, L. Sundermann, L. Garzon-Tovar, Y. Lebedev, S. Durini, R. Ahmad, S. M. Kozlov, G. Shterk, M. Karunakaran, I. D. Carja, D. Simic, I. Weilert, M. Kluppel, U. Giese, L. Cavallo, M. Rueping, M. Eddaoudi, J. Caro and J. Gascon, Nat. Mater., 2020, 19, 1346–1353 CrossRef CAS PubMed.
- X. Tan, S. Robijns, R. Thur, Q. Ke, N. De Witte, A. Lamaire, Y. Li, I. Aslam, D. Van Havere, T. Donckels, T. Van Assche, V. Van Speybroeck, M. Dusselier and I. Vankelecom, Science, 2022, 378, 1189–1194 CrossRef CAS PubMed.
- K. Duan, J. Wang, Y. Zhang and J. Liu, J. Membr. Sci., 2019, 572, 588–595 CrossRef CAS.
- Y. Cheng, B. Joarder, S. J. Datta, N. Alsadun, D. Poloneeva, D. Fan, R. Khairova, A. Bavykina, J. Jia, O. Shekhah, A. Shkurenko, G. Maurin, J. Gascon and M. Eddaoudi, Adv. Mater., 2023, 35, e2300296 CrossRef PubMed.
- A. Knebel and J. Caro, Nat. Nanotechnol., 2022, 17, 911–923 CrossRef CAS PubMed.
- M. Rezakazemi, A. Ebadi Amooghin, M. M. Montazer-Rahmati, A. F. Ismail and T. Matsuura, Prog. Polym. Sci., 2014, 39, 817–861 CrossRef CAS.
- J. Dechnik, J. Gascon, C. J. Doonan, C. Janiak and C. J. Sumby, Angew. Chem., Int. Ed., 2017, 56, 9292–9310 CrossRef CAS PubMed.
- A. J. Gosselin, C. A. Rowland and E. D. Bloch, Chem. Rev., 2020, 120, 8987–9014 CrossRef CAS PubMed.
- W. Drozdz, A. Ciesielski and A. R. Stefankiewicz, Angew. Chem., Int. Ed., 2023, 62, e202307552 CrossRef CAS PubMed.
- J. R. Li and H. C. Zhou, Nat. Chem., 2010, 2, 893–898 CrossRef CAS PubMed.
- T. Grancha, A. Carne-Sanchez, F. Zarekarizi, L. Hernandez-Lopez, J. Albalad, A. Khobotov, V. Guillerm, A. Morsali, J. Juanhuix, F. Gandara, I. Imaz and D. Maspoch, Angew. Chem., Int. Ed., 2021, 60, 5729–5733 CrossRef CAS PubMed.
- S. Mollick, S. Mukherjee, D. Kim, Z. Qiao, A. V. Desai, R. Saha, Y. D. More, J. Jiang, M. S. Lah and S. K. Ghosh, Angew. Chem., Int. Ed., 2019, 58, 1041–1045 CrossRef CAS PubMed.
- A. Khobotov-Bakishev, L. Hernandez-Lopez, C. von Baeckmann, J. Albalad, A. Carne-Sanchez and D. Maspoch, Adv. Sci., 2022, 9, e2104753 CrossRef PubMed.
- S. Mollick, S. Fajal, S. Mukherjee and S. K. Ghosh, Chem.–Asian J., 2019, 14, 3096–3108 CrossRef CAS PubMed.
- M. Sohail, H. An, W. Choi, J. Singh, K. Yim, B.-H. Kim, Y. C. Park, J. S. Lee and H. Kim, J. Membr. Sci., 2021, 620, 118826 CrossRef CAS.
- Y. N. Yun, M. Sohail, J. H. Moon, T. W. Kim, K. M. Park, D. H. Chun, Y. C. Park, C. H. Cho and H. Kim, Chem.–Asian J., 2018, 13, 631–635 CrossRef CAS PubMed.
- E. M. El-Sayed, Y. D. Yuan, D. Zhao and D. Yuan, Acc. Chem. Res., 2022, 55, 1546–1560 CrossRef CAS PubMed.
- J. Liu, W. Duan, J. Song, X. Guo, Z. Wang, X. Shi, J. Liang, J. Wang, P. Cheng, Y. Chen, M. J. Zaworotko and Z. Zhang, J. Am. Chem. Soc., 2019, 141, 12064–12070 CrossRef CAS PubMed.
- Z. Yang, G. Liu, Y. D. Yuan, S. B. Peh, Y. Ying, W. Fan, X. Yu, H. Yang, Z. Wu and D. Zhao, J. Membr. Sci., 2021, 636, 119564 CrossRef CAS.
- G. A. Taggart, A. M. Antonio, G. R. Lorzing, G. P. A. Yap and E. D. Bloch, ACS Appl. Mater. Interfaces, 2020, 12, 24913–24919 CrossRef CAS PubMed.
- T. Liu, R. Zhang, G.-R. Si, B. Liu, Y. Xie, L.-H. Xie and J.-R. Li, ACS Sustainable Chem. Eng., 2022, 10, 13534–13544 CrossRef CAS.
- H. Furukawa, J. Kim, K. E. Plass and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 8398–8399 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, J. B. Wachter, H. K. Chae, M. O’Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 4368–4369 CrossRef CAS PubMed.
