
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlleviating the volume expansion of silicon anodes by constructing a high-strength ordered multidimensional encapsulation structure†
Yun
Yu
,
Haiqiang
Gong
,
Xinyou
He
,
Lei
Ming
 *,
Xiaowei
Wang
* and
Xing
Ou
*
*,
Xiaowei
Wang
* and
Xing
Ou
*
National Energy Metal Resources and New Materials Key Laboratory, School of Metallurgy and Environment, Central South University, Changsha 410083, P. R. China. E-mail: minglei666@126.com; yjywxw@csu.edu.cn; ouxing@csu.edu.cn
First published on 5th September 2024
Abstract
The application of silicon-based nanomaterials in fast-charging scenarios is hindered by volume expansion during lithiation and side reactions induced by surface effects. Constructing a robust encapsulation structure with high mechanical strength and conductivity is pivotal for optimizing the electrochemical performance of nanostructured silicon anodes. Herein, we propose a multifaceted hierarchical encapsulation structure featuring excellent mechanical strength and high conductivity by sequentially incorporating SiOx, hard carbon, and closed-pore carbon layers around silicon quantum dots, thereby enabling stable cycling at high current densities. In this structure, the ultra-thin SiOx layer strengthens the Si–C interface, while the outermost carbon matrix with closed pores functions both as a conductive network and a barrier against electrolyte intrusion. Notably, the synthesized material exhibits a specific capacity of 1506 mA h g−1 with 90.17% retention after 300 cycles at 1.0 A g−1. After 500 cycles at 5.0 A g−1, it retains 640.4 mA h g−1, over 70% of its initial capacity.
Introduction
As electric vehicle technology advances, enhancing the driving range, reducing charging time, and ensuring battery longevity are pivotal goals in the development of lithium-ion power batteries.1,2 While the development of cathode materials for lithium-ion batteries has reached maturity, optimizing anode materials with greater potential for enhancing overall performance has become paramount.3 Silicon (Si) has emerged as a promising candidate due to its high energy density, abundant resources, and environmental sustainability.4,5 The high specific capacity of Si enables electrode thickness reduction without compromising overall energy density, thereby mitigating concentration and potential gradient effects during high-rate charging. However, challenges such as silicon's inherent low electrical conductivity and substantial volume expansion, and the resulting material pulverization and solid electrolyte interphase (SEI) layer formation remain significant barriers to its widespread adoption, despite its advantages for fast charging.6–8To effectively mitigate Si volume expansion, size control emerges as a highly effective strategy. Research indicates that reducing Si nanocrystals to the quantum dot (QD) scale (<20 nm) effectively mitigates particle pulverization under high stress and strain conditions.9 The inherent surface effects of QD structures facilitate efficient surface modification, reducing Si volume expansion during lithiation and shortening lithium-ion diffusion paths, thereby optimizing electrode performance.10,11 However, high specific surface area silicon nanoparticles (SiNPs) undergo continuous reactions with electrolytes, leading to persistent SEI layer formation that deteriorates the electrode structure and electrochemical performance.12,13
Core–shell structures, composite materials, and specialized encapsulations have proven effective in mitigating electrolyte-related side reactions and Si volume expansion at high rates.14–16 Combining hard carbon with Si enhances lithium-ion kinetics, improves material conductivity, and mitigates Si volume expansion.17,18 Despite achieving good electrochemical performance, the Si–C interface is prone to fracturing during long-term cycling, severely impacting battery stability.19 Thick encapsulation layers can also restrict rapid lithium-ion transport, affecting reaction kinetics.20 Therefore, designing rational hierarchical structures and components to simultaneously protect silicon quantum dot (SiQD) stability, enhance electron conductivity, and optimize interface stability is critical for advancing SiQD materials.21
Here, we propose a gradient and high-strength ordered hierarchical encapsulation strategy based on SiQDs. This encapsulation includes an ultra-thin in situ SiOx layer, a hard carbon (HC) layer from resin-based carbonization and a carbonaceous matrix with a closed pore (CPC) layer. The developed material, dual carbon encapsulated SiOx-enhanced SiQDs (CCOS), with a hard carbon@SiOx@Si structure exhibits a gradient and mechanically strength ordered distribution. The 2–3 nm SiOx layer enhances HC-SiQD interface adhesion, alleviating Si volume expansion and optimizing transport kinetics.22,23 This tight bonding structure facilitates efficient charge and ion transport between the two phases through its ultra-thin design, and significantly minimizes electronic and lithium-ion transport impedances at the Si–C interface, thereby ensuring stable operation of CCOS under high-performance demands. The HC layer enhances electron conductivity and mechanical stability, with SiOx crosslinking increasing encapsulation density to effectively isolate SiQDs from electrolytes and promote stable SEI layer formation.24,25 The interconnected CPC layer further enhances CCOS conductivity. Additionally, the CPC layer with closed pores effectively prevents the electrolyte from coming into contact with the SiQDs.26 With this composite hierarchical encapsulation featuring a strong bonding interface, Si and C are effectively connected, resulting in SiQD-based electrodes that exhibit excellent rate capability and cycling stability.27
Results and discussion
To achieve encapsulation, we designed the following scheme (Fig. 1a). (i) SiQDs prepared via the liquid-phase method are surface modified with citrate ester groups, forming Si–O bonds, and are subsequently encapsulated with resin (Fig. S1†). (ii) The resin together with an ester group forms single carbon encapsulated SiOx-enhanced SiQD (COS) materials via pre-curing and high-temperature carbonization. During the carbonization process (Fig. S2†), ester bonds undergo thermal decomposition converting alkyl chains into carbon. Then the remaining O reacts with Si, forming a SiOx layer on the surface of SiQDs, which contributes to the final structure of the material. Simultaneously, O and Si, as well as Si and C, react to form a covalent Si–O–C bonding structure, which enhances the bonding strength between Si and C. (iii) The resin solution was mixed with ethanol in a certain ratio, to which COS powder was subsequently added and mixed, and the heating and pressurization process was completed using a high-pressure vessel, followed by calcination to obtain a hard carbon matrix with closed-pores to complete the third encapsulation (Fig. S3†).28 Ethanol evaporates and forms bubbles during curing, creating closed pores within the resin that are preserved after high-temperature carbonization. TEM images illustrate that the SiQD materials have a particle size distribution of 10–20 nm (Fig. S4a†), while the particle size of the completed single layer carbon coated material (COS) is 30–40 nm (Fig. S4b†). This strategy mitigates the absolute volume effect of SiQDs during alloying and the side reactions caused by SEI fracture and recombination. Multilayer encapsulation and the SiQD structure buffer volume changes and isolate the electrolyte, indirectly enhancing reaction kinetics, making it an optimal host for SEI stabilization. The efficient carbon-based conductive network, together with the HC layer ensures high electron transfer. Based on the high specific surface area of SiQDs and their short ion/electron transport pathways, combined with the SiOx/HC/CPC encapsulation layer, fast and stable ion/electron dual transfer during (de)lithiation is enabled (Fig. 1b). The uneven lithiation of the SiNP electrode during rapid lithiation leads to the formation of cracks in the SiNPs (Fig. 1c), adversely affecting its electrochemical performance.29 The prepared CCOS anode exhibits outstanding rate performance and cycling stability, significantly improving the Si volume effect during cycling.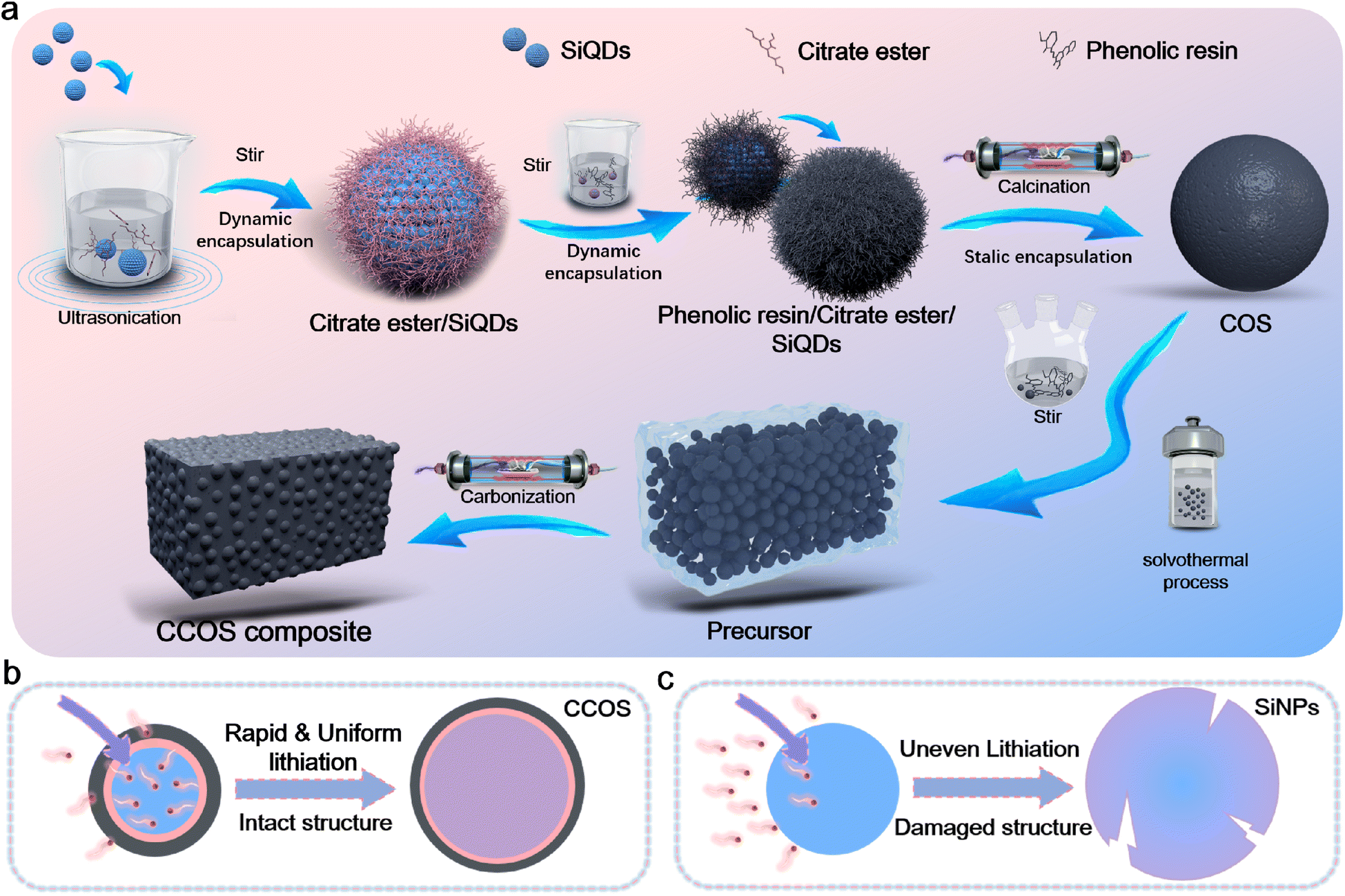 | ||
| Fig. 1 (a) Schematic illustration of the CCOS design strategy. Schematic illustration of the structural evolution of (b) CCOS and (c) SiNPs during rapid lithiation. | ||
The X-ray diffraction (XRD) patterns of CCOS (Fig. 2a) match the diffraction peaks of elemental Si (JCPDS no. 27-1402) with a broad peak between 20 and 28°, indicating an amorphous structure of carbon and SiOx, confirming successful synthesis.30 Thermogravimetric analysis (TGA) confirms 25.47% carbon content (Fig. S5†). The peak at 550 °C indicates the existence of SiOx (SiOx → SiO2). X-ray photoelectron spectrometry (XPS) spectrum of CCOS (Fig. 2b) displays the peaks at 99.7 eV, 100.2 eV, 100.6 eV, and 103.7 eV, corresponding to Si 2p3/2, Si 2p1/2, Si–C, and SiOx, respectively.31,32 The appearance of the SiOx peak indicates successful surface oxidation of SiQDs achieved during the carbonization process, with citrate-modified Si leading to the in situ formation of a SiOx protective layer. The Raman spectrum of CCOS shows two discernible peaks at 1327 and 1583 cm−1 (Fig. 2c), corresponding to the characteristic D band and G band of carbon, with a calculated ID/IG ratio of ∼1.2, indicating a high degree of structural disorder and defects.33 The peaks representing Si and Si–O are located at 490 and 880 cm−1, respectively.34 The FTIR spectra (Fig. S6†) of COS materials before and after carbonization reveal key changes. Initially, the COS precursor shows a broad –OH peak at 3300–3500 cm−1, aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–H vibrations at 1607.89 cm−1 and 1472.23 cm−1, and a C–O–C peak at 1209.37 cm−1. After carbonization, the disappearance of the –OH peak indicates dehydration, while the loss of aromatic vibrations suggests disruption of the aromatic structure. The reduced C–O–C peak intensity reflects ether bond cleavage. These changes confirm the successful formation of a uniform carbonized encapsulation layer on the SiQDs.
C and C–H vibrations at 1607.89 cm−1 and 1472.23 cm−1, and a C–O–C peak at 1209.37 cm−1. After carbonization, the disappearance of the –OH peak indicates dehydration, while the loss of aromatic vibrations suggests disruption of the aromatic structure. The reduced C–O–C peak intensity reflects ether bond cleavage. These changes confirm the successful formation of a uniform carbonized encapsulation layer on the SiQDs.
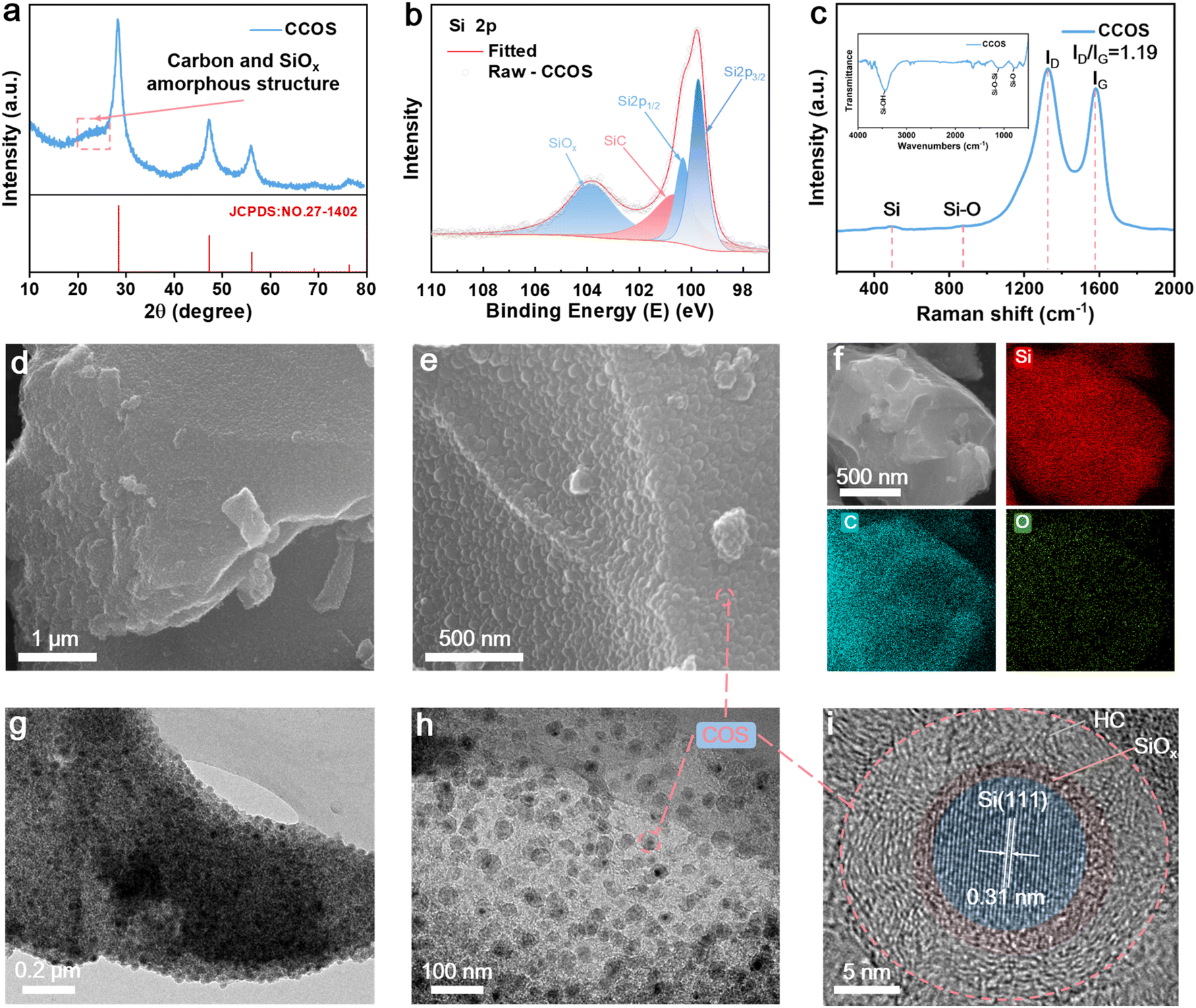 | ||
| Fig. 2 Structure and composite characterization of CCOS. (a) XRD pattern. (b) The Si 2p high-resolution XPS spectrum. (c) Raman and FTIR (inset) spectra. (d and e) SEM images at different magnifications. (f) SEM-based EDS mapping of Si, C, and O. (g and h) TEM images at different magnifications. (i) HRTEM image. | ||
However, due to the presence of the carbon encapsulation layer, their signals appear relatively weak. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) are performed to further investigate the microstructure and lattice structure of CCOS. The SEM images reveal that CCOS exhibits a rough surface (Fig. 2d), with the main body consisting of a carbon matrix and Si–C composite nanospheres (COS) (Fig. 2e and g). Energy dispersive spectrometry (EDS) mappings (Fig. 2f) have demonstrated the uniform distribution of C and Si elements on the surface area of the material. The TEM images further confirm the existence of the SiOx encapsulation layer and hard carbon encapsulation structure, illustrating two layers of amorphous structures on the outer part of the material (Fig. 2h and i), sequentially and completely encapsulating the ordered SiQD lattice structure. To further investigate the encapsulation state of the in situ SiOx layer, we etched the CCOS material using HF acid to dissolve the SiOx on the Si surface. As displayed in Fig. S7a–f,† combined TEM-based EDS mapping with O distribution and the etched CCOS material with clear voids confirm the presence of SiOx. The TEM image (Fig. S7f†) reveals a distinct void between the SiQD core and the carbon layer, with a thickness of the gap ∼3 nm, confirming the presence of the ultra-thin in situ SiOx layer. Additionally, the outermost encapsulating carbon layer contains closed cavities with pore sizes smaller than 5 nm (Fig. S7g†), formed by disordered graphite sheets facilitated by EtOH, maintaining a low specific surface area of ∼7.38 m2 g−1 (Fig. S8†) while providing a high buffering capacity against SiQD expansion.35 Under high-rate conditions, these tiny pores can serve as temporary storage sites for lithium-ions, preventing lithium deposition on the CPC layer.36
To elucidate the impact of multilayer encapsulation and the structural design of quantum dots (QDs) on Si, an in-depth investigation into the reaction kinetics is conducted through various electrochemical tests. SiNPs with a particle size distribution of 20–60 nm are prepared as electrodes for comparison (Fig. S4c†). Fig. S9a and b† display the cyclic voltammetry (CV) curves of the CCOS and SiNP electrodes for the first three cycles at 0.1 mV s−1 (in the voltage range of 0.01–3.0 V). The CV profile of the SiNP electrode shows cathodic peaks at 0.19 V and anodic peaks at 0.36 V and 0.52 V, which are relevant to the characteristics of Si–Li reactions.37,38 The large peak intensity variations in SiNPs suggest substantial Si exposure to the electrolyte due to volume effects. In contrast, CCOS displays minimal variation during cycling, indicating effective alleviation of Si volume expansion through the incorporation of SiQDs, the SiOx layer, the HC layer, and the CPC layer.
The CV curves of CCOS and SiNPs at different scan rates (Fig. 3a and S9c†) further demonstrate the stability of the CCOS interface structure. The calculated b values (ESI Note 1†) of the reduction peak for both materials (Fig. S10a and b†) reveal a lower b value for CCOS (0.61) compared to SiNPs (0.68), implying a predominance of diffusion-controlled processes in CCOS facilitated by multi-carbon encapsulation and SiOx layer encapsulation.39 The pseudocapacitive contribution of CCOS increases slowly, reaching 56.28% at 2.0 mV s−1 (Fig. 3b), while that of SiNPs increases to 68.92% (Fig. 3c), highlighting the efficient alloying reactions in CCOS.40 Moreover, Fig. S10c and d† display the relative electrochemical active surface areas (ECSAs) of CCOS and SiNPs, calculated using the Randles–Sevcik equation (ESI Note 2†).41 The results show that the relative ECSA of CCOS (1.42) is smaller than that of SiNPs (2.66).
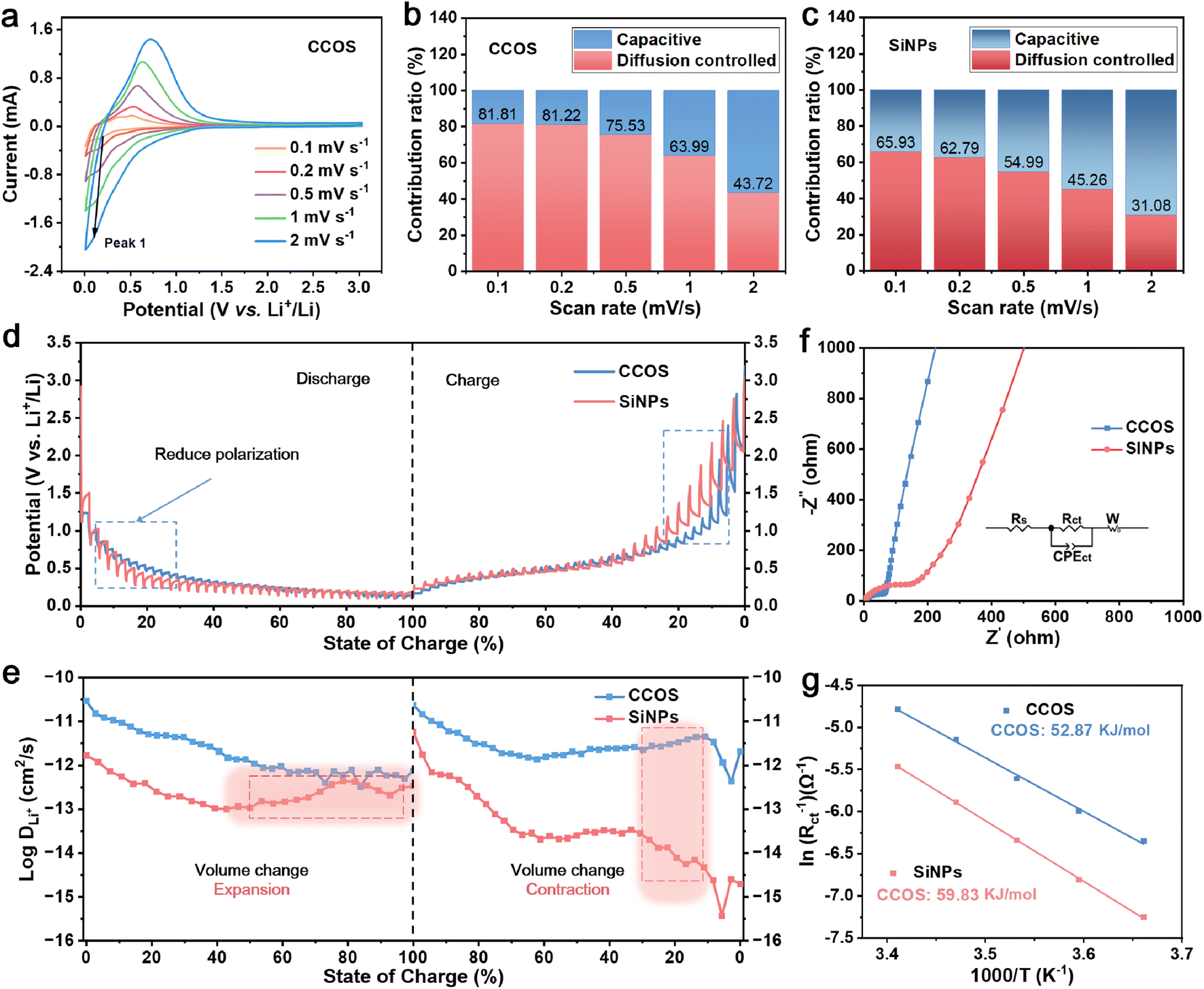 | ||
| Fig. 3 (a) CV curves at different sweep rates. The proportion of capacity contributions of (b) CCOS and (c) SiNPs. (d) GITT profiles. (e) The calculated lithium-ion diffusion coefficient. (f) EIS curves and fitting results. (g) Ea derived from Rct. | ||
To further comprehend the diffusion mechanism of Li+ at different (de)lithiation states, the galvanostatic intermittent titration technique (GITT) is employed to investigate the enhancement of lithium-ion diffusion kinetics. Fig. 3d illustrates the voltage variation with the degree of lithium insertion during the GITT testing process. At different lithiation depths, the CCOS electrode exhibits low polarization, indicating that the multi-layer encapsulation and SiQD structure effectively enhance lithium-ion transport. The values of DLi+ calculated by using the simplified Fick's second law (ESI Note 3†) are shown in Fig. 3e, suggesting that CCOS exhibits higher lithium-ion diffusion coefficients. It is noteworthy that during the lithiation process, the lithium-ion diffusion coefficient of CCOS gradually decreases, whereas for SiNPs, it initially decreases and then increases. This behavior is ascribed to the significant volume changes caused by the Si transition from a crystalline to an amorphous state during the lithiation reaction, which results in the fragmentation of SiNPs and structural degradation. Different trends are observed during delithiation, which are similarly correlated with the significant volume changes in Si-based materials during cycling and the associated interfacial side reactions. Electrochemical impedance spectroscopy (EIS) tests of CCOS and SiNPs at 25 °C (Fig. 3f) show that CCOS has a lower charge-transfer resistance (Rct) of 66.87 Ω compared to SiNPs (146.73 Ω), attributed to improved electron conductivity.42 Fig. S11a and b† illustrate the charge-transfer resistances generated by CCOS and SiNPs at various temperatures, indicating that the CCOS electrode exhibits lower charge-transfer resistance, primarily attributed to the constructed hard carbon encapsulation and porous structure effectively reducing the resistance to lithium-ion diffusion. The calculated activation energy (Ea) for CCOS (ESI Note 4†) is smaller (52.87 kJ mol−1) than that of SiNPs (59.83 kJ mol−1), suggesting a lower energy barrier for lithium-ion transport and rapid lithium storage capability (Fig. 3g).43 These results demonstrate that the multi-layer encapsulation and SiQD structure effectively enhance lithium storage performance, making CCOS an optimal host for SEI stabilization and high-rate cycling.
Moreover, in situ EIS measurement has been conducted to further investigate lithium-ion diffusion kinetics on the electrode. The Rct values at different voltages during the (dis)charge processes are shown in Fig. S12a–c,† indicating similar trends in electrochemical impedance for both materials. Initially, when discharging from 3.0 to 0.1 V, there is an increase in Rct due to the accumulation of electrolyte additives on the electrode surface. Subsequently, during discharge, two semicircles appear in the high-frequency region, corresponding to SEI layer formation, with a gradual decrease in Rct attributed to electrode material fracture reducing lithium-ion transport pathways.44,45 Besides, compared to SiNPs, CCOS exhibits lower Rct during the (dis)charge processes, indicating favorable reaction kinetics for CCOS.46 EIS tests of SiNPs and CCOS at high rates of 5.0 A g−1 (Fig. S12d and e†) further reveal that CCOS consistently has a lower charge transfer resistance (Rct) than that of SiNPs, which suggests its better ion transport and diffusion kinetics, thus contributing to its superior capacity retention at high rates. Thus, multiple encapsulation layers and a porous structured matrix effectively reduce the resistance encountered by lithium ions during transport, thereby achieving efficient lithium storage performance for the material during the fast-charging process.
