
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecoupled tin–silver batteries with long cycle life and power output stability based on dendrite-free tin anode and halide insertion cathode chemistry†
Feifei
Shi‡
a,
Yudong
Wu‡
a,
Binyan
Wang
b,
Jiawei
Bai
b,
Yihan
Ren
b,
Tao
Yang
 c,
Funian
Mo
*d,
Derek
Ho
ef and
Haibo
Hu
*ab
c,
Funian
Mo
*d,
Derek
Ho
ef and
Haibo
Hu
*ab
aSchool of Materials Science and Engineering, Anhui University, Hefei 230601, China. E-mail: haibohu@ahu.edu.cn
bStony Brook Institute at Anhui University, Anhui University, Hefei 230039, China
cDepartment of Mechanical Engineering, Centre for Mechanical Technology and Automation, University of Aveiro, Aveiro 3810-193, Portugal
dSchool of Future Technology, Shenzhen Technology University, Shenzhen 518055, China. E-mail: mofunian@sztu.edu.cn
eDepartment of Materials Science and Engineering, City University of Hong Kong, Kowloon, Hong Kong 999077, China
fHong Kong Center for Cerebro-Cardiovascular Health Engineering, Hong Kong 999077, China
First published on 28th August 2024
Abstract
Conventional Ag–Zn batteries have historically faced the challenge of poor cycling stability, rooting in issues associated with Ag cathode dissolution and Zn anode dendrites. Herein, we present a pioneering decoupled Sn–Ag cell, which features two chambers separated by a cation-exchange membrane, containing a dendrite-free Sn metal anode immersed in an alkaline anolyte, and an Ag nanowires/carbon nanotube 3D thick-network cathode in a neutral catholyte. Benefiting from the achieved high electroplating/stripping stability of the metallic Sn anode in the alkaline electrolyte and the electrochemical reversibility of the Ag/AgCl cathode redox couple in the neutral electrolyte, the assembled decoupled Sn–Ag cell demonstrates superior cycling stability, retaining 82.4% of its initial capacity even after 4000 cycles (2 mA cm−2), significantly outperforming both the contrastive decoupled Ag–Zn cell (1500 cycles) and conventional alkaline Ag–Zn batteries (<100 cycles). Furthermore, through the integration of the decoupled Sn–Ag battery with solar cells and power management circuits, an intelligent power system of photovoltaic charging and energy storage was designed, demonstrating its practical viability through maintenance-free charging–discharging during day–night cycles. This research not only significantly increases the lifespan of Ag-batteries with an ultra-flat voltage platform but also opens avenues for the decoupled design of a wide variety of aqueous battery systems.
Introduction
Conventional alkaline silver–zinc (Ag–Zn) batteries, renowned for their high energy density and stable power output over a century, face the notorious obstacle of poor cycling performance (<100 cycles).1–3 This limitation primarily stems from two factors: (i) dendrite formation in the Zn anode, posing a deadly short circuit risk,4–7 and (ii) the dissolution phenomenon of the Ag cathode during cycling in a highly concentrated alkaline electrolyte.8,9 To tackle these issues, anionic insertion/extraction chemistry has been proposed by introducing a novel interaction between negative charge carriers and electrode materials, leveraging cations in electrolytes as common charge carriers.10–12 For instance, to extend the cycle lifespan of Ag–Zn batteries, researchers have developed mild Ag halide–Zn batteries to address the challenges encountered in alkaline battery systems.13 By introducing anionic chloride ions into a mild electrolyte to replace OH−, a reversible oxidation of Ag to Ag halide occurs under an anodic potential, leading to the formation of insoluble AgCl. This mechanistic alteration effectively mitigates the Ag cathode dissolution phenomenon in alkaline batteries, resulting in long-term cycling stability exceeding 1000 cycles for the obtained mild battery.Although the dissolution problem of Ag electrodes can be alleviated by introducing anionic halides into the electrolyte to form insoluble Ag halides, the more arduous challenge of dendrite formation and side reactions persists with zinc electrodes in aqueous electrolytes, dramatically impacting the cycling performance.14–19 Dendrite proliferation, a critical concern for Zn anodes, propagates perpendicular to the substrate as an anisotropic crystal growth while working in aqueous electrolytes, leading to an irregular surface morphology (Fig. 1a).20–24 This not only increases the volume of the Zn anode but also provides more sites for corrosion and water-triggered side reactions (such as the hydrogen evolution reaction, HER) depleting the electrolyte and zinc metal (Fig. 1a). Moreover, sheet-like or needle-like Zn dendrites have weak contact with the substrate, leading to detachment and the formation of “dead zinc”. The loss of active material and electrolytes due to these side reactions, accompanied by the formation of “dead zinc”, seriously diminishes the coulombic efficiency (CE) and capacity of Zn batteries.25,26
 | ||
| Fig. 1 Schematic illustration of the design principles of the decoupled Sn–Ag cell. (a) Challenges of the metallic Zn electrode. (b) Advantages of the metallic Sn electrode. (c) Assembly of the decoupled Sn–Ag cell and its associated functional benefits. | ||
Based on the background mentioned above, to maximize the advantages of anionic insertion/extraction chemistry in aqueous electrolytes for further enhancing the cycling stability of Ag batteries, selecting a matching metal anode with superior electrochemical stability and reversibility is imperative. It is also noteworthy that the redox potential of the targeted metal anode should also surpass that of the HER. Considering these criteria, metallic tin (Sn) emerges as a rational choice due to its noble reaction potential (−0.921 V vs. standard hydrogen electrode (SHE)) and intrinsically high tolerance to alkaline electrolytes.27 Under an electric field, Sn metal anodes normally exhibit isotropic crystal growth with minimal polarization and high deposition–dissolution efficiency owing to the accelerated kinetics in alkaline environments, resulting in highly reversible and dendrite-free electroplating/stripping behavior (Fig. 1b). Moreover, the high overpotential for the hydrogen evolution side reaction on metallic tin electrodes further positions them as an attractive anode candidate for aqueous-based battery systems with an exceptional safety profile.28
In this work, we introduce a novel decoupled Sn–Ag cell design, whose configuration features two chambers explicitly separated by a cation-exchange membrane (CEM), containing a Sn foil anode immersed in an alkaline solution (1 M K2Sn(OH)6 with 0.5 M KOH) as the anolyte, and an Ag nanowires (AgNWs)/carbon nanotube (CNT) 3D thick-network cathode in a neutral solution (2.5 M KCl) as the catholyte. This decoupled structure significantly facilitates efficient and reversible deposition–dissolution processes at the Sn metal anode under an electric field, while enabling reversible solid-to-solid conversion between Ag and AgCl at the AgNWs/CNT thick-network hybrid cathode. The overall electrochemical reaction processes and the corresponding equations are depicted in Fig. 1c. This configuration offers multiple advantages, including: (i) a low-surface-energy attribute, which results in a smooth and dendrite-free morphology of the Sn(OH)62−/Sn anode, (ii) the halide insertion–extraction solid-to-solid conversion chemistry occurring at the AgNWs/CNT hybrid cathode effectively eliminates the dissolution issue of the Ag electrode during the repeated charging/discharging process, (iii) and the AgNWs/CNT 3D thick-network electrode design with a high-permeability and low-tortuosity structure leads to kinetic pathways of fast charge and electron transport. As a result, the assembled decoupled Sn–Ag cell exhibited a maximal areal/specific capacity of 1.58 mA h cm−2/198.49 mA h g−1 (normalized to AgNWs) at 0.5 mA cm−2 and superior cycling stability, retaining 82.4% of its initial capacity even after 4000 cycles at 2 mA cm−2, significantly outperforming both the contrastive decoupled Ag–Zn cell (1500 cycles) and conventional alkaline Ag–Zn battery (<100 cycles). This work marks a significant leap in advancing sustainable energy storage technologies intended for long-term use, such as those in static grid storage. It highlights the decoupled Sn–Ag cell, renowned for its long cycle lifespan and power output stability, as a pioneering solution to overcome the long-standing cycling stability challenges of conventional Ag battery systems.
Results and discussion
Assessment of the electrochemical reversibility of diverse metal electrodes in different electrolytes
The reaction mechanism of both the Sn anode and Ag cathode were first analyzed through the cyclic voltammetry (CV) technique in a three-electrode system. We employed Sn foil and a AgNWs/CNT cathode as the working electrodes, respectively, a graphite rod as the counter electrode, and a saturated calomel electrode (SCE) as the reference electrode. The results revealed that the Sn foil exhibited a distinct pair of redox peaks in the alkaline electrolyte, indicating its efficient deposition–dissolution process during the transformation between Sn and Sn(OH)62− (Fig. S1a†). Additionally, the Ag electrode in a Cl−-containing electrolyte exhibited a high conversion efficiency of 99.3%, as determined by the near-symmetric anodic and cathodic peaks (0.22 C cm−2). This symmetry suggests that the charge–discharge processes of the cathode are fully reversible, with no significant parasitic reactions occurring concurrently (Fig. S1b†). To assess the stability of the metallic Sn electrode, 0.1 mm thick Sn foil was utilized to assemble a Sn‖Sn symmetric cell with an alkaline electrolyte consisting of 1 M K2Sn(OH)6 and 0.5 M KOH. The efficiency of Sn ion deposition and dissolution on the metal electrode surface was systemically analyzed, with particular attention to side reactions such as corrosion, dendrite nucleation and growth. A comparative evaluation of the reversibility of Zn metal electrodes was also conducted by constructing a Zn‖Zn symmetric cell with neutral (1.25 M ZnCl2) and alkaline (0.5 M KOH and 0.025 M ZnO) electrolytes, respectively, utilizing Zn foil of the same thickness (0.1 mm). In Fig. 2a, operating under a moderate current density of 0.5 mA cm−2 and a deposition capacity of 0.5 mA h cm−2, the Zn‖Zn symmetric cell in an alkaline electrolyte experienced rapid short-circuiting, evident from a substantial voltage fluctuation within a mere 6 cycles (inset of Fig. 2a). In contrast, prolonged cycle life was observed in the case of the Zn‖Zn symmetric cell with the neutral electrolyte, achieving 124 cycles. This finding demonstrates that a neutral electrolyte environment can improve the electrochemical reversibility of zinc metal electrodes compared to an alkaline electrolyte environment. However, severe voltage fluctuations and subsequent rapid performance degradation were still observed in the case of the symmetric device thereafter (inset of Fig. 2a). These phenomena observed in both neutral and alkaline electrolytes were primarily attributed to the continual accumulation of undesirable dendrites (Fig. 2b and f) and by-products (Fig. 2c and g) on the Zn surface, resulting in a poor CE. Most notably, the Sn‖Sn symmetric cell in the alkaline electrolyte exhibited significantly extended electroplating/stripping lifespans exceeding 200 cycles, as indicated by far more stable voltage–time curves without notable fluctuations (inset of Fig. 2a). This duration markedly surpassed that of the contrastive Zn‖Zn symmetric cell, emphasizing the considerably enhanced reversibility of the metallic Sn electrode in the alkaline electrolyte. At a high current density of 1 mA cm−2 and a deposition capacity of 1.0 mA h cm−2, the Zn foil in an alkaline Zn‖Zn symmetric cell also failed after only a few cycles, accompanied by pronounced fluctuations. Similarly, Zn in a neutral electrolyte short-circuited after approximately 70 hours, also falling short of expectations. In sharp contrast, the Sn‖Sn symmetric cell demonstrated remarkable stability, sustaining operation for over 200 hours under alkaline conditions before encountering short-circuit failure (Fig. S2†). The failure of Sn metal could be attributed to the following factors. Sn metal has a body-centered tetragonal crystal structure, with similar surface energies across different crystal planes, leading to a tendency for isotropic deposition. However, the large polyhedral Sn grains have limited contact with the electrode, and as deposition capacity increases, they are more likely to detach from the electrode and become “dead Sn”. The continuous formation of dead Sn in the electrolyte and the subsequent depletion of Sn ions can degrade battery performance, ultimately leading to cell failure.29–31 Similar phenomena are also observed in the cycling performance of Sn‖Cu and Zn‖Cu asymmetrical half-cells, which are conducted at a current density of 0.5 mA cm−2 and with a cutoff capacity of 0.25 mA h cm−2, furthermore verifying the superior electrochemical stability of the metallic Sn anode, which benefits from the much higher CE compared to that of the Zn anode (Fig. S3†).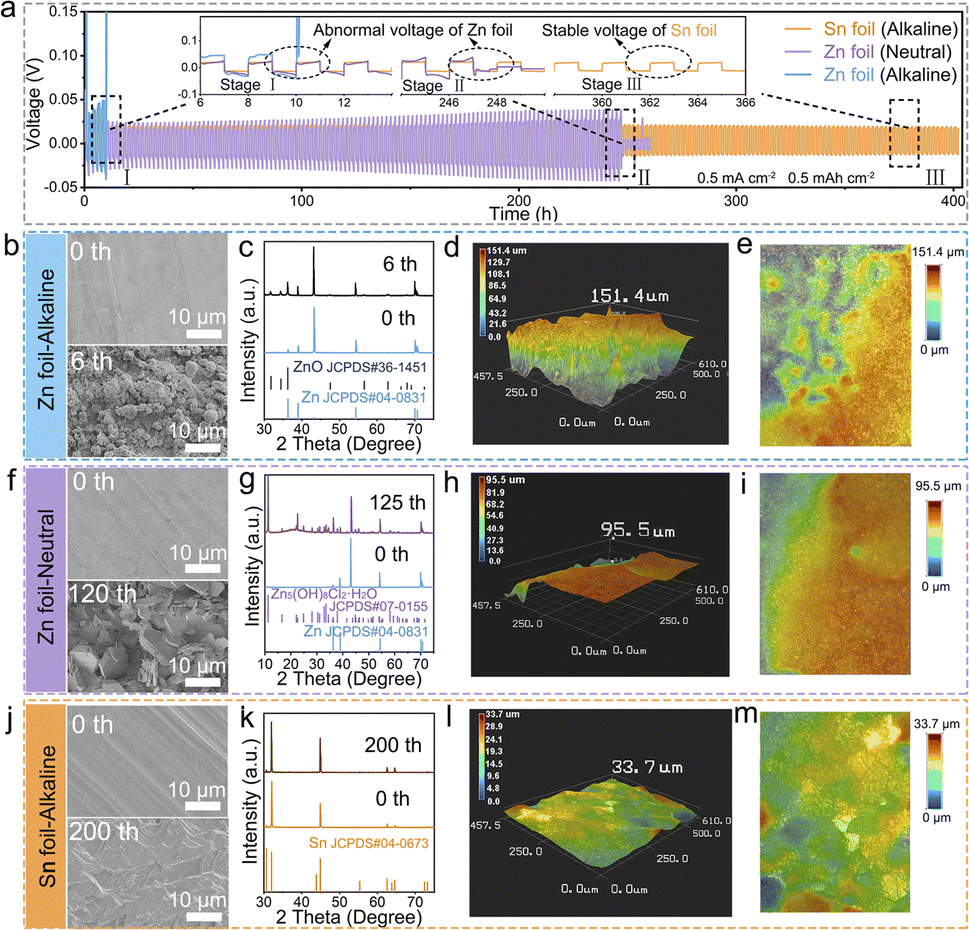 | ||
| Fig. 2 (a) Comparison of cycling performance between Sn‖Sn and Zn‖Zn symmetric coin-type cells with neutral and alkaline electrolytes, respectively. (b, f and j) Ex situ top-view SEM images of the surface morphology of metallic Zn and Sn electrodes in symmetrical cells respectively employing neutral and alkaline electrolytes after various specific cycles (scale bar: 10 μm). (c, g and k) The corresponding XRD patterns, (d, h and l) 3D microscope and (e, i and m) top-view of 3D microscope images of the metallic Zn and Sn electrodes in Zn‖Zn and Sn‖Sn symmetric cells respectively employing neutral and alkaline electrolytes after operation. | ||
The evolving surface morphology and phase composition of the metallic electrodes in the Zn‖Zn and Sn‖Sn symmetrical cells employing alkaline electrolytes were differentially analyzed through ex situ scanning electron microscopy (SEM) and X-ray diffraction (XRD) measurements. Fig. 2b, f and j illustrate the morphological evolution of metallic Zn and Sn electrodes during cycling in neutral and alkaline electrolytes, respectively, revealing the impact of the corrosive alkaline electrolyte on the electrochemical stability of different metallic electrodes. Compared to the smooth surface of the pristine Zn foil with few inherent scratches (Fig. 2b) and negligible height fluctuation (Fig. S4†), numerous microparticles rapidly emerged on the electrode surface after experiencing only 2 cycles in the alkaline electrolyte, leading to significant height fluctuation (Fig. S5†). Over successive cycles, these protruding microparticles continued to aggregate caused by surface defects. After merely 6 cycles, the micro-protrusions on the electrode surface gradually transformed into an unordered and loosely flake-like morphology with more drastic height fluctuation (Fig. 2d, e and S6†). Ultimately, this phenomenon may puncture the separator, inducing cell malfunction. In addition, the rapid emergence of Zn dendrites signifies irregular zinc electrodeposition behavior in the alkaline electrolyte, accompanied by other complex parasitic reactions. XRD analysis further confirms that the predominant phase composition of the proliferative substances observed during the cycling process is ZnO (Fig. 2c). With an increasing number of cycles, the gradual intensification of peak intensity indicates the continuous accumulation of these by-products on the Zn electrode surface. These by-products exhibit poor conductivity and inactivity, ultimately resulting in the passivation of the metallic Zn electrode. While the employment of a neutral electrolyte instead of an alkaline electrolyte can somewhat enhance the cycle life of Zn electrodes, serious dendritic growth (Fig. S7a, S8a† and 2f), by-product generation (Fig. 2g), and significant fluctuation in height (Fig. S7b, S7c, S8b, S8c,†2h and i) were still evident on the zinc metal surface as the cycle progresses compared to fresh Zn foil (Fig. 2b–e and S4†), ultimately leading to device failure.
In sharp contrast, the metallic Sn electrode in the Sn‖Sn symmetrical cell utilizing a highly corrosive alkaline electrolyte displayed a significantly smoother surface (Fig. 2j), with considerably reduced micro-protrusions and slight height fluctuation even after 200 cycles (Fig. S9–S11†), providing strong evidence for the effectiveness of uniform Sn ion electrodeposition in the alkaline electrolyte. XRD analysis indicated that there are no impurities generated on the surface of the Sn electrode after cycling (Fig. 2k). Additionally, 3D microscope images further verified the morphological changes of the metallic electrodes after the cycling process. The uneven height and irregular rough appearance of the loose deposition layer indicated the non-uniform zinc deposition and severe corrosion on the surface of the Zn electrode whether in alkaline or neutral electrolytes (Fig. 2d, e, h and i). In contrast, a flat surface morphology was observed on the metallic Sn electrode after cyclic operation, indicating uniform Sn deposition behavior on the Sn electrode (Fig. 2i and m).
Assembly and morphology characterization of the AgNWs/CNT hybrid film electrode
As shown in Fig. 3a, the 3D thick-network AgNWs/CNT hybrid film electrode was further fabricated based on AgNWs and CNTs employing an easily manipulated vacuum assisted self-assembly technique. Subsequently, the decoupled Sn–Ag cell was assembled by pairing the laser-cut AgNWs/CNT thick film electrode as the cathode with pristine metallic Sn foil serving as the anode. This specialized Sn–Ag cell is designed with two chambers partitioned by a CEM, each containing distinct electrolytes. A detailed microstructural analysis elucidating the composition of the AgNWs/CNT hybrid film was conducted through SEM and transmission electron microscopy (TEM), respectively. As illustrated in Fig. S12,† the utilization of carbon nanotubes (CNTs) with an average diameter of 40 nm served as the foundational conducting framework for the 3D net-structured assembly. In addition, AgNWs with a diameter of 100 nm and a high aspect ratio over 200 were synthesized through a polyol method, as detailed in our previous work (Fig. S13†).32 These AgNWs served a dual role,13 which not only functioned as supplementary conductive pathways, enhancing electron transmission within the 3D network skeleton but also contributed as electrochemically active materials, providing charge storage capacity. As depicted in the inset of Fig. 3b, the aqueous suspension of AgNWs and CNTs after mixing demonstrates a uniform dispersion. The as-fabricated 3D thick-network AgNWs/CNT hybrid thick film electrode, as illustrated in the top-view SEM images in Fig. S14,† exhibits a uniformly distributed 3D porous and interlocking network consisting of AgNWs and CNTs. As has been previously shown,12 the use of hybrid nanowires with large aspect ratios can facilitate the construction of a sturdy percolation network within the thick film electrode through mechanical interlocking, enabling the formation of free-standing electrodes without additional polymeric binders. Moreover, this porous structure can enhance electrolyte infiltration and accelerate ion transport.33,34 Elemental mapping images in both top-view and cross-sectional perspectives serve to affirm the homogeneous distribution of carbon and silver elements within the hybrid electrode, which intuitively exhibit exceptional uniformity in the interweaving of these nanowire components throughout the electrode structure (Fig. S14b and S15†). It is noteworthy that the thickness of the resultant AgNWs/CNT hybrid thick film electrode is approximately 125 μm, as evidenced by the side-view of the SEM images (Fig. 3c), significantly surpassing that of conventional electrodes, which typically measure less than 90 μm as compared in Fig. 3e.35–45 A large electrode thickness is beneficial for a high mass loading of active substances and reveals the great potential of the hybrid electrodes for achieving enhanced areal capacity. | ||