- T. Grancha, A. Carne-Sanchez, L. Hernandez-Lopez, J. Albalad, I. Imaz, J. Juanhuix and D. Maspoch, J. Am. Chem. Soc., 2019, 141, 18349–18355 CrossRef CAS PubMed.
- E. J. Gosselin, G. E. Decker, A. M. Antonio, G. R. Lorzing, G. P. A. Yap and E. D. Bloch, J. Am. Chem. Soc., 2020, 142, 9594–9598 CrossRef PubMed.
- D. Nam, J. Huh, J. Lee, J. H. Kwak, H. Y. Jeong, K. Choi and W. Choe, Chem. Sci., 2017, 8, 7765–7771 RSC.
- G. Liu, Y. Di Yuan, J. Wang, Y. Cheng, S. B. Peh, Y. Wang, Y. Qian, J. Dong, D. Yuan and D. Zhao, J. Am. Chem. Soc., 2018, 140, 6231–6234 CrossRef CAS PubMed.
- X. Guo, S. Xu, Y. Sun, Z. Qiao, H. Huang and C. Zhong, J. Membr. Sci., 2021, 632, 119354 CrossRef CAS.
- G. Liu, M. Zhou, K. Su, R. Babarao, D. Yuan and M. Hong, CCS Chem., 2021, 3, 1382–1390 CrossRef CAS.
- J. Liu, T. Luo, Y. Xue, L. Mao, P. J. Stang and M. Wang, Angew. Chem., Int. Ed., 2021, 60, 5429–5435 CrossRef CAS PubMed.
- L. Cao, P. Wang, X. Miao, Y. Dong, H. Wang, H. Duan, Y. Yu, X. Li and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 7005–7011 CrossRef CAS PubMed.
- Y. Zeng, J. Shi, K. Li, J. Li, H. Yu, F. Fang, X.-Q. Hao, H. Zhang and M. Wang, Chem. Synth., 2022, 2, 12 CrossRef CAS.
- H. J. Choi, S. Hong, Y. Son, K. T. Kim, C. Kim, M. Yoon and M. Kim, Mol. Syst. Des. Eng., 2023, 8, 598–603 RSC.
- P. H. M. Andrade, N. Henry, C. Volkringer, T. Loiseau, H. Vezin, M. Hureau and A. Moissette, ACS Appl. Mater. Interfaces, 2022, 14, 29916–29933 CrossRef CAS PubMed.
- K. Li, Y. Zhao, P. Zhang, C. He, J. Deng, S. Ding and W. Shi, Appl. Surf. Sci., 2016, 390, 412–421 CrossRef CAS.
- G. Rajendra Kumar, A. Dennyson Savariraj, S. N. Karthick, S. Selvam, B. Balamuralitharan, H. J. Kim, K. K. Viswanathan, M. Vijaykumar and K. Prabakar, Phys. Chem. Chem. Phys., 2016, 18, 7284–7292 RSC.
- A. Telfah, M. A. Al-Akhras, K. A. Al-Izzy, A. A. Ahmad, R. Ababneh, M. J. A. Ahmad, C. J. Tavares and R. Hergenröder, Polym. Bull., 2022, 79, 3759–3778 CrossRef CAS.
- H. A. Patel and C. T. Yavuz, Chem. Commun., 2012, 48, 9989–9991 RSC.
- M. Liu, M. D. Nothling, S. Zhang, Q. Fu and G. G. Qiao, Prog. Polym. Sci., 2022, 126, 101504 CrossRef CAS.
- Y. Han and W. S. W. Ho, J. Polym. Sci., 2020, 58, 2435–2449 CrossRef CAS.
- H. Sanaeepur, A. Ebadi Amooghin, S. Bandehali, A. Moghadassi, T. Matsuura and B. Van der Bruggen, Prog. Polym. Sci., 2019, 91, 80–125 CrossRef CAS.
- Z. Wang, H. Ren, S. Zhang, F. Zhang and J. Jin, J. Mater. Chem. A, 2017, 5, 10968–10977 RSC.
- M. Chawla, H. Saulat, M. Masood Khan, M. Mahmood Khan, S. Rafiq, L. Cheng, T. Iqbal, M. I. Rasheed, M. Z. Farooq, M. Saeed, N. M. Ahmad, M. B. Khan Niazi, S. Saqib, F. Jamil, A. Mukhtar and N. Muhammad, Chem. Eng. Technol., 2020, 43, 184–199 CrossRef CAS.
- P. Zhang, C. Zhang, L. Wang, J. Dong, D. Gai, W. Wang, T. S. Nguyen, C. T. Yavuz, X. Zou and G. Zhu, Adv. Funct. Mater., 2023, 33, 2210091 CrossRef CAS.
- Z. Wang, X. Luo, Z. Song, K. Lu, S. Zhu, Y. Yang, Y. Zhang, W. Fang and J. Jin, Nat. Commun., 2022, 13, 4169 CrossRef CAS PubMed.
- Z. Wang, W. Wang, T. Zeng, D. Ma, P. Zhang, S. Zhao, L. Yang, X. Zou and G. Zhu, Adv. Mater., 2022, 34, e2104606 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05080k |
| This journal is © The Royal Society of Chemistry 2024 |