Therefore, for the CCOS and SiNP electrodes in half-cells after three activation cycles at 0.1 A g−1 rate, followed by (dis)charge cycling at 0.5, 1.0, and 5.0 A g−1 rates, it can be seen that the initial capacity increase of the CCOS is related to the participation of small particles and previously inactive Si in the reactions. After 300 cycles, the CCOS electrode maintains a high capacity retention rate of 96.5% and a specific capacity of 1689 mA h g−1 at 0.5 A g−1 (Fig. 4a). Remarkably, even after 500 cycles at a high current density of 5.0 A g−1, CCOS sustains a capacity retention rate of 70.5% (Fig. S13†). Conversely, the SiNP electrode shows poor cycling stability at high rates, with rapid capacity decay and no remaining capacity after 170 cycles. Fig. S14a† shows that although SiQDs have a high initial discharge capacity of 3952 mA h g−1, their low initial coulombic efficiency (69%) results in a reduced initial charge capacity (2731 mA h g−1), due to the side reactions with electrolyte. However, SiQDs benefit from minimal volume expansion during lithiation, improving cycling stability. After 175 cycles at 5.0 A g−1, SiQDs maintain a capacity of 372 mA h g−1 (Fig. S14b†), while the capacity of SiNPs has degraded to 0 mA h g−1 (Fig. S13†). Fig. S14c and d† demonstrate that while COS has a higher initial capacity, CCOS achieves better coulombic efficiency (84% vs. 79%) and long-term cycling performance due to the outermost CPC layer, which contains closed pores, effectively isolating the Si core from the electrolyte.
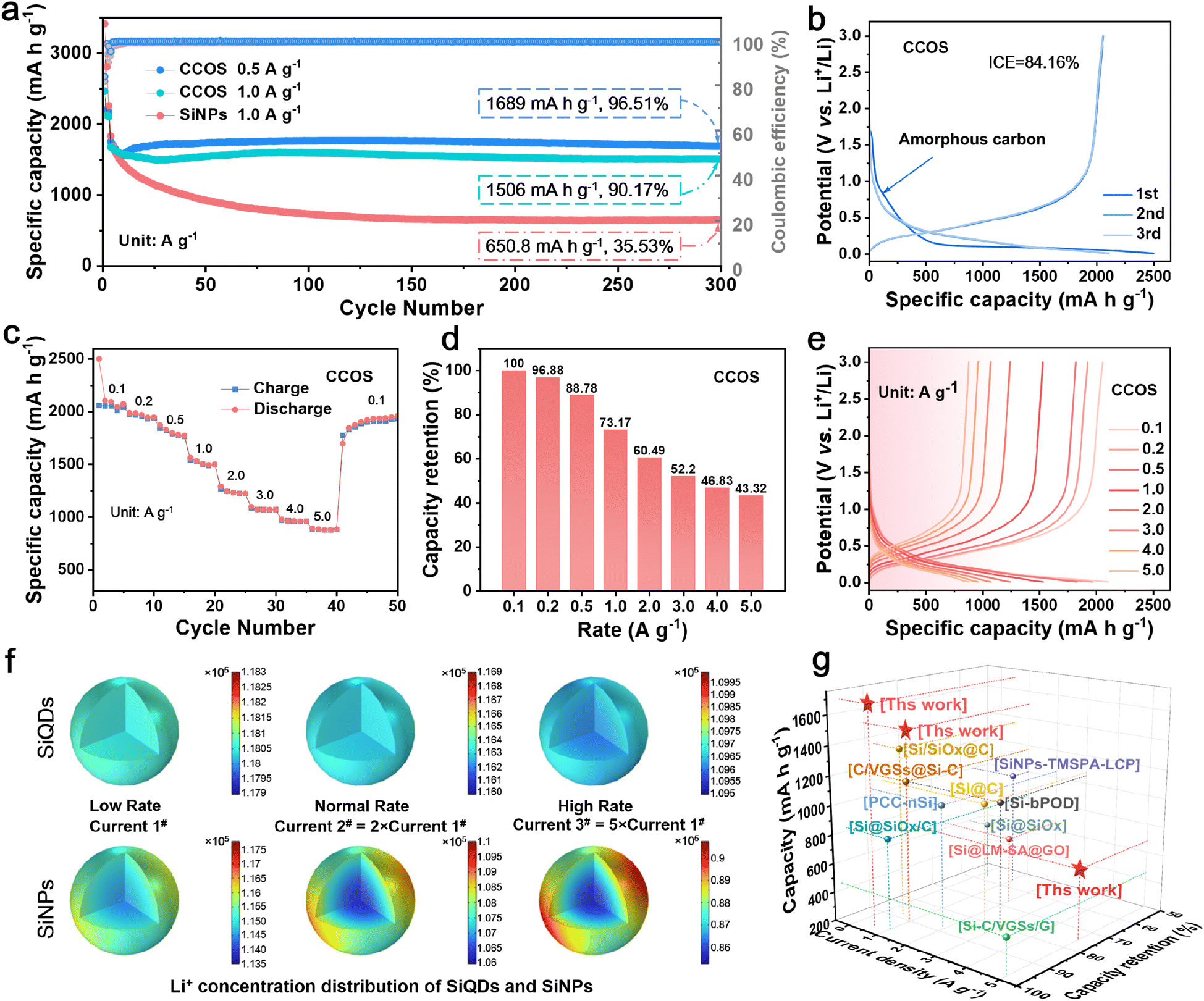 | ||
| Fig. 4 (a) Cycling performance of CCOS and SiNPs at 1.0 A g−1, and CCOS at 0.5 A g−1. (b) Initial (dis)charge curves. (c) Rate performance, (d) capacity retention and (e) (dis)charge curves at different rates. (f) COMSOL simulation diagrams of the Li+ concentration distribution of SiQDs and SiNPs during the lithiation process at different currents. (g) Ragone plots comparing CCOS with reported silicon-based anodes.22,39,S1–S8† | ||
Fig. 4b and S15a† illustrate (dis)charge curves of CCOS and SiNP electrodes in half-cells during the first three activation cycles. The voltage plateau at 0.6–1.0 V in CCOS corresponds to lithium-ion intercalation into hard carbon, confirming successful carbon encapsulation.47 The distinct voltage plateau observed at 0.1 V corresponds to the lithiation process, during which the Si–Si bonds within crystalline Si gradually break, transforming large Si clusters into smaller rings and chains with star and boomerang structures. This marks the phase transition from crystalline Si to amorphous LixSi, which is a solid-solution transformation.27 The almost overlapping (dis)charge curves of the second and third cycles indicate a stable SEI layer under 0.1 A g−1 conditions, providing stable lithium-ion storage.48,49Fig. 4c shows the rate performance of the CCOS electrode. The CCOS anode exhibits specific capacities of 2079, 1515, 1071, 953, and 877 mA h g−1 at discharge rates of 0.1, 1.0, 3.0, 4.0, and 5.0 A g−1, respectively. Under the same test conditions, the SiNP electrode shows 2100 and about 1000 mA h g−1 at 1.0 and 5.0 A g−1, respectively (Fig. S15b†). However, the rate capability is determined by the capacity retention at different current densities. When using the discharge capacity at 0.1 A g−1 as a standard, the capacity retention at a high rate of 5.0 A g−1 for CCOS is 43.3% (Fig. 4d), which is significantly higher than the capacity retention of SiNPs (24.6%), confirming the superior rate tolerance for CCOS (Fig. S15c†). From the perspective of long-term high-rate cycling (Fig. S13†), the initial capacity of SiNPs is indeed higher than that of CCOS in the first few cycles. However, it rapidly declines within 170 cycles, indicating the poor stability of high-rate retention.
These results indicate that CCOS with a multi-layer encapsulated SiQD structure exhibits outstanding high-rate lithium storage performance. The initial charging polarization of SiNPs at 5.0 A g−1 is 0.45 V (Fig. S15d†), while CCOS polarization is 0.26 V (Fig. 4e), much less than that of SiNPs. These issues severely impact the fast-charging applications of SiNPs, whereas the multi-layer encapsulation and SiQD design effectively reduce material polarization, ensuring excellent high-rate performance. To further investigate the mechanism of QD design for the lithiation process in the CCOS material, we develop a comprehensive COMSOL model. This model simulates the (dis)charging processes of both SiQD and SiNP electrodes under varying current conditions, analysing the distribution of Li+ at a voltage of 0.25 V (Fig. 4f). Clearly, the cycling stability of CCOS is better than that of most recently reported Si/C anode materials (Fig. 4g and Table S1†). Simulating the uncoated SiQD electrode helps to elucidate the influences of silicon quantum dots on structure ion transport and overall electrochemical performance. Current 1# serves as the baseline with a low rate, while currents 2# and 3# are 2 and 5 times current 1#, representing the normal rate and high rate, respectively. Under different current conditions, the SiQD electrode consistently exhibits highly uniform Li+ concentration distribution, superior Li+ diffusion kinetics, and higher Li+ concentration. In contrast, the SiNP material not only demonstrates lower Li+ concentration but also shows uneven lithiation, even under low current conditions, with significantly lower Li+ concentration in the Si core compared to the edge regions. As the current density increases, the Li+ concentration gradient between the edge and core of the Si particles in the SiNP electrode increases, indicating an exacerbation of uneven lithiation. Additionally, the analysis of delithiation reveals a similar phenomenon, showing that SiQDs achieve rapid and uniform delithiation compared to SiNPs (Fig. S16†).
Furthermore, the electrode expansion under different working conditions is analysed to verify the effectiveness of multi-layer encapsulation and SiQD design in alleviating the volume effect and enhancing lithium-ion diffusion kinetics. The cross-sections of the electrodes at different states of charge (SOC), current rates, and cycles are characterized by SEM. Cross-sectional images at 1.0 A g−1 on charging to 80% (Fig. 5a), 90% (Fig. 5b), and 105% (Fig. 5c) SOC after 100 cycles revealed a positive correlation between the electrode thickness and lithiation states. Even under over-lithiation conditions, the expansion of CCOS electrodes is slight. As shown in Fig. 5d3, after 5 cycles at 0.2 A g−1, the CCOS electrode thickness does not show obvious changes, indicating that CCOS particles effectively buffer the volume expansion of SiQDs.50 TEM images of the CCOS electrode after 100 and 300 cycles at 1.0 A g−1 (Fig. 5e1 and f1) reveal that the COS particles grow and the HC layers diminish. After 300 cycles, some small cavities appear, primarily caused by the expansion of SiQDs during lithiation. Comparing the cross-sectional images of electrodes after 50 (Fig. S17c†), 100 (Fig. 5e2), and 300 cycles (Fig. 5f2) at 1.0 A g−1, we observe that the thickness increases rapidly during the first 100 cycles compared to the subsequent 200 cycles. This rapid increase is due to the gradual reaction of small particles and initially unreacted Si at higher rates, where the stress from rapid lithium-ion insertion/extraction cannot be entirely relieved, leading to continuous particle expansion and repeated SEI film reconstruction.51 As reactions stabilize, the expansion tends to become steady, correlating with the capacity increase in the initial cycles. Fig. S18† illustrates that the Si core of SiQDs is smooth and tightly encapsulated by the carbon layer with no electrolyte intrusion, while the SEI film is formed on the outside of the carbon layer.
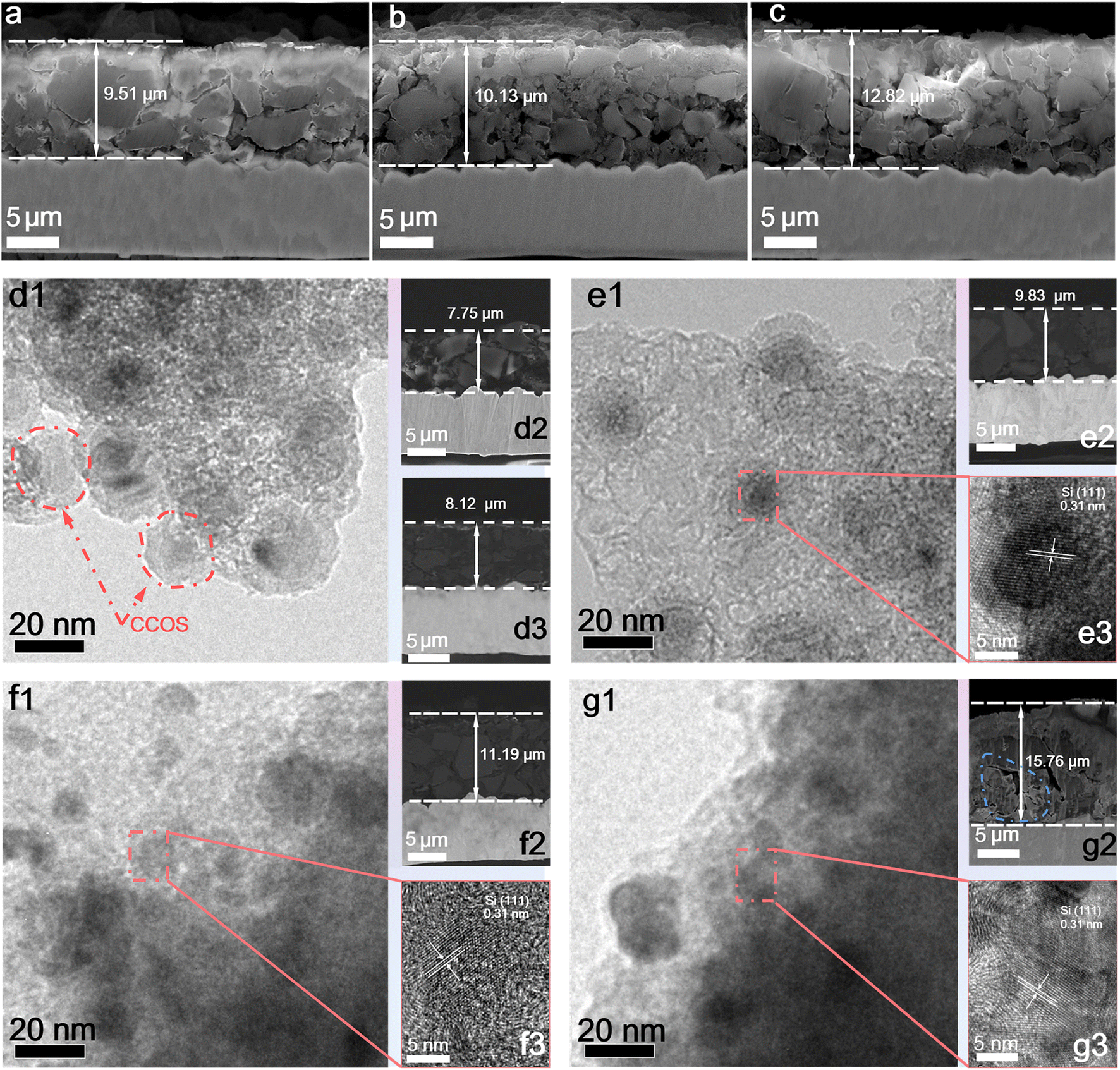 | ||
| Fig. 5 Cross-section images of the CCOS electrode after 100 cycles at (a) 80% SOC, (b) 90% SOC and (c) 105% SOC. (d1) TEM image before cycling and cross-section images of the electrode (d2) before cycling and (d3) after 5 cycles at 0.2 A g−1. TEM images, cross-section images, and HRTEM images of CCOS after (e) 100 cycles and (f) 300 cycles at 1.0 A g−1, and (g) 500 cycles at 5.0 A g−1. | ||
Moreover, HRTEM images of the CCOS electrode after 100 and 300 cycles at 1.0 A g−1 (Fig. 5e3 and f3) reveal that the internal SiQDs retain some smaller intact crystals compared to the pristine SiQDs, demonstrating a well-preserved morphology and structure. TEM images of completely delithiated SiNP electrodes reveal no discernible lattice patterns (Fig. S19a–c†), indicating complete structural disorder. Besides, the interior SiQDs in CCOS also become amorphous during lithiation without discernible lattice patterns (Fig. S19e and f†), which corresponds to the formation of the intermediate LixSi alloy. However, after complete delithiation, the CCOS particles maintain an ordered internal Si lattice (Fig. S19g†). The ordered lattice patterns observed are the result of recrystallization during the dealloying (delithiation) phase of Si, facilitated by this unique multidimensional encapsulation design. The hard carbon and SiOx layers stabilize the SiQDs, ensuring the uniform Li+ concentration and compressive stress, which can promote homogeneous LixSi nucleation and facilitate Si atom migration. This process results in a compact structure with nanocrystalline clusters during delithiation, overcoming the kinetic barriers to form ordered crystalline silicon clusters (5–10 nm).27,52 Besides, it suppresses the volume changes in Si particles during cycling and offers superior Li+ diffusion kinetics with lower electrode expansion compared to SiNP electrodes. The uniform gaps observed in the outer layer of the material particles result from the isotropic encapsulation, effectively relieving expansion stress. When the mechanical stress generated by lithiation exceeds the bonding strength between the encapsulation layer and SiQDs, fractures occur, creating voids and cracks. After 500 cycles at 5.0 A g−1 (Fig. 5g), the expansion of SiQDs becomes more extensive at high rates, with voids and cracks appearing, but the electrode structure remains robust for continued cycling. Moreover, even under high-rate conditions, ordered Si crystal clusters can still form (Fig. S20a and d†). However, the uneven lithium concentration distribution induces uneven volume expansion, which compromises the integrity of the SiOx and HC encapsulation layers. This damage results in insufficient compressive stress from the encapsulation layers to adequately support Si atoms in the LixSi phase to overcome the crystallization activation energy barrier, thereby reducing the formation of ordered crystal clusters compared to low-rate conditions.52
Conclusions
In summary, this gradient and mechanically strength ordered multidimensional encapsulation strategy effectively alleviates the volume expansion and balances lithium-ion concentration gradients of Si during (de)lithiation. The SiQD core enhances surface activity, shortens lithium-ion transport paths, and ensures efficient ion diffusion. Electrochemical investigations show that the CCOS electrode maintains rapid lithium-ion diffusion and uniform Li+ concentration even at high rates. This impressive lithium storage behavior is attributed to the encapsulation strategy with a QD core, leveraging their size and surface modification benefits. Benefiting from the abundant closed-pore structure in the outermost CPC layer, it not only isolates electrolyte contact with the SiQD core but also provides additional lithium-ion reaction sites. As a result, the composite material (CCOS) maintains 70.50% capacity retention and 640.4 mA h g−1 specific capacity after 500 cycles at 5.0 A g−1, demonstrating superior fast-charging capability and structural stability. These results affirm that the integrated SiOx, HC, and CPC layers, combined with the SiQD core, significantly enhance the performance of Si-based anodes by alleviating volume effects and optimizing lithium-ion transport. This study expands the potential for performance improvement by offering new design principles for balancing long lifespan, fast charging, and high energy density through a specialized encapsulation structure.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Yun Yu: experimental work, writing original draft, writing – review & editing; Haiqiang Gong: some experimental data analysis; Xinyou He: COMSOL simulation; Lei Ming: supervision, writing – review & editing, funding acquisition; Xiaowei Wang: writing – review & editing, funding acquisition; Xing Ou: supervision, writing-review & editing, funding acquisition. All the authors discussed the experimental results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52202338 and 52070194), the Natural Science Foundation of Hunan Province (2022JJ20069), and the Science and Technology Innovation Program of Hunan Province (2023RC3041).Notes and references
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich and S. Lambert, Nature, 2019, 575, 75–86 CrossRef CAS PubMed.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- G. G. Eshetu, H. Zhang, X. Judez, H. Adenusi, M. Armand, S. Passerini and E. Figgemeier, Nat. Commun., 2021, 12, 5459 CrossRef CAS PubMed.
- J. bSung, N. Kim, J. Ma, J. H. Lee, S. H. Joo, T. Lee, S. Chae, M. Yoon, Y. Lee and J. Hwang, Nat. Energy, 2021, 6, 1164–1175 CrossRef.
- S. Chae, S. Park, K. Ahn, G. Nam, T. Lee, J. Sung, N. Kim and J. Cho, Energy Environ. Sci., 2020, 13, 1212–1221 RSC.
- C. Cao, I. I. Abate, E. Sivonxay, B. Shyam, C. Jia, B. Moritz, T. P. Devereaux, K. A. Persson, H. G. Steinrück and M. F. Toney, Joule, 2019, 3, 762–781 CrossRef CAS.
- Y. Jin, B. Zhu, Z. Lu, N. Liu and J. Zhu, Adv. Energy Mater., 2017, 7, 1700715 CrossRef.
- Y. Ren, L. Xiang, X. Yin, R. Xiao, P. Zuo, Y. Gao, G. Yin and C. Du, Adv. Funct. Mater., 2022, 32, 2110046 CrossRef CAS.
- M. Aghajamali, H. Xie, M. Javadi, W. P. Kalisvaart, J. M. Buriak and J. G. Veinot, Chem. Mater., 2018, 30, 7782–7792 CrossRef CAS.
- H. Zhao, J. Li, Q. Zhao, X. Huang, S. Jia, J. Ma and Y. Ren, Electrochem. Energy Rev., 2024, 7, 11 CrossRef CAS.
- J. Tao, L. Lu, B. Wu, X. Fan, Y. Yang, J. Li, Y. Lin, Y. Y. Li, Z. Huang and J. Lu, Energy Storage Mater., 2020, 29, 367–376 CrossRef.
- H. Wang, M. Miao, H. Li, Y. Cao, H. Yang and X. Ai, ACS Appl. Mater. Interfaces, 2021, 13, 22505–22513 CrossRef CAS PubMed.
- M. Khan, S. Yan, M. Ali, F. Mahmood, Y. Zheng, G. Li and Y. Wang, Nano-Micro Lett., 2024, 16, 179 CrossRef CAS PubMed.
- Y. An, Y. Tian, C. Liu, S. Xiong, J. Feng and Y. Qian, ACS Nano, 2022, 16, 4560–4577 CrossRef CAS PubMed.
- Y. Chen, L. Liu, J. Xiong, T. Yang, Y. Qin and C. Yan, Adv. Funct. Mater., 2015, 25, 6701–6709 CrossRef CAS.
- W. Zeng, L. Wang, X. Peng, T. Liu, Y. Jiang, F. Qin, L. Hu, P. K. Chu, K. Huo and Y. Zhou, Adv. Funct. Mater., 2018, 8, 1702314 Search PubMed.
- M. Ge, C. Cao, G. M. Biesold, C. D. Sewell, S. M Hao, J. Huang, W. Zhang, Y. Lai and Z. Lin, Adv. Mater., 2021, 33, 2004577 CrossRef CAS PubMed.
- M. Y. Yan, G. Li, J. Zhang, Y. F. Tian, Y. X. Yin, C. J. Zhang, K. C. Jiang, Q. Xu, H. L. Li and Y. G. Guo, ACS Appl. Mater. Interfaces, 2020, 12, 27202–27209 CrossRef CAS PubMed.
- H. Li, Z. Chen, Z. Kang, W. Liu and Y. Chen, Energy Storage Mater., 2023, 56, 40–49 CrossRef.
- Y. Lu, Z. Ye, Y. Zhao, Q. Li, M. He, C. Bai, X. Wang, Y. Han, X. Wan, S. Zhang, Y. Ma and Y. Chen, Carbon, 2023, 201, 962–971 CrossRef CAS.
- J. Tao, Z. Yan, J. Yang, J. Li, Y. Lin and Z. Huang, Carbon Energy, 2022, 4, 129–141 CrossRef CAS.
- R. Yu, Y. Pan, Y. Jiang, L. Zhou, D. Zhao, G. Van Tendeloo, J. Wu and L. Mai, Adv. Mater., 2023, 35, 2306504 CrossRef CAS PubMed.
- B. Jiang, S. Zeng, H. Wang, D. Liu, J. Qian, Y. Cao, H. Yang and X. Ai, ACS Appl. Mater. Interfaces, 2016, 8, 31611–31616 CrossRef CAS PubMed.
- B. Wang, X. Li, B. Luo, L. Hao, M. Zhou, X. Zhang, Z. Fan and L. Zhi, Adv. Mater., 2015, 27, 1526–1532 CrossRef CAS PubMed.
- F. Wang, G. Chen, N. Zhang, X. Liu and R. Ma, Carbon Energy, 2019, 1, 219–245 CrossRef CAS.
- F. Fu, X. Wang, L. Zhang, Y. Yang, J. Chen, B. Xu, C. Ouyang, S. Xu, F. D and W. E, Adv. Funct. Mater., 2023, 33, 2303936 CrossRef CAS.
- Z. Liu, R. Hu, R. Yu, M. Zheng, Y. Zhang, X. Chen, L. Shen and Y. Xia, Nano Lett., 2024, 24, 1908–4916 Search PubMed.
- Q. Meng, Y. Lu, F. Ding, Q. Zhang, L. Chen and Y. S. Hu, ACS Energy Lett., 2019, 4, 2608–2612 CrossRef CAS.
- J. Ryu, T. Bok, H. Joo, S. Yoo, G. Song, H. Kim, S. Choi, H. Y. Jeong, M. G. Kim and S. Park, Energy Storage Mater., 2021, 36, 139–146 CrossRef.
- Z. Zhang, H. Ying, P. Huang, S. Zhang, Z. Zhang, T. Yang and W. Q. Han, Chem. Eng. J., 2023, 451, 138785 CrossRef CAS.
- F. Hong, R. Zhou, C. Gao, Y. Liu, Z. Sun and Y. Jiang, J. Alloys Compd., 2023, 947, 169511 CrossRef CAS.
- J. Zhang, S. Li, F. Xi, X. Wan, Z. Ding, Z. Chen, W. Ma and R. Deng, Chem. Eng. J., 2022, 447, 137563 CrossRef CAS.
- X. Xue, B. Lou, C. Wu, W. Pang, J. Zhang, N. Shi, Z. Men, F. Wen, X. Yang, J. Wu, L. Tian and D. Liu, Chem. Eng. J., 2024, 488, 150936 CrossRef CAS.
- S. Ji, R. Song, H. Yuan, D. Lv, L. Yang, J. Luan, D. Wan, J. Liu and C. Zhong, J. Electroanal. Chem., 2024, 959, 118141 CrossRef CAS.
- D. Yang, M. Chen, R. Han, Y. Luo, H. Li, Z. Kang, Y. Chen, J. Fu, N. Iqbal and W. Liu, Nano Energy, 2024, 127, 61109744 Search PubMed.
- A. Vu, Y. Qian and A. Stein, Adv. Energy Mater., 2012, 2, 1056–1085 CrossRef CAS.
- J. Hong, J. Zhang, X. Li, Y. F. Guo, X. F. Zhou and Z. P. Liu, ACS Appl. Nano Mater., 2023, 6, 10138–10147 CrossRef CAS.
- Q. Shi, J. Zhou, S. Ullah, X. Yang, K. Tokarska, B. Trzebicka, H. Q. Ta and M. H Rümmeli, Energy Storage Mater., 2021, 34, 735–754 CrossRef.
- Z. Li, M. Han, P. Yu, Q. Wu, Y. Zhang and J. Yu, J. Energy Storage, 2023, 67, 107582 CrossRef.
- S. Huang, L. Liu, Y. Zheng, Y. Wang, D. Kong, Y. Zhang, Y. Shi, L. Zhang, O. G. Schmidt and H. Y. Yang, Adv. Mater., 2018, 30, 1706637 CrossRef PubMed.
- L. Hu, M. Jin, Z. Zhang, H. Chen, F. Boorboor Ajdari and J. Song, Adv. Funct. Mater., 2022, 32, 2111560 CrossRef CAS.
- C. Zhong, S. Weng, Z. Wang, C. Zhan and X. Wang, Nano Energy, 2023, 117, 108894 CrossRef CAS.
- H. Q. Gong, X. Y. Wang, L. Ye, B. Zhang and X. Ou, Tungsten, 2024, 6, 574–584 CrossRef.
- X. Chen, C. Chen, Y. Zhang, X. Zhang, D. Yang and A. Dong, Nano Res., 2019, 12, 631–636 CrossRef CAS.
- D. Liu, C. Chen, Y. Hu, J. Wu, D. Zheng, Z. Xie, G. Wang, D. Qu, J. Li and D. Qu, Electrochim. Acta, 2018, 273, 26–33 CrossRef CAS.
- M. Choi, E. Lee, J. Sung, N. Kim and M. Ko, Nano Res., 2024, 17, 5270–5277 CrossRef CAS.
- L. Xie, C. Tang, Z. Bi, M. Song, Y. Fan, C. Yan, X. Li, F. Su, Q. Zhang and C. Chen, Adv. Energy Mater., 2021, 11, 2101650 CrossRef CAS.
- X. Wang, B. Zhang, Z. Xiao, L. Ming, M. Li, L. Cheng and X. Ou, Chin. Chem. Lett., 2023, 34, 107772 CrossRef CAS.
- L. Gu, J. Han, M. Chen, W. Zhou, X. Wang, M. Xu, H. Lin, H. Liu and J. Chen, Energy Storage Mater., 2022, 52, 547–561 CrossRef.
- M. Zhao, J. Zhang, X. Zhang, K. Duan and H. Dong, Energy Storage Mater., 2023, 61, 102857 CrossRef.
- X. Zhang, D. Kong, X. Li and L. Zhi, Adv. Funct. Mater., 2019, 29, 1806061 CrossRef.
- L. Li, C. Fang, W. Wei, L. Zhang, Z. Ye, G. He and Y. Huang, Nano Energy, 2020, 72, 104651 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04751f |
| This journal is © The Royal Society of Chemistry 2024 |