DOI:
10.1039/D4SC04853A
(Edge Article)
Chem. Sci., 2024,
15, 18000-18007
Role of ancillary ligands in S-nitrosothiol and NO generation from nitrite–thiol interactions at mononuclear zinc(II) sites†
Received
22nd July 2024
, Accepted 2nd October 2024
First published on 3rd October 2024
Abstract
Generation of S-nitrosothiol (RSNO) and nitric oxide (NO) mediated by zinc(II) coordination motifs is of prime importance for understanding the role of zinc(II)-based cofactors in redox-signalling pathways. This study uniquely employs a set of mononuclear [L2ZnII]2+ cores (where L = Me4PzPz/Me2PzPy/Me2PzQu) for introducing subtle alterations of the primary coordination sphere and investigates the role of ligand tuning in the transformation of NO2− in the presence of thiols. Single crystal X-ray diffraction (SCXRD) analyses on [L2ZnII–X](X) (where X = perchlorate/triflate) illustrate consistent changes in the bond distances, thereby showing variations of the metal–ligand interactions depending on the nature of the heterocyclic donor arms (pyrazole/pyridine/quinoline). Moreover, such tuning of the ligands affects the Lewis-acidity of the [L2ZnII]2+ cores as evaluated by 31P NMR and SCXRD studies on the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acid–base adducts [L2ZnII(OPEt3)]2+. Crystallographic and 15N NMR spectroscopic analyses on the nitrite complexes [L2ZnII(κ2-nitrite)](ClO4) reveal that the chemical environments of the nitrite anions in these complexes are nearly identical, despite the dissimilarity in the Lewis-acidity of the [L2ZnII]2+ cores. Interestingly, RSNO and NO generation from the reactions of [L2ZnII(κ2-nitrite)](ClO4) with 4-tert-butylbenzylthiol (tBuBnSH) exhibits that the [(Me2PzQu)2ZnII]2+ core is the most efficient in promoting nitrite–thiol interactions due to the ease of available hemilabile coordination sites at the Lewis acidic [ZnII]. Detailed UV-vis studies in tandem with computational investigation, for the first time, provide an unambiguous demonstration of the nitrous acid (HNO2) intermediate generated through an intramolecular proton-transfer from thiol to nitrite at zinc(II).
:1 acid–base adducts [L2ZnII(OPEt3)]2+. Crystallographic and 15N NMR spectroscopic analyses on the nitrite complexes [L2ZnII(κ2-nitrite)](ClO4) reveal that the chemical environments of the nitrite anions in these complexes are nearly identical, despite the dissimilarity in the Lewis-acidity of the [L2ZnII]2+ cores. Interestingly, RSNO and NO generation from the reactions of [L2ZnII(κ2-nitrite)](ClO4) with 4-tert-butylbenzylthiol (tBuBnSH) exhibits that the [(Me2PzQu)2ZnII]2+ core is the most efficient in promoting nitrite–thiol interactions due to the ease of available hemilabile coordination sites at the Lewis acidic [ZnII]. Detailed UV-vis studies in tandem with computational investigation, for the first time, provide an unambiguous demonstration of the nitrous acid (HNO2) intermediate generated through an intramolecular proton-transfer from thiol to nitrite at zinc(II).
Introduction
S-Nitrosothiol (RSNO) is an incredibly important functional group in nitric oxide (NO) signalling events encompassing neurotransmission, vasodilation, immune response and many more.1,2 While the nitric oxide synthase (NOS) activity of heme-Fe sites primarily serves as the NO-source through an oxidative transformation of L-arginine in the presence of dioxygen,3 the NOS activity is inhibited under hypoxia. Hence, the NOS-independent generation of NO is critical for the NO-bioactivities under hypoxic conditions. It is noteworthy that nitrite (NO2−) and S-nitrosothiol (RSNO) serve as circulators and sources of NO under hypoxic conditions, while both can also act as sinks for NO at a higher flux.4,5 One-electron reductive transformation of nitrite-to-NO, known as nitrite reductase activity (NiR) (Fig. 1A), is mediated by the reduced state of various heme-Fe sites (in deoxymyoglobin, deoxyhemoglobin, cytochrome c oxidase), Cu-containing nitrite reductase, and Mo-site of xanthine oxidoreductase.6 Surprisingly, a redox-inactive His3ZnII active site of carbonic anhydrase (CA) has been reported to bind the nitrite anion and transform to NO through nitrite anhydrase activity (Fig. 1B).7 However, a number of later studies suggest that an external reductant, such as thiol, is essential for the conversion of nitrite-to-NO at the redox-inactive zinc(II) site.8
 |
| | Fig. 1 (A and B) Transformations of the nitrite anion relevant to NO generation. (C) Mechanisms for the reactions of thiol with zinc(II)–nitrite leading to RSNO. (D) Schematic representations of the zinc(II)–nitrite complexes employed in this study. | |
Modelling of Fe/Cu mediated NiR activities in the presence of H+/e− has been demonstrated by utilizing a number of Fe– and Cu–nitrite complexes in the previous literature.9–11 In contrast, the examples of nitrite reduction involving zinc(II)–nitrite are limited. As the redox-active Fe/Cu-based active sites often engage in complex reactions with NO gas,11,12 insights into the NiR process at a redox-inactive metal site may serve critical for designing synthetic catalysts and understanding the role of zinc-containing enzymes in NO signalling. A prior study by Caulton and co-workers uniquely demonstrated nitrite-to-NO transformation utilizing a mononuclear zinc(II) core [(btzp)Zn]2+ featuring tetrazene-based redox-active moieties capable of managing up to 4 equivalents of protons and electrons through the btzp ⇄ H4btzp equilibrium (where H4btzp = bisdihydro-tetrazenepyridine).13 Warren and co-workers reported the first example of thiol reaction with a tris(pyrazolyl)borate supported tripodal [ZnII](κ2-nitrite) complex where 4-methylbenzenethiol (ArSH) smoothly reacts to provide the corresponding thiolate complex [ZnII]–SAr, disulfide (ArSSAr) and NO, via S-nitrosothiol (ArSNO). In contrast, an alkyl thiol, namely benzylthiol (BnSH), has been demonstrated to react sluggishly with the [ZnII]–nitrite complex.14 The differential reactivity of aromatic versus aliphatic thiols towards [ZnII]–nitrite suggests that the pKa of thiol plays an important role in these reactions. Two mechanistic possibilities for the reactions of thiol (RSH) and nitrite at [ZnII] have been considered (Fig. 1C): (a) a proton-transfer assisted concerted transnitrosation involving RSH and the nitrite anion in [ZnII](κ2-nitrite) resulting in zinc(II)-hydroxide and directly formed S-nitrosothiol (RSNO); (b) an acid–base exchange reaction between thiol and zinc-bound nitrite leading to the formation of zinc(II)-thiolate and nitrous acid (HNO2), a potent nitrosating agent. Some of our previous studies show that persulfidation of thiol in the presence of elemental sulfur enhances the efficiency of NO generation from a mononuclear [ZnII](κ2-nitrite).15 Moreover, the reactivity of thiol and nitrite in the presence of zinc(II) salts has been illustrated to provide good yields of NO via RSNO, while the nature of the coordination environment of the zinc(II) site is not clear. On the other hand, a phenolate bridged dinuclear {[ZnII](κ1-ONO)}2 core has been demonstrated to efficiently react with alkyl thiol, such as 4-tert-butylbenzylthiol (tBuBnSH), in comparison to the reactions of the previously reported mononuclear [ZnII](κ2-nitrite) complexes.14–16 It has been speculated earlier that the Lewis acidity of the zinc(II) site is the key factor for the activation of nitrite by rendering it more electrophilic.16 However, an experimental validation of the underlying mechanism and the factors influencing the thiol reactivity of mononuclear zinc(II)–nitrite remained poorly understood.
Herein, we employ a number of crystallographically characterized mononuclear complexes [L2ZnII(ClO4)](ClO4) (1-Pz/1-Py) and [L2ZnII(OTf)](OTf) (3-Pz/3-Py/3-Qu) to understand the role of heterocyclic ligand donors, such as pyrazole (Pz), pyridine (Py), quinoline (Qu), in modulating the properties of the zinc(II) sites (Fig. 2). Subsequently, the objective of this work is to provide experimental demonstration along with theoretical support for pinpointing the underlying mechanism and factors controlling interactions between thiol and nitrite at zinc(II) prior to the generation of RSNO/NO. Spectroscopic demonstration of the reactive intermediate such as nitrous acid (HNO2) and comparison of the rate of RSNO formation during the reactions of [L2ZnII(κ2-nitrite)](ClO4) complexes (5-Pz/5-Py/5-Qu) with less acidic alkyl thiol tBuBnSH17 have been investigated for the first time (Fig. 1D and 2). A combination of experimental and computational studies imply that both the hemilability of the ancillary ligand and the Lewis acidity of the [L2ZnII]2+ cores influence the generation of HNO2 through an intramolecular proton-transfer, thereby discerning the mechanistic insights into the thiol reactivity of zinc(II)–nitrite complexes 5-Pz/5-Py/5-Qu.
 |
| | Fig. 2 Synthesis of zinc(II) complexes. The dashed circle represents the variable heterocycle dimethylpyrazole/pyridine/quinoline. Inset shows the X-ray crystal structures of the cationic parts of complexes 1-Pz and 1-Py. The crystal structures of all isolated complexes at the 30% probability level of thermal ellipsoids are shown in the ESI.† | |
Results and discussion
Synthesis and characterization of zinc(II) complexes
Reactions of the bidentate ligand Me4PzPz/Me2PzPy (2.0 equiv.) and Zn(ClO4)2·6H2O (1.0 equiv.) in methanol provide the corresponding complexes [L2ZnII(ClO4)](ClO4) (1-Pz in 90% yield) and (1-Py in 78% yield) (Fig. 2 and S1–S8†). Single crystal X-ray diffraction (SCXRD) analysis on 1-Pz depicts a hexa-coordinated zinc site coordinating to two bidentate Me4PzPz ligands and one perchlorate anion bound in a κ2-O,O′ fashion (Zn1–O1 2.474(3) and Zn1–O4 2.548(4) Å). SCXRD analysis on 1-Py shows a distorted square pyramidal (τ5 = 0.30)18 zinc site bound to two Me2PzPy ligands and one κ1-OClO3 anion (Zn1–O1 2.450(2) Å). While the X-ray structures of the previously reported isoelectronic copper(I) complexes of the comparable ligands [(iPr4PzPz)2CuI](CuCl2) and [(Me2PzPy)2CuI](ClO4) show tetrahedral geometry of the copper(I) sites,12,19 the deviation from the tetrahedral geometric preference of the d10 system at the expense of interacting with the weakly coordinating perchlorate anion in the present zinc(II) complexes 1-Pz/1-Py may be attributed to the Lewis acidic nature of the [L2ZnII]2+ core (vide infra).
Unlike the metalation of Me4PzPz/Me2PzPy ligands, the quinoline substituted ligand Me2PzQu (2.0 equiv.) reacts with Zn(ClO4)2·6H2O (1.0 equiv.) in methanol to provide an off-white solid of [{(Me2PzQu)2ZnII}2(μ-OH)](ClO4)3 (2-Qu) in 62% yield (Fig. 2, S9 and S10†). The FTIR spectrum of complex 2-Qu exhibits a distinct feature for ν(OH) at 3645 cm−1 (Fig. S11†), consistent with the ν(OH) reported for the bridging hydroxide complexes in the previous literature.20 The molecular structure of complex 2-Qu obtained from SCXRD analysis portrays a dinuclear {[ZnII]–(μ-OH)–[ZnII]}3+ core (Fig. 3 and S12†). In contrast to the geometric deviations observed for the zinc(II) sites in 1-Pz/1-Py, the zinc(II) sites in complex 2-Qu exhibit a tetrahedral geometry (Zn1–O1 1.889(2), Zn1–N1 1.964(4), Zn1–N4 2.062(5), Zn1–N6 2.212(9) Å) with a geometry index τ4 = 0.81.21 A relatively longer interatomic distance Zn1⋯N3 2.720(9) Å renders one of the quinoline arms hemilabile. This may be attributed to the fact that a relatively strong interaction between the zinc site and the highly basic hydroxide ligand weakens the interaction with the less basic heterocyclic ligand Qu. We hypothesize that the hydroxide ligand in 2-Qu originates from the deprotonation of a coordinated water molecule at the Lewis acidic zinc(II) site of the transient complex [(Me2PzQu)2ZnII(OH2)](ClO4)2 (Fig. 2).
 |
| | Fig. 3 X-ray crystal structure of the cationic part [{(Me2PzQu)2ZnII}2(μ-OH)]3+ in complex 2-Qu. The atom sites labelled with prime are symmetry generated. | |
Aiming to understand the nature of the [L2ZnII]2+ core, we turned to isolate a comparable set of zinc(II) complexes of Me4PzPz/Me2PzPy/Me2PzQu ligands from the metalation reactions using anhydrous zinc(II)–triflate salt. Graciously, 2:1 reactions of ligand L (where L = Me4PzPz/Me2PzPy/Me2PzQu) and Zn(OTf)2 in acetonitrile resulted in the corresponding zinc(II)–triflate complexes [L2ZnII(OTf)](OTf) (3-Pz/3-Py/3-Qu) in good yields (Fig. 2 and S13–S24†). SCXRD analyses on 3-Pz/3-Py/3-Qu show the presence of two bidentate ligands L along with one triflate anion in the first coordination sphere of zinc. The geometry indices (τ5) are calculated as 0.27 (for 3-Pz), 0.01 (for 3-Py) and 0.46 (for 3-Qu) (Fig. 4).18 Notably, Zn–N(pyrazole) bond distances in all three complexes are comparable and range between 2.025(2) Å and 2.096(5) Å. Zn–N(pyridine) distances in 3-Py (Zn1–N3 2.069(2) and Zn1–N6 2.056(2) Å) are also comparable with the Zn–N(pyrazole) distances. In contrast, Zn–N(quinoline) distances in 3-Qu (Zn1–N3 2.245(2) and Zn1–N6 2.212(2) Å) are significantly longer relative to the Zn–N(pyrazole) distances (Zn1–N1 2.039(2) and Zn1–N4 2.025(2) Å). Interestingly, the Zn–O(triflate) bond distance of 2.098(2) Å in 3-Qu is the shortest in comparison to Zn1–O1 2.178(4) Å (in 3-Pz) and Zn1–O1/O2 2.542(2)/2.621(2) Å (in 3-Py) (Fig. 4). This trend of the Zn–O distances between the Lewis acidic zinc(II) site and weakly coordinating triflate anion perhaps suggests that the [(Me2PzQu)2ZnII]2+ core is the most Lewis acidic in nature as compared to [(Me4PzPz)2ZnII]2+ and [(Me2PzPy)2ZnII]2+.
 |
| | Fig. 4 X-ray crystal structures of the mono-cationic parts of complexes 3-Pz (A), 3-Py (B), and 3-Qu (C). The bond distances tabulated in Å. | |
Probing the Lewis acidity of zinc(II) sites
Equimolar reactions of [L2ZnII(ClO4)](ClO4) (where L = Me4PzPz/Me2PzPy) (1-Pz/1-Py) with triethyl phosphine oxide (Et3PO) in acetonitrile provide the corresponding Lewis acid–base adduct complexes [L2ZnII(OPEt3)](ClO4)2 (4-Pz/4-Py) (Fig. S25–S29†). SCXRD analyses on the single crystals of 4-Pz/4-Py confirm the formation of 1:1 adduct complexes (Fig. 5, S27 and S30†). X-ray structures of both the complexes 4-Pz/4-Py exhibit distorted penta-coordinated geometry halfway between square-pyramidal and trigonal bipyramidal with (τ5) 0.61 (for 4-Pz) and 0.74 (for 4-Py).18 The Zn1–O1 and O1–P1 distances associated with the [ZnII]–O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PEt3 moieties in 4-Pz/4-Py are comparable to that of the previously reported 1:1 Lewis acid–base adduct.22 As several attempts to isolate the analogous adduct complex [(Me2PzQu)2ZnII(OPEt3)]2+ were unsuccessful, [(Me2PzQu)2ZnII(OPEt3)](OTf)2 (4-Qu) was generated in situ by adding Et3PO (1 equiv.) to 3-Qu (Fig. S31 and S32†). We then probed the Lewis acidity of the [L2ZnII]2+ cores experimentally by monitoring the shifts in the 31P NMR resonances for these adduct complexes.23 The 31P NMR spectrum of in situ generated 4-Qu in CD3CN shows a resonance at 72.68 ppm, while the peak for an authentic sample of Et3PO in CD3CN appears at 49.78 ppm (Fig. 5B).1531P NMR spectra of the isolated samples of 4-Pz and 4-Py exhibit chemical shifts at 69.88 and 67.12 ppm, respectively. These 31P chemical shifts correspond to the acceptor numbers as 63.8 (for 4-Pz), 57.7 (for 4-Py), and 70.0 (for 4-Qu).22,23 These findings are comparable to those of the previously reported cationic zinc complexes.22 The acceptor numbers not only indicate the significant Lewis acidic nature of the [L2ZnII]2+ cores but also establish a trend of the Lewis acidity on varying the nature of the heterocyclic ligands: [(Me2PzQu)2ZnII]2+ > [(Me4PzPz)2ZnII]2+ > [(Me2PzPy)2ZnII]2+.
PEt3 moieties in 4-Pz/4-Py are comparable to that of the previously reported 1:1 Lewis acid–base adduct.22 As several attempts to isolate the analogous adduct complex [(Me2PzQu)2ZnII(OPEt3)]2+ were unsuccessful, [(Me2PzQu)2ZnII(OPEt3)](OTf)2 (4-Qu) was generated in situ by adding Et3PO (1 equiv.) to 3-Qu (Fig. S31 and S32†). We then probed the Lewis acidity of the [L2ZnII]2+ cores experimentally by monitoring the shifts in the 31P NMR resonances for these adduct complexes.23 The 31P NMR spectrum of in situ generated 4-Qu in CD3CN shows a resonance at 72.68 ppm, while the peak for an authentic sample of Et3PO in CD3CN appears at 49.78 ppm (Fig. 5B).1531P NMR spectra of the isolated samples of 4-Pz and 4-Py exhibit chemical shifts at 69.88 and 67.12 ppm, respectively. These 31P chemical shifts correspond to the acceptor numbers as 63.8 (for 4-Pz), 57.7 (for 4-Py), and 70.0 (for 4-Qu).22,23 These findings are comparable to those of the previously reported cationic zinc complexes.22 The acceptor numbers not only indicate the significant Lewis acidic nature of the [L2ZnII]2+ cores but also establish a trend of the Lewis acidity on varying the nature of the heterocyclic ligands: [(Me2PzQu)2ZnII]2+ > [(Me4PzPz)2ZnII]2+ > [(Me2PzPy)2ZnII]2+.
 |
| | Fig. 5 (A) X-ray crystal structure of [(Me2PzPy)2ZnII(OPEt3)]2+ in complex 4-Py. (B) 31P{1H} NMR spectra of 4-Py, 4-Pz, and an in situ generated sample of 4-Qu in CD3CN at room temperature. | |
Synthesis and characterization of zinc(II)–nitrite complexes
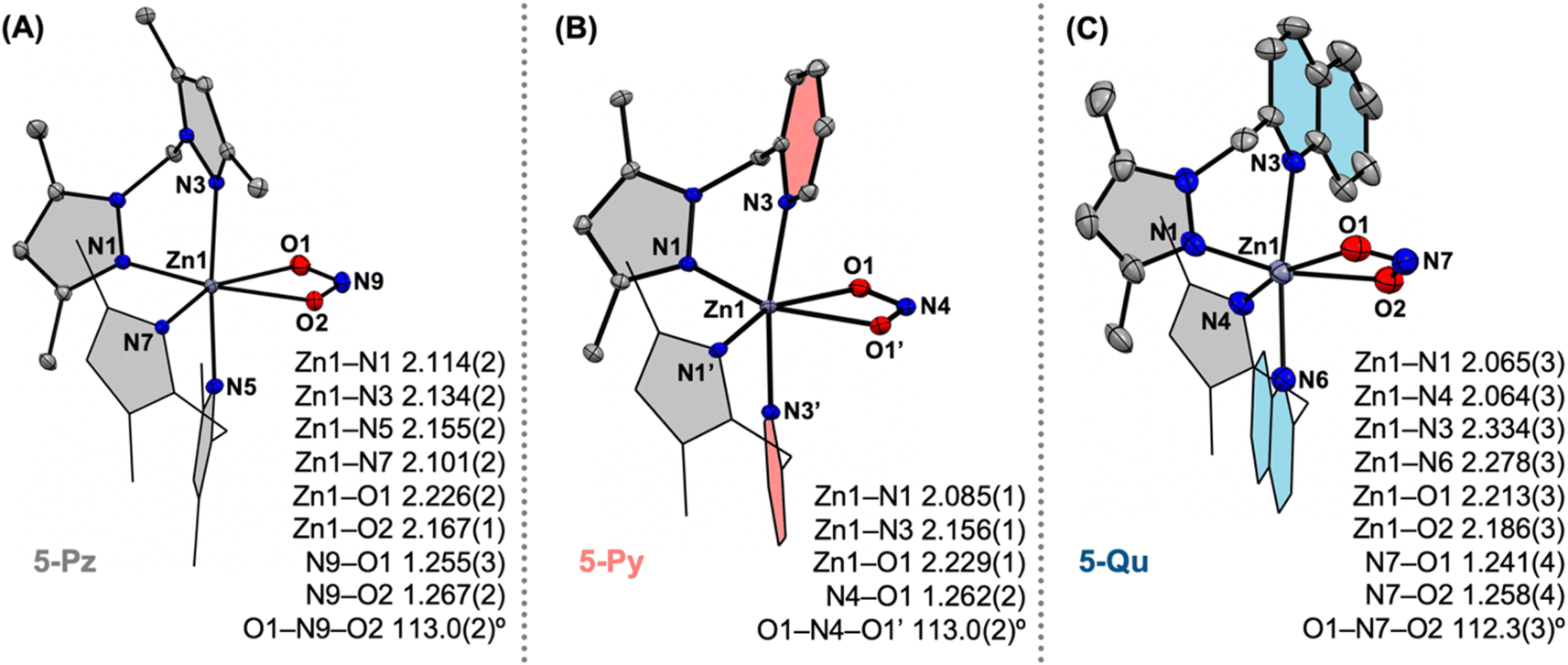
We then turned to understand the structure and properties of the nitrite anion coordinated to the electronically tuned [L2ZnII]2+ sites. 2:1 stoichiometric reactions of the ligands L (where L =Me4PzPz/Me2PzPy/Me2PzQu) and Zn(ClO4)2·6H2O in the presence of NaNO2 in methanol result in [L2ZnII(κ2-nitrite)](ClO4) complexes (5-Pz/5-Py/5-Qu) (Fig. 2). Single crystal X-ray diffraction analyses on each of these complexes reveal the presence of the hexa-coordinated zinc site with two ancillary ligands and one κ2-O,O-nitrite anion (Fig. 6). While the Zn–N(Pz/Py/Qu) bond distances in 5-Pz/5-Py/5-Qu vary as described for 3-Pz/3-Py/3-Qu (Fig. 6), Zn–O(nitrite) distances and metric parameters associated with nitrite (N–O distances and O–N–O angles) are comparable for all these complexes. Utilization of 15N-enriched Na15NO2 for the syntheses of the zinc–nitrite complexes affords corresponding isotopologues 5-Pz-15N/5-Py-15N/5-Qu-15N. FTIR analyses on 5-Pz (5-Pz-15N), 5-Py (5-Py-15N), 5-Qu (5-Qu-15N) show isotope sensitive bands at 1203 (1180), 1273 (1244), and 1269 (1240) cm−1, respectively (Fig. S37, S43 and S49†). These observed vibrational features associated with the nitrite anion in [L2ZnII(κ2-nitrite)](ClO4) are consistent with those of the previously reported analogous nickel(II)–/copper(II)–nitrite complexes supported by the respective ligands.12,2415N NMR spectra of 5-Pz-15N, 5-Py-15N, and 5-Qu-15N in dimethylsulfoxide-d6 show chemical shifts δ(15N) at 589.66, 590.71, and 589.57 ppm (Fig. S35, S41 and S47†), respectively, with reference to liquid ammonia. Notably, δ(15N) for Na15NO2 appears around 609.50 ppm.25 Thus, these comparable upfield shifts of δ(15N) for 5-Pz-15N, 5-Py-15N, and 5-Qu-15N (relative to Na15NO2) suggest similar chemical environments of the nitrogen site in the nitrite moieties. Although some of our prior report hypothesize that the Lewis acidic [ZnII] perturbs the nature of the coordinated nitrite anion,15 the present X-ray crystallographic and 15N NMR data strongly suggest that the Lewis acidity of the [L2ZnII]2+ core does not significantly influence structural and electronic environments of the κ2-nitrite moiety coordinated at the zinc(II) centres.
 |
| | Fig. 6 X-ray crystal structures of the mono-cationic parts of complexes 5-Pz (A), 5-Py (B), and 5-Qu (C). The bond distances tabulated in Å. | |
Reactivity of zinc(II)–nitrite complexes with thiols
Reactions of the zinc(II)–nitrite complex 5-Qu with 4-tert-butylbenzylthiol (tBuBnSH) (1 equiv.) in acetonitrile at room temperature provide S-nitrosothiol in 40% yield as assessed by 1H NMR analysis on the organic end product. Analysis of the head space using (TPP)Co (where TPP = tetraphenyl porphyrin) as the NO-trap provides 49% yield of NO gas, which generates from the decomposition of RSNO to disulfide RSSR and NO gas. Consistent with the near quantitative (89%) yield of NO-equivalents (i.e. combined yields of RSNO 40% and NO gas 49%) from the reaction of 5-Qu and tBuBnSH (Fig. 7A and Table S1†), FTIR analysis on the resultant crude shows a near complete disappearance of the nitrite vibrational band at 1269 cm−1 (Fig. S53†). In contrast, the reactions of 5-Pz/5-Py with tBuBnSH (1 equiv.) show lower yields of NO-equivalents 61% and 49%, respectively (Table S1†). Moreover, FTIR analyses on the crude reaction mixture indicate the presence of unreacted nitrite (Fig. S53†). Attempts to improve the efficiency of the reactions of 5-Pz/5-Py and tBuBnSH in the presence of two equivalents of thiol do not improve the yields of NO-equivalents 62% and 57%, respectively. Recrystallizations of the crude inorganic products obtained from the 1:2 stoichiometric reactions of 5-Pz/5-Py and tBuBnSH provide unreacted zinc(II)–nitrite along with the corresponding [L2ZnII(ClO4)](ClO4) complex 1-Pz/1-Py as assessed through unit cell analyses. In contrast, recrystallization of the inorganic end-product from the 1:1 stoichiometric reaction of 5-Qu and tBuBnSH results in [{(Me2PzQu)2ZnII}2(μ-OH)](ClO4)3 (2-Qu) in 88% yield (Fig. 7). It is noteworthy that the reactions of 5-Pz/5-Py/5-Qu with thiophenol derivative 4-methylbenzenethiol (ArSH) (2 equiv.) in acetonitrile at room temperature show 82%, 74%, and 99% yields of NO gas, respectively (Table S1†). Hence, these results with more reactive ArSH (pKa = 6.8 in H2O and 19.5 in MeCN), as compared to benzyl thiol (pKa = 9.4 in H2O, 22.1 in MeCN),17,26 show consistently higher yields of NO gas and the demarcation in the ArSH reactivity of 5-Qu is less notable in comparison to that of 5-Pz/5-Py.
 |
| | Fig. 7 (A) Schematic representation for the reaction of 5-Qu and tBuBnSH. (B) Changes in the UV-vis absorption spectrum of 5-Qu (black trace) upon addition of tBuBnSH (2 equiv.) in acetonitrile at room temperature. Red trace shows the spectrum recorded immediately after the thiol addition, whereas the green and blue traces represent the spectra of the same solution collected after 1 minute and 30 minutes, respectively. (C) Time trace (absorbance at 550 nm versus time) plots for the 1:2 reactions of 5-Pz/5-Py/5-Qu and tBuBnSH at room temperature. | |
UV-vis monitoring of the reaction of the zinc(II)–nitrite complexes 5-Pz/5-Py/5-Qu with two equivalents of tBuBnSH in acetonitrile at 25 °C shows development of a distinct band at 550 nm, which is congruent with the formation of tBuBnSNO (Fig. S67†).15 A comparison of absorbance at 550 nm (A550) versus time plot for the above-mentioned reactions clearly depicts that the rate of formation of tBuBnSNO follows the order 5-Qu ≫ 5-Py ≈ 5-Pz (Fig. 7C and S68†). Thus, the rate of RSNO formation and the relative yield of NO-equivalents suggest that the reactions of tBuBnSH with 5-Qu are distinctly efficient in comparison to that of 5-Pz/5-Py. A careful inspection of the UV-vis spectrum obtained immediately upon addition of thiol to the solution of zinc(II)–nitrite shows the formation of sharp features at 358, 372, and 388 nm prior to the generation of S-nitrosothiol (Fig. 7B). It is noteworthy that such finger-like distinct features at 336, 347, 358, 372, and 388 nm originate from nitrous acid (HNO2) as observed from the UV-vis analysis on an authentic sample of HNO2 (Fig. S69†).27 This finding indicates that HNO2, generated in situ from the interaction of zinc(II)–nitrite and tBuBnSH, serves as the obligatory intermediate in the transformation of nitrite to S-nitrosothiol/NO (Fig. 7). Moreover, based on the UV-vis monitoring of the reactions between tBuBnSH and 5-Pz/5-Py/5-Qu, the NO/RSNO generation proceeds through the same mechanism involving HNO2 (Fig. S68†). Notably, a control reaction between n-tetrabutylammonium nitrite [TBA+][NO2−] and tBuBnSH under analogous reaction conditions does not yield S-nitrosothiol as assessed by UV-vis spectroscopic analysis (Fig. S67†), thereby highlighting the importance of the zinc(II) site.
Understanding of the alkyl thiol reactivity of zinc(II)–nitrite prompted us to investigate the efficiency of such zinc sites in catalysing the transformations of the nitrite to RSNO/NO. Interestingly, a 1:2 reaction of [TBA+][NO2−] and tBuBnSH in the presence of 10 mol% of [(Me2PzQu)2ZnII(OTf)](OTf) (3-Qu) in acetonitrile at room temperature provides NO gas and RSNO in 54% and 46% yields, respectively, with respect to the nitrite anion as the limiting reagent. Notably, screening of [{(Me2PzQu)2ZnII}2(μ-OH)](ClO4)3 (2-Qu) in the analogous catalytic transformation shows 72% and 26% yields of NO gas and RSNO, respectively. As expected from the stoichiometric reactions, the catalytic efficiency of 3-Pz/3-Py is poorer in comparison to 3-Qu (Table S1†). Thus, this report illustrates a proof-of-concept catalytic transformation of nitrite-to-RSNO/NO utilizing Lewis-acidic activation of alkyl thiol tBuBnSH in the presence of a catalytic amount of zinc(II).
Theoretical insights into the metal–ligand coordination
Aiming to understand the origin of the more efficient interaction of thiol and nitrite at [(Me2PzQu)2ZnII]2+, we turned to density functional theory (DFT) calculations at the B3LYP level of theory.28 The metrical parameters obtained from the DFT optimized structures of the cationic moieties [L2ZnII(κ2-nitrite)]+ in 5-Pz/5-Py/5-Qu agree well with those of the SCXRD analyses (Table S2†), thereby verifying the reliability of the employed computational method. The second-order perturbation energy (E2) obtained through natural bond orbital (NBO) analyses on [L2ZnII(κ2-nitrite)]+ provided the extent of the electron donation ability of the ligand. The overall stabilization of the zinc(II) sites due to the donation of the nitrogen lone pairs of L to the lone vacant (LV) orbitals of zinc(II) ion has been estimated as 139.3, 127.5, and 119.4 kcal mol−1 for [(Me4PzPz)2ZnII(κ2-nitrite)]+, [(Me2PzPy)2ZnII(κ2-nitrite)]+, and [(Me2PzQu)2ZnII(κ2-nitrite)]+ complexes, respectively (Fig. S74†). These lone vacant (LV) orbitals of the zinc(II) metal ion can be assigned as the vacant 4s orbital. Thus, this result suggests that the sigma donation ability of the ligands follows the trend as Me4PzPz > Me2PzPy > Me2PzQu. It is noteworthy that the second-order perturbation energy (E2) calculated for the donation of the oxygen lone pairs of the nitrite anion to the LV orbital of zinc(II) is comparable regardless of the nature of the ancillary ligand (Fig. S74†). Hence, these findings correlate well with the nearly identical binding of the nitrite anion as observed from the X-ray crystal structures of 5-Pz/5-Py/5-Qu (Fig. 6).
Mechanism of thiol–nitrite interactions at zinc(II)
Following the formation of the initial reactant complex (RC) involving zinc(II)–nitrite and tBuBnSH, two mechanistic routes have been computationally assessed for the generation of S-nitrosothiol (Fig. 8 and S75–S77†). The transition state (TS) for the direct formation of tBuBnSNO through an intermolecular proton-transfer assisted S-nitrosation is considered as a four-membered concerted transition state (TS-1) with a Gibbs free energy barrier (ΔG1‡) of 29.6 kcal mol−1 for 5-Qu (Fig. 8). Alternatively, the reactant complex RC is proposed to proceed through an interaction of thiol at the zinc(II) site followed by an intramolecular proton-transfer from the coordinated thiol to nitrite (Int-1 → Int-2) via a transition state TS-2 with an overall barrier (ΔG2‡) of 17.7 kcal mol−1 (Fig. 8). Consistent with the difference in the transition state barriers (ΔG2‡ < ΔG1‡) and UV-vis spectroscopic observation of HNO2 species during the course of the reaction (Fig. 7 and 8), the acid–base exchange route viaTS-2 appears to be the preferred route. Moreover, tBuBnSH (pKa = 9.4 in H2O, 22.1 in MeCN)17,26 is not sufficiently acidic in comparison to the free nitrite anion (pKa = 3.4 in H2O, 16.1 in MeCN),17,26 and thus a possibility of an intermolecular proton-transfer can be ruled out. Polarization of the RS–H bond upon interacting with the Lewis-acidic zinc(II)-site is deemed to be essential for the facile intramolecular proton-transfer. As the initial zinc(II)–nitrite complex 5-Qu is coordinatively saturated, the hemilability of the quinoline arm of the ligand Me2PzQu is important for the formation of Int-1. Subsequent to the formation of the transient species Int-2, free HNO2 and [ZnII]–thiolate complex Int-3 are proposed to be generated prior to the S-nitrosation resulting in tBuBnSNO and a mono-nuclear zinc(II)-hydroxo intermediate (Int-4). Finally, Int-4 rapidly transforms to a highly thermodynamically favoured dinuclear [{(Me2PzQu)2ZnII}2(μ-OH)]3+ complex 2-Qu with a net driving force of 14.6 kcal mol−1. It is noteworthy that both the transition state barriers corresponding to TS-1 and TS-2 for the reaction of 5-Pz and 5-Py are higher as compared to that of 5-Qu (Fig. S75†), thereby rationalizing the slower reactivity of 5-Pz/5-Py. While the Lewis acidity follows the trend as [(Me4PzPz)2ZnII]2+ > [(Me2PzPy)2ZnII]2+, the rates of RSNO formation from the thiol reactivity of 5-Pz and 5-Py are comparable perhaps due to the fact that both the Zn–N(pyrazole) and Zn–N(pyridine) bonds are kinetically inert in comparison to the Zn–N(quinoline) bonds in 5-Qu. Thus, the initial interactions of the thiol at the coordinatively saturated zinc sites in 5-Pz and 5-Py are hindered.
 |
| | Fig. 8 (A) DFT calculated Gibbs free energy profile for the concerted transnitrosation and acid–base exchange routes for the reaction of [(Me2PzQu)2ZnII(κ2-nitrite)]+ and tBuBnSH at 298.15 K utilizing the B3LYP-D3BJ/[6-311++G(d,p)+LANL2TZ]/SMD(acetonitrile)//B3LYP-D3BJ[6-31G(d)+LANL2DZ] level of theory. (B) Schematic representations of the key intermediates and the transition state proposed for the acid–base exchange route. See ESI† for the optimized structures. | |
Conclusion
Systematic tuning of the structural and electronic properties of a set of zinc(II) complexes has been achieved by employing combinations of pyrazole, pyridine, and quinoline based ligand donors. The metrical parameters obtained from the X-ray crystallographic analyses on the [L2ZnII]2+ cores (where L = Me4PzPz/Me2PzPy/Me2PzQu) coordinated to the weakly coordinating anions (e.g. perchlorate and triflate) suggest the Lewis-acidic nature of the zinc(II) site. X-ray crystallographic and 31P NMR investigations on the 1:1 acid–base adduct complexes [L2ZnII(OPEt3)]2+ validate the trend of Lewis acidity as [(Me2PzQu)2ZnII]2+ > [(Me4PzPz)2ZnII]2+ > [(Me2PzPy)2ZnII]2+. In-depth characterization (SCXRD, FTIR, 15N NMR) of the isolated [L2ZnII(κ2-nitrite)](ClO4) complexes (5-Pz/5-Py/5-Qu) supported by the set of systematically tuned ligands unambiguously suggests that the nature of the ancillary ligand neither affects binding of the nitrite anion at the zinc(II) site nor alters the chemical environment of the nitrite anion, even though the [L2ZnII]2+ cores feature Lewis acidity variation. However, the distinct roles of ancillary ligands in the transformation of nitrite to RSNO/NO at the zinc(II) coordination sites in the presence of alkyl thiol have been demonstrated for the first time. Utilization of the relatively less acidic alkyl thiol tBuBnSH rather than aromatic thiol ArSH in the present study closely models biological thiols such as cysteine and glutathione. Furthermore, UV-vis spectroscopic monitoring of these thiol reactions of [L2ZnII(κ2-nitrite)]+ cores allows the experimental validation of the nitrous acid (HNO2) intermediate as the in situ generated active nitrosating agent. The comparison of the experimentally established trends in the rate of RSNO formation (5-Qu ≫ 5-Py ≈ 5-Pz) and the Lewis-acidity order of [L2ZnII]2+ cores unfolds the importance of an available hemilabile coordination site for the interaction of thiol at zinc(II) and subsequent Lewis acidic polarization of the RS–H bond. The relatively poorer reactivity of alkyl thiol with 5-Py/5-Pz in comparison to 5-Qu is attributed to the less facile interaction of thiol with the zinc(II) sites in 5-Py/5-Pz due to the presence of stronger σ-donor Py/Pz moieties, while the role of such Lewis acidic polarization of the ArS–H bond is less prominent for the reactions of aromatic thiol and 5-Pz/5-Py/5-Qu due to the inherent more acidic nature of the aromatic thiol relative to the alkyl thiol. Computational studies complement the experimental evidence and elucidate the stepwise mechanism for the thiol–nitrite interaction at zinc(II). Taken together, this present study demonstrates the potential role of zinc(II) based metalloenzymes (such as carbonic anhydrase) in influencing the cross-talk between thiol and nitrite through the intermediacy of reactive species like HNO2. In addition, HNO2 generation from the interaction between [ZnII]–nitrite and the weakly acidic substrate could represent a new way for the HNO2-mediated oxidative modifications (e.g. nitration of amino acid residues) under physiological pH conditions.29 Thus, this work underscores that the interactions between biological thiols and redox-inactive zinc(II) sites are capable of regulating complex redox-signalling pathways.30
Data availability
The data supporting this article have been included in the ESI. CCDC depositions 2355797 (for 1-Py), 2355798 (for 1-Pz), 2355799 (for 2-Qu), 2355800 (for 3-Py), 2355801 (for 3-Pz), 2355802 (for 3-Qu), 2355803 (for 4-Py), 2355804 (for 4-Pz), 2355805 (for 5-Py), 2355806 (for 5-Pz), and 2355807 (for 5-Qu) can be accessed free of charge from The Cambridge Crystallographic Data Centre.†
Author contributions
SK and BSA conceived the project. BSA and NRN performed the experiments. JR performed the computational analysis. JAB carried out the analysis of crystallographic data. SK and BM supervised the experimental and computational studies, respectively. SK, BSA, and BM contributed to the manuscript preparation. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
SK gratefully acknowledges the Core Research Grant (CRG/2021/001174) from SERB. JR thanks the Ministry of Education, Govt. of India for the research fellowship. The High-Performance Computing (HPC) and PARAM Himalaya computing facility at IIT Mandi are acknowledged for providing high-end computational resources. Support from Ms Nandha K during the initial synthesis of some of the complexes is appreciated.
Notes and references
- C. Zhang, T. D. Biggs, N. O. Devarie-Baez, S. Shuang, C. Dong and M. Xian, Chem. Commun., 2017, 53, 11266–11277 RSC.
- E. Culotta and D. E. J. Koshland, Science, 1992, 258, 1862–1865 CrossRef PubMed.
- U. Förstermann and W. C. Sessa, Eur. Heart J., 2012, 33, 829–837 CrossRef PubMed.
-
(a) J. S. Stamler, O. Jaraki, J. Osborne, D. I. Simon, J. Keaney, J. Vita, D. Singel, C. R. Valeri and J. Loscalzo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 7674–7677 CrossRef PubMed;
(b) Y. Zhang and N. Hogg, Free Radical Biol. Med., 2005, 38, 831–838 CrossRef.
-
(a) J. O. Lundberg, E. Weitzberg and M. T. Gladwin, Nat. Rev. Drug Discovery, 2008, 7, 156–167 CrossRef;
(b) S. Shiva, X. Wang, L. a Ringwood, X. Xu, S. Yuditskaya, V. Annavajjhala, H. Miyajima, N. Hogg, Z. L. Harris and M. T. Gladwin, Nat. Chem. Biol., 2006, 2, 486–493 CrossRef.
- L. B. Maia and J. J. G. Moura, Chem. Rev., 2014, 114, 5273–5357 CrossRef PubMed.
- R. Aamand, T. Dalsgaard, F. B. Jensen, U. Simonsen, A. Roepstorff and A. Fago, Am. J. Physiol.: Heart Circ. Physiol., 2009, 297, 2068–2074 CrossRef.
- L. Wang, C. E. Sparacino-Watkins, J. Wang, N. Wajih, P. Varano, Q. Xu, E. Cecco, J. Tejero, M. Soleimani, D. B. Kim-Shapiro and M. T. Gladwin, Br. J. Pharmacol., 2020, 177, 898–911 CrossRef PubMed.
-
(a) B. C. Sanders, S. M. Hassan and T. C. Harrop, J. Am. Chem. Soc., 2014, 136, 10230–10233 CrossRef;
(b) C.-C. Tsou, W.-L. Yang and W.-F. Liaw, J. Am. Chem. Soc., 2013, 135, 18758–18761 CrossRef.
-
(a) A. Mondal, K. P. Reddy, J. A. Bertke and S. Kundu, J. Am. Chem. Soc., 2020, 142, 1726–1730 CrossRef PubMed;
(b) M. Kumar, N. A. Dixon, A. C. Merkle, M. Zeller, N. Lehnert and E. T. Papish, Inorg. Chem., 2012, 51, 7004–7006 CrossRef PubMed;
(c) Y. L. Chang, Y. F. Lin, W. J. Chuang, C. L. Kao, M. Narwane, H. Y. Chen, M. Y. Chiang and S. C. N. Hsu, Dalton Trans., 2018, 47, 5335–5341 RSC.
-
(a) A. J. Timmons and M. D. Symes, Chem. Soc. Rev., 2015, 44, 6708–6722 RSC;
(b) N. Lehnert, E. Kim, H. T. Dong, J. B. Harland, A. P. Hunt, E. C. Manickas, K. M. Oakley, J. Pham, G. C. Reed and V. S. Alfaro, Chem. Rev., 2021, 121, 14682–14905 CrossRef PubMed.
- B. S. Anju, N. R. Nair and S. Kundu, Angew. Chem., Int. Ed., 2023, e202311523 Search PubMed.
- D. M. Beagan, N. A. Maciulis, M. Pink, V. Carta, I. J. Huerfano, C. H. Chen and K. G. Caulton, Chem.–Eur. J., 2021, 27, 11676–11681 CrossRef.
- A. J. P. Cardenas, R. Abelman and T. H. Warren, Chem. Commun., 2014, 50, 168–170 RSC.
- T. Sahana, A. K. Valappil, A. S. P. R. Amma and S. Kundu, ACS Org. Inorg. Au, 2023, 3, 246–253 CrossRef PubMed.
- G. Kolliyedath, T. Chattopadhyay, A. Mondal, A. Panangattu, G. Muralikrishnan and S. Kundu, Chem.–Eur. J., 2023, e202301409 CrossRef PubMed.
- M. M. Kreevoy, E. T. Harper, R. E. Duvall, S. Wilgus III and L. T. Ditsch, J. Am. Chem. Soc., 1960, 82, 4899–4902 CrossRef.
- The limiting values of the τ5 index as 0 and 1 for penta-coordinated metal sites refer to the perfect square pyramidal and trigonal bipyramidal geometries, respectively. A. G. Blackman, E. B. Schenk, R. E. Jelley, E. H. Krenske and L. R. Gahan, Dalton Trans., 2020, 49, 14798–14806 RSC.
-
(a) K. Fujisawa, Y. Noguchi, Y. Miyashita, K. I. Okamoto and N. Lehnert, Inorg. Chem., 2007, 46, 10607–10623 CrossRef;
(b) B. Herzigkeit, B. M. Flöser, T. A. Engesser, C. Näther and F. Tuczek, Eur. J. Inorg. Chem., 2018, 3058–3069 CrossRef.
- A. Saju, A. Mondal, T. Chattopadhyay, G. Kolliyedath and S. Kundu, Inorg. Chem., 2020, 59, 16154–16159 CrossRef PubMed.
- The geometry index values (τ4) = 0 and 1 for tetra-coordinate metal sites correspond to square planar and tetrahedral geometries, respectively. See L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- K. Huse, C. Wölper and S. Schulz, Organometallics, 2021, 40, 1907–1913 CrossRef.
- V. Gutmann, Coord. Chem. Rev., 1976, 18, 225–255 CrossRef.
- K. Fujisawa, T. Kataoka, K. Terashima, H. Kurihara, F. de Santis Gonçalves and N. Lehnert, Molecules, 2023, 28, 6206 CrossRef.
- S. Kundu, W. Y. Kim, J. A. Bertke and T. H. Warren, J. Am. Chem. Soc., 2017, 139, 1045–1048 CrossRef PubMed.
- The pKa values in the MeCN solvent system have been calculated empirically as described elsewhere. See E. Rossini, A. D. Bochevarov and E. W. Knapp, ACS Omega, 2018, 3, 1653–1662 CrossRef PubMed.
- J. Stutz, E. S. Kim, U. Platt, P. Bruno, C. Perrino and A. Febo, J. Geophys. Res.: Atmos., 2000, 105, 14585–14592 CrossRef.
- See ESI† for the details of computational methods.
- G. Ferrer-Sueta, N. Campolo, M. Trujillo, S. Bartesaghi, S. Carballal, N. Romero, B. Alvarez and R. Radi, Chem. Rev., 2018, 118, 1338–1408 CrossRef PubMed.
- C. Hübner and H. Haase, Redox Biol., 2021, 41, 101916 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Neeraja R.
Nair
a,
Janavi
Rajput
b,
Jeffery A.
Bertke
a,
Neeraja R.
Nair
a,
Janavi
Rajput
b,
Jeffery A.
Bertke