
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoconfined tandem three-phase photocatalysis for highly selective CO2 reduction to ethanol†
Hailing
Huo
a,
Ting
Hu
 a,
Zhiqing
Zhong
a,
Cheng
Zhan
a,
Chengxi
Huang
a,
Qiang
Ju
a,
Liang
Zhang
a,
Fang
Wu
b,
Erjun
Kan
*a and
Ang
Li
*a
a,
Zhiqing
Zhong
a,
Cheng
Zhan
a,
Chengxi
Huang
a,
Qiang
Ju
a,
Liang
Zhang
a,
Fang
Wu
b,
Erjun
Kan
*a and
Ang
Li
*a
aMIIT Key Laboratory of Semiconductor Microstructure and Quantum Sensing, Engineering Research Center of Semiconductor Device Optoelectronic Hybrid Integration in Jiangsu Province, Nanjing University of Science and Technology, Nanjing 210094, P. R. China. E-mail: liang2100@njust.edu.cn; ekan@njust.edu.cn
bCollege of Information Science and Technology, Nanjing Forestry University, Nanjing 210037, P. R. China
First published on 28th August 2024
Abstract
The conversion of CO2 and H2O into ethanol with high selectivity via photocatalysis is greatly desired for effective CO2 resource utilization. However, the sluggish and challenging C–C coupling hinders this goal, with the behavior of *CO holding the key. Here, a nanoconfined and tandem three-phase reaction system is established to simultaneously enhance the *CO concentration and interaction time, achieving an outstanding ethanol selectively of 94.15%. This system utilizes a tandem catalyst comprising an Ag core and a hydrophobic Cu2O shell. The hydrophobic Cu2O shell acts as a CO2 reservoir, effectively overcoming the CO2 mass-transfer limitation, while the Ag core facilitates the conversion of CO2 to CO. Subsequently, CO undergoes continuous reduction within the nanoconfined mesoporous channels of Cu2O. The synergy of enhanced mass transfer, nanoconfinement, and tandem reaction leads to elevated *CO concentrations and prolonged interaction time within the Cu2O shell, significantly reducing the energy barrier for *CO–*CO coupling compared to the formation of *CHO from *CO, as determined by density functional theory calculations. Consequently, C–C coupling preferentially occurs over *CHO formation, producing excellent ethanol selectivity. These findings provide valuable insights into the efficient production of C2+ compounds.
Introduction
The urgent need for sustainable development in response to the challenges posed by the greenhouse effect and dwindling fossil resources has led to the exploration of eco-friendly strategies.1,2 Among these strategies, the photocatalytic CO2 reduction reaction (PCRR) has emerged as a promising approach for converting CO2 and H2O into chemicals and fuels.3,4 During the PCRR, solar energy is transformed into chemical energy, and stored in the chemical bonds of resulting compounds containing one (C1) or multiple (C2+) carbon atoms. Importantly, the production of C2+ products is particularly desirable due to their higher energy density and economic potential.5 Furthermore, ethanol, a liquid C2 fuel, has garnered significant research attention owing to its ease of storage and transport.6 Nevertheless, ethanol production faces challenges in terms of low selectivity, largely attributed to the challenging C–C coupling process necessary for generating C2+ products.7The C–C coupling process, which is sluggish and possesses a high energy barrier, is significantly influenced by the adsorbed carbon monoxide (*CO) on the catalyst surface.7,8 Typically, following the conversion of a CO2 molecule to *COOH, the resulting *CO governs the direction of the reaction towards the production of either C1 compounds (e.g., methanol) or C2+ compounds (e.g., ethanol).9 Specifically, the hydrogenation of *CO into its derived intermediates, such as *CHO, facilitates the formation of C1 products, while the dimerization of *CO or the polymerization of *CO and its derived intermediates to proceed with C–C coupling initiates the C2 pathway.9 Furthermore, according to Le Chatelier's principle, an elevated concentration of *CO is generally beneficial for the PCRR10 and the C–C coupling process.11 Since *CO is a downstream intermediate of CO2 protonation, researchers have strived to increase the surface *CO concentration to promote the formation of C2+ products by enhancing the local concentration of CO2.8,12 However, in aqueous solutions, the PCRR encounters the mass-transfer limitation of CO2 due to its poor solubility and diffusion coefficient, resulting in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1300 ratio between CO2 and H2O molecules at 1 atm pressure. This limitation further leads to a significant scarcity of diffusing CO2 molecules reaching the catalyst surface.13 Researchers have proposed that this mass-transfer limitation can be overcome by adjusting the catalyst surface to be hydrophobic, which can promote the enrichment of gaseous CO2 molecules on the catalyst surface. This adjustment creates abundant three-phase (gaseous CO2, liquid water, and solid catalysts) reaction sites for enhanced PCRR.13,14 Our previous study has developed a hydrophobic cuprous oxide (Cu2O) hollow structure to mitigate the CO2 mass-transfer limitation, achieving enhanced ethanol production.5 Nonetheless, its selectivity remains unsatisfactory. One possible reason is the limited increase in *CO concentration, while another could be the insufficient interaction time between C1 intermediates necessary for effective C–C coupling, which could not align with the sluggishness of this process.15
:1300 ratio between CO2 and H2O molecules at 1 atm pressure. This limitation further leads to a significant scarcity of diffusing CO2 molecules reaching the catalyst surface.13 Researchers have proposed that this mass-transfer limitation can be overcome by adjusting the catalyst surface to be hydrophobic, which can promote the enrichment of gaseous CO2 molecules on the catalyst surface. This adjustment creates abundant three-phase (gaseous CO2, liquid water, and solid catalysts) reaction sites for enhanced PCRR.13,14 Our previous study has developed a hydrophobic cuprous oxide (Cu2O) hollow structure to mitigate the CO2 mass-transfer limitation, achieving enhanced ethanol production.5 Nonetheless, its selectivity remains unsatisfactory. One possible reason is the limited increase in *CO concentration, while another could be the insufficient interaction time between C1 intermediates necessary for effective C–C coupling, which could not align with the sluggishness of this process.15
Herein, a nanoconfined and tandem three-phase reaction system is constructed, simultaneously improving the *CO concentration and interaction time between C1 intermediates, thereby elevating the selectivity of ethanol production to 94.15%. Specifically, the tandem three-phase PCRR employs a tandem catalyst comprising silver (Ag) particles covered by a mesoporous shell of hydrophobic Cu2O. It is well-known that Ag exhibits high activity and selectivity for CO formation in the CO2 reduction reaction,16,17 with a relatively weak CO binding strength on its surface.18 Moreover, Cu2O contains CuI centers, which are considered effective catalytic sites for CO2 conversion,19 facilitating CO adsorption and enhancing *CO binding for subsequent coupling reactions with protons and electrons.20,21 Thus, when this tandem catalyst is constructed in a core–shell structure, denoted as O-Ag@Cu2O, the surface of Ag particles serves as the reactive sites for converting CO2 to CO, constituting the first step of the tandem reaction, as depicted in Scheme 1a.17 Then, the generated CO desorbs from the Ag particles, diffuses along the mesoporous channels, and re-adsorbs onto the hydrophobic Cu2O reactive sites for subsequent reduction, constituting the second step of the tandem reaction. During the reaction process, the hydrophobic Cu2O surface facilitates the capture and enrichment of CO2 molecules, ensuring a concentrated source of CO2 for efficient transport to the reactive sites. Upon exposure to visible light, the high concentration of CO2 can undergo direct reduction on the Cu2O surface. Moreover, CO2 can diffuse through the mesoporous Cu2O shell to interact with the inner Ag particles, where it is converted to CO (Scheme 1a). As CO diffuses outward along the mesoporous channels (Scheme 1b), the nanoconfinement effect of these channels increases the likelihood of collisions between CO molecules and the channel walls, as well as among the CO molecules themselves.22 This increased collision probability raises diffusion resistance, thereby extending the residence time of CO. This phenomenon not only leads to the aggregation of CO within the channels, increasing the *CO concentration, but also provides sufficient time for *CO molecules to interact with each other. Consequently, the designed system achieves a synergistic effect of mass transfer enhancement, nanoconfinement, and tandem reaction, resulting in high *CO concentrations at active sites and prolonged interaction time. Density functional theory (DFT) calculations demonstrate that the elevated *CO concentration significantly lowers the energy barrier for *CO–*CO coupling compared to the formation of *CHO from *CO. This results in the preferential formation of C2+ products over C1 products, thereby enhancing ethanol selectivity. This strategy achieves effective CO2-to-ethanol conversion with high selectivity and offers guidance for producing C2+ products efficiently.
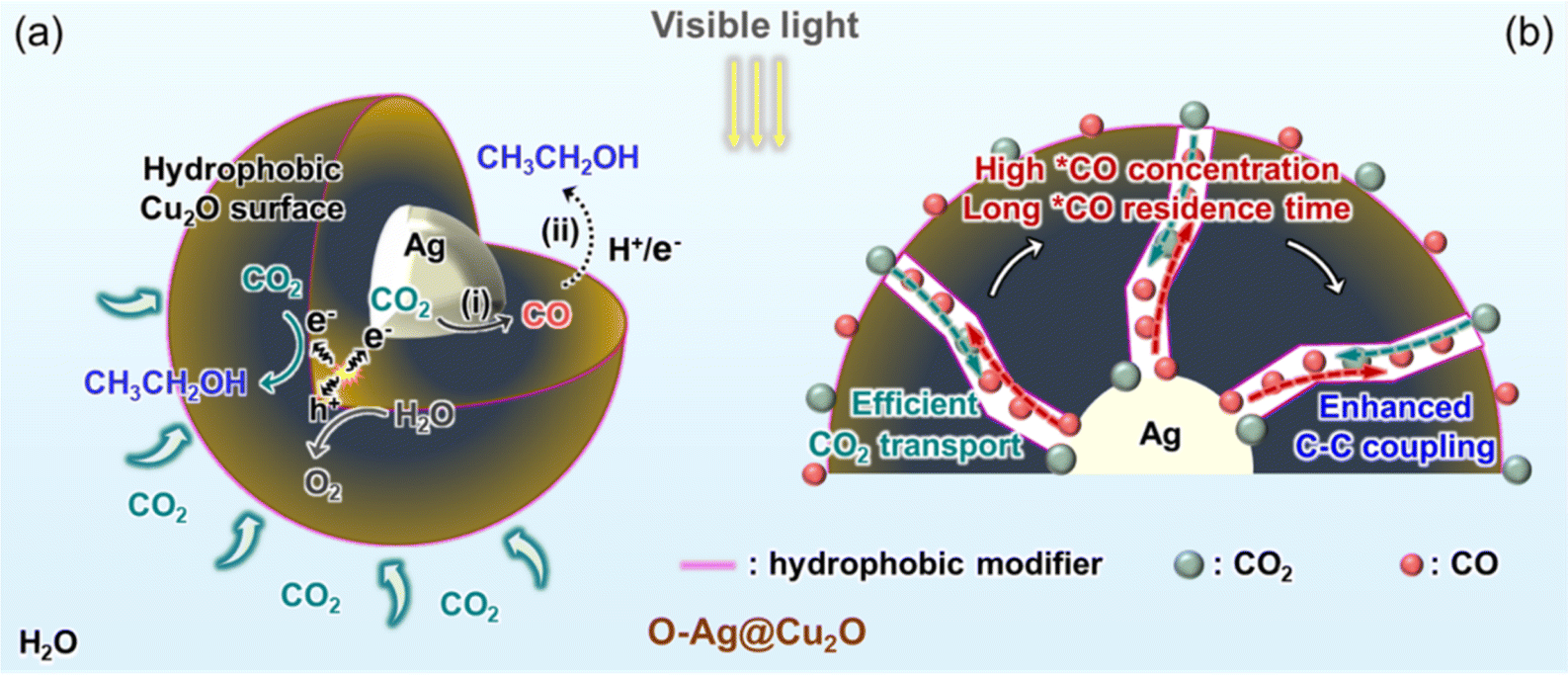 | ||
| Scheme 1 Photocatalytic CO2 reduction reaction (PCRR) over O-Ag@Cu2O. (a) PCRR mechanisms at the active sites of Ag and hydrophobic Cu2O surfaces. The hydrophobic surface serves as a reservoir for CO2 molecules, where they can also undergo reduction. (i) and (ii) Refer to the first and second steps of the tandem reaction, respectively. (b) Gas transport processes within the mesoporous channels. The hydrophobic Cu2O channels facilitate the transportation of CO2 to the inner Ag surface, where it is converted into CO. The nanoconfinement effect of the channels results in an extended residence time for CO. | ||
Results and discussion
Morphological characterization
The synthesis of O-Ag@Cu2O commences with the preparation of Ag particles derived from a silver chloride (AgCl) colloid. These prepared Ag particles are nearly spherical, with an average size of approximately 40 nm (Fig. S1†). Subsequently, the Ag particles are coated with Cu2O to establish the core–shell structure of Ag@Cu2O (Fig. S2a†). Following this, Ag@Cu2O is hydrophobically modified using minute quantities of 1-dodecanethiol (DDT) to form the target catalyst O-Ag@Cu2O (Fig. S2b†). To assess the impact of hydrophobicity on O-Ag@Cu2O, Ag@Cu2O is utilized as a control sample. Additionally, to assess the role of Ag particles within the catalyst, bare Cu2O (denoted as b-Cu2O) and its hydrophobically modified counterpart (denoted as O-b-Cu2O) are synthesized for comparison (Fig. S2c and d†). Furthermore, to investigate the impact of nanoconfinement effects in O-Ag@Cu2O, the catalyst with Ag particles loaded onto the external surface of Cu2O (denoted as Ag/Cu2O) and its hydrophobic variant (denoted as O-Ag/Cu2O) are prepared as control samples (Fig. S2e and f†). In this case, the spatial distribution of Ag particles on the external surface of Cu2O means that the CO generated on the surface of Ag particles does not need to diffuse out through the mesoporous Cu2O. Therefore, the nanoconfinement effect of the mesoporous channel does not impact the generated CO.The successful synthesis of the Ag@Cu2O structure is evident from the transmission electron microscope (TEM) images (Fig. 1a and S3†). The core–shell configuration of Ag@Cu2O, with Ag serving as the core and mesoporous Cu2O as the shell, exhibits a water contact angle (WCA) of 34.83°, indicative of its inherent hydrophilicity. Additionally, the mesoporous characteristic of the Cu2O shell is substantiated by the N2 adsorption–desorption measurement, which displays a type IV curve with an H3 hysteresis loop (Fig. S4a†). Following DDT modification, the core–shell structure of O-Ag@Cu2O remains intact. Notably, a transition to hydrophobic behavior is observed, marked by a significant rise in the WCA to 132.53° (Fig. 1b). The magnified TEM image of O-Ag@Cu2O further illustrates its mesoporous structure (Fig. 1c), featuring a pore diameter of approximately 19 nm (Fig. S4b†). Compared to microporous structures, which tend to impede molecular transport due to their smaller pore sizes (<2 nm),23 this remarkable mesoporous structure facilitates the mass transport of molecules.22 Moreover, the lattice fringe of 0.246 nm shown in Fig. 1d aligns with the (111) plane of Cu2O, suggesting the Cu2O composition of the shell. Furthermore, the energy-dispersive X-ray spectroscopy (EDS) mapping and line scan results presented in Fig. 1e–k verify the spatial distribution relationship between Ag and Cu2O within the core–shell structure of O-Ag@Cu2O.
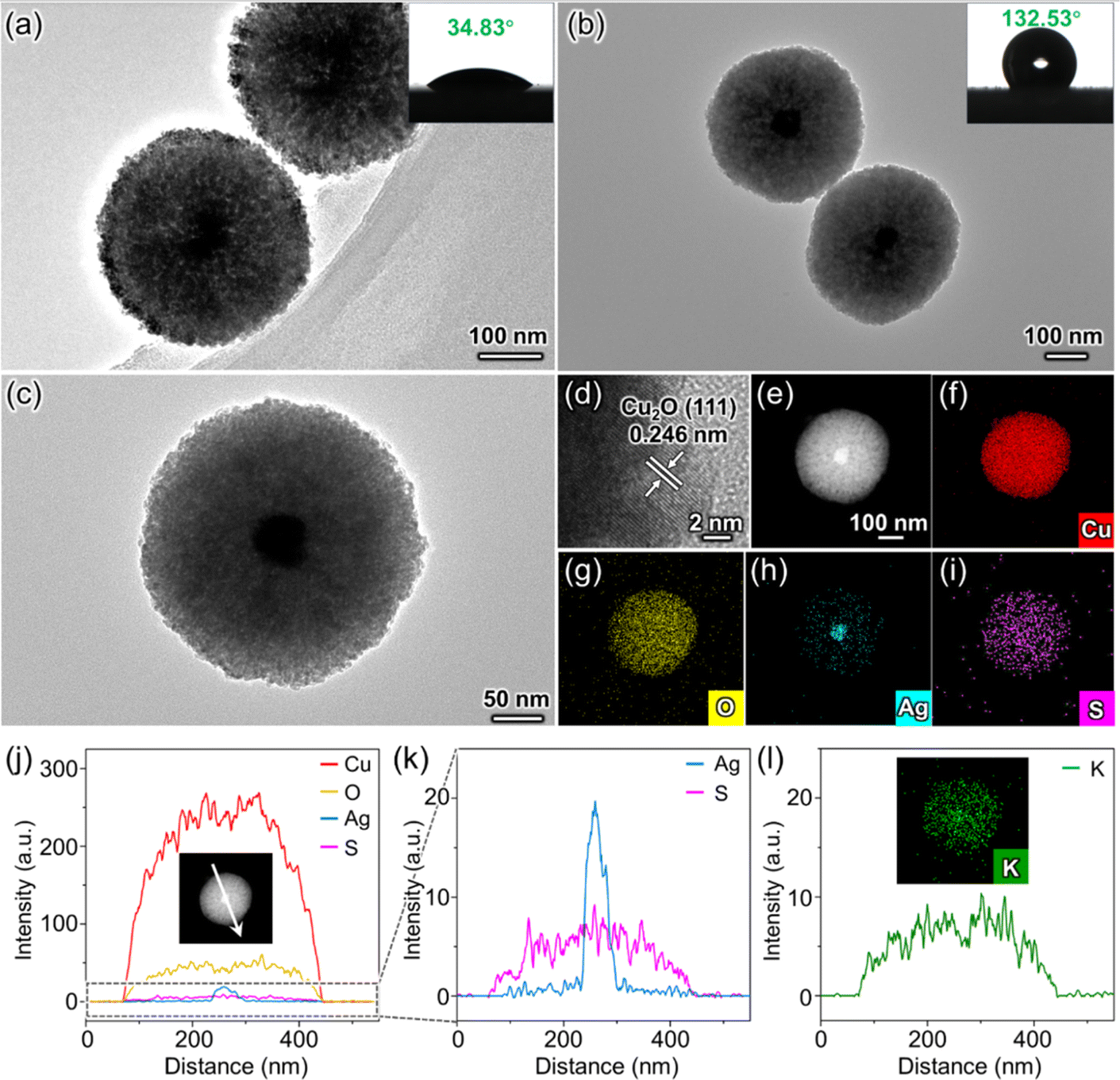 | ||
| Fig. 1 (a and b) TEM images of Ag@Cu2O and O-Ag@Cu2O, respectively. Insets in (a and b) show the WCAs of the corresponding samples. (c) TEM image of a single O-Ag@Cu2O particle. (d) HRTEM image of O-Ag@Cu2O. (e) HAADF-STEM image of O-Ag@Cu2O and corresponding EDS mapping profiles of Cu (f), O (g), Ag (h), and S (i). (j) EDS line scan of O-Ag@Cu2O. The inset displays the position and direction of the line scan. (k) Enlarged view of the region outlined by the dashed box in (j). (l) EDS line scan and mapping results from a K+ tracing experiment. The line scan position and direction are the same as that in the inset of (j). | ||
More importantly, the EDS line scan result of sulfur (S) element originating from DDT shows that DDT is evenly distributed throughout the entire mesoporous structure of O-Ag@Cu2O (Fig. 1k). This observation suggests that DDT permeates the mesoporous channels to some extent, increasing the hydrophobicity of the channels, thereby facilitating the CO2 transport within them. To assess whether the presence of DDT in mesoporous channels impedes water infiltration, a potassium ion (K+) tracing experiment was conducted (for details see Experimental Procedures in ESI†).24 The experimental procedure involved immersing the sample in a potassium bicarbonate solution and subsequently analyzing the distribution pattern of K+. EDS mapping combined with EDS line scan results clearly illustrate that K+ is evenly distributed throughout the entire sphere, indicating that water is capable of penetrating the mesoporous channels. This observation ensures the accessibility of subsequent CO2 and water molecules into the sphere through the mesoporous channels, where they can react on the surface of Ag particles. To further elucidate that the presence of DDT on the surface of O-Ag@Cu2O does not hinder the migration of charge carriers to the catalyst surface, Ag+ probe experiments for O-Ag@Cu2O and Ag@Cu2O are compared (Fig. S5 and S6,† for details see Experimental Procedures in ESI†).5 The atomic percentages of Cu and Ag elements in O-Ag@Cu2O (Fig. S5†) are found to be comparable to those in Ag@Cu2O (Fig. S6†), indicating that the quantity of Ag resulting from the reduction of adsorbed Ag+ on O-Ag@Cu2O surface by photogenerated electrons is analogous to that on Ag@Cu2O surface. Therefore, the surface hydrophobic modification does not significantly affect the interaction between photogenerated electrons migrating to the surface and reactants adsorbed on the surface. To further explore the impact of different synthesis scales on O-Ag@Cu2O and to preliminarily assess the potential for large-scale production, TEM and WCA characterizations were performed on samples obtained at various scales (Fig. S7†). The results reveal no significant differences in morphology or hydrophobicity across the different scales, indicating that the designed catalyst may be suitable for scaling up. This finding provides a basis for considering future large-scale production.
Additionally, control samples including Ag/Cu2O, O-Ag/Cu2O, b-Cu2O, and O-b-Cu2O are successfully synthesized and confirmed through TEM and WCA characterizations (Fig. S8 and S9†). Moreover, the comparable specific surface areas and pore diameter distributions for the synthesized samples render the variation in specific surface areas and pore size negligible (Fig. S4 and S10†). In addition, the Ag contents (wt%) in O-Ag@Cu2O and O-Ag/Cu2O are quantified as 2.032% and 2.027%, respectively, using inductively coupled plasma-optic emission spectroscopy (Table S1†). This similarity in Ag content between the two samples enables a comparison of the effects of distinct spatial distributions of Ag particles without the complicating factor of varying amounts of Ag.
Structural characterization
X-ray diffraction (XRD) analysis unveils the composition of the synthesized samples, showcasing a well-matched cubic phase Cu2O (JCPDS card no. 78-2076), alongside a weak peak corresponding to metallic Ag (Ag0) particles observed in O-Ag@Cu2O, Ag@Cu2O, O-Ag/Cu2O, and Ag/Cu2O (Fig. 2a). X-ray photoelectron spectroscopy (XPS) further confirms the chemical composition of the synthesized samples. The high-resolution Cu 2p XPS spectra of O-Ag@Cu2O, O-Ag/Cu2O, and O-b-Cu2O exhibit identical peak positions at 932.3 and 952.3 eV for Cu 2p3/2 and Cu 2p1/2, respectively (Fig. 2b), indicating consistent valence states of Cu (CuI), as confirmed by Cu LMM Auger spectra (Fig. S11a†).25 The weak peaks at 933.6 and 953.5 eV attributed to CuII indicate the slight oxidation of the surface Cu2O. Similarly, their corresponding samples without hydrophobic modification show comparable Cu 2p XPS spectra, indicating their similar composition (Fig. S11b†). Additionally, the XPS surveys of O-Ag@Cu2O, O-Ag/Cu2O, Ag@Cu2O, and Ag/Cu2O all exhibit discernible Ag signals (Fig. S11c and d†). In the cases of O-Ag@Cu2O and O-Ag/Cu2O, the high-resolution Ag 3d XPS spectra display consistent peak positions, indicating a uniform chemical state of Ag across these catalysts. Furthermore, the binding energies for Ag 3d5/2 and Ag 3d3/2 are located at 368.1 and 374.1 eV, respectively (Fig. 2c), consistent with the characteristic signals of Ag0 and in agreement with the XRD results. In the cases of Ag@Cu2O and Ag/Cu2O, although the Ag 3d peak positions are consistent, indicating the uniform chemical state of Ag in these two catalysts, the binding energies are slightly shifted to 368.3 eV for Ag 3d5/2 and 374.3 eV for Ag 3d3/2 (Fig. S11e†). While the signals typical for AgI are at 368.4 eV for Ag 3d5/2 and 374.4 eV for Ag 3d3/2,26 this suggests a coexistence of Ag0 and AgI on the surface of Ag particles in Ag@Cu2O and Ag/Cu2O,27 implying a slight oxidation of the Ag particles for Ag@Cu2O and Ag/Cu2O. Additionally, as demonstrated in Fig. S12,† DDT exhibits the ability to interact with Ag particles. Consequently, when DDT is utilized to modify Ag@Cu2O and Ag/Cu2O, AgI on the Ag particle surface may be reduced due to the reducibility of DDT,28 leading to the conversion of the chemical state of Ag in O-Ag@Cu2O and O-Ag/Cu2O to Ag0. This finding further supports the notion that DDT can penetrate the mesoporous Cu2O shell in O-Ag@Cu2O, thereby accessing the surface of internal Ag particles.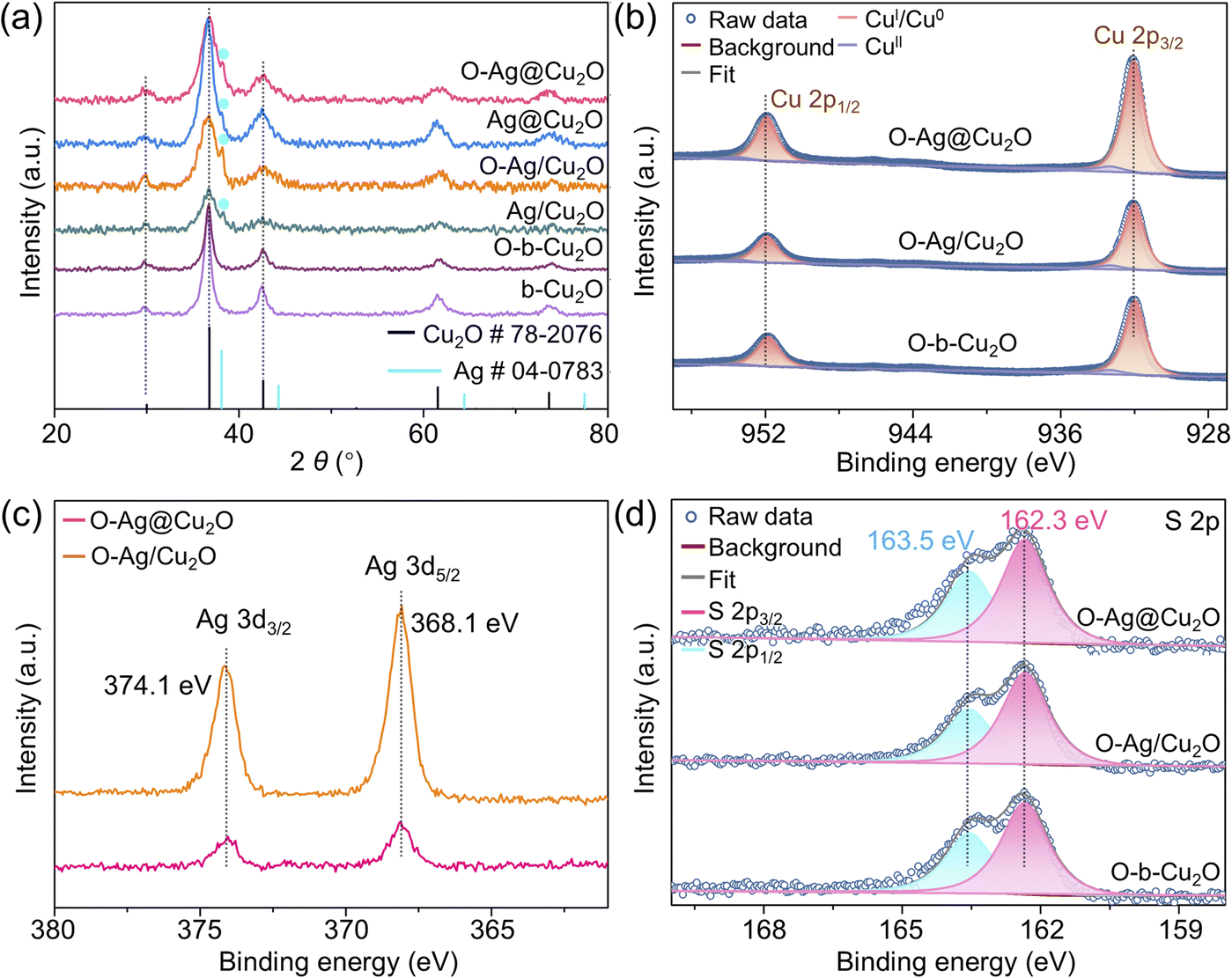 | ||
| Fig. 2 Structural characterizations. (a) XRD patterns of b-Cu2O, O-b-Cu2O, Ag/Cu2O, O-Ag/Cu2O, Ag@Cu2O, and O-Ag@Cu2O. (b) High-resolution Cu 2p XPS spectra of O-Ag@Cu2O, O-Ag/Cu2O, and O-b-Cu2O. (c) High-resolution Ag 3d XPS spectra of O-Ag@Cu2O and O-Ag/Cu2O. (d) High-resolution S 2p XPS spectra of O-Ag@Cu2O, O-Ag/Cu2O, and O-b-Cu2O. | ||
Additionally, S signals are detected in the XPS surveys of O-Ag@Cu2O, O-Ag/Cu2O, and O-b-Cu2O (Fig. S11d†), indicating the presence of DDT on the catalyst surface, consistent with observations from Fourier-transform infrared spectroscopy (FTIR) (Fig. S13†). Importantly, the high-resolution S 2p XPS spectra display consistent peak positions, suggesting a comparable state of DDT across the surfaces of all three samples (Fig. 2d and S11f†). Furthermore, the binding energy of S 2p3/2 at 162.3 eV corresponds to surface thiolate species signals, implying the formation of –S–Cu by cleaving the sulfhydryl group (–S–H) in DDT.29 This observation indicates the occurrence of chemical adsorption between DDT and the Cu2O surface, which is further supported by Raman spectra (Fig. S14†). This chemical adsorption enables DDT to stably bind to the catalyst surface, preserving its stable hydrophobic properties essential for the catalytic reaction.
Subsequently, UV-vis diffuse reflectance spectroscopy was utilized to assess the light absorption ability and bandgap of the synthesized catalysts (Fig. S15†). All samples display a similar visible-light absorption range (Fig. S15a†), and the estimated bandgaps based on the Tauc plots are uniformly approximately 2.18 eV across all samples (Fig. S15b†). Additionally, after DDT modification, the absorbance intensity of the samples slightly decreases, which theoretically is unfavorable for the catalytic reaction.1 However, subsequent photocatalytic performance tests demonstrate that the DDT-modified samples generally perform better than their unmodified counterparts. This indicates that variations in the catalyst's absorbance intensity are not crucial factors contributing to the difference in performance. The charge separation efficiency is also compared by photoluminescence (PL) spectra and transient photocurrent response (Fig. S16†). As charge carrier recombination can release fluorescence, higher PL spectra intensity generally correlates with reduced charge separation efficiency.30 Moreover, in transient photocurrent response, lower transient photocurrent intensity is indicative of inferior charge separation efficiency.30 Remarkably, the PL and photocurrent intensity of O-Ag@Cu2O, Ag@Cu2O, O-Ag/Cu2O, and Ag/Cu2O do not differ significantly. Following DDT modification, a slight increase in PL intensity indicates a slight decrease in carrier separation efficiency, aligning with the results of photocurrent testing, which might have had some detrimental effects on catalyst activity.31 However, subsequent evaluation of photocatalytic performance reveals that the DDT-modified sample outperforms its unmodified counterpart. This discrepancy implies that the minor variation in carrier separation efficiency is not the primary determinant of the disparity in catalyst performance. Additionally, the decreased PL intensity and increased photocurrent intensity observed in O-Ag@Cu2O, O-Ag/Cu2O, Ag@Cu2O, and Ag/Cu2O compared to O-b-Cu2O and b-Cu2O suggest enhanced efficiency in charge separation. This improvement can be ascribed to the presence of Ag particles, which can trap the photogenerated electrons.32 As illustrated in Fig. S17,† when Ag particles come into contact with Cu2O, the difference in their Fermi levels induces a redistribution of interfacial charges.33 Under illumination, electrons in the conduction band of Cu2O at the interface ultimately transfer to the Ag particles, while the holes in the valence band remain within Cu2O.34,35 Consequently, the presence of Ag particles suppresses the recombination of electrons and holes in Cu2O to some extent, thereby enhancing their separation efficiency.
PCRR performance
The photocatalytic performance of O-Ag@Cu2O and relevant reference catalysts for CO2 reduction with H2O was evaluated in an aqueous solution under visible light (λ > 420 nm). As depicted in Fig. 3a, all Cu2O-based catalysts demonstrate activity in producing liquid alcohols. However, the C2 product of ethanol is the main carbonaceous product for O-Ag@Cu2O, with an ethanol generation rate of 450.19 μmol g−1 h−1 and an ethanol selectivity of up to 94.15%, as evident from the chromatogram of the original product detection (Fig. S18†). In terms of both ethanol selectivity and generation rate, this performance ranks among the top levels observed in photocatalytic CO2 reduction systems over the past five years (a detailed comparison is presented in Table S2†). In contrast, methanol is the predominant carbonaceous product for the reference catalysts of Ag@Cu2O, O-Ag/Cu2O, Ag/Cu2O, O-b-Cu2O, and b-Cu2O. Specifically, O-Ag@Cu2O exhibits a 10.89-fold increase in ethanol selectivity compared to Ag@Cu2O. Additionally, the calculation of effective photogenerated electrons utilized in the PCRR reveals that O-Ag@Cu2O employs a 6.65-fold greater amount of electrons compared to Ag@Cu2O, with a significant portion contributing to ethanol formation (Fig. 3b). These findings suggest the promotive effect of hydrophobicity on the PCRR. This effect can be further corroborated by comparing the photocatalytic performance of O-Ag/Cu2O and Ag/Cu2O, O-b-Cu2O and b-Cu2O, with more carbonaceous product generation and greater utilization of electrons for the hydrophobic ones. Moreover, compared to O-Ag/Cu2O with Ag particles loaded on the outer surface of Cu2O, O-Ag@Cu2O with Ag particles encapsulated within Cu2O exhibits a 6.59-fold increase in ethanol selectivity and a 1.86-fold increase in electron utilization. Additionally, no CO is detected for O-Ag@Cu2O, whereas a small amount of CO is observed for O-Ag/Cu2O. To further rule out the possibility that the significant performance difference between O-Ag/Cu2O and O-Ag@Cu2O arises from the different sizes of Ag particles in the catalysts, the Ag particle size in the comparative catalyst O-Ag/Cu2O was adjusted to match that in O-Ag@Cu2O. Performance tests reveal that even with identical Ag particle sizes, O-Ag/Cu2O still exhibits significantly lower performance than O-Ag@Cu2O (Fig. S19†). In addition, compared with O-b-Cu2O, both O-Ag@Cu2O and O-Ag/Cu2O exhibit promoted CO2-to-alcohol conversion performance, closely related to the role of Ag particles.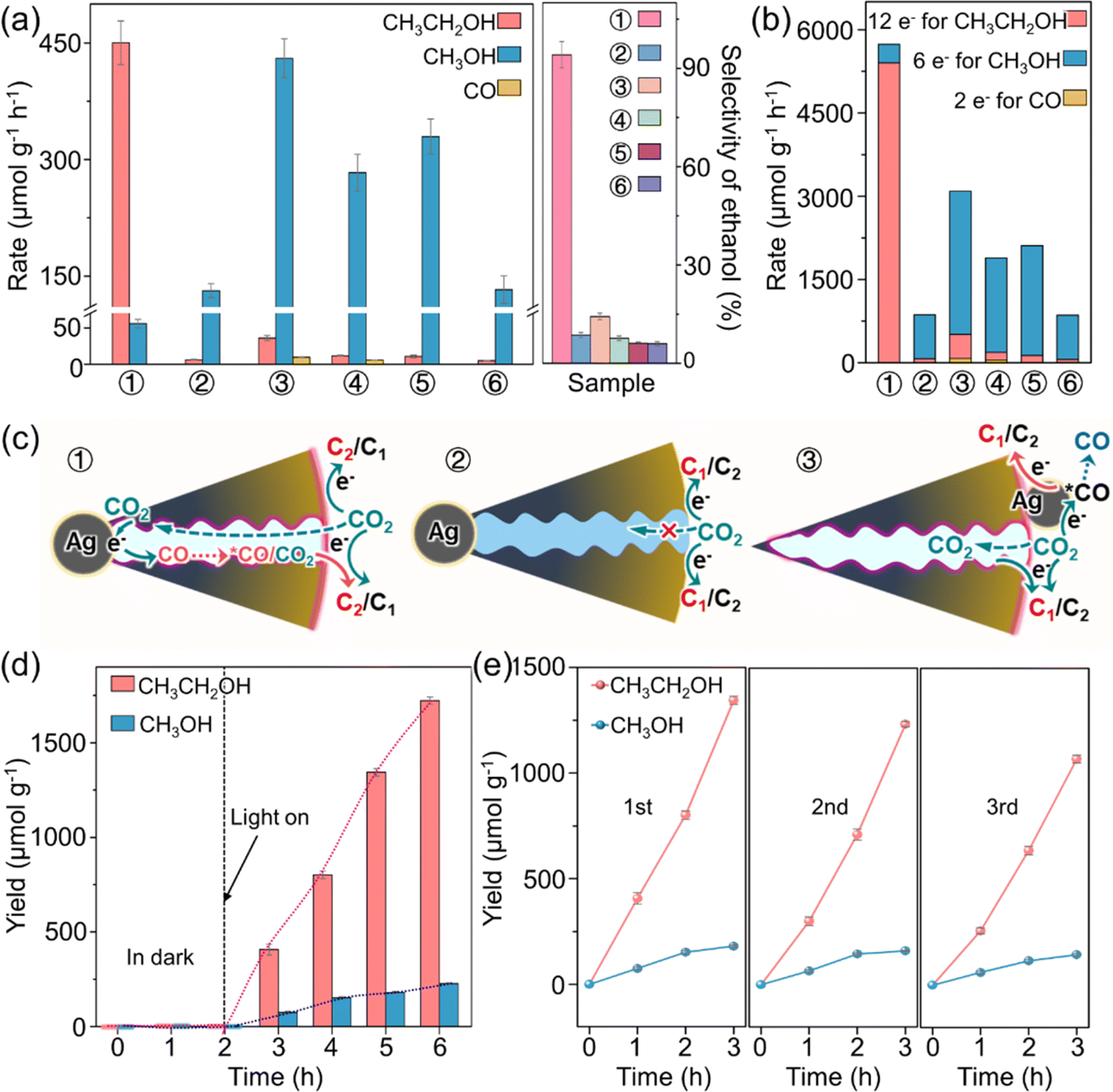 | ||
| Fig. 3 PCRR performance. Generation rates of carbonaceous products and selectivity towards ethanol (a) and amount of effective photogenerated electrons utilized in the PCRR (b) for ① O-Ag@Cu2O, ② Ag@Cu2O, ③ O-Ag/Cu2O, ④ Ag/Cu2O, ⑤ O-b-Cu2O, and ⑥ b-Cu2O. The error bar in (a) represents the standard deviation of three measurements. (c) Schemes of the PCRR in the structures of ① O-Ag@Cu2O, ② Ag@Cu2O, and ③ O-Ag/Cu2O. The pink color on the inner wall of the pores and the outer surfaces of O-Ag@Cu2O and O-Ag/Cu2O represents DDT. (d) Ethanol and methanol yields over time for O-Ag@Cu2O. (e) Recycle experiments for O-Ag@Cu2O. | ||
In light of these experimental findings, potential reasons for the performance differences observed among various catalysts are proposed. As illustrated in Fig. 3c, the hydrophobic modification imparts hydrophobic characteristics to both the external surface and mesoporous channels of the catalyst, facilitating the enrichment and capture of CO2. Consequently, concerning the disparity between O-Ag@Cu2O and Ag@Cu2O, the hydrophobic properties of O-Ag@Cu2O enable CO2 accumulation on both the inner and outer surfaces of O-Ag@Cu2O. Moreover, CO2 can migrate towards the surface of the Ag particles enclosed within O-Ag@Cu2O, allowing Ag to actively participate in the PCRR.36 Conversely, it is challenging for CO2 to accumulate on the hydrophilic surface and enter the mesoporous channels of Ag@Cu2O, rendering the Ag particles ineffective. This observation is consistent with the PCRR results showing that the photocatalytic performance of Ag@Cu2O is similar to that of b-Cu2O (Fig. 3a), indicating the inefficacy of the Ag particles (Fig. 3c and S20†). Furthermore, the difference between O-Ag@Cu2O and O-Ag/Cu2O stems from the distinct positions of the Ag particles within the catalyst. In the case of O-Ag@Cu2O, once the Ag particles inside the catalyst generate CO during PCRR, CO molecules need to diffuse out through the mesoporous channels. During this diffusion process, the nanoconfinement effect of the channels causes CO to aggregate within them. This phenomenon increases the concentration of *CO on the active Cu2O sites and ample duration for *CO molecules to engage in mutual interactions. This promotes the C–C coupling process and thereby accelerates ethanol production. In contrast, in the case of O-Ag/Cu2O, Ag particles are positioned on the external surface of the catalyst. Following CO generation, some of it may re-adsorb onto nearby active Cu2O sites, enhancing the PCRR, while the remainder may diffuse directly into the surrounding environment, leading to the production of gaseous CO. This direct overflow of CO limits the increase in *CO concentration on the Cu2O active sites, resulting in a reduced ethanol production capacity compared to O-Ag@Cu2O.
To further evaluate the solar energy utilization efficiency of O-Ag@Cu2O, the solar-to-fuel conversion efficiency (STF) and apparent quantum efficiency (AQE) are calculated (Fig. S21 and Table S3 and S4†). The STF and AQE (λ = 420 nm) are estimated to be 0.215% and 3.44%, respectively. These values surpass the typical records in the literature for liquid alcohol production, which range from 0.005% to 0.186% for STF and 0.37% to 1.5% for AQE.5,13,37–43
The exceptional photocatalytic performance of O-Ag@Cu2O can be further demonstrated by the time-dependent generation of ethanol and methanol (Fig. 3d). Under dark conditions, no discernible products are observed. However, upon exposure to visible light irradiation, the yields of ethanol and methanol progressively increase with prolonged reaction time, with ethanol exhibiting a notably higher yield compared to methanol. This nearly linear growth trend suggests the good stability of O-Ag@Cu2O, which can be further supported by recycling experiments. Notably, after three cycles, O-Ag@Cu2O retains approximately 80% of its initial activity (Fig. 3e), which is commensurate with the enhanced stability reported for Cu2O-based catalysts in previous studies.5,13,44–46 Moreover, a comprehensive post-reaction characterization analysis of O-Ag@Cu2O reveals minimal alterations in its morphology and structure (Fig. S22†), providing preliminary evidence of its commendable stability.
To further investigate the long-term stability of O-Ag@Cu2O, the PCRR was extended to 16 hours under continuous operation (Fig. S23a†). As the reaction progresses, the production of ethanol and methanol continues to increase, indicative of the catalyst's sustained effectiveness, albeit with a gradually diminishing growth rate. This slowdown in production growth may be attributed to two primary factors. Firstly, while the primary Cu-containing component in O-Ag@Cu2O remains Cu2O after the 16 hours reaction, a portion of its surface is oxidized to CuII by its photogenerated holes (Fig. S23b–e†),47 leading to a slight decrease in photocatalytic performance. Secondly, the generated methanol and ethanol may undergo re-oxidation by the photogenerated holes of Cu2O,48 thereby reducing their net yield. The re-oxidation of methanol and ethanol allows them to act as sacrificial agents for hole consumption during the PCRR, thereby partially suppressing the self-oxidation of Cu2O in O-Ag@Cu2O. Overall, despite experiencing some oxidation, O-Ag@Cu2O exhibits commendable stability.
To further confirm that CO2 is the carbon source for ethanol production, an isotope labeling experiment using 13CO2 was conducted. The gas chromatography-mass spectrometry (GC-MS) analysis of ethanol reveals a series of peaks at mass-to-charge ratios (m/z) ranging from 43 to 48 (Fig. S24a†), providing qualitative evidence for the formation of 13CH313CH2OH. Specifically, the m/z values of 47 and 48 correspond to the fragment ion of 13C2H4O+ and the molecular ion of 13C2H5OH+, respectively.49 Additionally, the most intense peak at m/z = 32 is attributed to the cleavage of the C–C bond, forming 13CH2OH+.49 Combined with the control experiments (Fig. S24b†), it can be concluded that the PCRR process is driven by light, with CO2 serving as the carbon source. To further ensure the accuracy of ethanol selectivity, detailed analysis of the CO2 reduction products was conducted using 1H nuclear magnetic resonance (NMR) spectroscopy. This analysis aimed to identify any other carbon-containing compounds, such as formic acid. The result indicates that ethanol and methanol are the only carbon-containing products detected (Fig. S24c†).
Additionally, the PCRR conducted using only CO2 and H2O as the reactants confirms the concurrent production of oxygen (O2). The band structure deduced from valence-band spectra (Fig. S25a†) and UV-vis diffuse reflectance spectroscopy (Fig. S15b†) provides evidence that the designed Cu2O-based catalyst is thermodynamically suitable for H2O oxidation reaction (Fig. S25b†). Furthermore, the oxidation of H2O to O2 involves the transfer of four holes (2H2O + 4h+ → O2 + 4H+), which are generated within the valence band (VB) of the material. In Cu2O, the VB is derived from the hybridization of Cu 3d and O 2p orbitals.50 Thus, both Cu and O atoms could theoretically serve as active sites for O2 production. However, since Cu atoms are primarily recognized as the active sites for CO2 reduction, it is more likely that O atoms serve as the active sites for O2 evolution.48,51 Additionally, the O2 generation rate of O-Ag@Cu2O reaches 375.77 μmol g−1 h−1 (Fig. S25c†), which is much higher than that of other catalysts, indicating the excellent photocatalytic performance of O-Ag@Cu2O.
Mechanism study
In situ FTIR technique was employed to identify reaction intermediates. To elucidate the differences caused by hydrophobicity, in situ FTIR spectra for O-Ag@Cu2O and Ag@Cu2O were compared (Fig. 4a and b). Furthermore, to investigate the influence of the spatial distribution of Ag particles, in situ FTIR spectra for O-Ag@Cu2O and O-Ag/Cu2O were analyzed (Fig. 4a and c). Notably, the signal at 1545 cm−1, corresponding to *COOH,52 indicates that CO2 is chemically adsorbed on the catalyst surface and interacts with protons. Moreover, the signals for monodentate carbonate (m-CO32−) at 1458, 1508, 1538, and 1556 cm−1, and bidentate bicarbonates (b-CO32−) at 1620 and 1650 cm−1,53 suggest interactions between the chemically adsorbed CO2 and H2O molecules on the catalyst surface. Therefore, the presence of *COOH, m-CO32−, and b-CO32− signals are indicative of effective CO2 adsorption.9 It is evident that the intensity of effective CO2 adsorption for O-Ag@Cu2O and O-Ag/Cu2O is significantly higher than that for Ag@Cu2O, implying a greater accumulation of CO2 molecules on the surfaces of O-Ag@Cu2O and O-Ag/Cu2O compared to Ag@Cu2O. This observation further demonstrates the enhanced CO2 accumulation facilitated by hydrophobic properties. As the reaction progresses, the appearance of the stretching vibration peak of the aldehyde carbonyl group at 1733 cm−1 implies the formation of *CHO,54 which is a crucial intermediate for C1 compound formation. Moreover, the symmetric and antisymmetric stretching vibration peaks of –CH3 at 2885 and 2975 cm−1,55 along with the C–OH stretching vibration peak of alcohols at 1060 cm−1,55 reveal the formation of alcohols across all three catalysts. However, an evident peak at 760 cm−1, corresponding to the rocking vibration of –CH2–,55 is exclusively observed for O-Ag@Cu2O, aligning with the significant ethanol production associated with O-Ag@Cu2O. Additionally, for O-Ag/Cu2O, a discernible signal at 2134 cm−1 can be attributed to *CO,56 demonstrating the formation of *CO on the outer surface of O-Ag/Cu2O. This finding corroborates the proposed hypothesis that CO generated on Ag particles located on the Cu2O outer surface of O-Ag/Cu2O can re-adsorb onto the Cu2O outer surface. In contrast, CO generated on Ag particles located in the core of O-Ag@Cu2O experiences diffusion through the mesoporous channels within the Cu2O shell. Due to the prolonged residence time within the pores, *CO intermediates interact with each other, resulting in a relatively low amount of CO reaching the outer surface, thereby no obvious signal being detected.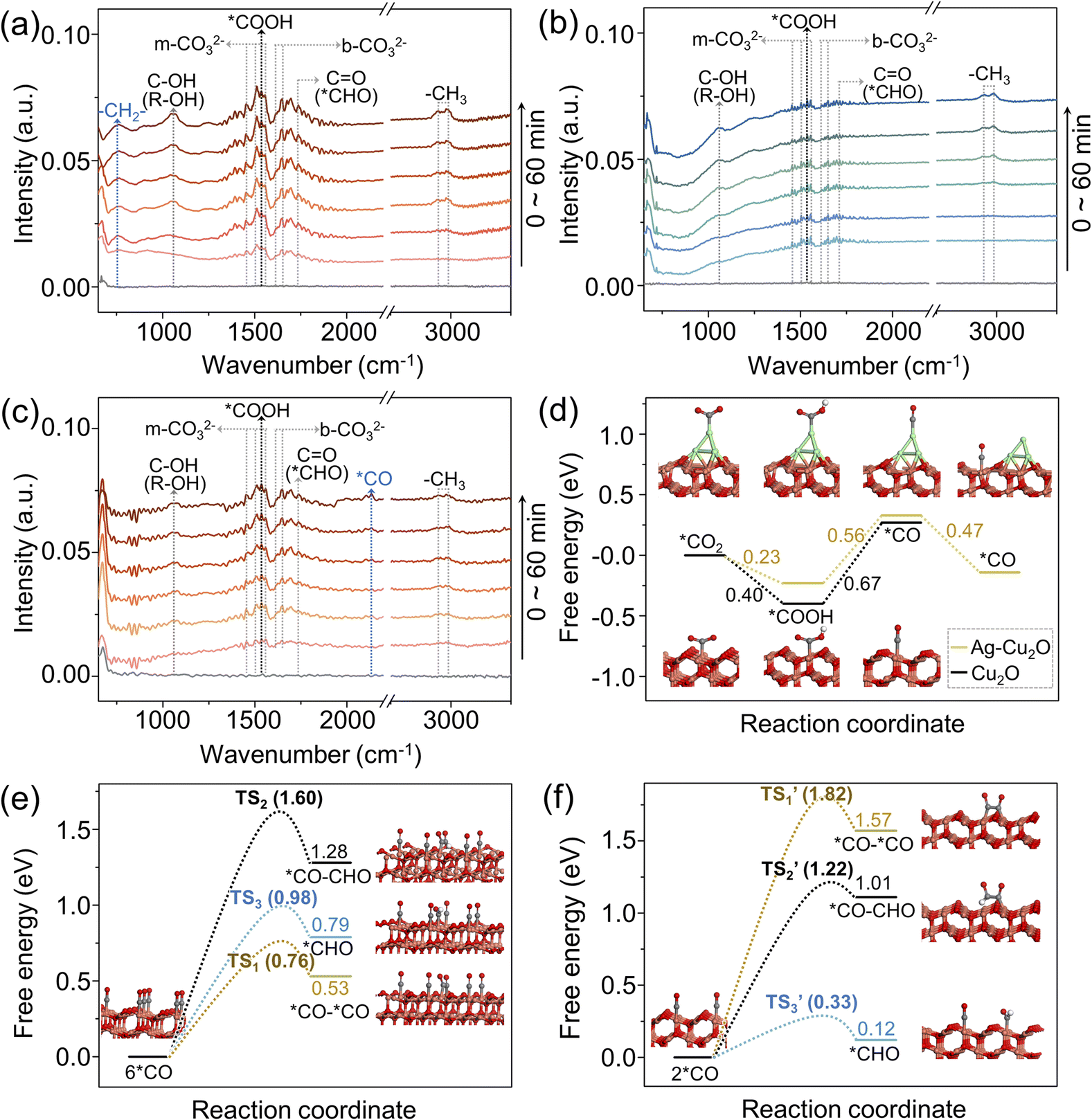 | ||
| Fig. 4 (a–c) In situ FTIR spectra obtained under different irradiation times for O-Ag@Cu2O, Ag@Cu2O, and O-Ag/Cu2O, respectively. (d) Gibbs free energy diagrams for *CO formation under the models of Ag–Cu2O and Cu2O. (e and f) Gibbs free energy diagrams for three potential reaction pathways after *CO formation at elevated and low *CO concentrations, respectively. The brown, gray, red, and green balls represent Cu, C, O, and Ag atoms, respectively. TS1, TS2, and TS3 represent the transition states of *CO–*CO, *CO–CHO, and *CHO, respectively, at elevated *CO concentration. TS1’, TS2’, and TS3’ represent the transition states of *CO–*CO, *CO–CHO, and *CHO, respectively, at low *CO concentration. | ||
To gain a deeper insight into the enhanced ethanol selectivity observed in O-Ag@Cu2O, DFT calculations were conducted. For clarity, the CO2-to-ethanol conversion process is delineated and analyzed in three stages:57 initially, the reduction of CO2 molecules to *CO; followed by the protonation of *CO and the C–C coupling process; and finally, the subsequent proton–electron transfer leading to the formation of ethanol. To reveal the working principle of Ag, the model of the Cu2O (111) surface with Ag atoms atop (designated as Ag-Cu2O) and the model of the bare Cu2O (111) surface (designated as Cu2O) were compared (Fig. S26†). As shown in the configurations inserted in Fig. 4d, for Ag-Cu2O, when a CO2 molecule is adsorbed onto the Ag atom, it undergoes protonation to form *COOH, followed by further protonation to produce *CO. Subsequently, the *CO desorbs from the Ag atom and re-adsorbs onto the adjacent Cu atom. While for Cu2O, the adsorbed CO2 molecule on the Cu atom is protonated into *COOH and *CO in order. The energy barrier of the uphill conversion of *COOH to *CO decreases by 0.11 eV over Ag-Cu2O, implying that the presence of Ag is conducive to *CO formation. More importantly, the desorption of *CO from Ag atoms and its subsequent adsorption onto Cu atoms is an energetically favorable, spontaneous process, suggesting that *CO generated on Ag can readily re-adsorb onto nearby Cu atoms. Therefore, for O-Ag@Cu2O, aided by the hydrophobic property and nanoconfinement effect, the Ag core produces more CO, resulting in a greater accumulation of *CO within the mesoporous channels of the Cu2O shell.
To investigate the impact of *CO concentration on the Cu2O surface on ethanol formation, simulations were performed with six and two *CO molecules adsorbed on Cu atoms, representing elevated *CO concentration and low *CO concentration scenarios, respectively (Fig. 4e and f). After *CO formation, three potential reaction pathways were considered: the hydrogenation to form *CHO, the dimerization of *CO (*CO–*CO coupling), and the polymerization of *CO and *CHO (*CO–CHO coupling). To gain deeper insight into the interaction of *CO in these three different reaction pathways, the transition state energies of *CO in these processes under different *CO concentrations were quantified,58 and the configurations of each transition state are presented (Fig. S27†). As depicted in Fig. 4e, at elevated *CO concentration, the energy barrier for the transition state (TS1) associated with the *CO–*CO coupling process is the lowest among the three investigated pathways, with a value of 0.76 eV, reduced by 0.22 and 0.84 eV compared to those of *CHO formation (TS3) and *CO–CHO coupling (TS2), respectively. In contrast, at low *CO concentration, *CHO formation is the most favorable, with its transition state (TS3’) energy barrier being the lowest, at 0.33 eV (Fig. 4f). These findings suggest that *CO–*CO coupling is preferred over *CHO formation at elevated *CO concentration, whereas *CHO formation is favored over C–C coupling at lower *CO concentration. Given that *CHO is a key intermediate for C1 compound formation and C–C coupling is essential for C2 compound formation, increased *CO concentration enhances ethanol selectivity. Combining these insights with possible CO2-to-ethanol and CO2-to-methanol conversion pathways proposed based on DFT calculations (Fig. S28†), elevated *CO concentration not only promotes ethanol formation but also methanol formation, albeit with a significantly greater effect on ethanol formation. Thus, increasing *CO concentration enhances both PCRR efficiency and ethanol selectivity, corroborating experimental results.
Conclusions
In summary, a nanoconfined and tandem three-phase reaction system has been developed to simultaneously address the limited *CO concentration and inadequate interaction time between C1 intermediates. Consequently, efficient ethanol production is achieved with a selectivity of 94.15% and a generation rate of 450.19 μmol g−1 h−1. This system is based on a tandem catalyst, O-Ag@Cu2O, comprising an Ag core covered by a mesoporous hydrophobic Cu2O shell. The hydrophobic Cu2O shell can alleviate the poor CO2 mass transfer, ensuring sufficient CO2 supply to the Ag core for conversion to CO. Subsequently, CO undergoes continuous reduction within the nanoconfined mesoporous channels of Cu2O. This process unfolds in three parts: (i) hydrophobicity guarantees ample CO2 supply, enabling Ag to generate substantial CO; (ii) nanoconfinement of CO within the mesoporous channels of the Cu2O shell leads to its aggregation and prolonged residence time; and (iii) the tandem reaction of CO2-to-CO and CO-to-C2 products is enhanced due to the substantial CO2 and CO present during this process. The synergy of improved mass transfer, nanoconfinement, and tandem reaction results in high *CO concentrations at active sites and prolonged interaction time, further promoting C–C coupling and aligning the reaction time with that of C–C coupling. Furthermore, the elevated *CO concentration makes the energy barrier for *CO–*CO coupling much lower than that for *CO forming *CHO, thereby prioritizing the occurrence of C–C coupling. This results in outstanding photocatalytic performance for ethanol production, particularly in terms of ethanol selectivity. Additionally, the elevated *CO concentration enhances the PCRR efficiency, achieving an STF of 0.215%. This strategy presents a promising avenue for the efficient production of C2+ compounds.Data availability
The authors declare that the data supporting the findings of this study are available within the article and its ESI.†Author contributions
Conceptualization: Hailing Huo, Ang Li, formal analysis: Hailing Huo, Ting Hu, Chengxi Huang, Ang Li, funding acquisition: Erjun Kan, Ang Li, investigation: Hailing Huo, Zhiqing Zhong, Qiang Ju, Liang Zhang, methodology: Hailing Huo, Ting Hu, Cheng Zhan, Chengxi Huang, Fang Wu, Ang Li, Erjun Kan, supervision: Ang Li, Erjun Kan, validation: Hailing Huo, Zhiqing Zhong, Qiang Ju, Liang Zhang, visualization: Hailing Huo, writing – original draft: Hailing Huo, Ang Li, writing – review & editing: Hailing Huo, Ang Li, Erjun Kan.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22378204, 51790492), National Outstanding Youth Science Fund Project of National Natural Science Foundation of China (T2125004), and Funding of NJUST (No. TSXK2022D002).Notes and references
- H. Huo, F. Wu, E. Kan and A. Li, Chem.–Eur. J., 2023, 29, e202300658 CrossRef CAS PubMed
.
- Y. Ji, J. Du and A. Chen, Trans. Tianjin Univ., 2022, 28, 292–306 CrossRef CAS
- Y. Wang, C. Zhang and R. Li, Trans. Tianjin Univ., 2022, 28, 227–235 CrossRef CAS
- L. Jiang, H. Du, L. Li, X. Guan, Y. Zhang, L. Li, X. Liu, L. Li, Y. Tian, L. Zhang, S. Wang, J. Chen and S. Shen, Trans. Tianjin Univ., 2023, 29, 462–472 CrossRef CAS
- H. Huo, H. He, C. Huang, X. Guan, F. Wu, Y. Du, H. Xing, E. Kan and A. Li, Chem. Sci., 2024, 15, 1638–1647 RSC
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S. Z. Qiao, Nat. Commun., 2022, 13, 1–11 CAS
- C. Liu, M. Zhang, J. Li, W. Xue, T. Zheng, C. Xia and J. Zeng, Angew. Chem., Int. Ed., 2021, 61, e202113498 CrossRef PubMed
- H. Shi, H. Wang, Y. Zhou, J. Li, P. Zhai, X. Li, G. G. Gurzadyan, J. Hou, H. Yang and X. Guo, Angew. Chem., Int. Ed., 2022, 61, e202208904 CrossRef CAS PubMed
- S. Xie, W. Ma, X. Wu, H. Zhang, Q. Zhang, Y. Wang and Y. Wang, Energy Environ. Sci., 2021, 14, 37–89 RSC
- Q. Huang, B. Xia, M. Li, H. Guan, M. Antonietti and S. Chen, Nat. Commun., 2024, 15, 4157 CrossRef CAS PubMed
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS
- T. H. M. Pham, J. Zhang, M. Li, T. H. Shen, Y. Ko, V. Tileli, W. Luo and A. Züttel, Adv. Energy Mater., 2022, 12, 2103663 CrossRef CAS
- H. Huo, T. Hu, C. Huang, F. Wu, T. Wang, X. Liu, L. Zhang, Q. Ju, Z. Zhong, H. Xing, E. Kan and A. Li, J. Energy Chem., 2024, 93, 202–212 CrossRef CAS
- A. Li, Q. Cao, G. Zhou, B. V. K. J. Schmidt, W. Zhu, X. Yuan, H. Huo, J. Gong and M. Antonietti, Angew. Chem., Int. Ed., 2019, 58, 14549–14555 CrossRef CAS PubMed
- Z.-Z. Wu, X.-L. Zhang, Z.-Z. Niu, F.-Y. Gao, P.-P. Yang, L.-P. Chi, L. Shi, W.-S. Wei, R. Liu and Z. Chen, J. Am. Chem. Soc., 2021, 144, 259–269 CrossRef PubMed
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Joule, 2020, 4, 1688–1699 CrossRef CAS
- L. Zhang, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed
- J. Rosen, G. S. Hutchings, Q. Lu, S. Rivera, Y. Zhou, D. G. Vlachos and F. Jiao, ACS Catal., 2015, 5, 4293–4299 CrossRef CAS
- M.-M. Yang, J.-M. Cao, G.-D. Qi, X.-Y. Shen, G.-Y. Yan, Y. Wang, W.-W. Dong, J. Zhao, D.-S. Li and Q. Zhang, Inorg. Chem., 2023, 62, 15963–15970 CrossRef CAS PubMed
- L. Ding, N. Zhu, Y. Hu, Z. Chen, P. Song, T. Sheng, Z. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202209268 CrossRef CAS PubMed
- S. Wang, X. Bai, Q. Li, Y. Ouyang, L. Shi and J. Wang, Nanoscale Horiz., 2021, 6, 661–668 RSC
- C. H. Sharp, B. C. Bukowski, H. Li, E. M. Johnson, S. Ilic, A. J. Morris, D. Gersappe, R. Q. Snurr and J. R. Morris, Chem. Soc. Rev., 2021, 50, 11530–11558 RSC
- J.-H. Han, E. Lee, S. Park, R. Chang and T. D. Chung, J. Phys. Chem. C, 2010, 114, 9546–9553 CrossRef CAS
- D. McLaughlin, M. Bierling, R. Moroni, C. Vogl, G. Schmid and S. Thiele, Adv. Energy Mater., 2020, 10, 2000488 CrossRef CAS
- A. A. Saraev, A. Y. Kurenkova, D. D. Mishchenko, A. L. Trigub, E. Y. Gerasimov and E. A. Kozlova, Trans. Tianjin Univ., 2024, 30, 140–151 CrossRef CAS
- L. Liu, S. J. Zheng, H. Chen, J. Cai and S. Q. Zang, Angew. Chem., Int. Ed., 2024, 63, e202316910 CrossRef CAS PubMed
- Y. Q. Liang, Z. D. Cui, S. L. Zhu, Y. Liu and X. J. Yang, J. Catal., 2011, 278, 276–287 CrossRef CAS
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed
- Y. Li, D. Hui, Y. Sun, Y. Wang, Z. Wu, C. Wang and J. Zhao, Nat. Commun., 2021, 12, 123 CrossRef CAS PubMed
- R. Das, R. Paul, A. Parui, A. Shrotri, C. Atzori, K. A. Lomachenko, A. K. Singh, J. Mondal and S. C. Peter, J. Am. Chem. Soc., 2022, 145, 422–435 CrossRef PubMed
- T. Soltani, X. Zhu, A. Yamamoto, S. P. Singh, E. Fudo, A. Tanaka, H. Kominami and H. Yoshida, Appl. Catal., B, 2021, 286, 119899 CrossRef CAS
- Z. Zhang and J. T. Yates, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed
- S. Ghose, S. Singh and T. S. Bhattacharya, ACS Appl. Mater. Interfaces, 2020, 12, 7727–7735 CrossRef CAS PubMed
- H. Feng, W. Wang, W. Wang, M. Zhang, C. Wang, C. Ma, W. Li and S. Chen, J. Colloid Interface Sci., 2021, 601, 531–543 CrossRef CAS PubMed
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863–20868 CrossRef CAS PubMed
- A. Sabbah, I. Shown, M. Qorbani, F.-Y. Fu, T.-Y. Lin, H.-L. Wu, P.-W. Chung, C.-I. Wu, S. R. M. Santiago and J.-L. Shen, Nano Energy, 2022, 93, 106809 CrossRef CAS
- F. Almomani, R. Bhosale, M. Khraisheh, A. Kumar and M. Tawalbeh, Appl. Surf. Sci., 2019, 483, 363–372 CrossRef CAS
- J. Mao, T. Peng, X. Zhang, K. Li, L. Ye and L. Zan, Catal. Sci. Technol., 2013, 3, 1253–1260 RSC
- O. Ola and M. M. Maroto-Valer, Appl. Catal., A, 2015, 502, 114–121 CrossRef CAS
- O. Ola and M. M. Maroto-Valer, J. Catal., 2014, 309, 300–308 CrossRef CAS
- L. Tang, L. Kuai, Y. Li, H. Li, Y. Zhou and Z. Zou, Nanotechnology, 2018, 29, 064003 CrossRef PubMed
- J.-P. Zou, D.-D. Wu, J. Luo, Q.-J. Xing, X.-B. Luo, W.-H. Dong, S.-L. Luo, H.-M. Du and S. L. Suib, ACS Catal., 2016, 6, 6861–6867 CrossRef CAS
- D. Jiang, Y. Zhang and X. Li, Chin. J. Catal., 2019, 40, 105–113 CrossRef CAS
- L. Yu, X. Ba, M. Qiu, Y. Li, L. Shuai, W. Zhang, Z. Ren and Y. Yu, Nano Energy, 2019, 60, 576–582 CrossRef CAS
- T. Wang, K. Deng, H. Huo, C. Huang, Y. Du, M. Yu, J. Ma, E. Kan and A. Li, ACS Catal., 2024, 14, 8748–8757 CrossRef CAS
- C. Y. Toe, J. Scott, R. Amal and Y. H. Ng, J. Photochem. Photobiol., C, 2019, 40, 191–211 CrossRef CAS
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C.-J. Sun, Z. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Liu and T. Rajh, Nat. Energy, 2019, 4, 957–968 CrossRef CAS
- S. Wang, B. Jiang, J. Henzie, F. Xu, C. Liu, X. Meng, S. Zou, H. Song, Y. Pan, H. Li, J. Yu, H. Chen and J. Ye, Nat. Commun., 2023, 14, 2534 CrossRef CAS
- M. Zhang, J. Wang, H. Xue, J. Zhang, S. Peng, X. Han, Y. Deng and W. Hu, Angew. Chem., Int. Ed., 2020, 59, 18463–18467 CrossRef CAS PubMed
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed
- W. Shangguan, Q. Liu, Y. Wang, N. Sun, Y. Liu, R. Zhao, Y. Li, C. Wang and J. Zhao, Nat. Commun., 2022, 13, 3894 CrossRef CAS
- H. Yu, J. Li, Y. Zhang, S. Yang, K. Han, F. Dong, T. Ma and H. Huang, Angew. Chem., Int. Ed., 2019, 58, 3880–3884 CrossRef CAS PubMed
- L. Xiong, X. Zhang, L. Chen, Z. Deng, S. Han, Y. Chen, J. Zhong, H. Sun, Y. Lian, B. Yang, X. Yuan, H. Yu, Y. Liu, X. Yang, J. Guo, M. H. Rümmeli, Y. Jiao and Y. Peng, Adv. Mater., 2021, 33, 2101741 CrossRef CAS
-
J. G. Speight, in Lange's handbook of chemistry, McGraw-Hill, New York, 2005, ch. 3, pp. 3.3–3.50 Search PubMed
- K. Hadjiivanov and H. Knözinger, Phys. Chem. Chem. Phys., 2001, 3, 1132–1137 RSC
- L. R. L. Ting, O. Piqué, S. Y. Lim, M. Tanhaei, F. Calle-Vallejo and B. S. Yeo, ACS Catal., 2020, 10, 4059–4069 CrossRef CAS
- K. Black, P. Liu, L. Xu, C. Doubleday and K. N. Houk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12860–12865 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04647a |
| This journal is © The Royal Society of Chemistry 2024 |