| Fig. 3 (a) Schematic illustration of the fabrication procedure of the AgNWs/CNT hybrid cathode, Sn anode, and the assembly of the decoupled Sn–Ag cell. (b) Optical photograph of the fabricated AgNWs/CNT hybrid film (inset is the suspension containing AgNWs and CNTs). (c) The corresponding cross-sectional SEM image of the AgNWs/CNT hybrid film electrode, and (d) the zoom-in view. (e) Comparison of the electrode thickness and the corresponding areal mass loading of active substances with recently developed advanced electrodes utilized in aqueous batteries. (f) XRD patterns of CNTs, Ag nanowires, and AgNWs/CNT hybrid films, respectively. (g) The real time observation of images showing how the water droplets disperse and spread across the surface of the AgNWs/CNT hybrid film electrode. | ||
X-ray diffraction (XRD) analyses further verified the compositions of the distinct materials constituting the 3D thick-network AgNWs/CNT hybrid film electrode (Fig. 3f), which are in good agreement with the previously discussed elemental mapping images (Fig. S14b and S15†). The as-prepared AgNWs/CNT hybrid thick film electrode exhibits exceptional hydrophilicity and rapid electrolyte infiltration, attributable to the porous 3D-network structure and its large specific surface area (Fig. 3d). As shown in Fig. 3g, the hydrophilicity is substantiated by the remarkably low static water contact angle of approximately 42.8° at the initial contact (0.05 s) of the water with the AgNWs/CNT hybrid electrode substrate. This expeditious infiltration is further demonstrated by the swift water adsorption, exemplified by the rapid reduction of the water contact angle to approximately 0° within a mere 60 seconds (Fig. 3g). This superior electrolyte penetration can not only enhance the energy efficiency of the full cell, but also improve the interfacial stability between the electrodes and electrolyte.
Initially, we performed cyclic voltammetry tests using a mixed electrolyte comprising 1 M K2Sn(OH)6, 0.5 M KOH, and 2.5 M KCl, devoid of any ion-exchange membrane (Fig. S16†). After merely six cycles, the resulting curves exhibited pronounced instability and erratic redox peaks. This phenomenon can be attributed to the intricate chemical interactions within the mixed electrolyte, coupled with the propensity for alkaline conditions to induce abundant side reactions at the Ag electrode. Consequently, to enhance the stability of the battery system, we employed a CEM to segregate the distinct electrolytes according to the specific electrochemical reactions of the anode and cathode, thereby establishing a stable decoupled cell configuration.
Electrochemical performance and analysis of the electrochemical mechanism of the decoupled Sn–Ag cell
To assemble the decoupled Sn–Ag full cell, the Sn foil and fabricated AgNWs/CNT hybrid thick film electrode were also laser-cut to achieve the desired shapes. The cell comprises two chambers separated by a CEM, containing distinct electrolytes. The cell structure is depicted in Fig. S17,† wherein Sn foil, an alkaline solution (1 M K2Sn(OH)6/0.5 M KOH), a neutral solution (2.5 M KCl), AgNWs/CNT hybrid film, and a titanium plate serve as the anode, anolyte, cathode, catholyte, and current collector, respectively. Both Sn foil and the AgNWs/CNT hybrid film electrode possess an effective area of 0.5 cm2 (Fig. S18†). Additionally, the initial pH of the electrolyte in both chambers was measured using a pH meter. The results revealed that the 2.5 M KCl solution exhibited a neutral pH of 7.0, while the mixed solution of 1 M K2Sn(OH)6/0.5 M KOH displayed a strongly alkaline pH of 13.7, as illustrated in Fig. S19.† For comparison, a decoupled Zn–Ag cell with a Zn anode and an AgNWs/CNT hybrid cathode was also fabricated as the control group. Synthesis and fabrication details are provided in the Experimental section of the ESI.†The electrochemical performance of the decoupled Sn–Ag cell was systematically investigated. Initially, cyclic voltammetry (CV) curves were recorded at various scan rates ranging from 1.0 to 5.0 mV s−1, obviously unveiling a symmetrical pair of redox peaks indicative of chloride insertion and extraction behavior (Fig. 4a). In the CV profile, the peak response (I) exhibits a correlation with the scan rate (ν) described by equation I = aνb, where a denotes a constant, and b signifies the coefficient that determines the electrochemical kinetics governed by a diffusion-controlled or non-diffusion-controlled mechanism.52,53 In particular, the b value in close proximity to 0.5 is indicative of battery-type behavior, whereas the value approaching 1.0 signifies dominant capacitor-type behavior on the electrochemical surface. In the case of the decoupled Sn–Ag cell, the ‘b’ values associated with the redox peaks are 0.558 and 0.531 respectively, closely aligning with 0.5, thereby indicating a pronounced battery-like insertion/extraction behavior of chloride ions at the AgNWs/CNT cathode, as illustrated in Fig. 4b. To delve deeper into the charge storage mechanism of the decoupled Sn–Ag cell and understand the reaction kinetics more comprehensively, we utilize the well-established equation i = k1v + k2v1/2,54,55 in which the response current i of the decoupled Sn–Ag cell with the AgNWs/CNT hybrid cathode at various voltage levels can be divided into two parts: (i) pseudocapacitive contribution (k1v), and (ii) diffusion-limited contribution (k2v1/2). At a relatively low sweep speed of 1.0 mV s−1, the pseudocapacitive component accounted for approximately 48.3% of the total current, and this proportion rises to 81.9% as the sweep speed increases to 5.0 mV s−1 (Fig. 4c). We think that the phenomenon could be attributed to the gradual transition of the dominant faradaic reaction of the conversion-type AgNW active materials for charge storage at low scan speeds to pseudocapacitive charge storage behavior at high scan speeds.
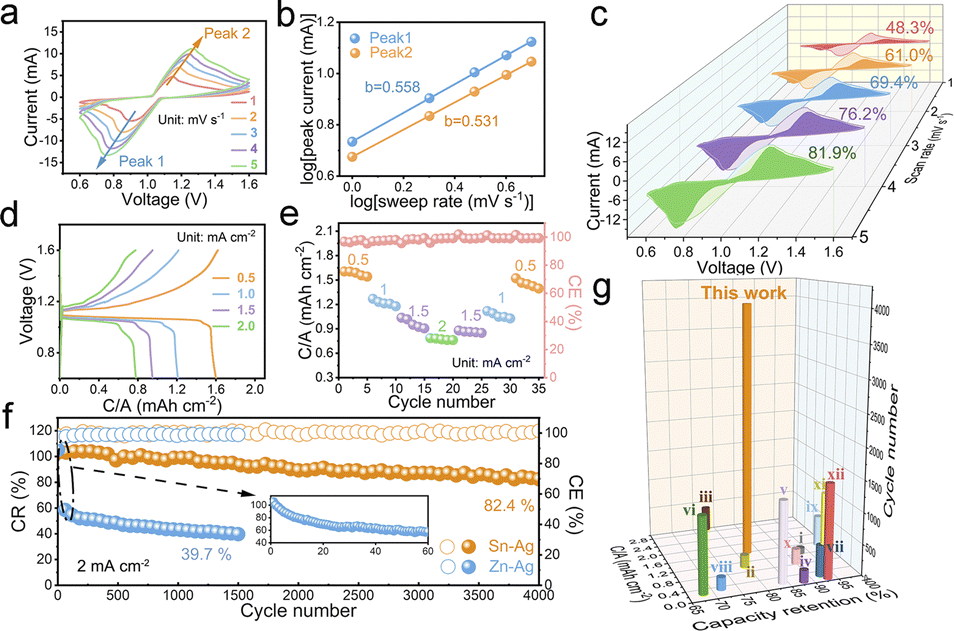 | ||
| Fig. 4 (a) CV curves at different scan rates. (b) b-Value corresponding to CV curves. (c) Contribution ratio from pseudocapacitive and diffusion-controlled charge storage behaviors at different scan rates. (d) GCD curves and (e) rate performance at different current densities. (f) Comparison of the cycling stability of the decoupled Sn–Ag cell and the comparative decoupled Zn–Ag cell. (g) Comparison of the areal capacity and capacity retention in long-term cycling measurement with other state-of-the-art Zn-ion batteries (i,46 ii,47 iii,48 iv,37 v,42 vi,45 vii,49 viii,35 ix,50 x,10 xi,13 xii51). | ||
Furthermore, the almost overlapping discharging plateau at approximately 1.1 V of the cell demonstrates an exceptionally flat profile even at varying discharging current densities within the range of 0.5 to 2.0 mA cm−1, as illustrated in Fig. 4d, indicative of its excellent power output stability. This is a desirable characteristic of energy storage devices intended for long-term use, such as those in static grid storage based on the purpose of “power grid peak shaving and valley filling”, enabling deeper penetration of intermittent renewable energies.56–59 The rate performance of the decoupled Sn–Ag cell indicates notable areal/specific capacities, averaging 1.58 mA h cm−2 (198.49 mA h g−1 normalized to AgNWs), 1.22 mA h cm−2 (153.27 mA h g−1), 0.97 mA h cm−2 (121.86 mA h g−1), and 0.77 mA h cm−2 (96.73 mA h g−1) at current densities of 0.5, 1.0, 1.5, and 2.0 mA cm−2, respectively (Fig. 4e). It is noteworthy that we also assembled a decoupled Sn-CNT cell to ascertain the capacity contribution of CNTs within the AgNWs/CNT hybrid film electrode (Fig. S20†). Only employing 100 mg of CNTs as the cathode active material, we performed charge–discharge tests on the decoupled Sn-CNT cell at current densities of 0.5, 1.0, 1.5, and 2.0 mA cm−2. The findings demonstrated that the exhibited capacities of the cell with the pure CNT electrode were 13.91, 11.54, 9.91, and 8.24 μA h cm−2, respectively. This indicates that within the Sn–Ag decoupled cell, the capacity is predominantly attributable to the AgNW active materials, with the contribution from CNTs being little.
In contrast, the assembled Zn–Ag battery featuring a decoupled design with an ion-exchange membrane (electrolytes: alkaline 0.5 M KOH + 0.025 M ZnO//0.5 M KCl) demonstrated capacities of 1.75, 1.31, 1.02, and 0.82 mA h cm−2 at current densities of 0.5, 1.0, 1.5, and 2.0 mA cm−2, respectively (Fig. S21†). Although these capacities are slightly higher than the values delivered in the decoupled Sn–Ag battery, the CV curves of the Zn–Ag decoupled cell exhibited significant instability at higher current densities. This instability suggests that its rechargeability under high current conditions will be inferior to that of the Sn–Ag decoupled battery. Additionally, we assessed the electrochemical performance of a neutral Zn–Ag battery with a single electrolyte (1.25 M ZnCl2) and without an ion-exchange membrane (Fig. S22†). The results demonstrated that at current densities of 0.5, 1.0, 1.5, and 2.0 mA cm−2, the Zn–Ag full cell achieved capacities of 1.98, 1.54, 1.22, and 0.91 mA h cm−2, respectively, the highest among all tested systems. However, the CV curves revealed substantial peak polarization at various currents, suggesting that the cycling stability of this system is compromised.
Moreover, the specific capacity of the Sn–Ag decoupled cell maintains its initial level when the current density reverted from 2.0 to 0.5 mA cm−2 (Fig. 4e), indicative of the outstanding electrochemical reversibility of the AgNWs/CNT hybrid electrode. In long-term cyclability assessment, the decoupled Sn–Ag cell demonstrates remarkable stability, retaining 82.4% of its initial capacity even after 4000 cycles at 2 mA cm−2 (Fig. 4f). Notably, the CE remains consistently close to 100% throughout the cycling process, suggesting the highly reversible conversion between metallic Ag and insulating AgCl. It should be noted that after thousands of continuous cycles, there still accumulates a portion of silver chloride that cannot be completely converted (Fig. S23†). Nevertheless, this exceptional stability is unparalleled when compared to the corresponding decoupled Zn–Ag cell, which exhibits a retention rate of only 39.7% after 1500 cycles. Moreover, the neutral Zn–Ag battery only utilizing 1.25 M ZnCl2 without an ion-exchange membrane demonstrated a cycle life of merely 275 cycles at a current density of 2.0 mA cm−2, with an initial capacity retention of 75.4%. By 300 cycles, this retention had diminished sharply to only 16.3% (Fig. S22c†). The difference in cycling performance primarily arises from the significant differences in the electrochemical performances of various metallic electrodes in different electrolytes, highlighting the effectiveness of the demonstrated decoupled configuration based on a dendrite-free tin anode and halide insertion cathode chemistry for enhancing the cycle life of the silver metal batteries. In contrast to limitations posed by side reactions such as dendrite formation and hydrogen evolution in the zinc metal anode, the tin metal anode exhibits greater stability during the electrochemical cycling process (Fig. S24†). This stability contributes significantly to the outstanding cycling performance of the decoupled Sn–Ag cell. Furthermore, the long-cycle performances of decoupled Zn–Ag and Sn–Ag batteries at a high current density of 5 mA cm−2 were also evaluated, respectively (Fig. S25†). The decoupled Zn–Ag battery exhibited an initial capacity of 0.45 mA h cm−2, but its capacity retention plummeted to just 53.9% after 120 cycles. In sharp contrast, the decoupled Sn–Ag battery with a higher initial capacity of 0.5 mA h cm−2 maintained a capacity retention of 61.9% even after over 4000 cycles, indicating markedly superior cycling stability compared to the Zn–Ag decoupled cell counterpart. Similar results were observed at the lower current density of 1 mA cm−2, where the decoupled Sn–Ag battery retained 75.72% of its capacity after 3250 cycles. In contrast, the decoupled Zn–Ag battery began to exhibit signs of failure after just 40 cycles (Fig. S26†). In this innovative decoupled cell architecture proposed, which partially leverages the solid-to-solid conversion mechanism of the Ag/AgCl redox couple for charge storage, the attained cycle life has notably surpassed that of recently reported state-of-the-art Zn-ion batteries, as depicted in Fig. 4g.10,13,35,37,42,45–51 Additionally, the Ag/AgCl redox couple facilitates the storage of a substantial amount of chloride ions in the form of the electroneutral AgCl phase, which can effectively mitigate the cross-diffusion of charge carriers, thereby remaining unaffected by electropositive ions in the electrolyte. Consequently, the decoupled Sn–Ag cell presents a highly stable discharge curve over a duration of 100 hours, displaying remarkable capabilities in mitigating self-discharge behavior (Fig. S27†).
To further verify the existence of redox conversion charge storage behavior in the decoupled Sn–Ag cell, ex situ XRD analyses were conducted during the charge–discharge process. Fig. 5b and c depict the ex situ XRD results of the AgNWs/CNT hybrid film electrode within the decoupled Sn–Ag cell. These outcomes were derived from seven specific points representing different charge/discharge states during operation, corresponding to the charge/discharge curves illustrated in Fig. 5a. During the initial charging phase, peaks at approximately 38.3° and 44.5° are attributed to metallic Ag (111) and Ag (200) lattice planes, respectively. As the decoupled Sn–Ag cell undergoes charging, the intensities of the characteristic peaks (111) and (200) belonging to metallic Ag (JCPDS #04-0783) gradually decrease, while new peaks corresponding to AgCl (JCPDS #31-1238) emerge, exhibiting a progressive increase in intensity. The discharging process exhibits the opposite phenomenon. This observation indicates that, with the extensive insertion of Cl− into the AgNWs/CNT hybrid film electrodes during charging, the interaction between Cl− and the distributed 3D porous and interlocking network comprising AgNWs and CNTs can lead to the formation of AgCl within the decoupled Sn–Ag cell. In addition, in the ex situ XRD spectrum during the charging and discharging process of the Sn anode, the characteristic peaks of the Sn anode exhibit minimal variation even after numerous repetitive charging cycles, as evident in Fig. 5d and e. This demonstrates its effective deposition–dissolution efficiency and remarkable reversibility throughout the charging and discharging processes, thus solidifying its pivotal role in guaranteeing the superior cycling stability of the Sn-based decoupled full cell. In contrast, during the charging process of the battery, the zinc anode manifests a distinct characteristic peak corresponding to ZnO (JCPDS #36-1451) (Fig. 5f). However, even as the discharge process advances, the presence of ZnO (100) and (002) still persists (Fig. 5g). The formation of insoluble ZnO byproducts adheres to the surface of the zinc metal negative electrode, inducing passivation of its reactivity. This phenomenon is compounded by various intricate parasitic reactions. Moreover, in alkaline electrolytes, the Zn electrode is prone to the formation of pronounced zinc dendrites. The cross-sectional SEM images visually reveal the distinct morphological changes in metallic anodes after cycling. Uniform Sn deposition is evident in dense, smooth coatings, highlighting stable operation in alkaline electrolytes (Fig. S28†). In contrast, the irregular and rough sedimentary layers indicate uneven Zn deposition and severe corrosion on Zn electrode surfaces (Fig. S29†). Collectively, these factors significantly impede the cycling stability of the Zn-based decoupled full cell.
 | ||
| Fig. 5 (a) The charge/discharge curves of the decoupled Sn–Ag cell recording the different charge/discharge states. The corresponding ex situ XRD patterns and magnified ex situ XRD mapping of (b and c) the AgNWs/CNT hybrid cathode and (d and e) the metallic Sn anode in the decoupled Sn–Ag cell at different charge/discharge states. (f) Ex situ XRD patterns and (g) magnified ex situ XRD mapping of the metallic Zn anode in the decoupled Zn–Ag cell at different charge/discharge states. | ||
Demonstration of the integrated decoupled Sn–Ag cell for practical system-level applications
To highlight the practicality of the design, the decoupled Sn–Ag cell has been integrated into series and parallel configurations with two and even three cell units (Fig. 6a and c). This allows for the generation of customized voltages and currents, respectively. In terms of electrochemical performance, the GCD curves exhibit an increase in the output voltage plateau from 1.1 V for a single cell to 2.2 V when two cells are connected in series, and further to 3.3 V when three cells are connected in series (Fig. 6b). Concurrently, the discharge time of the decoupled Sn–Ag cell in parallel connection shows a linear increase, enabling a tripled output capacity of 1.98 mA h and a doubled output capacity of 1.36 mA h, in comparison to the 0.61 mA h output of a single cell (Fig. 6d). It is noteworthy that the interface impedance of the series-connected cell pack was evaluated through electrochemical impedance spectroscopy (EIS) to assess its integration viability. The outcomes reveal a linear increase in the interface impedance of series-connected cell packs with the growing number of cell units, as this impedance is directly linked to the reaction surface area (Fig. S30†). A parallel trend is also evident in parallel cell packs. The aforementioned findings indicate the high scalability and suitability of the decoupled Sn–Ag cell for large-scale production. | ||
| Fig. 6 (a) Digital images of a single cell, two cells in series, three cells in series, and the (b) corresponding discharge curves. (c) Digital images of a single battery, two batteries in parallel, three batteries in parallel, and (d) the corresponding discharge curves. (e and g) Demonstration of the intelligent power system by integrating a solar cell with the decoupled Sn–Ag cell based on (f) a power management circuit, and schematically depicting the potential application, simulating the operational scenarios of solar street lights during both day and night. (h) The voltage profile showing the charging under solar energy and subsequent nighttime discharge of the integrated cells. | ||
Based on a power management circuit (Fig. 6f), by further integrating the decoupled Sn–Ag cell, serving as an energy storage device, with the solar cell, functioning as an energy conversion device, we have successfully demonstrated an integrated plug-free power system for solar-powered, energy storage and output applications. This self-maintained system simulates the operational states of solar streetlights during both daytime and nighttime (Fig. 6e and g). It includes components (Fig. S31†) controlling the day–night circuit switching, making it adaptable to varying solar intensities to match the different voltage outputs of commercial solar cells. The integrated solar cell produced an output voltage of 1.99 V under illumination, compared to just 0.073 V in the absence of light. This demonstrates that the solar cell can provide a stable output voltage for charging the decoupled battery under illuminated conditions (Fig. S32†). The decoupled cell module charges during the day using solar energy converted by the solar cell module, and the voltage remain remarkably stable during nighttime discharge (Fig. 6h and Video S1†). This typical system-level integration capability further verifies the immense potential of the decoupled Sn–Ag cell as a commercially viable integrated power source, providing reliable and consistent power output throughout its operational lifespan.
Conclusions
The development of the decoupled Sn–Ag cell represents a significant leap towards overcoming the notorious challenges associated with cycling stability that are inherent in traditional Ag-based aqueous batteries. This work proposes a pioneering decoupled Sn–Ag cell design, which contains a Sn foil anode immersed in an alkaline anolyte, and a 3D thick network AgNWs/CNT hybrid film cathode in a neutral catholyte. This configuration realizes efficient and reversible deposition–dissolution processes at the Sn anode, while enabling reversible solid conversion of Ag to AgCl at the Ag cathode in the halide-containing electrode. Through a systematical investigation of electrochemical behavior, results elucidate a robust charge storage mechanism and dynamic structural changes during cycling. Benefitting from the superior electroplating/stripping reversibility of the metallic Sn anode, the Sn‖Sn symmetric cell exhibits an extended electroplating/stripping lifespan in an alkaline electrolyte, exceeding 200 cycles at 0.5 mA cm−2 and 0.5 mA h cm−2, offering valuable insights into the effectiveness of uniform Sn deposition–dissolution behavior. The constructed decoupled Sn–Ag cell achieves superior cycling stability, retaining 82.4% of the initial capacity after 4000 cycles at 2 mA cm−2 with the CE remaining consistently close to 100%, which is remarkable. Moreover, the practical integration of this innovative cell into a simulated solar streetlight substantiates its real-world viability through a stable power output during nighttime discharge. This work contributes towards overcoming cycling stability challenges in sustainable energy storage technologies, by demonstrating the potential and practicality of the decoupled architecture realized in the Sn–Ag cell as a groundbreaking solution.Data availability
The data that support the findings of this study are available within the paper and the ESI.† Other relevant data are available from the corresponding authors on request.Author contributions
F. F. Shi, Y. D. Wu: formal analysis, data curation, investigation, validation, writing – original draft. B. Y. Wang, J. W. Bai, Y. H. Ren: formal analysis, validation, visualization. T. Yang, D. Ho: writing – review & editing. F. N. Mo: formal analysis, funding acquisition, project administration, writing – review & editing. H. B. Hu: formal analysis, funding acquisition, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. S. and Y. W. contributed equally to this work. This work was financed by the National Natural Science Foundation of China (52471154 and 22309036), Excellent Youth Fund of Anhui Province (2108085Y17), Hundred-Talent Program of Anhui Province, Innovation and Entrepreneurship Support Plan of Anhui Province for Returned Personnels Studying Abroad (2022LCX001), the Major Project of Anhui Provincial University Science Research Program, Opening Foundation Program of Shanxi Provincial Key Laboratory for High Performance Battery Materials and Devices (2023HPBMD01001), Shenzhen Municipality under Shenzhen Science and Technology Program (RCBS20221008093222009) and the project of PTDC/EME-REN/1497/2021.References
- W. Shang, W. Yu, Y. Liu, R. Li, Y. Dai, C. Cheng, P. Tan and M. Ni, Energy Storage Mater., 2020, 31, 44 CrossRef.
- Y. Zhu, P. Guan, R. Zhu, S. Zhang, Z. Feng, M. Li, T. Wan, L. Hu, Y. Liu, Q. Li, J. Yu and D. Chu, J. Energy Chem., 2023, 87, 61 CrossRef CAS.
- J. Islam, R. Anwar, M. Shareef, H. M. Zabed, J. Sahu, X. Qi, M. U. Khandaker, A. Ragauskas, I. Boukhris and M. R. Rahman, J. Power Sources, 2023, 563, 232777 CrossRef CAS.
- J. F. Parker, C. N. Chervin, E. S. Nelson, D. R. Rolison and J. W. Long, Energy Environ. Sci., 2014, 7, 1117 RSC.
- D. Ozgit, P. Hiralal and G. A. J. Amaratunga, ACS Appl. Mater. Interfaces, 2014, 6, 20752 CrossRef CAS PubMed.
- Z. Cheng, K. Wang, J. Fu, F. Mo, P. Lu, J. Gao, D. Ho, B. Li and H. Hu, Adv. Energy Mater., 2024, 2304003, 1 Search PubMed.
- S. Jiao, J. Fu, M. Wu, T. Hua, H. Hu and M. Wu, ACS Nano, 2022, 16, 1013 CrossRef CAS.
- R. F. Amlie and P. Rüetschi, J. Electrochem. Soc., 1961, 108, 813 CrossRef CAS.
- A. M. Zamarayeva, A. E. Ostfeld, M. Wang, J. K. Duey, I. Deckman, B. P. Lechêne, G. Davies, D. A. Steingart and A. C. Arias, Sci. Adv., 2017, 3, 1 Search PubMed.
- Y. Zhu, J. Fan, S. Zhang, Z. Feng, C. Liu, R. Zhu, Y. Liu, P. Guan, M. Li and Z. Han, Chem. Eng. J., 2024, 480, 148334 CrossRef CAS.
- Y. Zhu, R. Zhu, F. Chen, S. Zhang, Y. C. Kuo, P. Guan, M. Li, Y. Liu, Z. Han and T. Wan, Energy Environ. Mater., 2024, 7, e12493 CrossRef CAS.
- Y. Wu, N. He, G. Liang, C. Zhang, C. Liang, D. Ho, M. Wu and H. Hu, Adv. Funct. Mater., 2024, 34, 2301734 CrossRef CAS.
- G. Liang, F. Mo, D. Wang, X. Li, Z. Huang, H. Li and C. Zhi, Energy Storage Mater., 2020, 25, 86 CrossRef.
- J. Hao, X. Li, X. Zeng, D. Li, J. Mao and Z. Guo, Energy Environ. Sci., 2020, 13, 3917 RSC.
- W. Lu, C. Zhang, H. Zhang and X. Li, ACS Energy Lett., 2021, 6, 2765 CrossRef CAS.
- C. Li, X. Xie, S. Liang and J. Zhou, Energy Environ. Mater., 2020, 3, 146 CrossRef CAS.
- Z. Zhao, Y. He, W. Yu, W. Shang, Y. Ma and P. Tan, Proc. Natl. Acad. Sci. U.S.A., 2023, 120, e2307847120 CrossRef CAS.
- Y. Cui, Y. He, W. Yu, Y. Ma, Z. Zhao, J. Yu and P. Tan, J. Power Sources, 2024, 601, 234294 CrossRef CAS.
- L. Wang, B. Zhang, W. Zhou, Z. Zhao, X. Liu, R. Zhao, Z. Sun, H. Li, X. Wang, T. Zhang, H. Jin, W. Li, A. Elzatahry, Y. Hassan, H. Fan, D. Zhao and D. Chao, J. Am. Chem. Soc., 2024, 146, 6199 CrossRef CAS PubMed.
- J. Yu, W. Yu, Z. Zhang and P. Tan, Chem. Eng. J., 2024, 481, 148556 CrossRef CAS.
- Y. Cui, Y. He, W. Yu, W. Shang, J. Yu and P. Tan, ACS Appl. Mater., 2023, 15, 3028 CrossRef CAS.
- Y. Hu, J. Fu, H. Hu, D. Ho and H. Hu, Energy Storage Mater., 2023, 55, 669 CrossRef.
- S. Jiao, J. Fu, Q. Yin, H. Yao and H. Hu, Energy Storage Mater., 2023, 59, 102774 CrossRef.
- Y. Gao, J. Fu, F. Mo, L. Zhang, D. Ho and H. Hu, Small, 2024, 2400085 CrossRef CAS PubMed.
- Y. Zuo, K. Wang, P. Pei, M. Wei, X. Liu, Y. Xiao and P. Zhang, Mater. Today Energy, 2021, 20, 100692 CrossRef CAS.
- Q. Yang, Q. Li, Z. Liu, D. Wang, Y. Guo, X. Li, Y. Tang, H. Li, B. Dong and C. Zhi, Adv. Mater., 2020, 32, 2001854 CrossRef CAS PubMed.
- Z. Yu, Q. Wang, Y. Li, F. Zhang, X. Ma, X. Zhang, Y. Wang, J. Huang and Y. Xia, Joule, 2024, 8, 1063 CrossRef CAS.
- W. Zhou, S. Ding, D. Zhao and D. Chao, Joule, 2023, 7, 1104 CrossRef CAS.
- Y. Yao, Z. Wang, Z. Li and Y. C. Lu, Adv. Mater., 2021, 33, 2008095 CrossRef CAS.
- H. Zhang, D. Xu, F. Yang, J. Xie, Q. Liu, D. J. Liu, M. Zhang, X. Lu and Y. S. Meng, Joule, 2023, 7, 971 CrossRef CAS.
- Z. Yu, Q. Wang, Y. Li, F. Zhang, X. Ma, X. Zhang, Y. Wang, J. Huang and Y. Xia, Joule, 2024, 8, 1063 CrossRef CAS.
- J. Zhang, K. Wang, P. Lu, J. Gao, Z. Cao, F. Mo, D. Ho, B. Li and H. Hu, Adv. Funct. Mater., 2024, 34, 2310775 CrossRef CAS.
- R. H. Baughman, A. A. Zakhidov and W. A. De Heer, Science, 2002, 297, 787 CrossRef CAS.
- J. Y. Cheong, S. H. Cho, J. Lee, J. W. Jung, C. Kim and I. D. Kim, Adv. Funct. Mater., 2022, 32, 2208374 CrossRef CAS.
- J. Shi, S. Wang, X. Chen, Z. Chen, X. Du, T. Ni, Q. Wang, L. Ruan, W. Zeng and Z. Huang, Adv. Energy Mater., 2019, 9, 1901957 CrossRef CAS.
- P. Zhang, Y. Li, G. Wang, F. Wang, S. Yang, F. Zhu, X. Zhuang, O. Schmidt and X. Feng, Adv. Mater., 2019, 31, 1806005 CrossRef PubMed.
- M. Zhu, Z. Wang, H. Li, Y. Xiong, Z. Liu, Z. Tang, Y. Huang, A. L. Rogach and C. Zhi, Energy Environ. Sci., 2018, 11, 2414 RSC.
- H. Wang, R. Guo, H. Li, J. Wang, C. Du, X. Wang and Z. Zheng, Chem. Eng. J., 2022, 429, 132196 CrossRef CAS.
- Z. Cui, S. Shen, J. Yu, J. Si, D. Cai and Q. Wang, Chem. Eng. J., 2021, 426, 130068 CrossRef CAS.
- S. Bi, F. Wan, S. Wang, S. Jia, J. Tian and Z. Niu, Carbon Energy, 2021, 3, 167 CrossRef CAS.
- H. Hong, L. Jiang, H. Tu, J. Hu, K. Moon, X. Yan and C. Wong, J. Mater. Sci. Technol., 2022, 101, 294 CrossRef CAS.
- X. Wang, S. Zheng, F. Zhou, J. Qin, X. Shi, S. Wang, C. Sun, X. Bao and Z. Wu, Natl. Sci. Rev., 2020, 7, 64 CrossRef CAS PubMed.
- C. Yan, X. Wang, M. Cui, J. Wang, W. Kang, C. Foo and P. Lee, Adv. Energy Mater., 2014, 4, 1301396 CrossRef.
- K. Yang, Y. Ying, L. Cui, J. Sun, H. Luo, Y. Hu and J. Zhao, Energy Storage Mater., 2021, 34, 203 CrossRef.
- R. Li, L. Li, R. Jia, K. Jiang, G. Shen and D. Chen, Small Methods, 2020, 4, 2000363 CrossRef CAS.
- C. Li, Q. Zhang, J. Sun, T. Li, S. E, Z. Zhu, B. He, Z. Zhou, Q. Li and Y. Yao, ACS Energy Lett., 2018, 3, 2761 CrossRef CAS.
- C. Li, Q. Zhang, S. E, T. Li, Z. Zhu, B. He, Z. Zhou, P. Man, Q. Li and Y. Yao, J. Mater. Chem. A, 2019, 7, 2034 RSC.
- Y. Zhu, R. Zhu, P. Guan, M. Li, T. Wan, L. Hu, S. Zhang, C. Liu, D. Su, Y. Liu, D. Liu, Q. Li, J. Yu and D. Chu, Energy Storage Mater., 2023, 60, 102836 CrossRef.
- H. Tang, Y. Yin, Y. Huang, J. Wang, L. Liu, Z. Qu, H. Zhang, Y. Li, M. Zhu and O. G. Schmidt, ACS Energy Lett., 2021, 6, 1859 CrossRef CAS.
- H. Li, Z. Liu, G. Liang, Y. Huang, Y. Huang, M. Zhu, Z. Pei, Q. Xue, Z. Tang, Y. Wang, B. Li and C. Zhi, ACS Nano, 2018, 12, 3140 CrossRef CAS.
- C. Li, W. Wang, X. Sun, H. Hou, C. Zheng, J. Zhang, F. Hou, D. Zhang, J. Du and Y. Yao, J. Mater. Chem. A, 2022, 10, 2470 CrossRef.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146 CrossRef CAS.
- Q. Yang, S. Cui, Y. Ge, Z. Tang, Z. Liu, H. Li, N. Li, H. Zhang, J. Liang and C. Zhi, Nano Energy, 2018, 50, 623 CrossRef CAS.
- C. Zhang, S.-H. Park, A. Seral-Ascaso, S. Barwich, N. McEvoy, C. S. Boland, J. N. Coleman, Y. Gogotsi and V. Nicolosi, Nat. Commun., 2019, 10, 849 CrossRef.
- S. Zheng, J. Ma, K. Fang, S. Li, J. Qin, Y. Li, J. Wang, L. Zhang, F. Zhou and F. Liu, Adv. Energy Mater., 2021, 11, 2003835 CrossRef CAS.
- C. Zhong, B. Liu, J. Ding, X. R. Liu, Y. W. Zhong, Y. Li, C. B. Sun, X. P. Han, Y. D. Deng, N. Q. Zhao and W. B. Hu, Nat. Energy, 2020, 5, 440 CrossRef CAS.
- C. Liu, X. W. Chi, Q. Han and Y. Liu, Adv. Energy Mater., 2020, 10, 1903589 CrossRef CAS.
- D. L. Chao, C. Ye, F. X. Xie, W. H. Zhou, Q. H. Zhang, Q. F. Gu, K. Davey, L. Gu and S. Z. Qiao, Adv. Mater., 2020, 32, 2001894 CrossRef CAS PubMed.
- B. Wang, W. Zhou, Y. Zhang, T. Zhang, X. Li, Y. Feng, R. Zhao, W. Li, A. Elzatahry, Y. Hassan, D. Zhao and D. Chao, Joule, 2024, 8, 2033 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04851b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |