
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nitrous oxide as diazo transfer reagent
Alexandre
Genoux
 and
Kay
Severin
*
and
Kay
Severin
*
Institut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: kay.severin@epfl.ch
First published on 14th August 2024
Abstract
Nitrous oxide, commonly known as “laughing gas”, is formed as a by-product in several industrial processes. It is also readily available by thermal decomposition of ammonium nitrate. Traditionally, the chemical valorization of N2O is achieved via oxidation chemistry, where N2O acts as a selective oxygen atom transfer reagent. Recent results have shown that N2O can also function as an efficient diazo transfer reagent. Synthetically useful methods for synthesizing triazenes, N-heterocycles, and azo- or diazo compounds were developed. This review article summarizes significant advancements in this emerging field.
 Alexandre Genoux | Alexandre Genoux received his MSc from Grenoble Alps University, including research at UC Santa Barbara with Prof. Liming Zhang and at the University of Cambridge with Prof. Robert Phipps. He earned his PhD from the University of Zurich in 2020 under the supervision of Prof. Cristina Nevado. From 2021 to 2023, he was a postdoctoral associate with Prof. Patrick L. Holland at Yale University, supported by the Center for Hybrid Approaches in Solar Energy to Liquid Fuels. Since 2024, he is a Marie Skłodowska-Curie Fellow with Prof. Kay Severin at the Ecole Polytechnique Fédérale de Lausanne (EPFL), exploring new synthetic avenues with nitrous oxide. |
 Kay Severin | Kay Severin is a professor of chemistry at the Ecole Polytechnique Fédérale de Lausanne (EPFL), Switzerland. His research group works on synthetic organic and inorganic chemistry, with projects ranging from small molecule activation (the chemistry of nitrous oxide) to the construction of functional nanostructures. |
1. Introduction
Nitrous oxide was brought to the public's attention by Sir Humphry Davy, an influential British chemist and inventor. In 1800, the 21-year-old Davy published a book entitled “Researches, Chemical and Philosophical; Chiefly Concerning Nitrous Oxide, or Dephlogisticated Nitrous Air, and its Respiration”.1 This 580-page monograph is divided into two parts. The first part provides a comprehensive review of the chemistry of nitrous oxide, summarizing the state of knowledge at the time. The second part explores the physiological effects of nitrous oxide with Davy giving detailed descriptions of the sensations caused by inhaling this gas. The book also summarizes the effects of nitrous oxide on various animals. Davy was fascinated by this gas, and his enthusiasm was contagious. As a result, nitrous oxide quickly became a popular recreational drug among the British upper class.The use of nitrous oxide as a drug continues to make headlines today,2 but other concerns have emerged. Nitrous oxide is a potent greenhouse gas (GWP = 300), contributing significantly to global warming.3 Furthermore, it is an ozone-depleting substance.4 Various anthropogenic sources contribute to N2O emissions, many of which are linked to agriculture.5 However, mitigation strategies have primarily focused on industrial processes, where nitrous oxide is formed as a side product.6 The largest amount of industrial N2O is generated during the production of nitric acid (Scheme 1a).7 N2O is produced alongside the desired NO during the catalytic oxidation of ammonia, with the amount of N2O depending on the process conditions. Plants without abatement technologies are estimated to emit between 4 and 19 kg of N2O per ton of HNO3 (100%).7b Another significant source of nitrous oxide is the production of adipic acid.8 Adipic acid is obtained through the catalytic oxidation of a mixture of cyclohexanone and cyclohexanol with nitric acid, resulting in the formation of approximately 300 kg of N2O per ton of adipic acid (Scheme 1b).
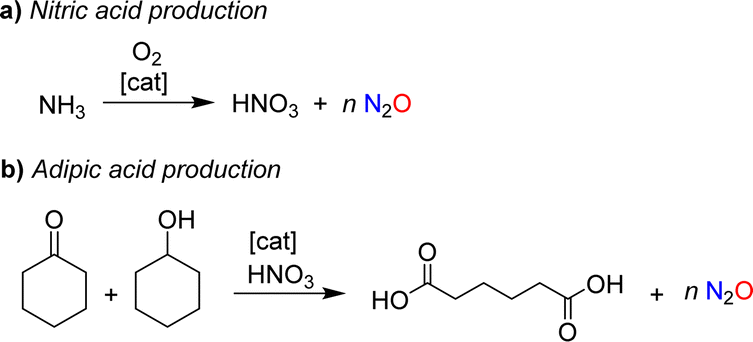 | ||
| Scheme 1 Nitrous oxide is formed as a side product during the industrial production of nitric acid (a) and adipic acid (b). | ||
While the formation of N2O during the production of nitric and adipic acid is a major concern, it also presents an opportunity. Some companies have developed processes that allow the isolation of N2O.9 Nitrous oxide can then be sold, or used as a reagent in downstream applications.10
The targeted synthesis of nitrous oxide is achieved through the thermal decomposition of a concentrated ammonium nitrate solution (Scheme 2a).11 This process is performed on an industrial scale. However, it is not ideal because the production of NH4NO3 involves a multi-step manufacturing route. An interesting alternative is the direct oxidation of ammonia with a catalytic system that provides high selectivity for N2O over NO and N2 (Scheme 2b).12 Pilot tests12 and advances in catalyst design13 suggest that ammonia oxidation could become a feasible method for large-scale industrial N2O production.
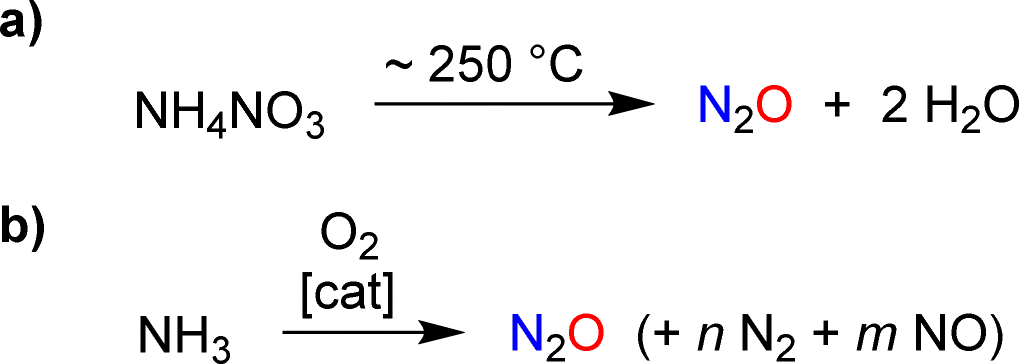 | ||
| Scheme 2 Synthesis of nitrous oxide by thermal decomposition of ammonium nitrate (a) or by catalytic oxidation of ammonia (b). | ||
The chemical valorization of nitrous oxide is traditionally achieved via oxidation reactions.12,14–16 N2O is a powerful oxidant from a thermodynamic standpoint,14 and the by-product, N2, is both easy to separate and harmless. Furthermore, N2O displays good solubility in organic solvents, enabling liquid-phase reactions in low-polarity media.15 A drawback of N2O as an oxidant is its kinetically inert nature. However, the high kinetic barrier can be overcome by using a catalyst and/or elevated temperatures and pressures.
An example of a catalytic process involving N2O is the hydroxylation of benzene (Scheme 3a).8b,12 This reaction is catalyzed by iron-containing zeolites. Nitrous oxide represents an interesting oxidant for this reaction because it provides phenol with high selectivity. Furthermore, one could couple the N2O-mediated phenol production with the N2O-liberating formation of adipic acid (phenol could be hydrogenated to give cyclohexanol, the precursor for adipic acid; see Scheme 1). The pilot-scale production of phenol using N2O as the oxidant has been realized by Solutia, together with the Boreskov Institute of Catalysis.8b,12 The process has not yet been commercialized due to economic reasons, and because a circular phenol/adipic acid production would require additional N2O.8b,12
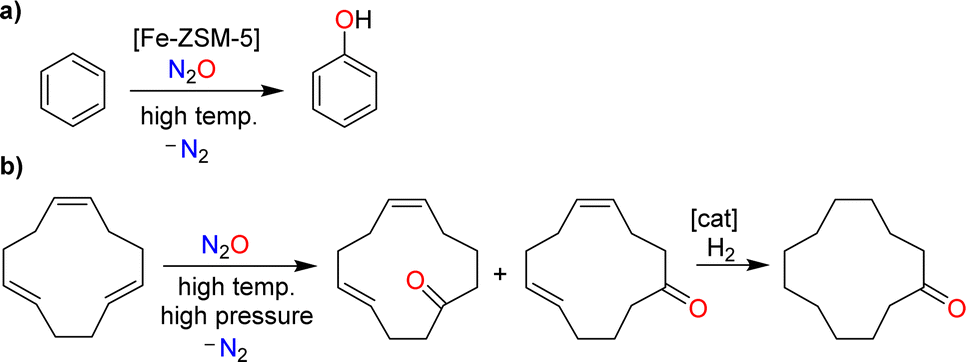 | ||
| Scheme 3 The use of nitrous oxide as an O-atom donor: synthesis of phenol by catalytic oxidation of benzene (a), and synthesis of cyclododecanone by non-catalytic oxidation of 1,5,9-cyclododecatriene, followed by hydrogenation (b). | ||
The non-catalyzed oxidation of olefins using N2O at elevated temperatures and pressures gives ketones alongside N2.15 This type of reactivity forms the basis for the industrial production of cyclododecanone, as developed by BASF.10 The process starts with the oxidation of 1,5,9-cyclododecatriene with N2O to give cyclododeca-4,8-dien-1-one as a mixture of isomers (Scheme 3b). Catalytic hydrogenation then provides the target cyclododecanone. It is worth noting that the N2O, which is used in this process, is obtained from the production of adipic acid.10
The reactions depicted in Scheme 3 demonstrate that nitrous oxide can be employed for the synthesis of bulk chemicals. However, many reports about the use of N2O as an oxidant pertain to small-scale syntheses, typically conducted in academic settings.17–22
For the oxidation of highly reactive compounds, the inert nature of N2O can be advantageous, as it prevents potential overoxidation reactions. For example, N2O is frequently used for the oxidation of reactive main-group element compounds.18 These reactions are typically performed in solution using atmospheric pressure of N2O.
The chemical activation of N2O under mild conditions can also be achieved with certain transition metal complexes.19,20 This capability has spurred efforts to develop reactions with N2O using homogeneous catalysts. Over the past few years, significant progress has been made in this field, with efficient catalysts being developed for a variety of oxidation reactions.21,22
Most of the reactions discussed thus far proceed via oxygen atom transfer and extrusion of dinitrogen. This review focuses on a different type of reactivity, namely the use of N2O as a diazo transfer reagent (Scheme 4). The formal by-product in these reactions is O2−, which is released in the form of hydroxide, alkoxide, oxide salts (MIOH, MIOR, MIIO), or water, depending on the substrate that was employed.
 | ||
| Scheme 4 Upon chemical activation, nitrous oxide typically acts as O-atom donor. This review focuses on reactions in which N2O functions as diazo transfer reagent. | ||
The use of N2O as a diazo transfer reagent was first demonstrated by Wislicenus in 1892.23 By subjecting NaNH2 to N2O at elevated temperatures, he was able to obtain NaN3. The ‘Wislicenus reaction’ is nowadays employed for the industrial production of NaN3.24 Despite this early success, N2O-based diazo transfer reactions have historically remained underdeveloped. However, significant progress has been made in recent years, resulting in numerous synthetically useful processes. This review summarized significant developments in this area. Before discussing these advancements, we will describe the covalent capture of intact N2O by (semi-)metal complexes, frustrated Lewis pairs (FLPs), and organic nucleophiles. Only a few of these adducts were used in productive diazo transfer reactions, but they provide valuable insights into the underlying reactivity of N2O.
2. Covalent capture of nitrous oxide
The covalent trapping of N2O by (semi-)metal complexes leverages metal–ligand cooperation.25–33 A common theme in these reactions is the formation of a covalent bond at the terminal N-atom of N2O along with a coordination bond to the other O/N-atom (Scheme 5a–f). Nitrous oxide can also act as a simple ligand for metal complexes (without concomitant formation of a covalent bond to a main group element), but these cases will not be discussed further in this review.34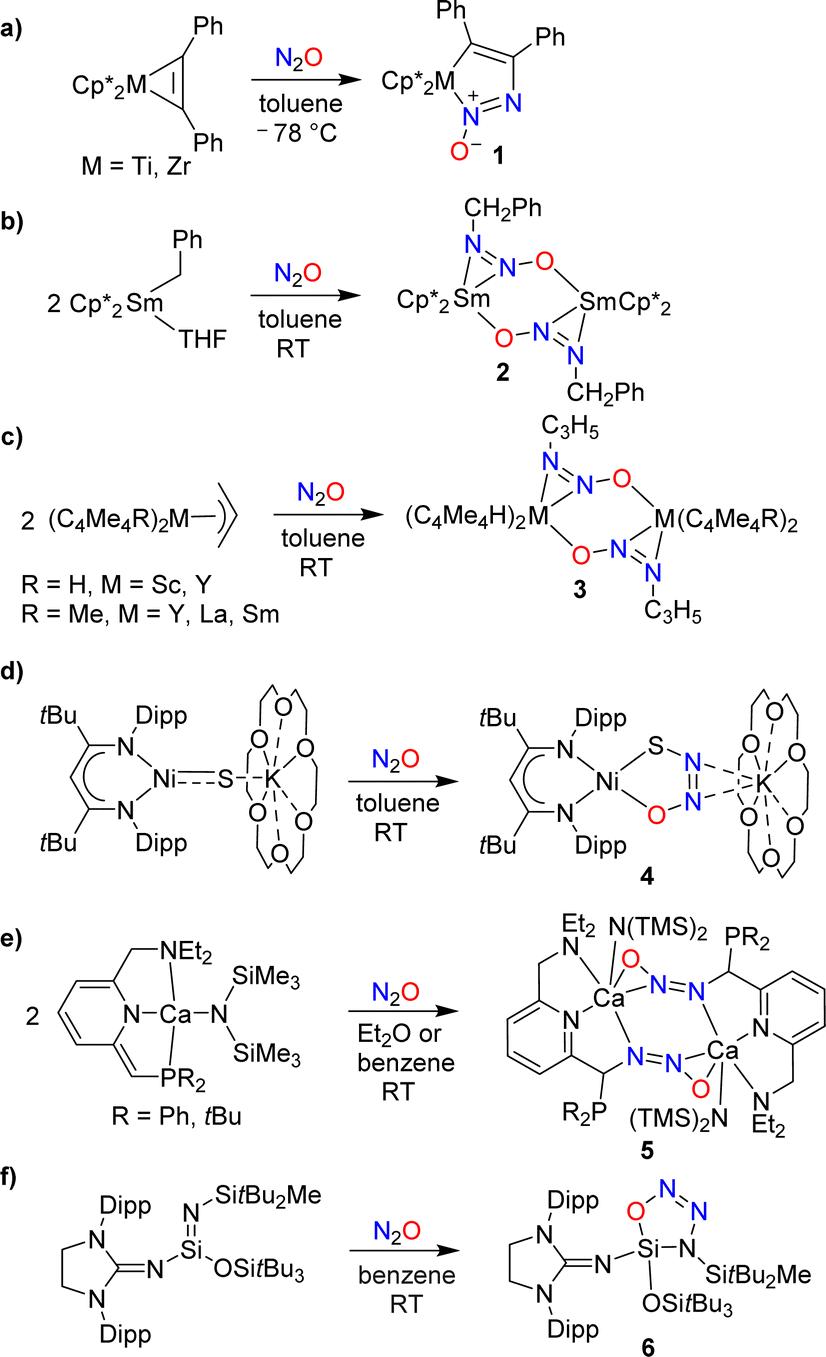 | ||
| Scheme 5 Cooperative covalent capture of N2O by (semi-)metal complexes (Dipp = 2,6-C6H3iPr2). | ||
Hillhouse and coworkers have investigated reactions of the diphenylacetylene complexes Cp*M(PhC2Ph) (M = Ti, Zr) with N2O.25 At low temperatures, azoxymetallacyclopentene complexes of type 1 were obtained (Scheme 5a). The zirconium complex was found to be thermally labile, undergoing extrusion of N2 upon warming to room temperature. The titanium complex was more stable, allowing for a crystallographic characterization. More recently, it was found that the zirconium complex can be stabilized by N-alkylation with MeOTf.26
Insertion of N2O into a metal–carbon bond was also observed for samarium complexes. When a solution of (Cp*)2SmBn(THF) in toluene was exposed to N2O, the dinuclear complex 2 was formed (Scheme 5b).27 Allyl complexes of the general formula (C5Me5R)2M(C3H5) (R = H, Me; M = Sc, Y, Sm, La) were found to display a similar reactivity (Scheme 5c).28
Hayton and coworkers reported that a “masked” terminal Ni(II) sulfide complex is able to react with N2O to give the thiohyponitrite complex 4 (Scheme 5d).29 Liberation of N2 was observed when a solution of complex 4 was heated in toluene at 45 °C for 6 days, resulting in the formation of a η2-SO complex as the main product.30 More recently, the Hayton group showed that a Zn(II) sulfide analogous to complex 4 is also converted to a thiohyponitrite complex when exposed to N2O.31
The cooperative metal–ligand activation of N2O is not restricted to transition metal complexes. Milstein and coworkers examined the reaction of N2O with dearomatized calcium pincer complexes supported by pyridine-based PNN-type ligands.32 A rapid transformation into dinuclear diazotate complexes (5) was observed at room temperature (Scheme 5e).
The reactions of low-valent silicon compounds with N2O typically proceed via O-atom transfer and liberation of dinitrogen.18 An exception to this reactivity pattern was reported by Inoue and coworkers. They showed that an oxatriazasilole, 6, is formed upon reaction of a silaimine with N2O (Scheme 5f).33 The reaction proceeds via a concerted 1,3-dipolar cycloaddition mechanism, first proposed by Wiberg,35 and later supported by computational studies.36 It is interesting to note that solutions of the cycloaddition product 6 are thermally very stable; no isomerization or decomposition was observed at temperatures up to 130 °C.33
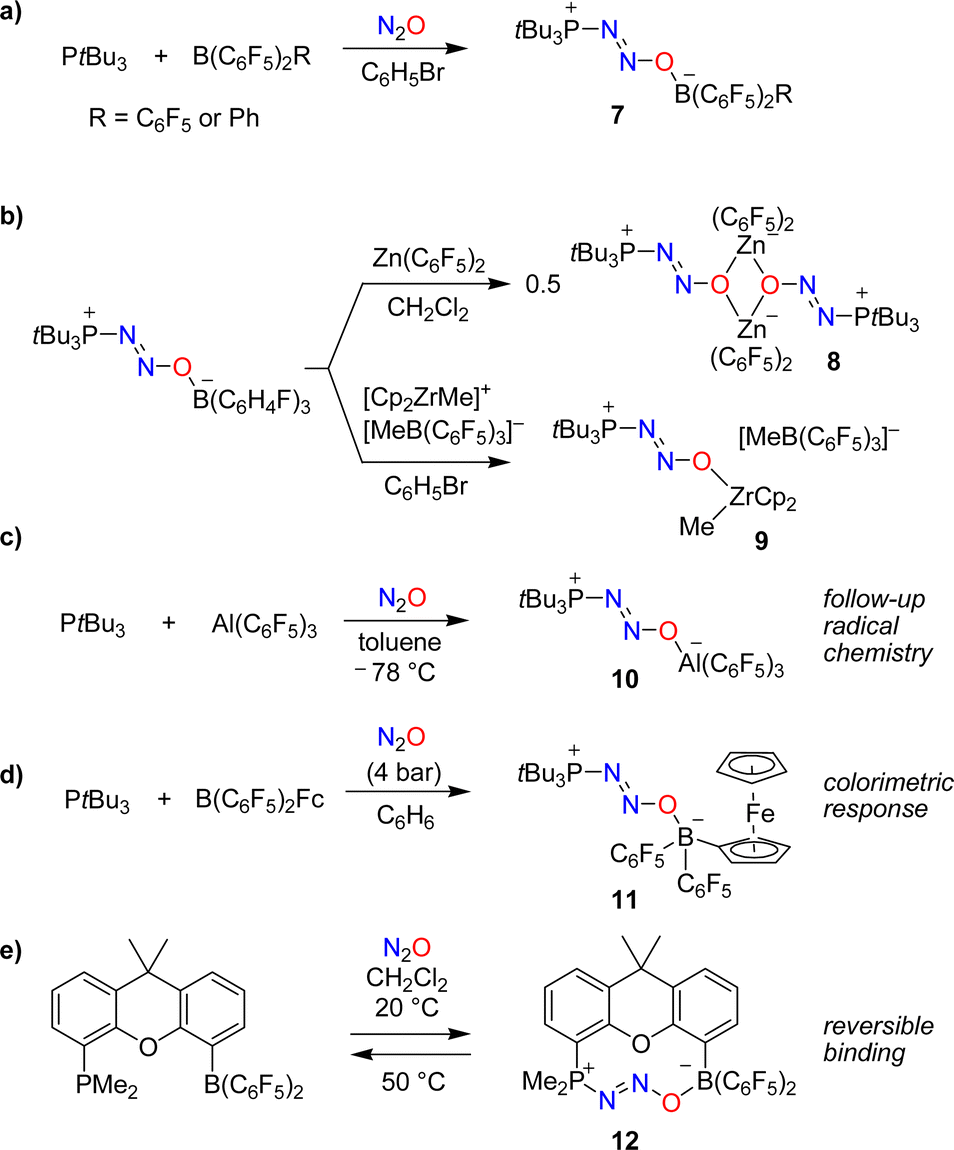
The utilization of FLPs for the capture of N2O was first investigated by Stephan and coworkers.37,38 When mixtures of the bulky phosphine PtBu3 and the Lewis acids B(C6F5)2R (R = C6F5 or Ph) were exposed to an atmosphere of N2O, zwitterionic adducts of type 7 with P–N2O–B linkages were obtained (Scheme 6a). The thermal or photochemical activation of 7 resulted in the formation of (tBu3PO)B(C6F5)2R along with the liberation of dinitrogen.
 | ||
| Scheme 6 Covalent capture of nitrous oxide by frustrated Lewis pairs (FLPs). | ||
In follow-up studies, the Stephan group showed that the boron-based Lewis acid can be varied widely to give adducts of the general formula ‘tBu3P(N2O)BR2R’.39–41 In contrast, phosphines with reduced steric hindrance or Lewis basicity do not form similar compounds. The borane can be exchanged for other Lewis acids. The adduct tBu3P(N2O)B(C6H4F)3 is particularly well suited for exchange reactions because it contains the relatively weak Lewis acid B(C6H4F)3. For example, the dinuclear complex 8 was obtained when adding one equivalent of Zn(C6F5)2,39 whereas exchange with [Cp2ZrMe][MeB(C6F5)3] gave complex 9 (Scheme 6b).40
The capture of N2O can also be achieved by using an alane. The adduct tBu3P(N2O)Al(C6F5)3 (10) was obtained by slow addition of N2O to a cooled solution containing PtBu3 (2 eq.) and Al(C6F5)3(toluene) (Scheme 6c).42 The reaction with additional Al(C6F5)3(toluene) resulted in N–O bond rupture, generating the highly reactive radical ion pair (tBu3P˙)[(C6F5)3Al(O˙)Al(C6F5)3] that can activate C–H bonds.
The colorimetric detection of N2O was realized using a borane with a ferrocenyl (Fc) substituent as the Lewis acid in an FLP.43 Exposing a mixture of PtBu3 and B(C6F5)2Fc to N2O resulted in the formation of the adduct tBu3P(N2O)B(C6F5)2Fc (11), accompanied by a color change from maroon to amber (Scheme 6d). A different UV-Vis-responsive FLP was created by using a phosphine containing a cycloheptatrienyl-cyclopentadienyl titanium sandwich complex as substituent.44
The use of a single-component FLP with a dimethylxanthene backbone allowed for the reversible binding of N2O.45 Exposing a solution of this FLP in dichloromethane to one atmosphere of N2O resulted in the slow (t1/2 ∼ 12 h) formation of the adduct 12 (Scheme 6e). Warming a solution of this adduct in dichloromethane to 50 °C for 2 h led to the quantitative removal of N2O.
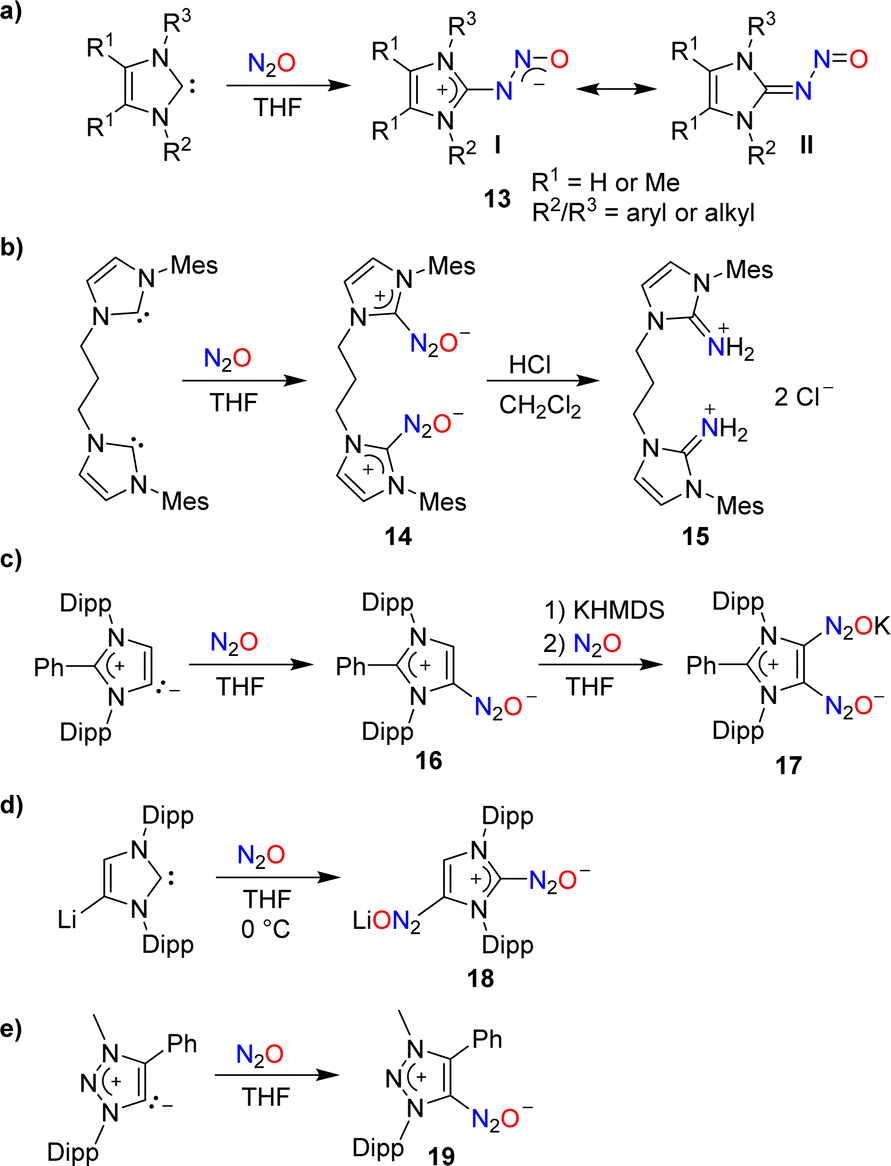
In 2012, our group demonstrated that N-heterocyclic carbenes (NHCs) can effectively capture N2O.46 When a solution of 1,3-dimesitylimidazol-2-ylidene (IMes) in THF was subjected to one atmosphere of N2O, the adduct IMes(N2O) was formed in high yield (90%). A similar compound was obtained using an imidazole-2-ylidene with Dipp wingtip groups (IPr). Subsequent studies by our group and by others showed that N2O adducts of the general formula NHC(N2O) (13) are accessible with a range of different substituents on the heterocycle (Scheme 7a).47–49 The adducts can be described as zwitterionic imidazolium diazotates (13, I) or as nitrosoimines (13, II). Crystallographic analyses revealed a preference for a trans configuration for the N–N bond, even though exceptions have been reported.48 NHC(N2O) adducts display good stability at room temperature. At elevated temperatures, the release of N2 and formation of the corresponding ureas was observed.
 | ||
| Scheme 7 Covalent capture of N2O by N-heterocyclic carbenes (NHCs). | ||
The addition of Brønsted acids to IMes(N2O) resulted in the rupture of the N–N bond and the formation of N-heterocyclic iminium salts.47 This type of reactivity was used by Dielmann and coworkers for the synthesis of the dimer 15 (Scheme 7b).50 The latter was employed as a precursor for the synthesis of a chelate ligand.
N–N bond cleavage was also observed when IMes(N2O) was combined with nickel(0)51 or cobalt(I)52 complexes. A different type of reactivity was noted in reactions with vanadium(III)53,54 and uranium(III)55 complexes. Here, NHC(N2O) adducts served as mild O-atom donors.
Similar to Arduengo-type NHCs, mesoionic carbenes derived from C2-arylated 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene are able to form adducts with N2O (Scheme 7c).56 Interestingly, it was possible to introduce a second N2O group by treating adduct 16 with first potassium hexamethyldisilazide (KHMDS) and then N2O.
A direct double functionalization with two N2O groups was observed when a solution of lithiated IPr in THF was subjected to an atmosphere of N2O (Scheme 7d).56
N2O capture can also be achieved by triazole-based carbenes: the triazolium diazotate 19 was isolated in 86% yield from a reaction of the corresponding carbene with N2O (Scheme 7e).57
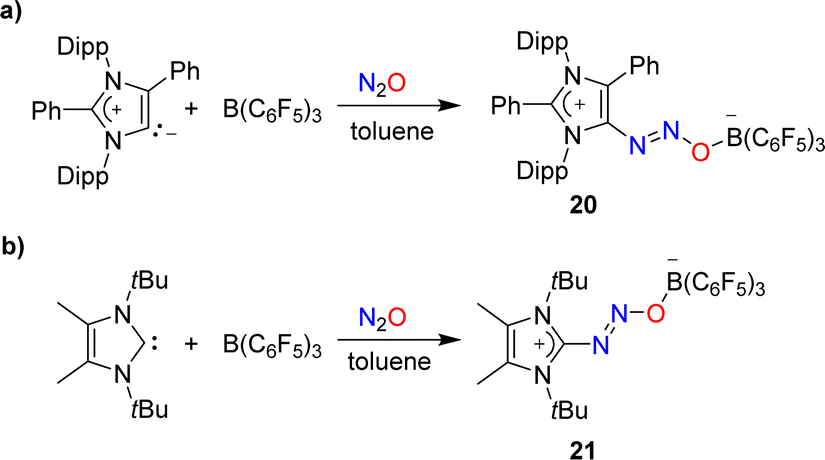
Attempts to capture N2O with a mesoionic carbene featuring phenyl substituents at the 2- and the 4-position were unsuccessful. However, in the presence of B(C6F5)3, the C–N2O–B-bridged adduct 20 was isolated (Scheme 8a).58 A similar situation was encountered with a carbene having tert-butyl wingtip groups and methyl substituents in 4/5-position: while direct N2O capture by the carbene could not be achieved, an adduct (21) was obtained in the presence of the Lewis acid B(C6F5)3 (Scheme 8b).49
 | ||
| Scheme 8 Covalent capture of N2O by mixtures of N-heterocyclic carbenes and B(C6F5)3. | ||
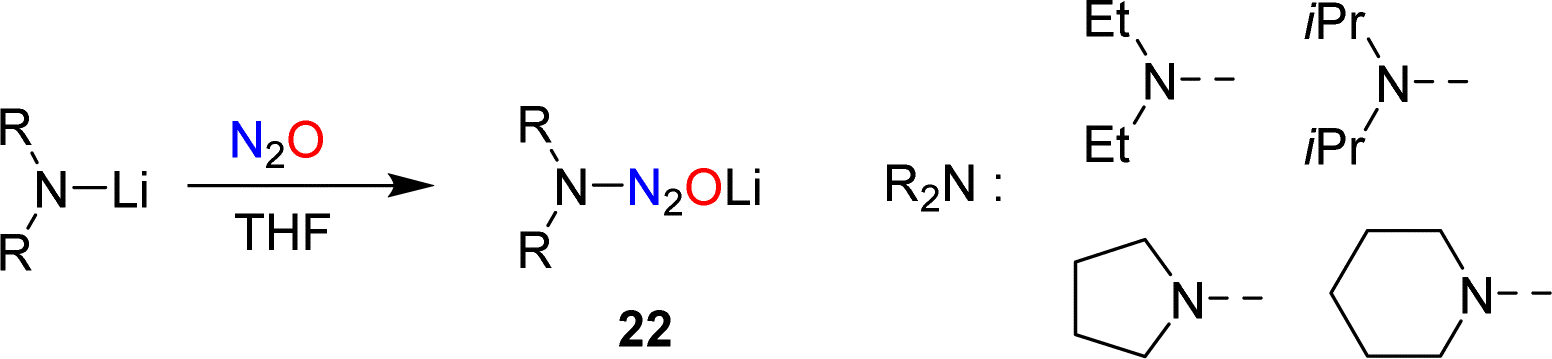
In 1953, Meier reported that lithiated amines react with N2O.59 In the case of Et2NLi, he was able to isolate tetraethyltetrazene, albeit in low yield. Meier proposed aminodiazotates as intermediates, but the isolation of these adducts was not attempted. Our group has re-investigated this type of reaction and found that aminodiazotates (22) are formed in good yields when solutions of lithium amides in THF are subjected to an atmosphere of N2O (Scheme 9).60 N2O adducts of type 22 can serve as precursors for the synthesis of triazenes, and more details about such transformations are given in Section 4.
 | ||
| Scheme 9 Covalent capture of N2O by lithium amides. | ||
3. Synthesis of azides
The standard procedure for the synthesis of NaN3 involves the reaction between NaNH2 and N2O (see Section 1).23,24 Meier showed that this chemistry can be extended to organic azides. He noted that a pale yellow oil, most likely phenyl azide, was formed in low yield when a solution of lithiated aniline in diethyl ether was exposed to N2O.59A more detailed investigation was conducted by Koga and Anselme in 1968.61 They showed that aryl azides (23) are formed by reactions of lithiated aromatic amines with N2O (Scheme 10). However, the yields of these diazo transfer reactions were poor (<35%). Significantly higher yields were obtained when silylated aryl amides were used as starting materials in NMR-scale reactions.62 When the reactions were performed on a preparative scale, an increased amount of side products was observed.
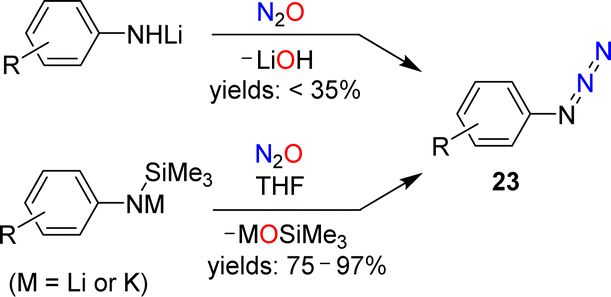 | ||
| Scheme 10 Synthesis of aryl azides. | ||
4. Synthesis of triazenes
Aromatic triazenes of the general formula (aryl)N3R2 have been investigated extensively in the context of synthetic organic chemistry.63 An important feature of aryl triazenes is the possibility to replace the N3R2 group under acidic conditions by a broad range of other functionalities. The substitution reactions proceed via diazonium compounds, and aryl triazenes are often referred to as “masked diazonium salts”.64Aryl triazenes of type (aryl)N3R2 are typically prepared by coupling of aryldiazonium salts with secondary amines.63 In 2015, our group reported an alternative synthetic procedure involving nitrous oxide.60 Solutions of lithium amides in THF were allowed to react with N2O, resulting in the formation of aminodiazotates (22). The latter were not isolated,65 but combined directly with aryl Grignard reagents to give aryl triazenes of type 24 (Scheme 11).
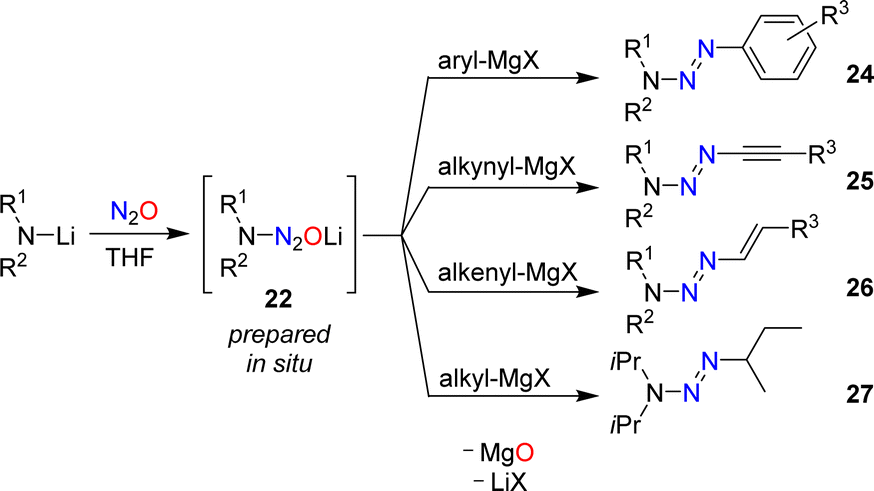 | ||
| Scheme 11 Synthesis of triazenes by reactions of N2O-derived aminodiazotates with Grignard reagents. | ||
A key advantage of the N2O-based methodology for synthesizing triazenes is that it can be extended to alkynyl (25) and alkenyl triazenes (26). These compounds are difficult to access by alternative procedures.66,67 Alkyl triazenes can also be prepared by this method, as illustrated by the synthesis of 1-isobutyl-3,3-diisopropyltriazene (27). However, the yield of 27 was low (12%).
Alkynyl triazenes are attractive starting materials for application in organic synthesis (Scheme 12).66 From a practical standpoint, it is worth noting that alkynyl triazenes are not particularly sensitive to air or moisture. Furthermore, they can be purified by chromatography, and they exhibit good thermal stability.
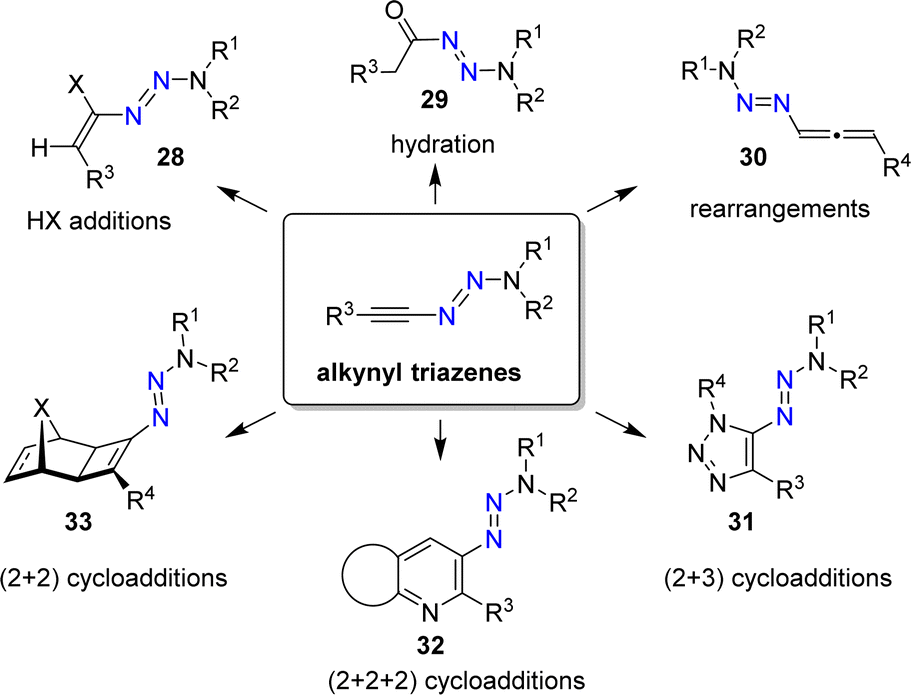 | ||
| Scheme 12 Alkynyl triazenes as versatile starting materials in synthetic organic chemistry. | ||
The N3R2 group is electron-donating, resulting in an ynamide-like reactivity for alkynyl triazenes. For example, it is possible to perform addition reactions with Brønsted acids to give alkenyl triazenes of type 28.68,69 The acid-catalyzed hydration of alkynyl triazenes provides acyl triazenes (29),70 and allenyl triazenes of type 30 are accessible by base-induced rearrangements.71
Alkynyl triazenes are suitable substrates for transition metal-catalyzed cycloaddition reactions. Cui and coworkers have prepared a wide range of triazoles (31) by Ir-catalyzed (2 + 3) cycloaddition reactions of alkynyl triazenes and organic azides (Scheme 12).72 In our group, we have used Ru-catalyzed cycloaddition reactions for synthesizing densely functionalized arenes and pyridines (32),73 as well as cyclobutenyl triazenes (33).74 Further transformations of alkynyl triazenes include Rh-catalyzed annulation reactions,75–77 Au-catalyzed cyclizations,78,79 Pd-catalyzed addition reactions,80 Ficini-type reactions,81 light-induced isomerizations,82 and electrophilic fluorinations.83
An interesting aspect of using alkynyl triazenes as substrates in these reactions is the possibility of performing post-synthetic substitution reactions. For example, the triazene group in pyridines of type 32 can be substituted by a wide range of nucleophiles, including fluoride.
The synthesis of alkynyl triazenes by coupling of lithium amides first with nitrous oxide and then with an alkynyl Grignard reagent restricts the functional groups that can be employed. To overcome this limitation, we have developed a synthetic route for a terminal alkynyl triazene, 35 (Scheme 13).84 Subsequent functionalization of 35 allowed to prepare alkynyl triazenes with a range of functional groups including esters, alcohols, cyanides, phosphonates, and amides.
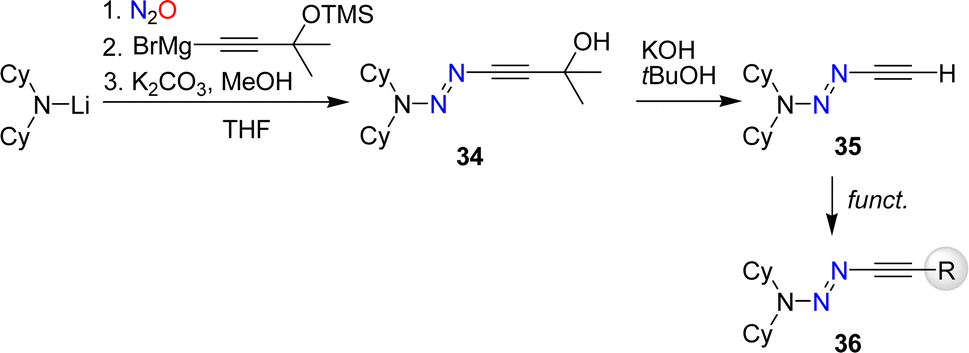 | ||
| Scheme 13 Synthesis of a terminal alkynyl triazene and its functionalization. | ||
5. Synthesis of N-heterocycles
The reactions of alkenes and alkynes with N2O generally proceed via O-atom transfer and liberation of dinitrogen.15 An interesting exception to this type of reactivity was reported by Banert and Plefka.85 When cyclooctyne or cycloocten-5-yne were treated with nitrous oxide (∼50 bar) in the presence of nucleophiles (amines or alcohols), the formation of pyrazoles of type 37 was observed (Scheme 14). The reactions were proposed to proceed via heterocycles of type 38. In the case of cyclooctyne, the intermediate could be isolated if the reaction was performed without nucleophiles.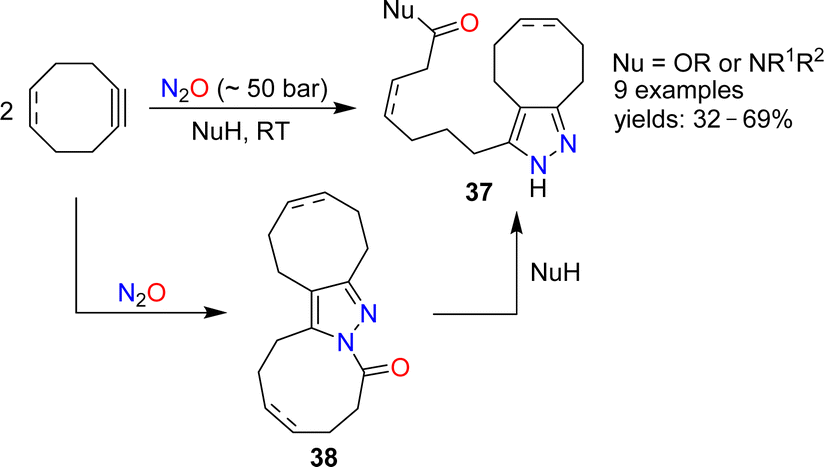 | ||
| Scheme 14 Synthesis of pyrazoles. | ||
Cui and coworkers have shown that nitrous oxide can be used for the synthesis of benzotriazines.86 Aromatic amides or sulfonamides were deprotonated with strong bases and then exposed to an atmosphere of N2O. After work-up, the heterocycles 39 or 40 were obtained (Scheme 15). The substrate scope for these transformations was found to be broad, and the heterocycles were obtained in synthetically useful yields. The authors propose that the reactions are initiated by the reaction of N2O with an aryl lithium species, leading to the formation of a diazotate. The products are then generated by N–N bond formation and liberation of Li2O.
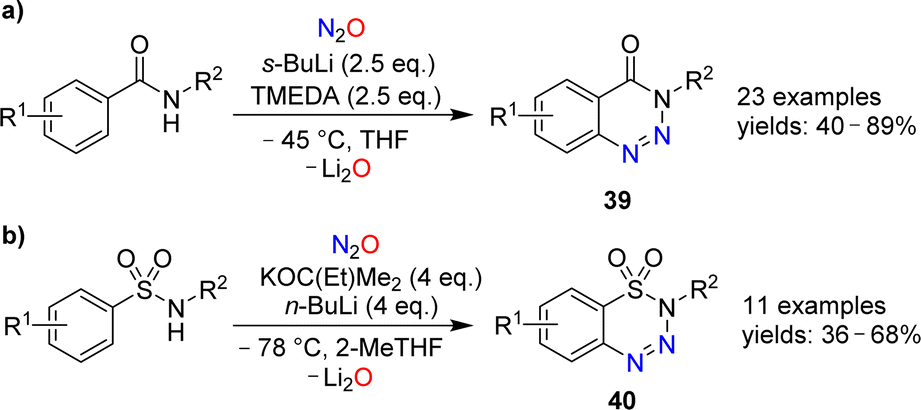 | ||
| Scheme 15 Syntheses of benzotriazines. | ||
Azobenzene can be obtained by reaction of phenylcalcium iodide and N2O (see Section 6). A related diazo transfer reaction was observed when a dimeric biphenylcalcium complex was mixed with N2O.87 Benzo[c]cinnoline (41) was obtained in 55% yield (Scheme 16).
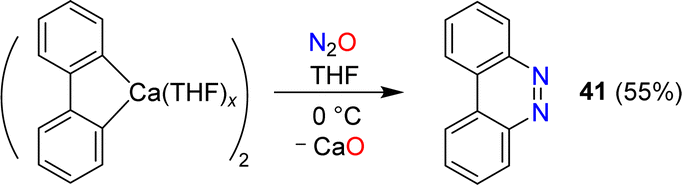 | ||
| Scheme 16 Synthesis of benzo[c]cinnoline. | ||
Triazolopyridines are valuable starting materials for heterocycle synthesis.88 Our group has shown that triazolopyridines can be prepared using nitrous oxide.89,90 A range of lithiated 2-benzylpyridines could be converted into triazolopyridines of type 42 upon reaction with N2O (Scheme 17a).
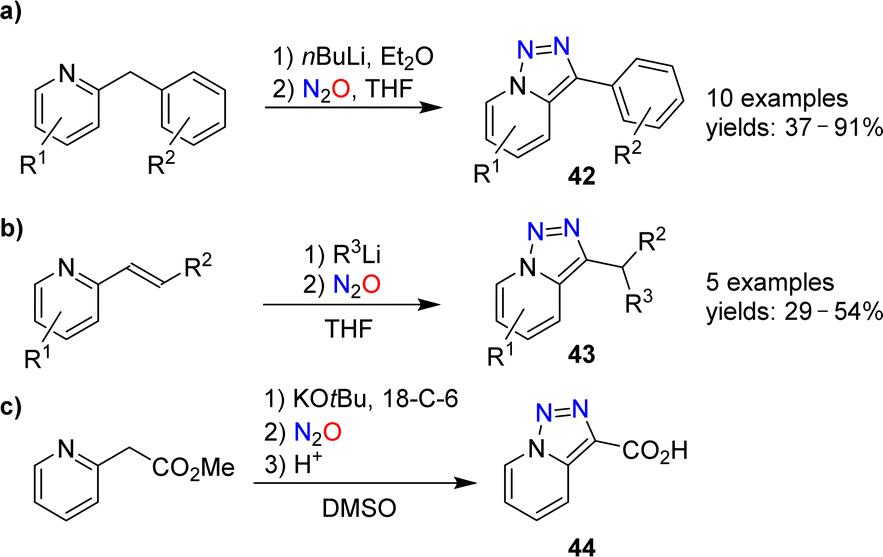 | ||
| Scheme 17 Syntheses of triazolopyridines. | ||
The diazo transfer reaction can be combined with a C–C bond-forming reaction. Heterocycles of type 43 were obtained by coupling of organolithium reagents with 2-vinylpyridines, followed by N2O-induced triazole formation (Scheme 17b). The carboxylic acid 44, on the other hand, was prepared in 89% yield by deprotonation of methyl 2-(pyridin-2-yl)acetate, followed by reaction with N2O and hydrolysis (Scheme 17c).
6. Synthesis of azo compounds
In 1953, Beringer, Farr and Sands published a study describing reactions of organolithium reagents with N2O.91 For phenyllithium, they observed a mixture of products, including biphenyl, triphenylhydrazine, phenol, and a small amount (7%) of azobenzene 45 (Scheme 18a). Similar products were found by Meier when using PhNa instead of PhLi. Meier also showed that PhCaI can be converted into azobenzene.92 In this context, it is worth noting that aryl Grignard reagents are largely inert towards N2O.92,93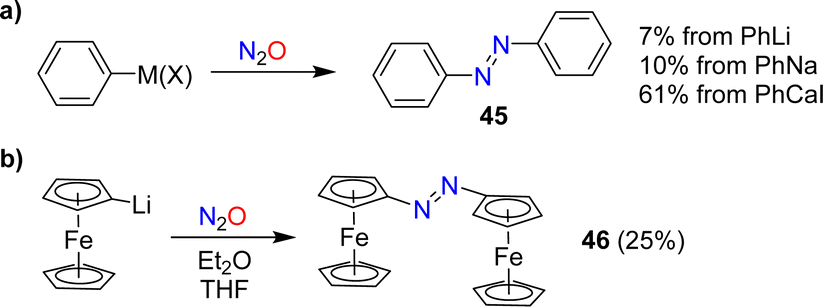 | ||
| Scheme 18 Synthesis of azobenzene and azo-bridged ferrocene. | ||
In 1995, the reaction between PhCaI and N2O was re-investigated by Hays and Hanusa.94 By optimizing the procedure, they were able to obtain azobenzene with a yield of up to 61% (Scheme 18a). However, they noted difficulties in obtaining reproducible results.
Azo-bridged ferrocene (46) was obtained in 25% yield by reaction of lithiated ferrocene with N2O (Scheme 18b).95 A related reaction was used to synthesize azo-bridged ferrocene oligomers.96
N-Heterocyclic carbenes are able to form stable covalent adducts with N2O (see Section 2 and Scheme 7). In the presence of AlCl3, adducts of type 13 can be coupled to arenes (Scheme 19a).97 The resulting azo compounds are of interest as dyes. They are produced industrially via different routes, and they have found diverse applications.98 The N2O-based methodology has a good scope with regard to the arene coupling partner, and NHC(N2O) adducts with alkyl or aryl wingtip groups can be employed in these reactions.
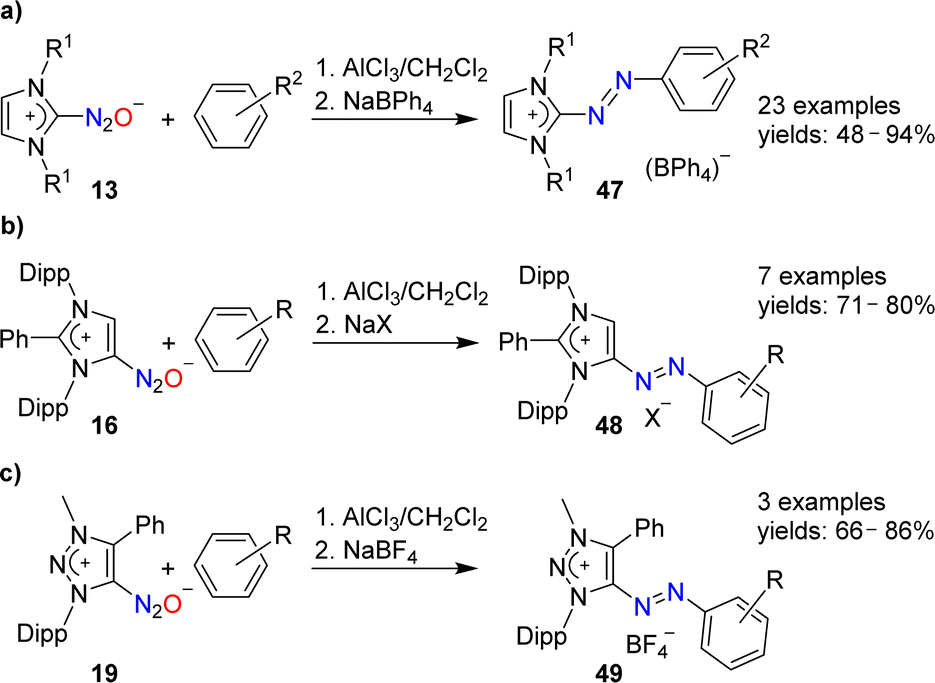 | ||
| Scheme 19 Synthesis of cationic azo dyes from NHC–N2O adducts. | ||
Azoimidazolium dyes with N-aryl substituents were found to display interesting chemistry. Upon reduction, stable aminyl radicals were formed.99 Moreover, they can be used as precursors for mesoionic carbene ligands.100
The AlCl3-mediated coupling chemistry can be extended to N2O adducts of mesoionic carbenes.101 Azoimidazolium salts of type 48 were formed by coupling of arenes with 16 (Scheme 19b), whereas azotriazolium salts (49) were obtained from 19 (Scheme 19c).
N-Heterocyclic olefins (NHOs) display a highly polarized exocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, making them strong bases and nucleophiles.102 In 2019, our group reported that NHOs with Dipp, mesityl or xylyl wingtip groups are able to activate N2O.103 When a solution of the respective NHO in acetonitrile was subjected to an atmosphere of N2O, azo-bridged dimers of type 50 were obtained (Scheme 20). The yields were not high (∼50%), but the products were easily isolated because they crystallized from solution. Reactions between NHOs and N2O can also give diazoolefins, and more details about these transformations are given in the next section.
C double bond, making them strong bases and nucleophiles.102 In 2019, our group reported that NHOs with Dipp, mesityl or xylyl wingtip groups are able to activate N2O.103 When a solution of the respective NHO in acetonitrile was subjected to an atmosphere of N2O, azo-bridged dimers of type 50 were obtained (Scheme 20). The yields were not high (∼50%), but the products were easily isolated because they crystallized from solution. Reactions between NHOs and N2O can also give diazoolefins, and more details about these transformations are given in the next section.
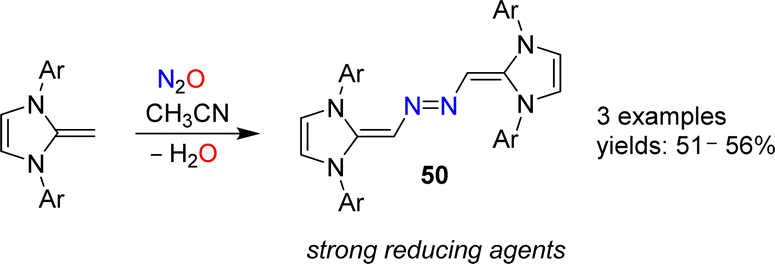 | ||
| Scheme 20 Synthesis of azo-bridged N-heterocyclic olefins. | ||
The dimers 50 were found to be very strong electron donors, with first oxidation potentials between −1.32 and −1.38 V (vs. Fc/Fc+). Upon reduction, stable radical cations or dicationic imidazolium salts were obtained.103
7. Synthesis of diazo compounds
The reaction of N2O with methyllithium was first investigated by Müller and coworkers.104 They found that diazomethane (51) was formed after basic workup (Scheme 21a). A yield of 70% was obtained under optimized conditions.105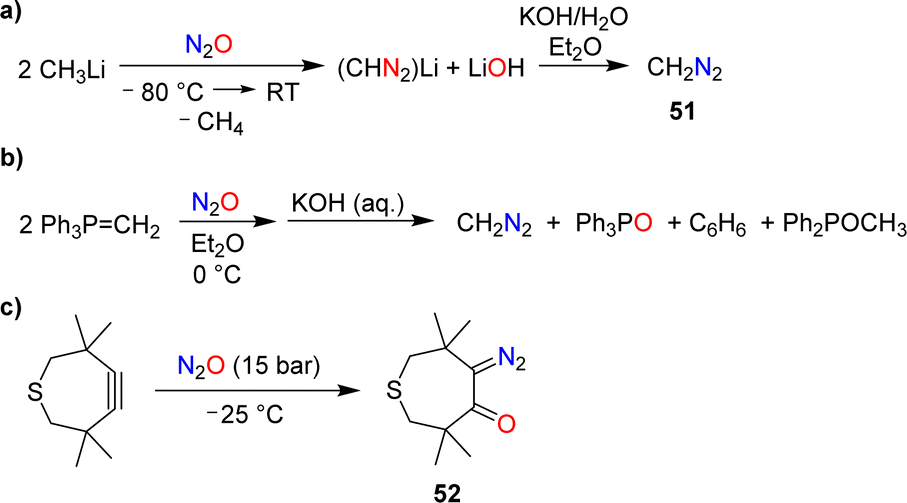 | ||
| Scheme 21 Synthesis of diazomethane (a and b) and addition of N2O to a cyclic alkyne (c). | ||
The formation of diazomethane was also evidenced in reactions of the ylide Ph3PCH2 with N2O (Scheme 21b).106 However, the yield of CH2N2 in this transformation was low (20–25%).
During their investigations about reactions of cyclic alkynes with N2O, Banert and Plefka were able to isolate the diazo compound 52 in 95% yield (Scheme 21c).85 Upon warming to room temperature, loss of dinitrogen was observed, resulting in a mixture of compounds.
Erker and coworkers have investigated the reactivity of a carbene-stabilized boraalkene.107 The reaction with N2O gave a mixture of the diazo compound 53 and the oxaborirane 54 (Scheme 22). The authors propose that the compounds are derived from the same intermediate, a (2 + 3) cycloaddition product of the starting material and N2O.
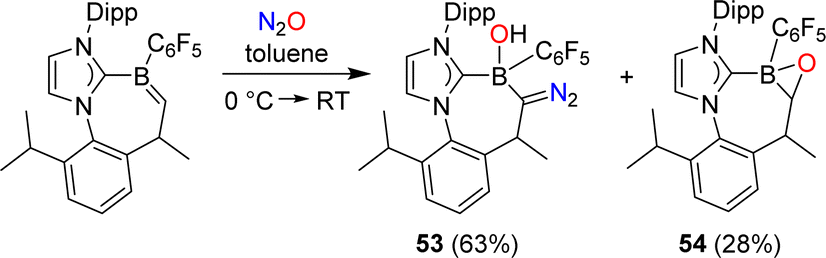 | ||
| Scheme 22 Synthesis of diazo compound 53. | ||
Diazoolefins of the general formula R1R2CCN2 (R1/2 = alkyl, aryl, or H) are highly reactive compounds, which rapidly lose N2.108 In 2021, the Hansmann group reported that N2O could be used for the synthesis of a room-temperature-stable diazoolefin (diazoalkene).109 The reaction of a mesoionic NHO110 with N2O gave diazoolefin 55 along with amide 56 (Scheme 23a). The diazoolefin could be isolated in 41% yield. A crystallographic analysis of 55 revealed a bent heterocumulene group. The unusual stability of 55 was attributed to both resonance stabilization and polarization of the C–CN2 bond.108a,109
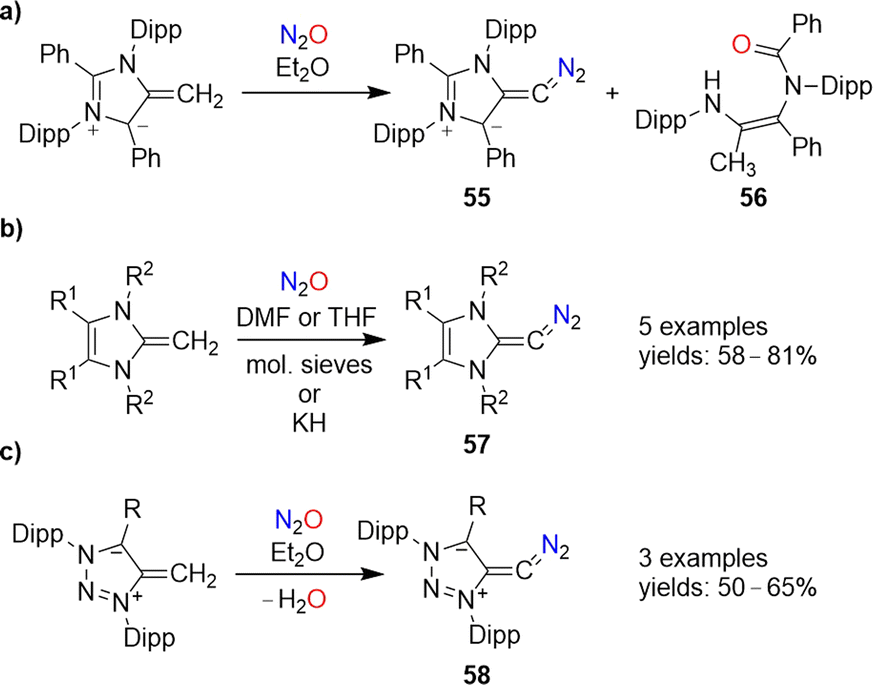 | ||
| Scheme 23 Synthesis of N-heterocyclic diazoolefins. | ||
‘Normal’ N-heterocyclic olefins can also react with N2O to give diazoolefins of type 57 (Scheme 23b). First examples were published by our group in 2021,111 and a new member of this compound class with R1 = R2 = Me was recently disclosed by Bismuto and coworkers.112
The use of triazole-based NHOs allowed access to diazoolefins of type 58 (Scheme 23d).113 It is worth noting that both 57 and 58 can also be prepared by using the more conventional diazo transfer reagent p-tosyl azide instead of N2O.114 While nitrous oxide is more atom-economical, the use of p-TsN3 avoids the formation of the potentially problematic side product water.
N-Heterocyclic diazoolefins display intriguing chemistry, as evidenced by recent studies (Scheme 24).108a They can be used as C-donor ligands for metal complexes110,111,115,116 and as precursors for N-heterocyclic vinylidenes (Scheme 24).112,117–119 The N2 group of N-heterocyclic diazoolefins can be exchanged for isocyanides or for CO to give novel heterocumulenes.110,113,120 Cycloaddition reactions with dipolarophiles give pyrazole derivatives,111,121 and methanol was found to promote the dimerization of N-heterocyclic diazoolefins.122
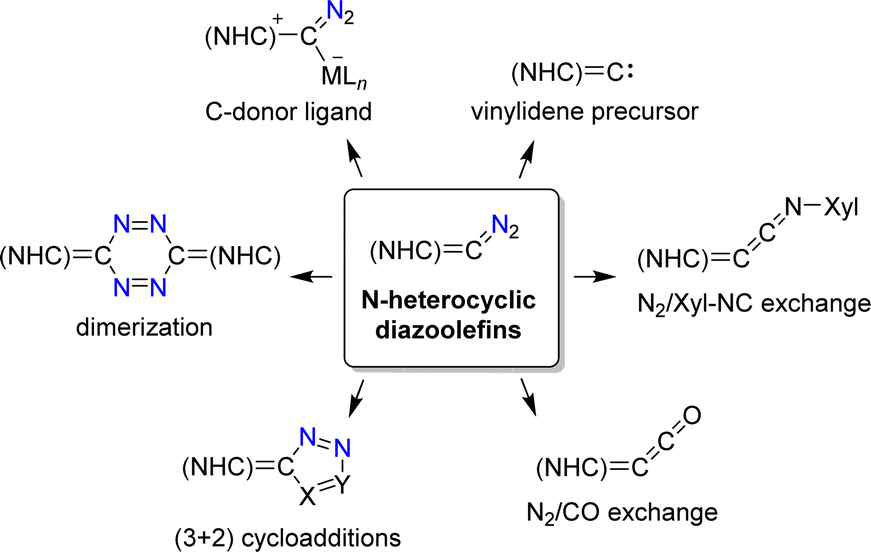 | ||
| Scheme 24 The multifaceted chemistry of N-heterocyclic diazoolefins. | ||
The conversion of NHOs to diazoolefins requires the presence of a terminal CH2 group. Gellrich and coworkers reported that a gem-dimethylated NHO was still able to activate N2O.123 They observed cleavage of the exocyclic double bond to give the urea 59 along with azine 60 (Scheme 25). The latter was formed by denitrogenative coupling of 2-diazopropane.
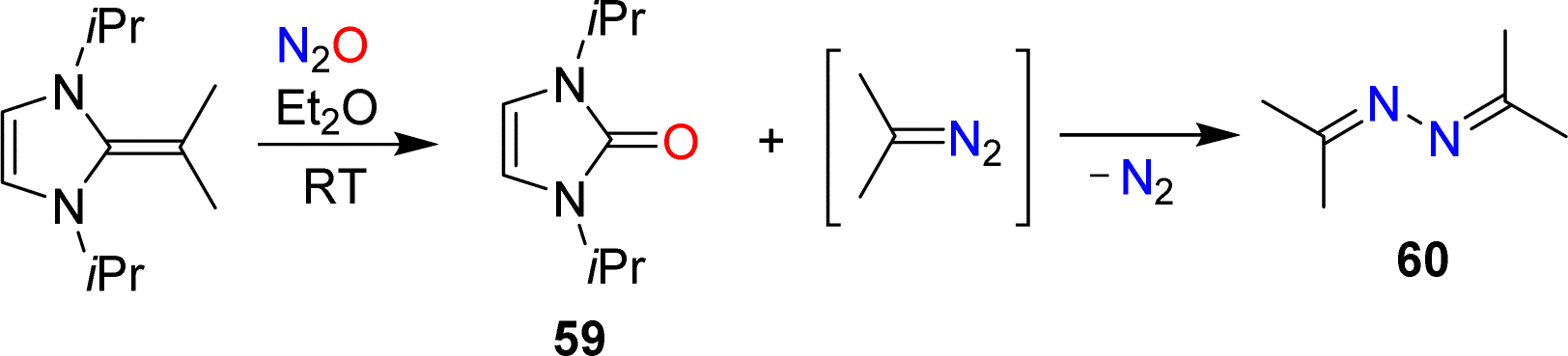 | ||
| Scheme 25 The reaction of a dimethylated N-heterocyclic olefin with nitrous oxide. | ||
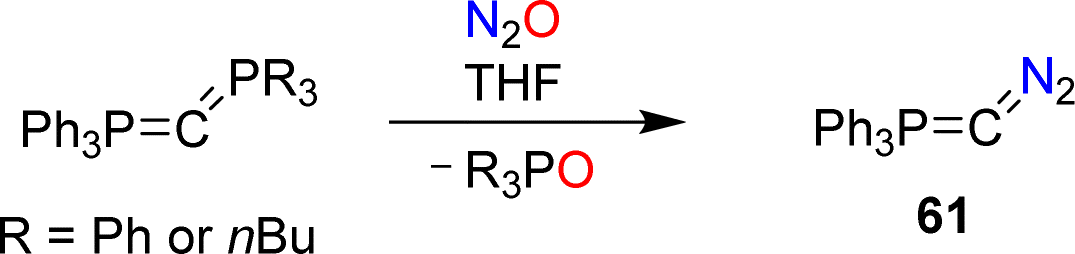
Recently, the Hansmann group reported the synthesis of the diazophosphorus ylide 61.124 The diazo compound was obtained by combining carbodiphosphoranes Ph3PCPR3 (R = Ph or nBu) with nitrous oxide (Scheme 26). The ylide serves as a selective transfer reagent for the fragments Ph3PC and CN2. Furthermore, carbon-atom transfer was observed in reactions of 61 with aldehydes and ketones.
 | ||
| Scheme 26 Synthesis of the diazophosphorus ylide 61. | ||
8. Conclusions
In synthetic chemistry, nitrous oxide is well known for its ability to act as an oxygen-atom transfer reagent. The present review highlights a distinct reactivity of N2O: diazo transfer. Although the application of N2O for diazo transfer dates back to the 19th century, it is only in recent years that these reactions have received increased interest.High-yielding diazo transfer reactions with N2O were realized with a range of compounds including lithium amides, metalated arenes and alkanes, N-heterocyclic carbenes, N-heterocyclic olefins, and carbodiphosphoranes. The reactions with these nucleophiles are likely initiated by an attack at the terminal nitrogen atom of N2O. In the case of carbenes and amides, the corresponding diazoates could be isolated and characterized. For other nucleophiles, spontaneous N–O bond rupture gave directly nitrogen-containing products.
Several of the compounds described in this review can be prepared by using alternative synthetic procedures. In this case, the advantages and disadvantages of the N2O-based methodology must be balanced considering specific constraints (yields, costs, time, availability of N2O, etc.). For some compounds, nitrous oxide remains the sole viable option for synthesis to date. Alkynyl triazenes, for example, can thus far only be accessed with N2O. These activated alkynes are very attractive starting materials for synthetic organic chemistry.66
Overall, we hope to have shown with this review that nitrous oxide is more than a simple O-atom donor. Efficient diazo transfer was observed in reactions with a range of carbon- and nitrogen-based nucleophiles. We are confident that there is significant room for further developments. Nitrous oxide has the potential to become a routinely used reagent in synthetic organic and inorganic chemistry.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
A. G. and K. S. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the European Union under the Marie Skłodowska-Curie fellowship HORIZON-TMA-MSCA-PF-GF No. 1011150286, by the Swiss National Science Foundation, and by the École Polytechnique Fédérale de Lausanne (EPFL).References
- H. Davy, Researches, Chemical and Philosophical; Chiefly Concerning Nitrous Oxide, or Dephlogisticated Nitrous Air, and its Respiration, Printed for J. Johnson, 1800, London, available at: https://wellcomecollection.org/works/wtdr8dvd.
- BBC News, Nitrous oxide: Laughing gas possession becomes illegal, retrieved June 6th, 2024, https://www.bbc.com/news/uk-67344299.
- R. L. Thompson, L. Lassaletta, P. K. Patra, C. Wilson, K. C. Wells, A. Gressent, E. N. Koffi, M. P. Chipperfield, W. Winiwarter, E. A. Davidson, H. Tian and J. G. Canadell, Nat. Clim. Change, 2019, 9, 993–998 CrossRef CAS.
- (a) D. Kanter, D. L. Mauzerall, A. R. Ravishankarac, J. S. Daniel, R. W. Portmann, P. M. Grabiel, W. R. Moomaw and J. N. Galloway, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4451–4457 CrossRef CAS; (b) A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123–125 CrossRef CAS PubMed.
- D. S. Reay, E. A. Davidson, K. A. Smith, P. Smith, J. M. Melillo, F. Dentener and P. J. Crutzen, Nat. Clim. Change, 2012, 2, 410–416 CrossRef CAS.
- (a) X. Wu, J. Du, Y. Gao, H. Wang, C. Zhang, R. Zhang, H. He, G. Lu and Z. Wu, Chem. Soc. Rev., 2024 10.1039/d3cs00919j; (b) Z. Zhuang, B. Guan, J. Chen, C. Zheng, J. Zhou, T. Su, Y. Chen, C. Zhu, X. Hu, S. Zhao, J. Guo, H. Dang, Y. Zhang, Y. Yuan, C. Yi, C. Xu, B. Xu, W. Zeng, Y. Li, K. Shi, Y. He, Z. Wei and Z. Huang, Chem. Eng. J., 2024, 486, 150374 CrossRef CAS; (c) L. Alves, L. I. V. Holz, C. Fernandes, P. Ribeirinha, D. Mendes, D. P. Fagg and A. Mendes, Renewable Sustainable Energy Rev., 2022, 155, 111916 CrossRef CAS; (d) U. Singh, M. Algren, C. Schoeneberger, C. Lavallais, M. G. O'Connell, D. Oke, C. Liang, S. Das, S. D. Salas and J. B. Dunn, iScience, 2022, 25, 105661 CrossRef CAS PubMed; (e) L. Li, J. Xu, J. Xu and J. Han, Environ. Sci. Technol., 2014, 48, 5290–5297 CrossRef CAS.
- (a) P. Capała, M. Ruszak, A. Rudawska, M. Inger and M. Wilk, Appl. Sci., 2023, 13, 7492 CrossRef; (b) J. Pérez-Ramírez, F. Kapteijn, K. Schöffel and J. A. Moulijn, Appl. Catal., B, 2003, 44, 117–151 CrossRef.
- (a) S. He, Y. Han and X. Qin, J. Environ. Sci., 2024 DOI:10.1016/j.jes.2024.03.014; (b) F. Cavani and H. Teles, ChemSusChem, 2009, 2, 508–534 CrossRef CAS.
- A Chinese Chemical Company Captures and Reuses 6000 Tons of a Super-Polluting Greenhouse Gas, Inside Climate News report, https://insideclimatenews.org/news/02102020/china-super-pollutants-nitrous-oxide-linggas-henan-shenma/, retrieved June 6th, 2024 Search PubMed.
- (a) F. Thrun, V. Hickmann, C. Stock, A. Schäfer, W. Maier, M. Breugst, N. E. Schlörer, A. Berkessel and J. H. Teles, J. Org. Chem., 2019, 84, 13211–13220 CrossRef CAS PubMed; (b) BASF Starts Upa New Production Facility for Intermediates, BASF news release, https://www.chemicalonline.com/doc/basf-starts-up-a-new-production-facility-for-0001 retrieved June 6th, 2024; (c) J. H. Teles, B. Roessler, R. Pinkos, T. Genger and T. Preiss, Method for producing a ketone, US Pat., US7449606, 2008 Search PubMed.
- K. O. Denisova, A. A. Ilyin, R. N. Rumyantsev, A. P. Ilyin and A. V. Volkova, Russ. J. Gen. Chem., 2019, 89, 1338–1346 CrossRef CAS.
- V. N. Parmon, G. I. Panov and A. S. Noskov, Catal. Today, 2005, 100, 115–131 CrossRef CAS.
- (a) I. Surin, Z. Tang, J. Geiger, S. Damir, H. Eliasson, M. Agrachev, F. Krumeich, S. Mitchell, V. A. Kondratenko, E. V. Kondratenko, G. Jeschke, R. Erni, N. López and J. Pérez-Ramírez, Adv. Mater., 2023, 35, 2211260 CrossRef CAS; (b) Q. Yang, I. Surin, J. Geiger, H. Eliasson, M. Agrachev, V. A. Kondratenko, A. Zanina, F. Krumeich, G. Jeschke, R. Erni, E. V. Kondratenko, N. López and J. Pérez-Ramírez, ACS Catal., 2023, 13, 15977–15990 CrossRef CAS PubMed; (c) Z. Tang, I. Surin, A. Rasmussen, F. Krumeich, E. V. Kondratenko, V. A. Kondratenko and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2022, 61, e202200772 CrossRef CAS.
- A. V. Leont’ev, O. A. Fomicheva, M. V. Proskurnina and N. S. Zefirov, Russ. Chem. Rev., 2001, 70, 91–104 CrossRef.
- K. A. Dubkov, G. I. Panov and V. N. Parmon, Russ. Chem. Rev., 2017, 86, 510–529 CrossRef CAS.
- A. M. Arinaga, M. C. Ziegelski and T. J. Marks, Angew. Chem., Int. Ed., 2021, 60, 10502–10515 CrossRef CAS PubMed.
- K. Severin, Chem. Soc. Rev., 2015, 44, 6375–6386 RSC.
- For selected recent publications describing the oxidation of main group element compounds with N2O, see: (a) M. Jörges, S. Mondal, M. Kumar, P. Duari, F. Krischer, J. Löffler and V. H. Gessner, Organometallics, 2024, 43, 585–593 CrossRef; (b) L. Groll, J. A. Kelly and S. Inoue, Chem.–Asian J., 2024, 19, e202300941 CrossRef CAS PubMed; (c) T. S. Koptseva, M. V. Moskalev, E. V. Baranov and I. L. Fedushkin, Organometallics, 2023, 42, 965–970 CrossRef CAS; (d) J. Schoening, C. Wölper and S. Schulz, Eur. J. Inorg. Chem., 2022, e202200638 Search PubMed; (e) P. Garg, D. Dange, Y. Jiang and C. Jones, Dalton Trans., 2022, 51, 7838–7844 RSC; (f) Y. Xiong, S. Yao, A. Ruzicka and M. Driess, Chem. Commun., 2021, 57, 5965–5968 RSC; (g) D. Dhara, P. K. Pal, R. Dolai, N. Chrysochos, H. Rawat, B. J. Elvers, I. Krummenacher, H. Braunschweig, C. Schulke, V. Chandrasekhar, U. D. Priyakumar and A. Jana, Chem. Commun., 2021, 57, 9546–9549 RSC; (h) N. Szynkiewicz, J. Chojnacki and R. Grubba, Inorg. Chem., 2020, 59, 6332–6337 CrossRef CAS PubMed; (i) L. Anthore-Dalion, E. Nicolas and T. Cantat, ACS Catal., 2019, 9, 11563–11567 CrossRef CAS; (j) A. V. Protchenko, P. Vasko, D. C. H. Do, J. Hicks, M. Á. Fuentes, C. Jones and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 1808–1812 CrossRef CAS PubMed.
- For reviews, see: (a) S. Sinhababu, Y. Lakliang and N. P. Mankad, Dalton Trans., 2022, 51, 6129–6147 RSC; (b) S. R. Pauleta, M. S. P. Carepo and I. Moura, Coord. Chem. Rev., 2019, 387, 436–449 CrossRef CAS; (c) S. R. Pauleta, S. Dell'Acqua and I. Moura, Coord. Chem. Rev., 2013, 257, 332–349 CrossRef CAS; (d) W. B. Tolman, Angew. Chem., Int. Ed., 2010, 49, 1018–1024 CrossRef CAS.
- For selected recent publications describing the activation of N2O by synthetic transition metal complexes, see: (a) J. Bermejo, I. Ortega-Lepe, L. L. Santos, N. Rendón, J. López-Serrano, E. Álvarez and A. Suárez, Chem. Commun., 2024, 60, 1575–1578 RSC; (b) N. P. Mankad, Chem. Sci., 2024, 15, 1820–1828 RSC; (c) L. M. A. Quintana, Y. Yang, A. Ramanathan, N. Jiang, J. Bacsa, L. Maron and H. S. La Pierre, Chem. Commun., 2021, 57, 6664–6667 RSC; (d) C.-W. Hsu, S. C. Rathnayaka, S. M. Islam, S. N. MacMillan and N. P. Mankad, Angew. Chem., Int. Ed., 2020, 59, 627–631 CrossRef CAS; (e) J. D. Smith, E. Chil, W. E. Piers and D. M. Spasyuk, Polyhedron, 2018, 155, 281–290 CrossRef CAS; (f) D. W. Beh, W. E. Piers, I. Del Rosal, L. Maron, B. S. Gelfand, C. Gendy and J.-B. Lin, Dalton Trans., 2018, 47, 13680–13688 RSC; (g) B. J. Johnson, W. E. Antholine, S. V. Lindeman, M. J. Graham and N. P. Mankad, J. Am. Chem. Soc., 2016, 138, 13017–13110 Search PubMed; (h) L. E. Doyle, W. E. Piers and J. Borau-Garcia, J. Am. Chem. Soc., 2015, 137, 2187–2190 CrossRef CAS PubMed; (i) S. I. Kalläne, T. Braun, M. Teltewskoi, B. Braun, R. Herrmann and R. Laubenstein, Chem. Commun., 2015, 51, 14613–14616 RSC; (j) U. Jayarathne, S. R. Parmelee and N. O. Mankad, Inorg. Chem., 2014, 53, 7730–7737 CrossRef CAS PubMed; (k) A. G. Tskhovrebov, E. Solari, R. Scopelliti and K. Severin, Organometallics, 2012, 31, 7235–7240 CrossRef CAS.
- For reviews, see: (a) K. Severin, Trends Chem., 2023, 5, 574–575 CrossRef CAS; (b) V. R. Landaeta and R. E. Rodríguez-Lugo, Inorg. Chim. Acta, 2015, 431, 21–47 CrossRef CAS.
- For selected recent publications, see: (a) V. I. Timokhin, R. D. Grigg and J. M. Schomaker, Eur. J. Inorg. Chem., 2024, 27, e202300782 CrossRef CAS; (b) S. Ni and J. Cornella, Tetrahedron, 2023, 145, 133602 CrossRef CAS; (c) H. Zhang, J. Rodrigalvarez and R. Martin, J. Am. Chem. Soc., 2023, 145, 17564–17569 CrossRef CAS PubMed; (d) J. Bösken, R. E. Rodríguez-Lugo, S. Nappen, M. Trincado and H. Grützmacher, Chem.–Eur. J., 2023, 29, e202203632 CrossRef; (e) J. S. Stanley, X. S. Wang and J. Y. Yang, ACS Catal., 2023, 13, 12617–12622 CrossRef CAS; (f) R. Deeba, S. Chardon-Noblat and C. Costentin, ACS Catal., 2023, 13, 8262–8272 CrossRef CAS; (g) F. Le Vaillant, A. Mateos Calbet, S. González-Pelayo, E. J. Reijerse, S. Ni, J. Busch and J. Cornella, Nature, 2022, 604, 677–683 CrossRef CAS; (h) S. Ni, F. Le Vaillant, A. Mateos-Calbet, R. Martin and J. Cornella, J. Am. Chem. Soc., 2022, 144, 18223–18228 CrossRef CAS; (i) I. Ortega-Lepe, P. Sánchez, L. L. Santos, P. Lara, N. Rendón, J. López-Serrano, V. Salazar-Pereda, E. Álvarez, M. Paneque and A. Suárez, Inorg. Chem., 2022, 61, 18590–18600 CrossRef CAS; (j) X. Chen, H. Wang, S. Du, M. Driess and Z. Mo, Angew. Chem., Int. Ed., 2022, 61, e202114598 CrossRef CAS PubMed; (k) P. Jurt, A. S. Abels, J. J. Gamboa-Carballo, I. Fernández, G. Le Corre, M. Aebli, M. G. Baker, F. Eiler, F. Müller, M. Wörle, R. Verel, S. Gauthier, M. Trincado, T. L. Gianetti and H. Grützmacher, Angew. Chem., Int. Ed., 2021, 60, 25372–25380 CrossRef CAS; (l) R. Zeng, M. Feller, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2018, 140, 7061–7064 CrossRef CAS; (m) R. Zeng, M. Feller, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2017, 139, 5720–5723 CrossRef CAS PubMed.
- W. Wislicenus and B. Dtsch, Chem. Ges., 1892, 25, 2084–2087 CrossRef.
- J. Haase, in Organic Azides, Syntheses and Applications, ed. S. Bräse and K. Banert, John Wiley & Sons, Weinheim, 2010, pp. 29–51 Search PubMed.
- (a) G. A. Vaughan, G. L. Hillhouse and A. L. Rheingold, J. Am. Chem. Soc., 1990, 112, 7994–8001 CrossRef CAS; (b) G. A. Vaughan, C. D. Sofield and G. L. Hillhouse, J. Am. Chem. Soc., 1989, 111, 5491–5493 CrossRef CAS.
- D. J. Mindiola, L. A. Watson, K. Meyer and G. L. Hillhouse, Organometallics, 2014, 33, 2760–2769 CrossRef CAS.
- T. Labahn, A. Mandel and J. Magull, Z. Anorg. Allg. Chem., 1999, 625, 1273–1277 CrossRef CAS.
- S. Demir, E. Montalvo, J. W. Ziller, G. Meyer and W. J. Evans, Organometallics, 2010, 29, 6608–6611 CrossRef CAS.
- N. J. Hartmann, G. Wu and T. W. Hayton, Angew. Chem., Int. Ed., 2015, 54, 14956–14959 CrossRef CAS PubMed.
- N. J. Hartmann, G. Wu and T. W. Hayton, Chem. Sci., 2018, 9, 6580–6588 RSC.
- M. Á. B. Cinco, G. Wu, N. Kaltsoyannis and T. W. Hayton, Angew. Chem., Int. Ed., 2020, 59, 8947–8951 CrossRef.
- Y. Liang, I. Efremenko, Y. Diskin-Posner, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2024, 63, e202401702 CrossRef CAS.
- D. Reiter, P. Frisch, D. Wendel, F. M. Hörmann and S. Inoue, Dalton Trans., 2020, 49, 7060–7068 RSC.
- For recent publications describing the coordination of N2O to metal complexes without the formation of an additional covalent bond, see: (a) B. M. P. Lombardi, C. Gendy, B. S. Gelfand, G. M. Bernard, R. E. Wasylishen, H. M. Tuononen and R. Roesler, Angew. Chem., Int. Ed., 2021, 60, 7077–7081 CrossRef; (b) C. C. Mokhtarzadeh, C. Chan, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2019, 141, 15003–15007 CrossRef CAS PubMed; (c) M. R. Gyton, B. Leforestier and A. B. Chaplin, Angew. Chem., Int. Ed., 2019, 58, 15295–15298 CrossRef CAS; (d) V. Zhuravlev and P. J. Malinowski, Angew. Chem., Int. Ed., 2018, 57, 11697–11700 CrossRef CAS; (e) N. A. Piro, M. F. Lichterman, W. H. Harman and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 2108–2111 CrossRef CAS.
- N. Wiberg, K. Schurz and G. Fischer, Angew. Chem., Int. Ed. Engl., 1985, 24, 1053–1054 CrossRef.
- C. B. Yildiz, Comput. Theor. Chem., 2018, 1134, 47–53 CrossRef CAS.
- E. Otten, R. C. Neu and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 9918–9919 CrossRef CAS.
- For a computational analysis of FLP-N2O adducts, see: T. M. Gilbert, Dalton Trans., 2012, 41, 9046–9055 RSC.
- R. C. Neu, E. Otten and D. W. Stephan, Angew. Chem., Int. Ed., 2009, 48, 9709–9712 CrossRef CAS.
- R. C. Neu, E. Otten, A. Lough and D. W. Stephan, Chem. Sci., 2011, 2, 170–176 RSC.
- For a review, see: D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625–2641 RSC.
- G. Ménard, J. A. Hatnean, H. J. Cowley, A. J. Lough, J. M. Rawson and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 6446–6449 CrossRef.
- M. J. Kelly, J. Gilbert, R. Tirfoin and S. Aldrigde, Angew. Chem., Int. Ed., 2013, 52, 14094–14097 CrossRef CAS.
- E. Theuergarten, A. C. T. Kuate, M. Freytag and M. Tamm, Isr. J. Chem., 2015, 55, 202–205 CrossRef CAS.
- Z. Mo, E. L. Kolychev, A. Rit, J. Campos, H. Niu and S. Aldridge, J. Am. Chem. Soc., 2015, 137, 12227–12230 CrossRef CAS.
- A. G. Tskhovrebov, E. Solari, M. Wodrich, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2012, 51, 232–234 CrossRef CAS PubMed.
- A. G. Tskhovrebov, B. Vuichoud, E. Solari, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2013, 135, 9486–9492 CrossRef CAS PubMed.
- M. Göhner, P. Haiss, N. Kuhn, M. Stöbele and K.-P. Zeller, Z. Naturforsch., 2013, 68, 539–545 CrossRef.
- E. Theuergarten, T. Bannenberg, M. D. Walter, D. Holschumacher, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2014, 43, 1651–1662 RSC.
- L. F. B. Wilm, P. Mehlmann, F. Buß and F. Dielmann, J. Organomet. Chem., 2020, 909, 121097 CrossRef CAS.
- A. G. Tskhovrebov, E. Solari, R. Scopelliti and K. Severin, Inorg. Chem., 2013, 52, 11688–11690 CrossRef CAS PubMed.
- C. Camp, L. C. E. Naested, K. Severin and J. Arnold, Polyhedron, 2016, 103, 157–163 CrossRef CAS.
- (a) T. D. Palluccio, E. V. Rybak-Akimova, S. Majumdar, X. Cai, M. Chui, M. Temprado, J. S. Silvia, A. F. Cozzolino, D. Tofan, A. Velian, C. C. Cummins, B. Captain and C. D. Hoff, J. Am. Chem. Soc., 2013, 135, 11357–11372 CrossRef CAS PubMed; (b) A. G. Tskhovrebov, E. Solari, M. D. Wodrich, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2012, 134, 1471–1473 CrossRef CAS PubMed.
- For a computational study, see: R. Robinson Jr., M. F. Shaw, R. Stranger and B. F. Yates, Dalton Trans., 2016, 45, 1047–1054 RSC.
- (a) R. A. K. Shivaraam, M. Keener, D. K. Modder, T. Rajeshkumar, I. Živković, R. Scopelliti, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2023, 62, e202304051 CrossRef; (b) M. Falcone, L. Barluzzi, J. Andrez, F. Fadaei Tirani, I. Zivkovic, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, Nat. Chem., 2019, 11, 154–160 CrossRef CAS PubMed.
- L. Y. M. Eymann, R. Scopelliti, F. T. Fadaei, G. Cecot, E. Solari and K. Severin, Chem. Commun., 2017, 53, 4331–4334 RSC.
- L. Y. M. Eymann, R. Scopelliti, F. Fadaei Tirani and K. Severin, Chem.–Eur. J., 2018, 24, 7957–7963 CrossRef CAS PubMed.
- A. Thakur, P. K. Vardhanapu, G. Vijaykumar, P. K. Hota and S. K. Mandal, Eur. J. Inorg. Chem., 2016, 913–920 CrossRef CAS.
- R. Meier, Chem. Ber., 1953, 86, 1483–1492 CrossRef CAS.
- G. Kiefer, T. Riedel, P. J. Dyson, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2015, 54, 302–305 CrossRef CAS.
- G. Koga and J.-P. Anselme, Chem. Commun., 1968, 446–447 RSC.
- Y. Liu, L. Y. M. Eymann, E. Solari, F. Fadaei Tirani, R. Scopelliti and K. Severin, Inorg. Chem., 2018, 57, 11859–11863 CrossRef CAS PubMed.
- For reviews, see: (a) W. Dong, Z. Chen, J. Xu, M. Miao and H. Ren, Synlett, 2016, 27, 1318–1334 CrossRef CAS; (b) Y. Zhang, D. Cao, W. Liu, H. Hu, X. Zhang and C. Liu, Curr. Org. Chem., 2015, 19, 151–178 CrossRef CAS; (c) H. Sun and Y. Huang, Synlett, 2015, 26, 2751–2762 CrossRef CAS; (d) D. K. Kölmel, N. Jung and S. Bräse, Aust. J. Chem., 2014, 67, 328–336 CrossRef; (e) D. B. Kimball and M. M. Haley, Angew. Chem., Int. Ed., 2002, 41, 3338–3351 CrossRef CAS.
- F.-X. Felpin and S. Sengupta, Chem. Soc. Rev., 2019, 48, 1150–1193 RSC.
- Lithium aminodiazotates should not be isolated, because they can be explosive in the dry state.
- A. A. Suleymanov and K. Severin, Angew. Chem., Int. Ed., 2021, 60, 6879–6889 CrossRef CAS PubMed.
- A. A. Suleymanov, R. Scopelliti, F. Fadaei Tirani and K. Severin, Org. Lett., 2018, 20, 3323–3326 CrossRef CAS.
- F. G. Perrin, G. Kiefer, L. Jeanbourquin, S. Racine, D. Perrotta, J. Waser, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2015, 54, 13393–13396 CrossRef CAS PubMed.
- The adducts of carboxylic acids and alkynyl triazenes can undergo subsequent reactions. See: (a) M. Yu, L. Zeng, G. Xu and S. Cui, J. Org. Chem., 2023, 88, 12150–12161 CrossRef CAS PubMed; (b) X. Bao, L. Zeng, J. Jin and S. Cui, J. Org. Chem., 2022, 87, 2821–2830 CrossRef CAS PubMed; (c) C. Wang, Z. Lai, H. Xie and S. Cui, Angew. Chem., Int. Ed., 2021, 60, 5147–5151 CrossRef CAS; (d) J.-F. Tan, C. T. Bormann, K. Severin and N. Cramer, Chem. Sci., 2021, 12, 9140–9145 RSC.
- I. R. Landman, E. Acuña-Bolomey, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Org. Lett., 2019, 21, 6408–6412 CrossRef CAS.
- L. N. Jeanbourquin, R. Scopelliti, F. Fadaei Tirani and K. Severin, Org. Lett., 2017, 19, 2070–2073 CrossRef CAS PubMed.
- L. Zeng, Z. Lai, C. Zhang, H. Xie and S. Cui, Org. Lett., 2020, 22, 2220–2224 CrossRef CAS PubMed.
- J.-F. Tan, C. T. Bormann, F. G. Perrin, F. M. Chadwick, K. Severin and N. Cramer, J. Am. Chem. Soc., 2019, 141, 10372–10383 CrossRef CAS PubMed.
- D. Kossler, F. G. Perrin, A. A. Suleymanov, G. Kiefer, R. Scopelliti, K. Severin and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 11490–11493 CrossRef CAS.
- T. Wezeman, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Adv. Synth. Catal., 2019, 361, 1383–1388 CrossRef CAS.
- J.-F. Tan, C. T. Bormann, K. Severin and N. Cramer, ACS Catal., 2020, 10, 3790–3796 CrossRef CAS.
- C. T. Bormann, F. G. Abela, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Eur. J. Org Chem., 2020, 2130–2139 CrossRef CAS.
- L. N. Jeanbourquin, R. Scopelliti, F. Fadaei Tirani and K. Severin, Helv. Chim. Acta, 2017, 100, e1700186 CrossRef.
- Z. Mao and H. Zeng, RSC Adv., 2022, 12, 24857–24860 RSC.
- (a) S. Xu, L. Zeng and S. Cui, Org. Lett., 2022, 24, 2689–2693 CrossRef CAS; (b) A. A. Suleymanov, R. Scopelliti, F. Fadaei Tirani and K. Severin, Adv. Synth. Catal., 2018, 360, 4178–4183 CrossRef CAS.
- C. T. Bormann, F. Fadaei-Tirani, R. Scopelliti and K. Severin, Org. Biomol. Chem., 2021, 19, 8113–8117 RSC.
- E. Michel, F. F. Grieser, A. V. Mackenroth, M. Schukin, P. Krämer, S. Tahir, F. Rominger, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2023, 62, e202309274 CrossRef CAS PubMed.
- J.-F. Tan, C. T. Bormann, K. Severin and N. Cramer, Chem. Sci., 2022, 13, 3409–3415 RSC.
- C. T. Bormann, C. Mathew, M. M. António, A. Trotti, F. Fadaei-Tirani and K. Severin, J. Org. Chem., 2022, 87, 16882–16886 CrossRef CAS PubMed.
- K. Banert and O. Plefka, Angew. Chem., Int. Ed., 2011, 50, 6171–6174 CrossRef CAS.
- Z. Lai, C. Wang, J. Li and S. Cui, Org. Lett., 2020, 22, 2017–2021 CrossRef CAS.
- B. Wei, W.-X. Zhang and Z. Xi, Dalton Trans., 2018, 47, 12540–12545 RSC.
- For reviews, see: (a) D. Yadagiri, M. Rivas and V. Gevorgyan, J. Org. Chem., 2020, 85, 11030–11046 CrossRef CAS PubMed; (b) I. P. Filippov, G. D. Titov and N. V. Rostovskii, Synthesis, 2020, 3564–3576 CrossRef CAS.
- I. R. Landman, F. Fadaei-Tirani and K. Severin, Chem. Commun., 2021, 57, 11537–11540 RSC.
- For a computational study, see: R. Monreal-Corona, E. Besalú, A. Pla-Quintana and A. Poater, Org. Chem. Front., 2022, 9, 4347–4357 RSC.
- F. M. Beringer, J. A. Farr and S. Sands, J. Am. Chem. Soc., 1953, 75, 3984–3987 CrossRef CAS.
- R. Meier and K. Rappold, Angew. Chem., 1953, 65, 560–561 CrossRef CAS.
- For metal-catalyzed coupling reactions of aryl Grignard reagents with N2O, see: G. Kiefer, L. Jeanborquin and K. Severin, Angew. Chem., Int. Ed., 2013, 52, 6302–6305 CrossRef CAS PubMed.
- M. L. Hays and T. P. Hanusa, Tetrahedron Lett., 1995, 36, 2435–2436 CrossRef CAS.
- A. N. Nesmeyanov, E. G. Perevalova and T. V. Nikitina, Dokl. Akad. Nauk SSSR, 1961, 138, 1118–1121 CAS.
- M. Kurusawa, T. Nankawa, T. Matsuda, K. Kubo, M. Kurihara and H. Nishihara, Inorg. Chem., 1999, 38, 5113 CrossRef.
- A. G. Tskhovrebov, L. C. E. Neasted, E. Solari, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2015, 54, 1289–1292 CrossRef CAS.
- R. Raue, Methine Dyes and Pigments, Ullman's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000, pp. 1–55 Search PubMed.
- L. Y. M. Eymann, A. G. Tskhovrebov, A. Sienkiewicz, J. L. Bila, I. Živković, H. M. Rønnow, M. D. Wodrich, L. Vannay, C. Corminboeuf, P. Pattison, E. Solari, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2016, 138, 15126–15129 CrossRef CAS PubMed.
- F. M. Chadwick, B. F. E. Curchod, R. Scopelliti, F. Fadaei Tirani, E. Solari and K. Severin, Angew. Chem., Int. Ed., 2019, 58, 1764–1767 CrossRef CAS PubMed.
- L. Y. M. Eymann, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Chem.–Eur. J., 2018, 24, 7957–7963 CrossRef CAS.
- For reviews, see: (a) G. Mahantesh, D. Sharma, R. Dandela and V. Dhayalan, Chem.–Eur. J., 2023, 29, e202302106 CrossRef PubMed; (b) S. Naumann, Chem. Commun., 2019, 55, 11658–11670 RSC; (c) M. M. D. Roy and E. Rivard, Acc. Chem. Res., 2017, 50, 2017–2025 CrossRef CAS PubMed.
- L. Y. M. Eymann, P. Varava, A. M. Shved, B. F. E. Curchod, Y. Liu, O. M. Planes, A. Sienkiewicz, R. Scopelliti, F. Fadaei Tirani and K. Severin, J. Am. Chem. Soc., 2019, 141, 17112–17116 CrossRef CAS PubMed.
- E. Müller, D. Ludsteck and W. Rundel, Angew. Chem., 1955, 67, 617 CrossRef.
- E. Müller and W. Rundel, Chem. Ber., 1957, 90, 1302–1306 CrossRef.
- W. Rundel and P. Kästner, Liebigs Ann. Chem., 1964, 686, 88–91 CrossRef.
- C. Chen, C. D. Daniliuc, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2021, 60, 19905–19911 CrossRef CAS PubMed.
- For reviews, see: (a) M. M. Hansmann, Angew. Chem., Int. Ed., 2023, 62, e202304574 CrossRef CAS PubMed; (b) R. Knorr, Chem. Rev., 2004, 104, 3795–3849 CrossRef CAS PubMed; (c) W. Kirmse, Angew. Chem., Int. Ed. Engl., 1997, 36, 1164–1170 CrossRef CAS; (d) D. Bourissou and G. Bertrand, C. R. Acad. Sci., Ser. IIb, 1996, 489–506 CAS.
- P. W. Antoni, C. Golz, J. J. Holstein, D. A. Pantazis and M. M. Hansmann, Nat. Chem., 2021, 13, 587–593 CrossRef CAS PubMed.
- M. M. Hansmann, P. W. Antoni and H. Pesch, Angew. Chem., Int. Ed., 2020, 59, 5782–5787 CrossRef CAS PubMed.
- P. Varava, Z. Dong, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Nat. Chem., 2021, 13, 1055–1060 CrossRef CAS PubMed.
- D. Meleschko, P. Palui, R. M. Gomila, G. Schnakenburg, A. C. Filippou, A. Frontera and A. Bismuto, Angew. Chem., Int. Ed., 2024, 63, e202405400 CrossRef CAS PubMed.
- P. W. Antoni, J. Reitz and M. M. Hansmann, J. Am. Chem. Soc., 2021, 143, 12878–12885 CrossRef CAS PubMed.
- J. Reitz, P. W. Antoni, J. J. Holstein and M. M. Hansmann, Angew. Chem., Int. Ed., 2023, 62, e202301486 CrossRef CAS PubMed.
- T. Eisner, A. Kostenko, F. J. Kiefer and S. Inoue, Chem. Commun., 2024, 60, 558–561 RSC.
- B. Kooij, Z. Dong, F. Fadaei-Tirani, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2023, 62, e202308625 CrossRef CAS PubMed.
- For the spectroscopic characterization of an N-heterocyclic vinylidene, see: Y. Kutin, J. Reitz, P. W. Antoni, A. Savitski, D. A. Pantazis, M. Kasanmaschef and M. M. Hansmann, J. Am. Chem. Soc., 2021, 143, 21410–21415 CrossRef CAS PubMed.
- For vinylidene complexes of type (NHC)CMLn, see:
(a) S. Kumar, L. R. Maurer, G. Schnakenburg, U. Das and A. C. Filippou, Angew. Chem., Int. Ed., 2024, 63, e202400227 CrossRef CAS PubMed;
(b) B. Kooij, P. Varava, F. Fadaei-Tirani, R. Scopelliti, D. A. Pantazis, G. P. Van Trieste III, D. C. Powers and K. Severin, Angew. Chem., Int. Ed., 2023, 62, e202214899 CrossRef CAS PubMed;
(c) B. Kooij, Z. Dong, P. Varava, F. Fadaei-Tirani, R. Scopelliti, L. Piveteau and K. Severin, Chem. Commun., 2022, 58, 4204–4207 RSC.
- For a metal-mediated vinylidene-carbene coupling, see: B. Kooij, D. Chen, F. Fadaei-Tirani and K. Severin, Angew. Chem., Int. Ed., 2024, e202407945 Search PubMed.
- W. Feuerstein, P. Varava, F. Fadaei-Tirani, R. Scopelliti and K. Severin, Chem. Commun., 2021, 57, 11509–11512 RSC.
- S. Hauer, J. Reitz, T. Koike, R. Wolf and M. Hansmann, Angew. Chem., Int. Ed., 2024, e202410107 Search PubMed.
- P. Varava, T. H. Wong, Z. Dong, A. Yu. Gitlina, A. Sienkiewicz, W. Feuerstein, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Angew. Chem., Int. Ed., 2023, 62, e202303375 CrossRef CAS PubMed.
- J. Ariai, J. Becker and U. Gellrich, Eur. J. Org Chem., 2024, 27, e202301252 CrossRef CAS.
- T. Koike, J.-K. Yu and M. M. Hansmann, Science, 2024, 385, 305–311 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |