
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The efficient synthesis of three-membered rings via photo- and electrochemical strategies
Xinyu
Han†
a,
Na
Zhang†
b,
Qiannan
Li†
a,
Yu
Zhang
 *ad and
Shoubhik
Das
*c
*ad and
Shoubhik
Das
*c
aShanghai Frontiers Science Center for Chinese Medicine Chemical Biology, Institute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, No. 1200, Cailun Road, Shanghai 201203, China. E-mail: yzhang@shutcm.edu.cn
bDepartment of Laboratory Medicine, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, China
cDepartment of Chemistry, University of Bayreuth, Bayreuth 95447, Germany. E-mail: Shoubhik.Das@uni-bayreuth.de
dSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, People's Republic of China
First published on 14th August 2024
Abstract
Three-membered rings, such as epoxides, aziridines, oxaziridines, cyclopropenes, vinyloxaziridines, and azirines, are recognized as crucial pharmacophores and building blocks in organic chemistry and drug discovery. Despite the significant advances in the synthesis of these rings through photo/electrochemical methods over the past decade, there has currently been no focused discussion and updated overviews on this topic. Therefore, we presented this review article on the efficient synthesis of three-membered rings using photo- and electrochemical strategies, covering the literature since 2015. In this study, a conceptual overview and detailed discussions were provided to illustrate the advancement of this field. Moreover, a brief discussion outlines the current challenges and opportunities in synthesizing the three-membered rings using these strategies.
1 Introduction
Three-membered rings, including cyclopropanes, epoxides, aziridines, oxaziridines, cyclopropenes, vinyloxaziridines, and azirines, play crucial roles as pharmacophores in medicinal chemistry, and are prevalent in natural products and drugs.1–5 They are also commonly present in pesticides, perfumes, and specialty chemicals.6–9 Additionally, these motifs are frequently identified in the top 200 highest-grossing pharmaceutical products and other approved drugs.10 Notably, cyclopropyl drugs such as the phytotoxin coronatine, anticancer agent ptaquiloside, and antiplatelet agent ticagrelor are well-known examples.11–14 Furthermore, diverse natural products with three-membered rings exhibit significant bioactive properties.15,16 For instance, mitomycin and azinomycin are noted for their antibiotic and antitumor activities, and azicemicin is effective against Gram-negative bacteria and mycobacteria (Fig. 1a).17–19 In addition to their prevalence in pharmaceuticals and natural products, three-membered rings are utilized as foundational components in synthetic chemistry.20–24 Moreover, achieving sustainable synthesis of structurally diverse three-membered rings has long been a goal in organic and medicinal chemistry, with numerous classic methods reported for their preparation (Fig. 1b).25–27 Initially, it was discovered that nature constructs cyclopropanes through biosynthetic pathways involving the ring closure of alkenes and carbocations, which inspired synthetic chemists to mimic these processes.28 Noteworthy synthetic routes include the Freund reaction, Kishner cyclopropane synthesis, the Kulinkovich reaction, and the Simmons–Smith reaction.29–33 In addition, intramolecular methods for constructing aziridines, such as the Wenker-type reaction of amino alcohols or their derivatives, have been reported.34–36 Catalytic alkene aziridination is another efficient method for the synthesis of aziridines.37 Nowadays, the established strategies for achieving aziridination have included the application of nitrenes, nitrogen-centered radicals, halonium ions, and oxaziridines.38–41 The epoxidation of alkenes typically necessitates strong oxidants such as hydrogen peroxide (H2O2), tert-butyl hydroperoxide (TBHP), hypervalent iodine, and ozone, or classic reactions including the Darzens condensation and Corey–Chaykovsky reaction.42–45 Transition-metal catalysis has been another prevalent method to afford epoxides efficiently.46,47 Furthermore, the synthesis of other three-membered rings, such as cyclopropenes, vinyloxaziridines, and azirines, has also been well-established.48–58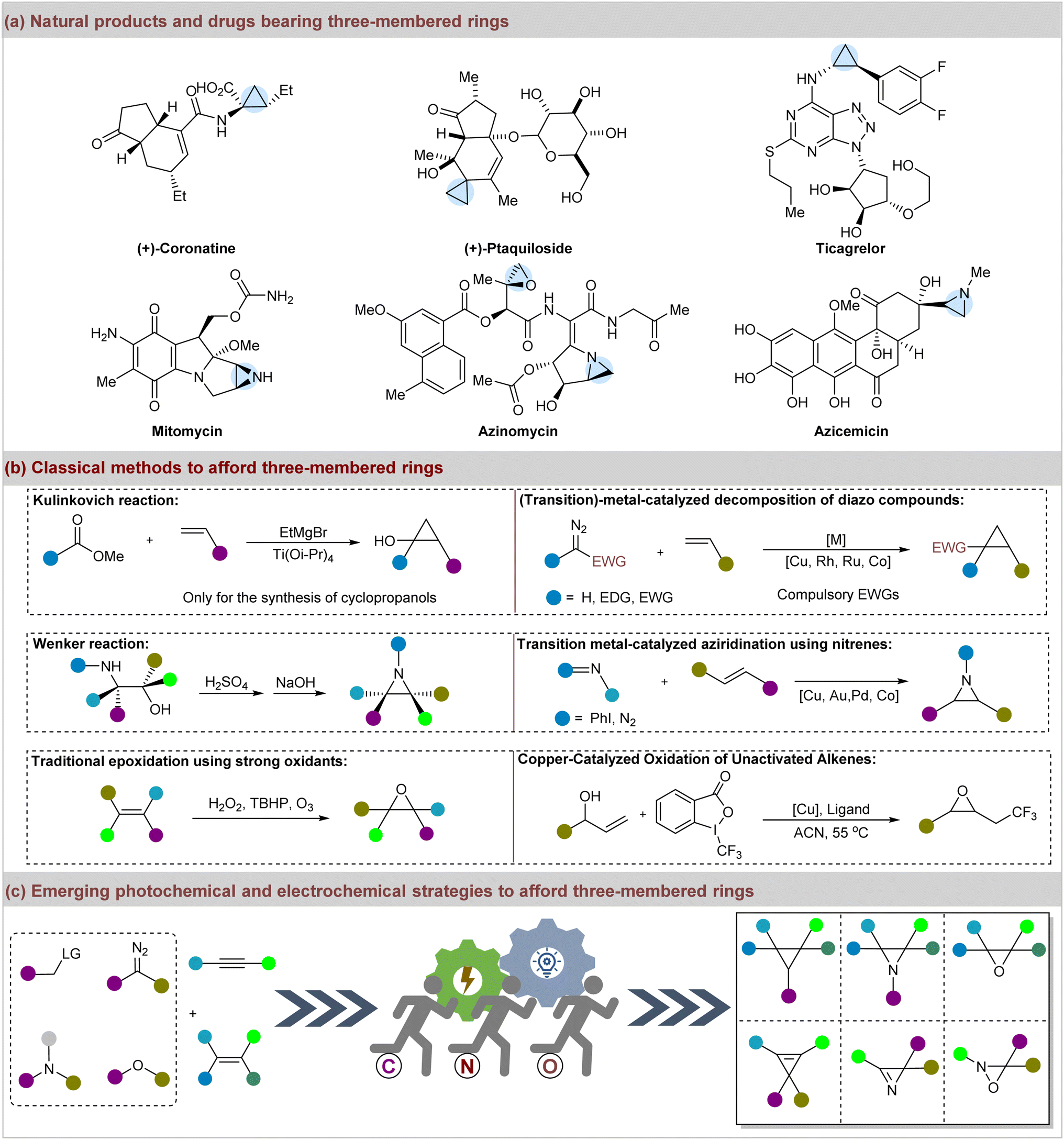 | ||
| Fig. 1 (a) Representative drugs and natural products bearing three-membered rings; (b) classical methods to afford cyclopropanes, aziridines, and epoxides. (c) Emerging visible-light mediated and electrochemical strategies to afford three-membered rings. | ||
Despite the advancements in obtaining three-membered rings, numerous strategies still involve high temperatures, external oxidants, and poor atom economy.59,60 In this regard, the visible-light-mediated and electrocatalytic syntheses of three-membered frameworks have flourished61,62 and have attracted growing interest as environmentally friendly and potent approaches (Fig. 1c).63,64 In fact, significant progress has been made in the construction of three-membered rings using these innovative approaches. However, there is a lack of focus on this topic.65–67 To address this gap, we aimed to provide a comprehensive and critical review of recent advancements in the synthesis of three-membered rings via visible-light-mediated and electrochemical strategies since 2015. By focusing on homogeneous systems, we anticipate that this review will be valuable to readers of organic synthesis, catalysis, and medicinal chemistry.
2 Conceptual overview of the synthesis of three-membered rings via photo/electrochemical strategies
To illustrate the formation of three-membered ring compounds via photo/electrochemical methods, Fig. 2 illustrates an overview of key mechanistic principles. There are generally two types of initiation pathways for the construction of three-membered ring compounds, including the donor-initiated and acceptor-initiated methods. The donor-initiated methods for synthesizing cyclopropanes typically involve the generation of carbon radicals or carbenes. Carbon radicals are generally derived from radical precursors containing leaving groups (such as halogens, carboxylic acids, and silicon reagents)68–72 that depart in the presence of catalysts, light, or electricity to form radicals. These radicals can further react with alkenes to produce cyclopropanes. On the other hand, carbenes are often produced from diazo esters73 and other diazo precursors such as N-tosylhydrazones to achieve cyclopropanation.74,75 In contrast, acceptor-initiated methods can activate alkenes through the formation of radical cations or diradicals under photo/electrocatalytic conditions to facilitate cyclopropanation.76–78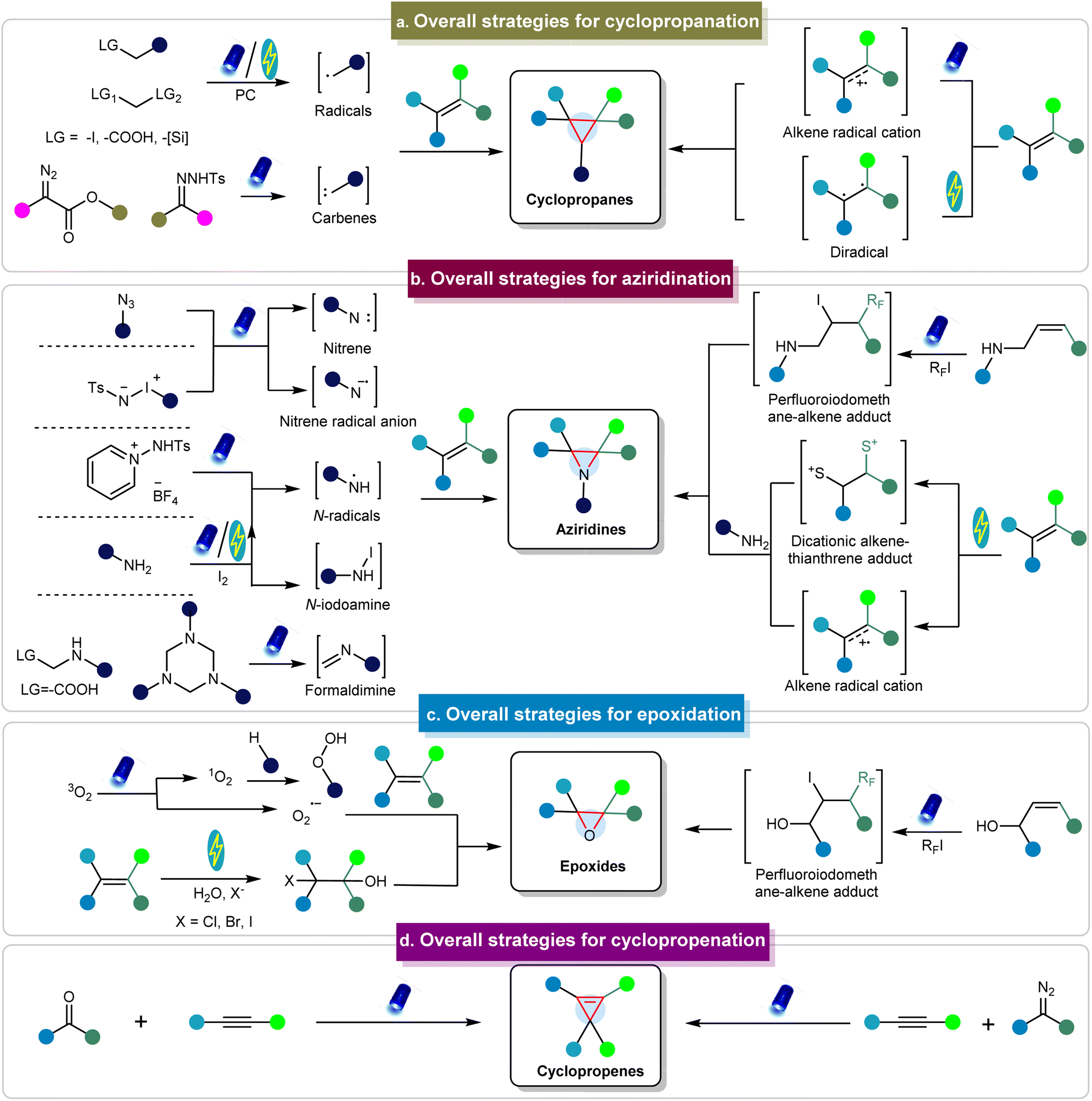 | ||
| Fig. 2 Conceptual overview of synthesizing three-membered rings via photo/electrochemical approaches. | ||
In terms of donor initiation methods for synthesizing aziridine compounds, several pathways involve the formation of nitrenes, nitrene radical anions, N-radicals, N-iodoamine, and formaldimine intermediates derived from various precursors, facilitated by photo/electrocatalysis (Fig. 2).79–83 These intermediates can subsequently react with alkenes to produce the corresponding aziridines. Acceptor-initiated methods involve the formation of alkene radical cations and dicationic alkene–thianthrene adducts.84 Moreover, alkenes can react with perfluoroalkyl iodides under photocatalytic conditions to generate perfluoroiodomethane–alkene adducts, which then react with amines to form aziridines.85 Epoxides are typically prepared via photochemical pathways involving O2, including the pathways using singlet oxygen (1O2) or superoxide radical anions (O2˙−).86 In addition, epoxides can be generated via perfluoroiodomethane–alkene adducts.87 The electrochemical synthesis of epoxides typically involves H2O and requires the halogen atom as the leaving group.88–90 Cyclopropenes are primarily synthesized through reactions between carbenes and alkynes.91,92 In addition to these methods, other mechanisms have also been employed to obtain three-membered rings.93–96 However, there are limited examples of the synthesis of other three-membered rings via photo/electrochemical methods, as detailed in the following sections. These methods are discussed in detail in the subsequent sections.
3 Synthesis of cyclopropanes by photo/electrocatalysis
As mentioned earlier, cyclopropanes are crucial structural elements in drug molecules. Various methods have been developed for the synthesis of cyclopropanes using diverse catalytic systems and specially designed substrates. Under mild photo/electrocatalytic reaction conditions, radical precursors can be derived from dichloromethanes, halogenated methyl silicates, or carboxylic acids;68–70 and diazo compounds,73 trifluoroacetyl silanes,74 and N-tosylhydrazones75 serve as the carbene sources. In addition, alkenes can be directly activated to form alkene radical cations or diradicals, leading to cyclopropane products.76–78 These advancements have significantly enhanced the efficient construction of structurally diverse cyclopropanes.3.1 Cyclopropanation through the construction of radical intermediates
The generation of radical intermediates to produce cyclopropanes has been a pivotal strategy over the past decade. In 2002, the Nédélec group introduced an electrocatalytic method for cyclopropanation, involving the reaction between reactive alkenes and activated 1,1,1-trichloro compounds.97 This method allowed the generation of a variety of chlorine-substituted cyclopropanes in acceptable yields (25–76%). Although the substrate scope of this reaction was limited with no entirely elucidated mechanism, it can be notable for its significant advantage of not requiring hazardous reagents such as diazo compounds or diazirines. Navarro et al. further investigated the mechanism through cyclic voltammetry and electrolysis in 2011.98 They discovered that cyclopropanation occurred via radical or anionic intermediates, and that subsequent iron catalysis facilitated the coupling reactions by preventing dimer formation in radical-mediated pathways. Conversely, in the anionic pathway, the electrocatalytic reduction of dimethyl itaconate to the corresponding carbanion, followed by nucleophilic addition, led to the formation of cyclopropane products.In 2015, Guo et al. pioneered a visible light-induced strategy for the cyclopropanation of dibromomalonates with 2-benzylidenemalononitrile. This method involved generating a carbanion through a double single-electron transfer (SET) mechanism (Scheme 1a).99 This strategy was feasible under sunlight in open air, highlighting its sustainability and ease of application. Initially, the Ru(bpy)32+ photocatalyst absorbed photons under visible-light irradiation, entering an excited state. Subsequently, this excited species captured an electron from the amine via a SET process, resulting in the formation of highly reduced Ru(bpy)3+. Subsequently, dibromomalonate (3) underwent the first electron transfer process, lost a bromide ion to generate a dibromomalonate radical (4) and regenerated the original photocatalyst. Subsequent catalytic cycles involved a second electron transfer process that converted the carbon-centered radical into the desired dibromomalonate anion (5). Michael addition occurred to an electron-deficient alkene (6), followed by intramolecular nucleophilic substitution, ultimately yielding cyclopropane (8) as the final product. Although this was a breakthrough to access cyclopropanes under photocatalytic conditions, this method was restricted to specific alkenes bearing electron-withdrawing groups, and thereby led to poor substrate scope.
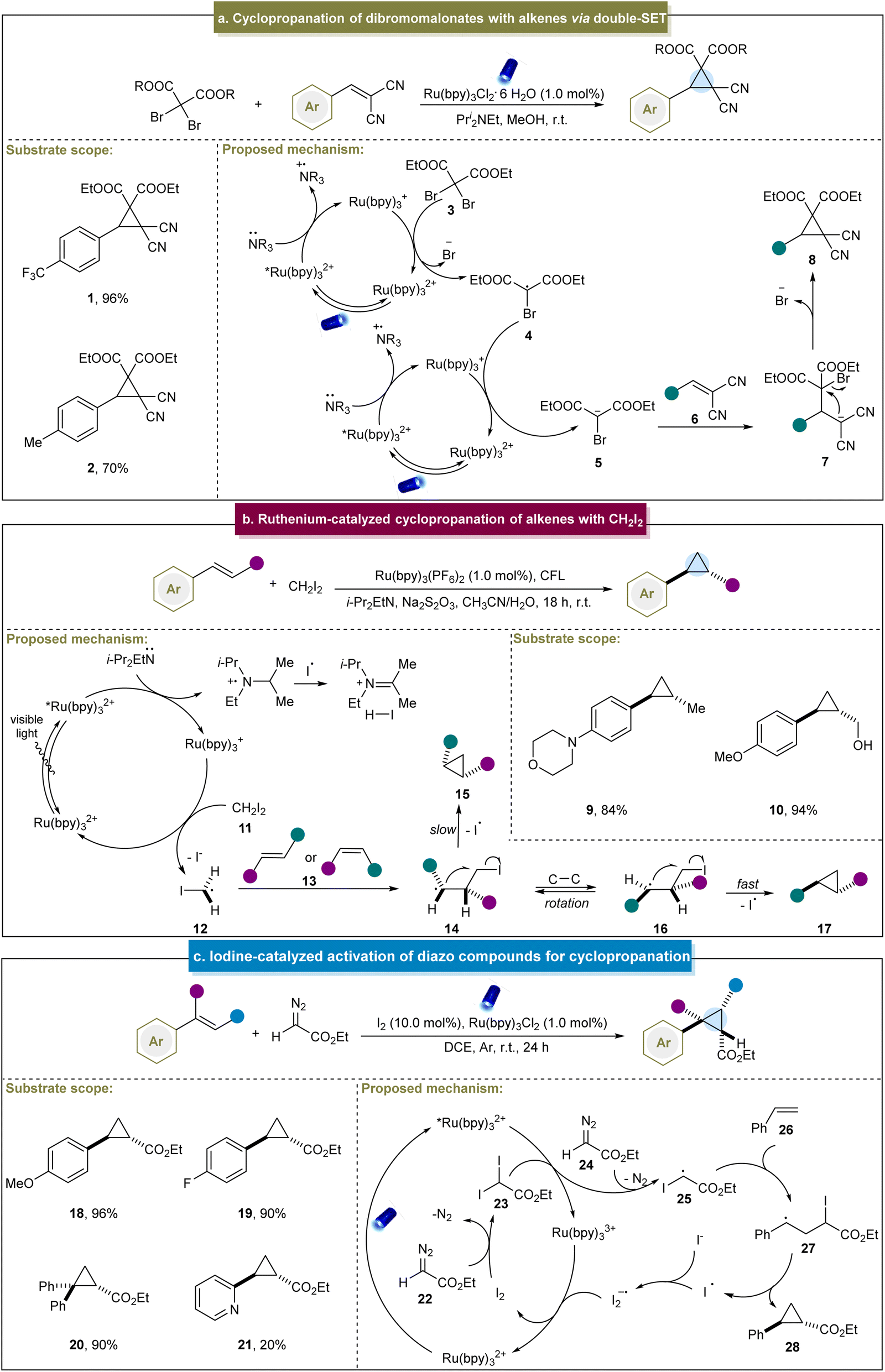 | ||
| Scheme 1 (a) Cyclopropanation of dibromomalonates with alkenes via double-SET. (b) Ruthenium-catalyzed cyclopropanation of alkenes. (c) Iodine-catalyzed activation of diazo compounds for cyclopropanation. | ||
Similarly, the group of Suero reported the stereoselective cyclopropanation of styrenes via photoredox catalysis.68 This method facilitated the selective formation of trans-cyclopropanes using both E- and Z-type anetholes and CH2I2. In particular, high yields were obtained from aromatic alkenes with unprotected groups such as hydroxyl (10). In their reaction, the photoexcited state *[Ru(bpy)3]2+ underwent the SET process with N,N-diisopropylethylamine to generate [Ru(bpy)3]+ (Scheme 1b). Subsequently, [Ru(bpy)3]+ donated an electron to CH2I2 (11) to produce an iodomethyl radical (12) and in the presence of E, Z-alkenes, was converted into intermediates (14) and (15). Finally, the cyclization step facilitated the homolytic substitution of intermediate (16), favoring a trans configuration of the R1 and R2 groups. This method enabled the selective formation of trans-cyclopropanes under mild conditions and effectively controlled the stereochemistry of the E- or Z-type aromatic alkenes. However, several challenges remain; for example, aromatic alkenes with moderate/strong electron-withdrawing groups exhibited lower yields. On this basis, Suero et al. extended their research by utilizing diiodomethane in the photocatalytic cyclopropanation of α,β-unsaturated carbonyl compounds.100
In 2018, the group of Li reported a photoinduced iodine-catalyzed diazo activation method for the synthesis of cyclopropanes.101 This approach proved effective for the arene-substituted alkenes bearing both electron-donating and electron-withdrawing groups, yielding the desired products in good to excellent yields (18–20). However, the yield decreased significantly for aromatic alkenes containing heteroatoms (21). Mechanistically, diazo compound (22) initially reacted with I2 to form diiodo compound (23), which converted *[Ru(bpy)3]2+ into [Ru(bpy)3]3+ (Scheme 1c). Subsequently, an active intermediate was formed when the diiodo compound reacted with 24, generating a carbon radical (25) through the SET process. This carbon radical (25) attacked the alkene to form another radical intermediate (27). Ultimately, cyclopropane was produced through the release of an iodine radical. However, it was challenging for aliphatic and other unactivated alkenes to undergo cyclopropanation through this strategy and diazo compounds are only applicable to diazo esters.
Based on the pioneering studies mentioned above, Ooi et al. utilized halogen activation to generate radical species, achieving boryl cyclopropanation with α-MIDA-boryl styrenes and diiodoborylmethane (MIDA: N-methyliminodiacetyl) under visible-light irradiation.102 This approach is similar to the previous halogen activation pathways but exhibited limited substrate compatibility and lacked detailed mechanistic insights, owing to insufficient experimental investigations. Furthermore, Suero et al. developed a photocatalyst-free method employing gem-diiodomethyl carbonyl reagents to access a variety of cyclopropanes.103 This protocol demonstrated the efficacy for various substituted styrenes. Moreover, the late-stage functionalization of bioactive molecule derivatives highlighted the synthetic versatility of this method. Unfortunately, styrenes containing electron-withdrawing groups (such as –CF3) and vinylpyridines were not suitable for this reaction. Tokuyama et al. (2021) developed an intramolecular cyclopropanation strategy using α-bromo-β-keto esters and alkenes.104 Under photocatalytic conditions, α-bromo-β-keto esters underwent bromide ion loss to generate a carbon radical intermediate. This intermediate participated in the intramolecular radical addition to the alkene, followed by cyclization to produce cyclopropane products. Additionally, Maji et al. introduced a concise method for the rapid and efficient synthesis of various monosubstituted 1,1- and 1,2-disubstituted cyclopropanes from unactivated 1,3-alkyl electrophiles.105 This mild approach exhibited tolerance towards diverse substrates that were previously ineffective, such as alkynes, free hydroxyls, and terminal alkenes. Inspired by this work, Liu et al. recently described photoredox-catalyzed boron cyclopropanation of diverse alkenes, including styrenes and aliphatic alkenes.106
Later, the group of Carreira carried out intermolecular cyclopropanation using α-bromo-β-ketoesters and α-bromo-maleic acid esters with unactivated alkenes in the presence of an organic photocatalyst.107 This reaction demonstrated broad functional group tolerance and excellent compatibility with mono-, di- and tri-substituted alkenes, yielding highly substituted cyclopropane compounds. Mechanistic studies indicated that the reaction proceeded primarily through catalyst reduction by α-bromo-β-ketoesters, generating a carbon-centered radical that engaged in subsequent reactions with the alkene to afford cyclopropane products. Recently, Xie and co-workers reported the gold-catalyzed cyclopropanation between styrenes and gem-dichloroalkanes (Scheme 2a).108 In this work, diverse gem-dichloroalkanes were used as chloroalkyl radical equivalent precursors for cyclopropanation with 1,1-disubstituted styrenes. Unfortunately, this method was not feasible for aliphatic alkenes, and thereby led to poor substrate scope. The mechanism of this reaction primarily involved the interaction of a photocatalyst and Hantzsch ester (HEH), forming a potentially weakly bound gold complex. This complex absorbs visible light to generate an excited-state species that coordinated with dichloromethane to produce an exciplex, thereby releasing chloromethyl radicals. These radicals could be rapidly introduced to the alkenes, facilitating the formation of cyclopropane products through radical 3-exo-tet cyclization. Additionally, Charette et al. developed a visible-light-mediated borosilylcyclopropanation method for styrene derivatives.109 Mechanistic studies have revealed that the cyclopropanation process can be stabilized by a four-membered-ring boronate anion. The transient radical anion decomposes into an iodo-boromethylsilane radical, which subsequently undergoes a series of reactions to produce cyclopropane. Furthermore, Vassilikogiannakis et al. employed dihaloalkanes as cyclopropanation reagents to achieve metal-free photocatalytic cyclopropanation through the XAT mechanism.110 Notably, complicated colibactin metabolite compounds were also accommodated in this system to furnish valuable natural product derivatives.
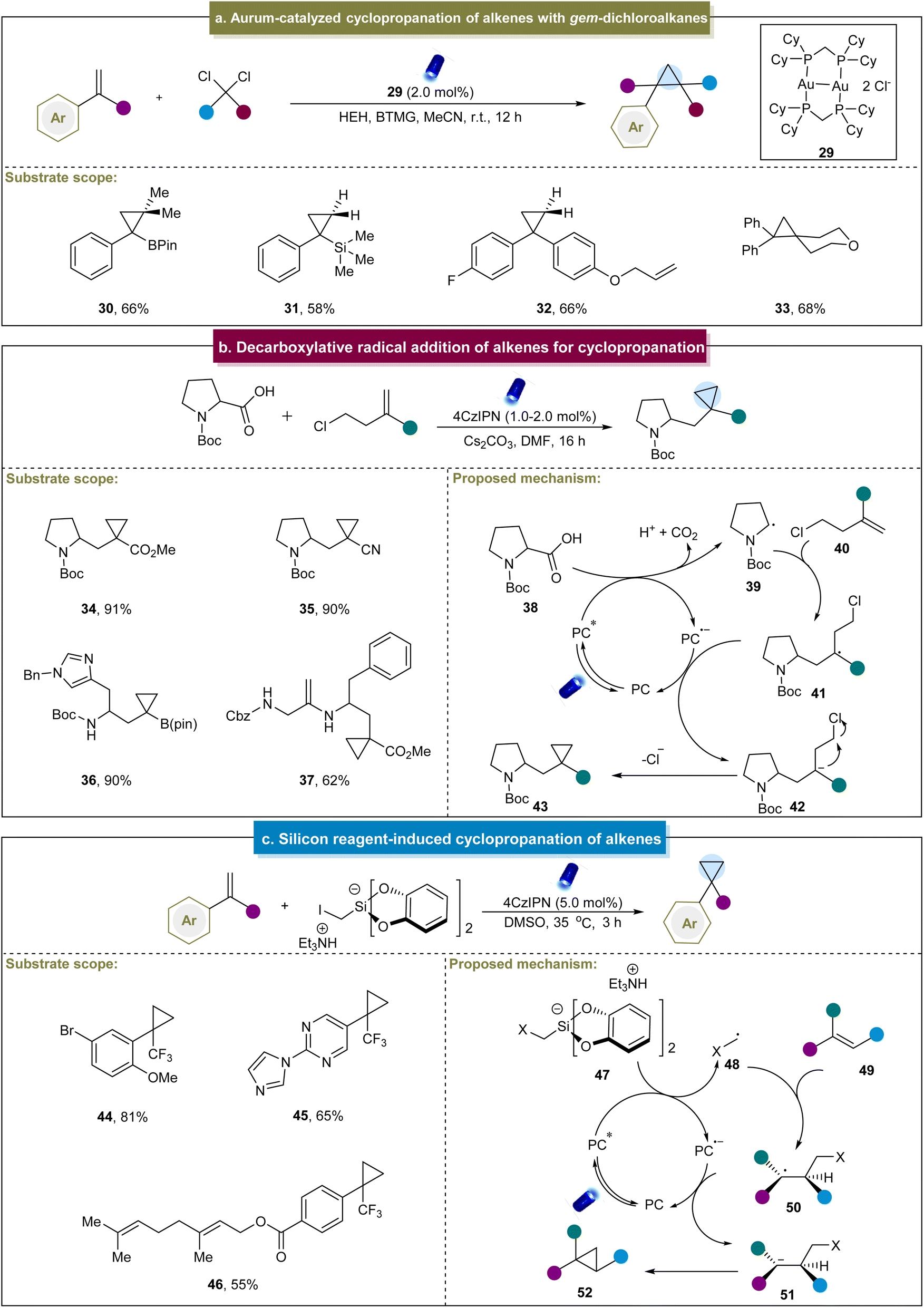 | ||
| Scheme 2 (a) Aurum-catalyzed cyclopropanation of alkenes with gem-dichloroalkanes. (b) Decarboxylative radical addition of alkenes for cyclopropanation. (c) Silicon reagent-induced cyclopropanation of alkenes. | ||
In addition to the radical formation via halogen elimination, Aggarwal et al. devised an elegant method for the synthesis of cyclopropanes. This method utilized carboxylic acids and chloroalkyl alkene substrates through a decarboxylative radical addition-polar cyclization cascade.69 The strategy was suitable for various aliphatic alkenes containing different electron-withdrawing groups and carboxylic acids with multiple functional groups. Initially, carboxylic acid (38) underwent the SET with an excited-state catalyst (Scheme 2b). This process led to the deprotonation of the carboxylic acid and liberation of CO2, generating a carbonyl radical (39). The radical attacked the homoallyl chloride (40), forming a stable alkyl radical (41), followed by the second SET with PC˙−. Finally, the resulting stable carbon anion (42) underwent polar 3-exo-tet cyclization to yield the cyclopropane product. Similarly, Sureshkumar et al. reported a method for the synthesis of 2-acyclic cyclopropane derivatives by coupling α-keto acids with cyclopropenes.111 The key step involved SET-mediated decarboxylation to generate the corresponding acyl radical, which was selectively added to one side of cyclopropene to form cyclopropanes. This study advanced the synthetic versatility of cyclopropenes, whereas the mechanistic details were already understood.
In addition to the aforementioned methods, Molander et al. pioneered the development of a novel bifunctional reagent, triethylammonium bis(catecholato)iodomethylsilicate, for cyclopropanation in 2018.70 This method facilitated the straightforward synthesis of cyclopropanes from α-trifluoromethyl styrenes, yielding moderate to excellent yields (44 and 45). Additionally, in substrates with multiple olefinic double bonds, cyclopropanation selectively occurred on the electron-deficient trifluoromethyl alkenes (46). A plausible mechanism was proposed (Scheme 2c), beginning with the formation of the excited state of 4CzIPN under visible light irradiation. Subsequent reductive quenching of 4CzIPN* by halomethyl silicate 47 generated the halomethyl radical 48, which underwent a Giese-type addition to form adduct 50. Further SET reduction of 50 by the reduced 4CzIPN produced anion 51, regenerating 4CzIPN to its ground state. Finally, anion 51 underwent 3-exo-tet cyclization to yield cyclopropane 52. Although this method was limited to specific α-trifluoromethyl styrenes, it provided a novel approach for the subsequent cyclopropane's construction efforts.
Building on this refined method, several subsequent studies have utilized similar bifunctional reagents. Ouyang et al. developed a comparable strategy for cyclopropanation using chloromethyl silicates as the methyl source.112 In addition to the α-substituted vinyl phosphonates, a range of Michael acceptors have been shown to be suitable substrates for photocatalytic cyclopropanation. Notably, vinyl phosphonate, a complex derivative of estrone, exhibited high reactivity, yielding the desired cyclopropane product. Continuing with this approach, the Molander group extended the application of this reagent, outlining a cyclization process to synthesize 1,1-disubstituted cyclopropanes from homoallylic tosylates.113 In this approach, cyclopropanes were generated by the reaction of alkyl radicals with alkenes, where homoallylic tosylate served as the leaving group. The method exhibited high tolerance for structural diversity in nucleophilic reagents and enabled the rapid and efficient synthesis of a broad array of cyclopropane products without requiring strongly polarized alkenes. However, it was limited to styrenes containing electron-withdrawing groups. Chen et al. reported the photoredox-catalyzed cyclopropanation of 1,1-disubstituted alkenes using halogens as the leaving groups.114 This reaction involved the alkyl and acyl radicals in a radical addition-anionic cyclization process, utilizing the bis-catecholato silicates as radical precursors.
In 2020, Zhang et al. introduced a protocol for the visible-light-induced cyclopropanation of alkenes using bromomethyl silicate as a methylene transfer reagent.115 This method enabled the cyclopropanation of terminal alkenes and demonstrated the feasibility of methyl transfer reactions to internal alkenes and inactivated 1,1-dialkyl ethylenes. Moreover, the synthesis of LG100268, a potent retinoid X receptor (RXR) agonist, validated the utility of this methylene transfer reaction. Subsequently, Du et al. reported the efficient cyclopropanation of alkenyl N-methylimino diacetyl boronates.116 Various MIDA cyclopropyl boronate derivatives were synthesized via photoredox-catalyzed cyclopropanation using chloro- or bromo-methyl silicate as the methylene source. Ohmiya et al. developed a method for the cyclopropanation of reactive alkenes by direct photoexcitation of borate under visible-light irradiation to generate iodomethyl radicals.117 This method offered a straightforward protocol for the cyclopropanation of dehydroamino acids (DHAAs), yielding the pharmacologically valuable cyclopropanyl amino acids. Recently, You et al. reported the efficient synthesis of methylene-unsubstituted cyclopropane-fused indolines via cyclopropanation between the indoles and iodomethylsilicates.118 However, the reaction was limited to N-protected and electron-deficient indoles, thus limiting its applicability. In 2022, Fang et al. reported the cyclopropanation of 1-alkyl-substituted ethenylphosphonates,119 which resulted in a series of alkyl phosphonate-substituted cyclopropane products. Fang et al. described a method for N-vinylimine cyclopropanation via photoredox catalysis.120 This cyclopropanation exhibited stereochemical control, and its synthetic utility was demonstrated through the deprotection of the amine protecting groups.
In addition to the bifunctional reagents developed by Molander et al.,70 in 2019, Murphy et al. achieved the synthesis of cyclopropanes by the direct irradiation of a mixture of iodonium ylides and aromatic alkenes using blue LEDs.71 This method efficiently yielded cyclopropanes from a variety of alkenes and iodonium ylides, with isolated yields ranging from excellent to good (53–55). Additionally, previously challenging aliphatic alkene substrates with medium reactivity were accommodated in this system (56). A one-pot method was developed using activated methylene ylide precursors. Furthermore, a plausible reaction mechanism was proposed based on the computational and experimental studies (Scheme 3a). The photoexcitation of iodonium ylide (57) induced the reversible formation of biradical intermediate (60), which then reacted with the alkene to form biradical (61). Cyclization resulted in the formation of iodocyclobutane (62), and subsequent reductive elimination of iodobenzene furnished the final cyclopropane product (63). Another plausible pathway involved the ground state of iodonium ylide, featuring an entirely positive charge on the iodine atom. This reaction was initiated by the alkene acting as a Lewis base towards the iodonium, thereby forming complex 59, which served as a precursor to form the biradical (61) under light irradiation. The subsequent cyclization and reductive elimination gave the final product.
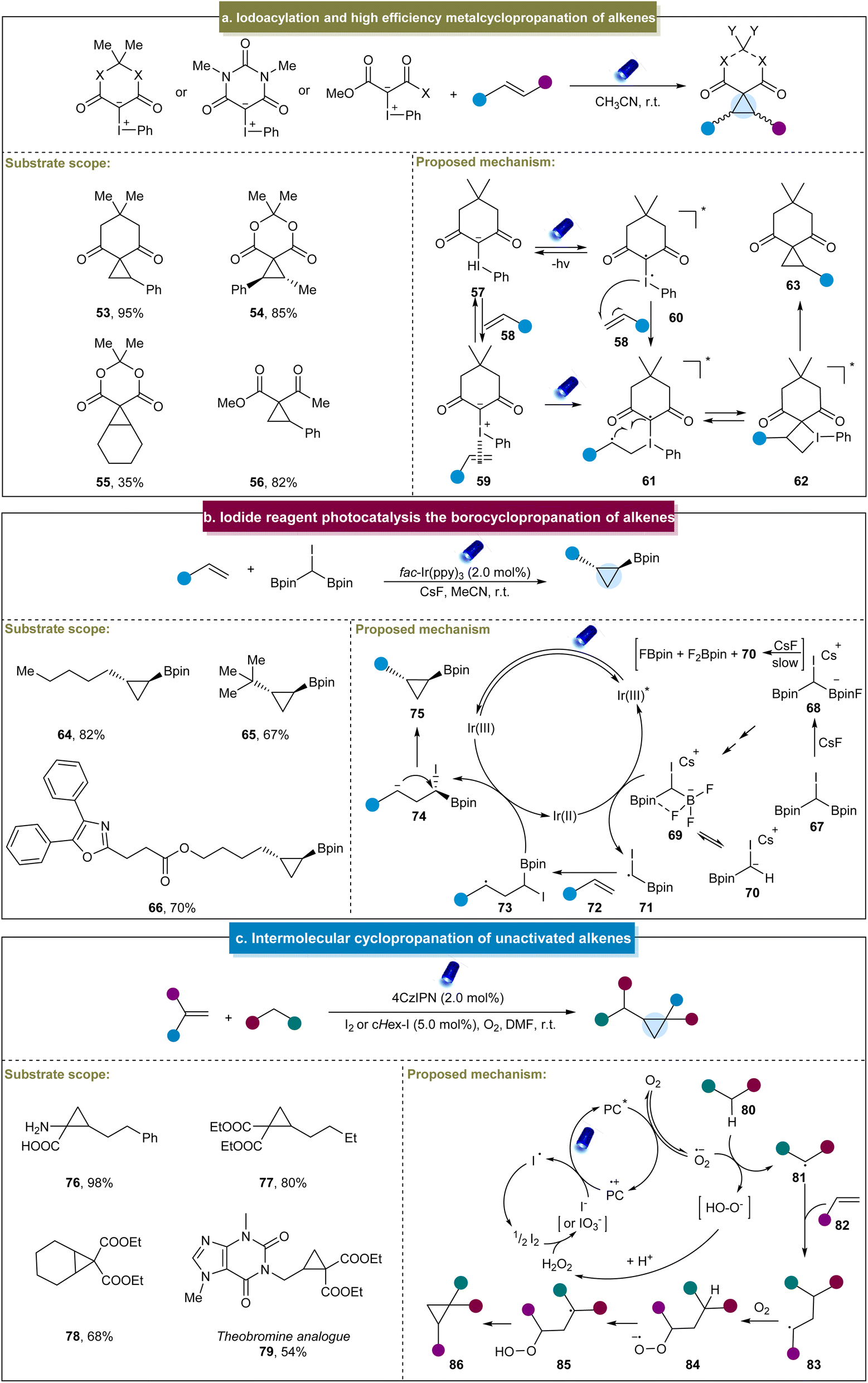 | ||
| Scheme 3 (a) Iodoacylation and high efficiency metalcyclopropanation of alkenes. (b) Iodide reagent photocatalysis and the borocyclopropanation of alkenes. (c) Intermolecular cyclopropanation of unactivated alkenes. | ||
Marder et al. developed a (bisborylmethyl)iodine reagent (CHI(Bpin)2) for the photoinduced cyclopropanation with aliphatic alkenes, yielding a range of 1,2-substituted cyclopropylboronates.121 This α-haloboronic ester easily synthesized from commercially available starting materials produced diverse cyclopropylboronates with high yields and excellent diastereoselectivity and demonstrated good tolerance towards various functional groups (64–66). However, this strategy was only applicable to terminal alkenes. The proposed mechanism for the reaction was deduced from the experimental data (Scheme 3b). Initially, the reaction of 67 with CsF produced a mixture of species, including trifluoroborate complex 69. Subsequently, the excited-state Ir(III)* catalyst was reductively quenched by 69, generating the carbon-centered radical CHI(Bpin) 71. The alkyl radical was then added to the alkene to form radical intermediate (73), followed by the electron transfer from Ir(II) to generate carbon anions (74) and restore the ground-state Ir(III) photocatalyst. Finally, polar 3-exo cyclization occurred, resulting in the formation of the borylcyclopropane product.
More recently, Giri et al. discovered a photoinduced cyclopropanation between EWG-containing aliphatic alkenes and reactive methylene compounds.72 Under the standard reaction conditions, the process efficiently constructed cyclopropanes using both terminal and non-terminal alkenes (76–79). Mechanistically, upon photoexcitation, PC* reduced O2 to form a peroxide ion, O˙−, which abstracted an α-H from the active methyl compound 80, generating an α-C radical, 81 (Scheme 3c). The α-C radical then underwent an addition reaction with the alkene to form a secondary C radical (83), which reacted with O2 to produce a peroxyl radical anion 84. This radical anion subsequently extracted the α-H to form another α-C radical (85), which underwent the radical 1,3-substitution reaction with the peroxide to generate a cyclopropane ring. A distinctive aspect of this reaction was the application of iodine as a co-catalyst, derived either from molecular iodine or generated in situ from alkyl iodides.
Besides the above photoinduced strategies, Xu et al. achieved an efficient intramolecular cyclopropanation of active methylene compounds through electrocatalysis.122 With RVC (reticulated vitreous carbon) and Pt as electrodes and under constant current conditions, this reaction was compatible with cyclic or acyclic alkenes, providing desirable products in moderate to excellent yields (88 and 89). Furthermore, other reactive methylene compounds such as γ-lactams and γ-lactones also underwent smooth cyclopropanation (90 and 91). Mechanistically, the methylene substrate (92) was oxidized by 87˙+ to form an electrophilic carbon radical intermediate (93) (Scheme 4a). Subsequently, this radical attacked the alkene to obtain the alkyl radical (94) through cyclization. The alkyl radical then underwent radical–radical coupling with 87˙+ to yield the sulfonium ion 95. In the final step, nucleophilic substitution triggered by CF3CH2O− in 95 formed the cyclopropanation product and regenerated 97. Unfortunately, this electrocatalytic system was only applicable to methylene compounds with an electron-withdrawing group such as the cyanide group.
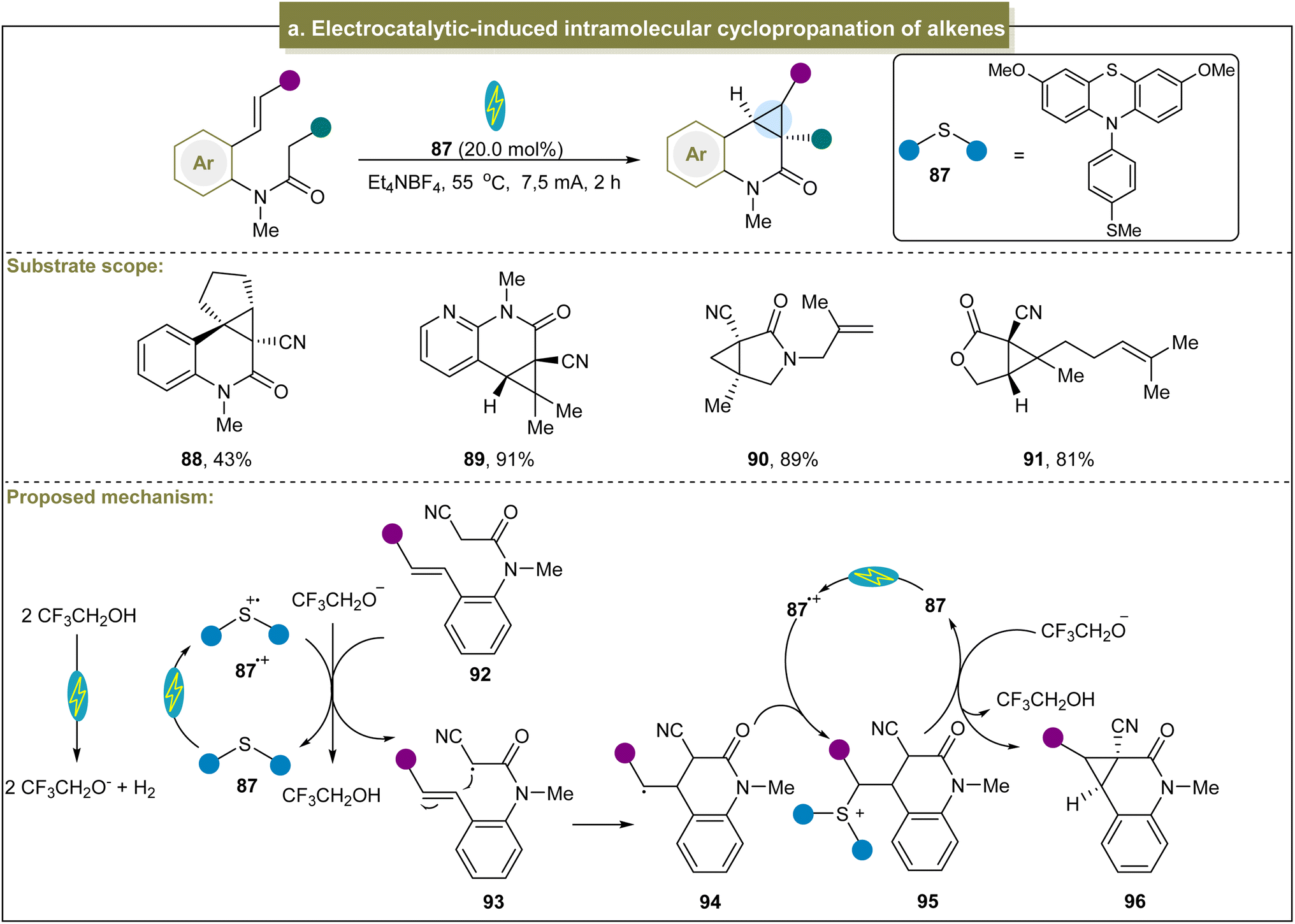 | ||
| Scheme 4 (a) Electrocatalytic-induced intramolecular cyclopropanation of alkenes. | ||
Later, Koenigs et al. reported a method for synthesizing dialkyl-substituted cyclopropanes by the in situ generation of dialkyl diazo compounds from the corresponding N-tosylhydrazones.123 Based on the experimental and computational studies, the key step in this strategy involved the generation of α-Co(III)-alkyl radicals from the activated dimethyl diazomethane. In addition, Pitre et al. described a method using vitamin B12 as a photocatalyst to facilitate cyclopropanation between dichloromethane and electron-deficient alkenes.124 This reaction was effective for a variety of functionalized, electron-deficient alkenes, yielding products in good to excellent yields under mild conditions. The method also utilized CD2Cl2 as a methyl source to prepare D2-cyclopropane products. Mechanistic studies have revealed that the Co(I) oxidation state of vitamin B12 can undergo SN2-type nucleophilic substitution with 1,1-dichloroalkane, followed by photolysis to generate a halogenated alkyl radical and a Co(II) radical. The subsequent steps included Giese addition, single-electron reduction, and 3-exo-tet cyclization to produce the desired cyclopropane product. Because vitamin B12 has excellent biocompatibility and water solubility, this method is promising for the direct cyclopropanation of biomolecules.
The development of radical species for alkene reactions has proven effective and powerful for accessing cyclopropanes. Current radical precursors used in photo- and electrocatalysis include dibromides, carboxylic acids, and silicates. Moreover, adjacent electron-withdrawing groups can enhance the stability of carbon radical intermediates. The radical intermediates can then react with the alkene receptors to produce cyclopropanes. A variety of cyclopropane products can be successfully synthesized using these methods. However, ongoing efforts have focused on developing new radical precursors, utilizing accessible raw materials, and minimizing the reliance on costly catalysts.
3.2 Cyclopropanation through the construction of carbene/carbyne intermediates
The carbene and carbyne intermediates have been widely adopted to synthesize cyclopropanes. In 2018, Suero et al. employed a photoredox strategy to generate diazomethyl radicals that served as direct carbyne equivalents (Scheme 5a).73 This approach facilitated the installation of tailored chiral centers at the carbon-hydrogen bonds, offering an efficient route to a potentially diverse library of bioactive molecules. However, this study was limited in substrate scope, focusing primarily on cyclopropanation, thereby highlighting further exploration in this field. Subsequently, Song et al. developed a method using aryl diazoacetates as carbene precursors under blue light irradiation, achieving the site-selective cyclopropanation of indole.125 This method accommodated a range of substituents on indoles and variations of diazo compounds. However, it did not apply to indoles with electron-donating properties or to unprotected indole compounds. Davies et al. developed a similar method enabling cyclopropanation with styrenes, indoles, pyrroles, and benzofurans, among others.126 They also utilized various aryl diazoacetates to yield cyclopropanes in moderate to excellent yields.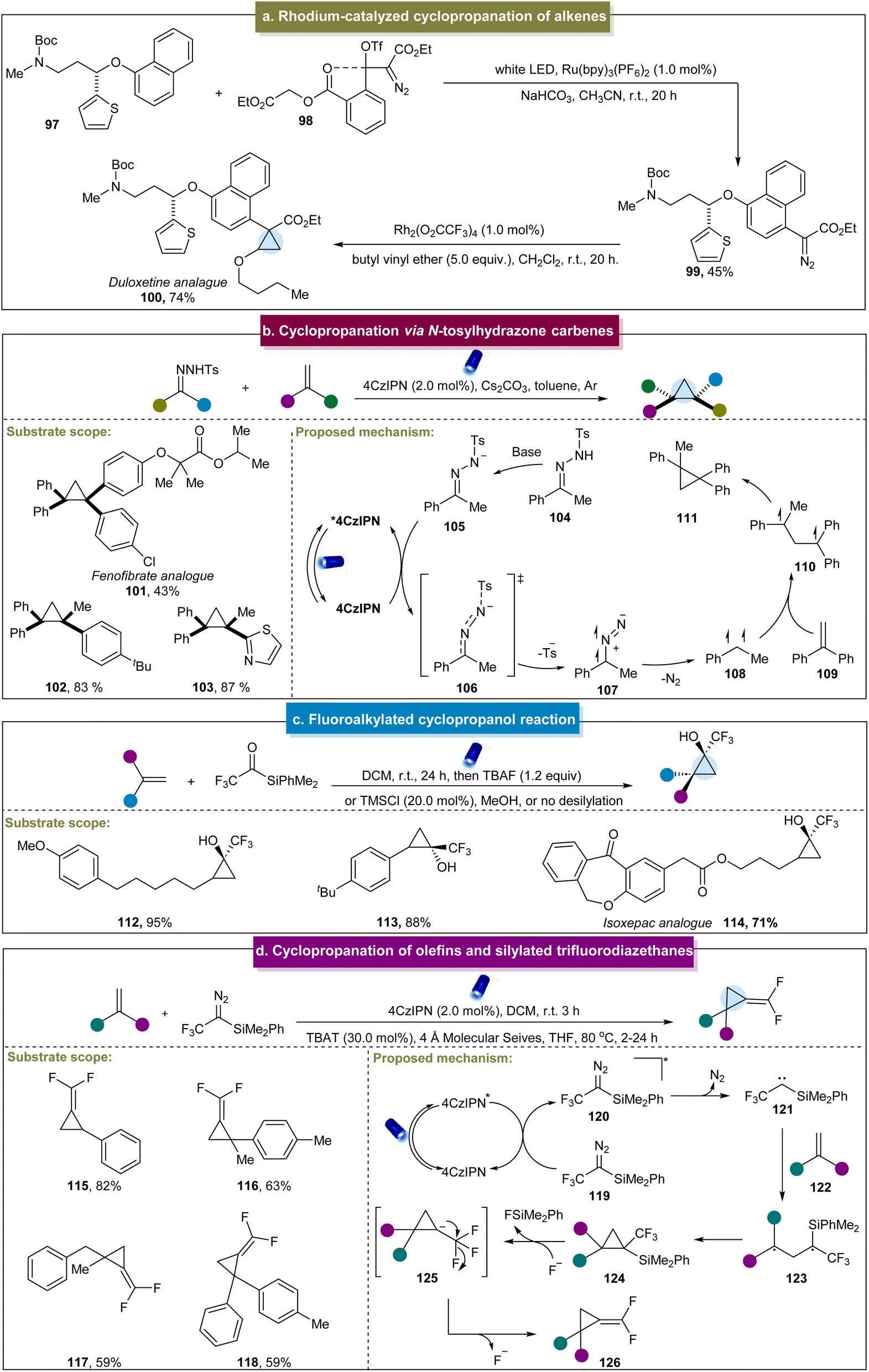 | ||
| Scheme 5 (a) Rhodium-catalyzed cyclopropanation of alkenes. (b) Cyclopropanation via N-tosylhydrazone carbene. (c) Fluoroalkylated cyclopropanol reaction. (d) Cyclopropane of alkenes and silylated trifluorodiazethane. | ||
In 2020, Koenigs et al. developed another cyclopropanation method using diazoesters as the carbene precursors,127 achieving moderate-to-high yields with various methyl-substituted aromatic and polycyclic aromatic compounds. However, this strategy was limited by the application of donor–acceptor diazoalkanes and was unsuitable for benzene rings containing electron-withdrawing groups. The research team continued their research by investigating the cyclopropanation reactions between N-tosylhydrazones and indoles or thiophenes under stoichiometric conditions.128 This approach relied heavily on the donor–acceptor N-tosylhydrazones containing the electron-withdrawing groups as the carbene precursors. In 2020, Sen and co-workers reported the cyclopropanation of tryptamine derivatives to yield polycyclic indoles.129 However, the reactivity of the reaction was generally low, requiring additional protection of the indole substrates. Later, Koenigs et al. developed a method using aryl diazoacetates to cyclopropanate cyclooctadiene and polyunsaturated cyclic alkenes.130 This approach demonstrated broad applicability with various aryl diazoacetates and (poly)unsaturated cyclic hydrocarbons, efficiently synthesizing the functionalized bicyclic cyclopropanes in high yields and stereoselectivities. Shortly thereafter, they reported a continuous-flow method for generating cyclopropanes from aryl diazoesters and styrenes under photochemical conditions.131 In 2021, Barham et al. introduced another continuous-flow approach for the cyclopropanation of heterocyclic compounds.132 Compared to the traditional synthetic methods, continuous flow conditions can significantly improve the production efficiency and process safety in the large-scale experiments. Recently, Lv et al. presented a method using benzyl-substituted diazo esters as carbene precursors for the cyclopropanation of alkenes,133 which demonstrated excellent reactivity towards both aromatic and aliphatic substrates. Additionally, Xuan et al. reported the cyclopropanation of NHC boranes with diazo esters,134 achieving successful reactions with imidazole heterocyclic alkenes, while diazo esters containing electron-withdrawing groups on the benzene ring were unsuitable for this process.
In 2020, Wu et al. developed a photocatalytic method for the cyclopropanation of N-tosylhydrazones and aromatic alkenes in the presence of triethylamine.135 However, this approach has limited substrate compatibility, and the specific reaction mechanism is not detailed. Recently, Zhang et al. reported a visible-light-mediated strategy for synthesizing highly hindered cyclopropanes via energy transfer utilizing stable N-tosylhydrazones as accessible carbene precursors without hazardous diazo compounds.75 This method facilitated the straightforward construction of cyclopropanes in complex drug molecules and natural products. Initially, N-tosylhydrazone 104 underwent deprotonation to form the N-tosylhydrazone anion 105 in the presence of a base (Scheme 5b). Subsequently, the anion engaged in energy transfer with the excited-state catalyst 4CzIPN* to reach the triplet state (106), followed by the release of the sulfonyl group and nitrogen to generate triplet carbene species 108. Finally, the carbene species reacted with aromatic alkenes to complete the cyclopropanation process. Unfortunately, this strategy was only applicable to aromatic alkenes. Subsequently, König et al. developed an efficient method for the synthesis of spirocyclopropanes.136 Various alkyl-substituted N-tosylhydrazones reacted effectively with the electron-deficient alkenes in this two-step process, which could be readily terminated. Subsequent studies have suggested an ester substitution pathway involving a pyrazolidine intermediate. Recently, Wu et al. reported a strategy for selectively cyclopropanating 2-pyridones, which demonstrated high site selectivity and good tolerance towards functional groups.137 Although the mechanism was understood, this method could offer a versatile platform for the synthesis of bridged heterocycles.
In addition to the application of typical diazo esters or N-tosylhydrazones, Itoh et al. developed a synthetic method to generate selenium ylides from readily available dibenzoselenophenes.138 These ylides were excited under visible light to produce carbenes without the need for photocatalysts. This approach enabled the cyclopropanation of various alkenes through the formation of carbene species. However, the reaction involved the drawback of relatively low product yields. In 2022, the Priebbenow group described a reaction that generated a carbene intermediate from a ketone, followed by intramolecular [2 + 1] cyclopropanation with alkenes.139 Due to the high nucleophilicity of the silicon-based carbene intermediate, this method exhibited good compatibility with electron-deficient alkenes and could form spirocyclic frameworks, even with inactivated alkenes. The advantage of this strategy included the rapid reaction completion within 10 min without the requirement of a catalyst, while it required multiple complex reaction steps.
Furthermore, Shen et al. developed a strategy to directly synthesize fluoromethylated cyclopropanols using tri(di)fluoroacetylsilanes as carbene precursors.74 This reaction was suitable for various aliphatic and aromatic alkenes (Scheme 5c, 112 and 113). Moreover, the gram-scale reaction and late-stage functionalization of complex molecules, such as isoxepac (114), indicated the synthetic capabilities of this method. The mechanism involved a three-linear-state carbene, rather than a synergistic mechanism with a single-linear-state carbene. Although the strategy was novel, this approach did not extend to non-terminal alkenes, and the selectivity of difluoromethyl acylsilane-derived carbenes was generally lower than that of trifluoromethyl acylsilane-derived carbenes in reactions. Later, they continued their exploration in this field and reported the synthesis of (difluoromethylene) cyclopropanes via the reaction of silylated trifluorodiazoethanes with aromatic alkenes.140 This one-pot strategy exhibited excellent functional group tolerance, with the ability to tolerate various aromatic and aliphatic alkenes, yielding a diverse range of (difluoromethylene) cyclopropanes (115–118) in moderate to excellent yields. Based on the detailed mechanistic experiments and computational data, a plausible mechanism was proposed (Scheme 5d). Initially, the excited state of the catalyst underwent energy transfer (EnT) with 119, generating 120 and subsequently releasing N2 to form triplet carbene 121. This triplet carbene was added to the alkene to form a diradical intermediate (123), which then underwent intramolecular cyclization to generate cyclopropane product 124. Fluoride then attacked the silyl group, generating unstable anionic intermediate (125), which finally eliminated fluoride to yield (difluoromethylene) cyclopropane. In 2023, Gryko et al. used 1,3,4-oxadiazole compounds as diazo precursors for the synthesis of spirocyclopropanes.141 The excited catalyst transferred the energy to the 1,3,4-oxadiazole compound in its triplet state. Subsequently, the cleavage of the C2–N3 bond in the oxadiazole led to the formation of a diazo group, which decomposed into the triplet carbene and diazoalkane. In the presence of alkenes, the triplet carbene formed a diradical intermediate followed by cyclopropane. Simultaneously, diazoalkanes underwent 1,3-dipolar cycloaddition with an alkene, resulting in the formation of pyrazoline. The pyrazoline could generate the spirocyclopropane via the diradical intermediate state under photosensitization. Although this strategy was effective for synthesizing various alkyl-substituted spirocyclopropanes, it was challenging to synthesize aromatic cyclopropanes that did not react with alkenes containing electron-donating groups.
Overall, photo/electrochemical methods have recently emerged as effective approaches for accessing carbene/carbyne intermediates. These mild and efficient techniques have enabled several carbene precursors, including diazo compounds, N-tosylhydrazones, and ylides to effectively facilitate cyclopropanation, significantly advancing the field of photocatalytic carbene chemistry. Despite the substantial progress made with these strategies, safer and more widely available carbene precursors still need to be designed, especially compared to cyclopropanation via radical processes.
3.3 Cyclopropanation through the activation of alkenes
In addition to the significant efforts to access cyclopropanes via radical or carbene pathways, the activation of alkenes to produce cyclopropanes is another crucial approach. In 2017, Ferreira et al. disclosed a visible light-mediated cyclopropanation between electron-rich aromatic alkenes and diazoesters.76 A range of aryl alkenes were successfully applied to cyclopropanation under standard conditions (127–129). For the cycloaddition reaction, the excited state Cr(III) catalyst initially induced a single-electron oxidation of alkene (130), and the resulting radical cation (131) reacted with the nucleophilic diazo compound (Scheme 6a). The loss of N2 provided a “long-bonded” radical cation intermediate 134. At this stage, electrons were transferred from either the reduced-state Cr complex or another equivalent of the alkene to produce the cyclopropane product. However, this approach still faced challenges, such as its inapplicability to tetrasubstituted alkenes and the nullification of the reaction by some arylalkyl alkenes, which requires further exploration.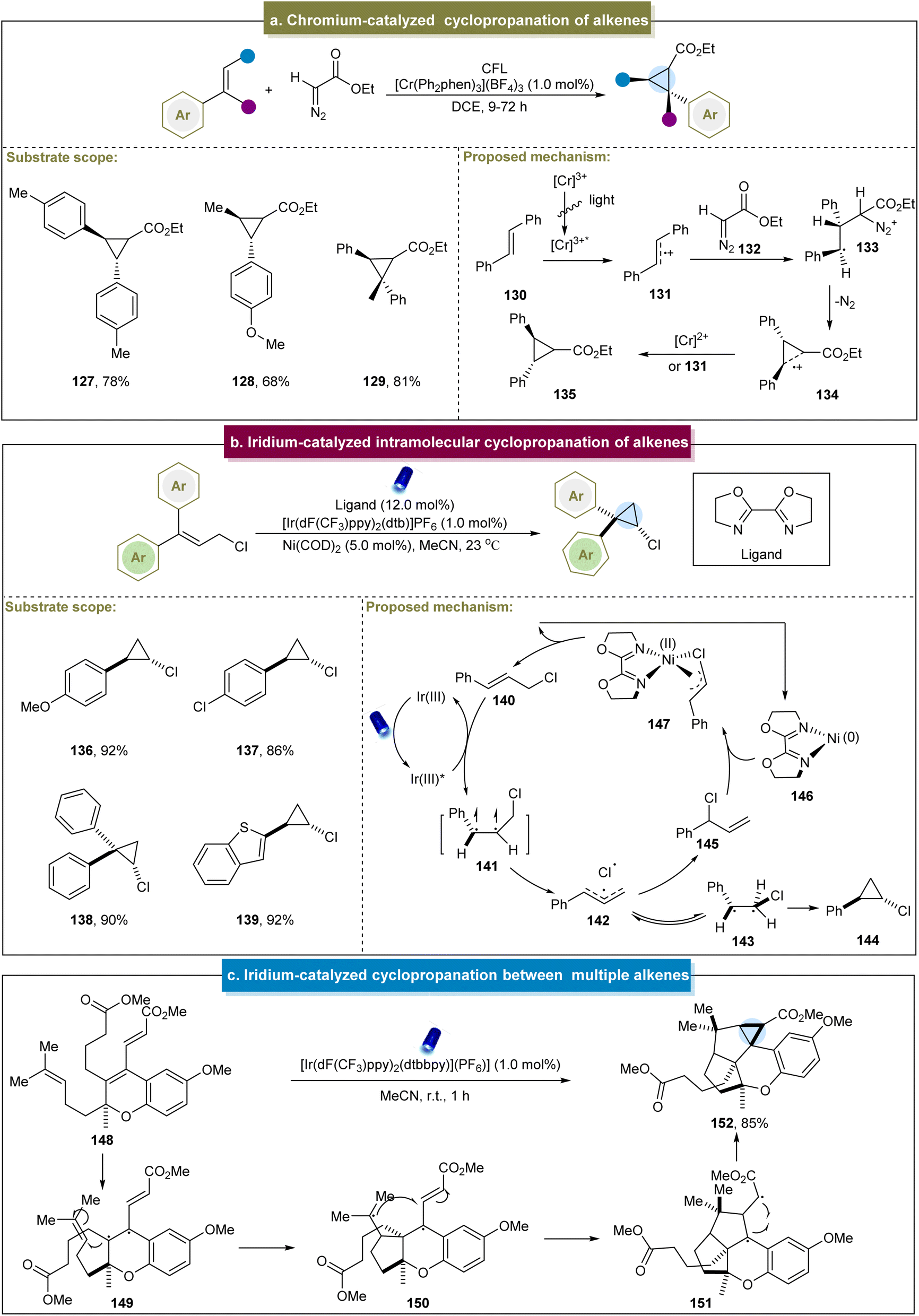 | ||
| Scheme 6 (a) Chromium-catalyzed cyclopropanation of alkenes. (b) Iridium-catalyzed intramolecular cyclopropanation of alkenes. (c) Iridium-catalyzed cyclopropanation between multiple alkenes. | ||
Later, Zeller et al. developed a photo-induced method for the intramolecular cyclopropanation of alkene–alkyne molecules.142 The initial step of this reaction involved the oxidation of the alkyne by an excited-state photocatalyst to form an ynamide radical cation, followed by a radical cascade process to complete the cyclopropanation. This strategy provided a valuable new approach for the synthesis of various 3-aza[n.1.0]bicycles.
In 2020, Tambar et al. reported the photocatalytic intramolecular cyclopropanation of acyclic cinnamyl chlorides.77 The reaction exhibited high tolerance towards the aromatic systems with various substituents and demonstrated excellent anti-selectivity and yields (136–138). This approach also indicated the tolerance to heterocycles such as benzothiophene (139). Upon irradiation, the energy transfer from the Ir(III) photocatalyst activated the cinnamyl chloride (140) to its excited state (141, Scheme 6b). Subsequently, a triallyl pair (142) containing a chlorine radical was formed from the excited complex. There were two pathways for intermediate 142. One path passed through triplet-β-Cl intermediate (143), generating the desired cyclopropanation product through radical coupling. The other pathway involved the reaction of Ni(0) complex (146) to form the nickel alkenyl complex (147), thus regenerating cinnamyl chloride. Unfortunately, this strategy was not applicable to cyclopropanation of terminal alkenes. Similarly, Schindler et al. reported a 14-step synthesis of (+)-cochlearol B, suggesting that the cyclopropane byproduct was obtained during the photochemical reaction (Scheme 6c).143 In this process, the alkene in 148 was initially excited to its photochemical state, generating the diradical 149. The resulting diradical reacted with the homoprenyl subunit to form ring B (150). Subsequently, addition to the electrophilic carbon of the methyl acrylate fragment occurred, resulting in a second five-membered ring (151). Ultimately, this led to the formation of cyclopropanes through radical recombination.
Recently, Glorius et al. reported an energy transfer-mediated cascade dearomatization [2 + 2] cycloaddition between the quinoline derivatives and aromatic alkenes, selectively yielding the corresponding cyclopropane.144 The method exhibited excellent tolerance towards diverse functionalized 2-chloropropene derivatives, providing the corresponding chlorinated cyclopropanes in good yields (153–155). A plausible mechanism was proposed (Scheme 7a). Initially, an energy transfer occurred between 156 and the catalyst, generating the excited state 158. Subsequently, 158 underwent [2 + 2] cycloaddition with 157 to form the cinnamyl chloride intermediate 159. Intermediate (159) underwent another energy transfer, leading to the cleavage of the C–Cl bond and generation of triplet radical pair 161. Finally, the Cl radical reacted with the intermediate radical to form the C–Cl bond, and the radicals at both ends couple to form the C–C bond, yielding the final cyclopropanes. This energy-transfer-mediated strategy demonstrated remarkable cis-selectivity, circumventing the general reactivity and selectivity issues associated with the photochemical [2 + 2] cycloadditions of various other aromatic substances.
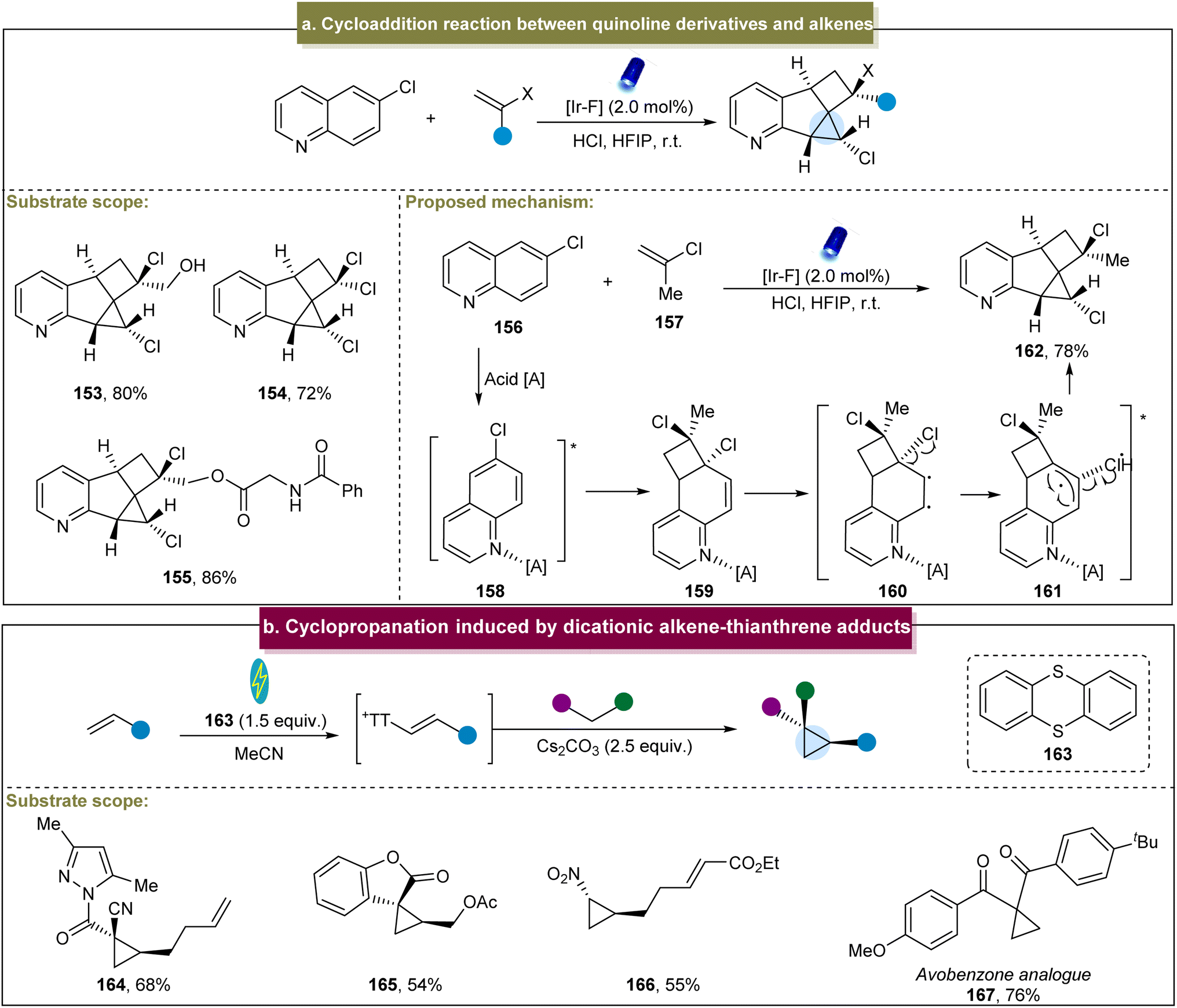 | ||
| Scheme 7 (a) Cycloaddition reaction between quinoline derivatives and alkenes. (b) Cyclopropanation induced by the dicationic alkene–thianthrene adduct. | ||
In 2023, Wickens and co-workers developed a modular method for the preparation of cyclopropanes.78 With RVC and Ni as electrodes and under constant current conditions, this method was scalable and applicable for the generation of diverse cyclopropanes and exhibited broad substrate tolerance including diverse alkenes and nucleophilic reagents (Scheme 7b, 164–167); however, this method was restricted to specific alkenes bearing electron-withdrawing groups. Mechanistic studies have indicated that the dicationic addition compound was rapidly eliminated under the reaction conditions to form the alkenyl thianthrenium salt, a crucial electrophilic intermediate in the cyclopropanation reaction. Compared with the traditional metal-catalyzed methods, this approach has expanded the utilization of inexpensive coupling agents and readily available reagents for constructing the cyclopropane building blocks.
In summary, strategies for activating alkenes in the synthesis of cyclopropanes through photo/electrocatalysis mainly involve generating the alkene radical cations or diradicals that efficiently enable diverse cyclopropane syntheses. However, the photoredox activation of alkenes is generally challenging because of their relatively high redox potentials. Therefore, the selectivity of alkene activation should be addressed when applying this method.
3.4 Cyclopropanation through other methods
In addition to the aforementioned methods for synthesizing cyclopropanes via donor- or acceptor-induced reactions, other methods can be triggered by the exceptional reactivity. For instance, Egorov et al. developed an electrochemical method for the synthesis of cyclopropanes from alkylidene malononitriles and malononitriles.145 This method differed from the radical generation through halogen departure. The presence of pyridine facilitated the generation of carbanion intermediates upon the departure of the bromine atom.In 2021, Hayashi et al. introduced a photosynthetic method that directly coupled the cyanoalkanes with electron-accepting C1 carbon units to produce cyclopropanes.93 This method exhibited excellent chemical selectivity for both electron-rich and electron-poor para-substituted phenyl groups, yielding cyclopropanes in good yields. Subsequently, a possible mechanism for this strategy was proposed (Scheme 8a). First, under the influence of a base, the cyanoalkane underwent deprotonation to generate the potassium salt 173. The α-anion (173) formed an electron donor/acceptor complex with N-hydroxyphthalimide ester (174). Under light irradiation, this complex generated the β-acidic α-radical (175) and radical anion (177). The radical anion (177) fragmented into a C1 α-radical (178) by releasing CO2 and N-phthalimide anions. The α-radical (175) lost the β-proton to obtain the α/β-radical anion (176). The intermolecular homogeneous coupling between the radical anion and the o-stabilized radical then formed a new Csp3–Csp3 bond at the β-position, resulting in the cyclization of the α-anion (179) to provide the corresponding cyclopropane. However, this strategy had some limitations; for example, the redox-active ester had poor tolerance towards aromatic rings, and the cyano group and strong electron-withdrawing groups were also necessary. Similarly, Kawashita et al. developed an electrochemical transformation method to convert alkyl 2-chloroacetates into cyclopropanes.146 Although the substrate range for this reaction was relatively limited, it offered a new approach for preparing 1,2,3-trisubstituted cyclopropane derivatives under electrochemical conditions.
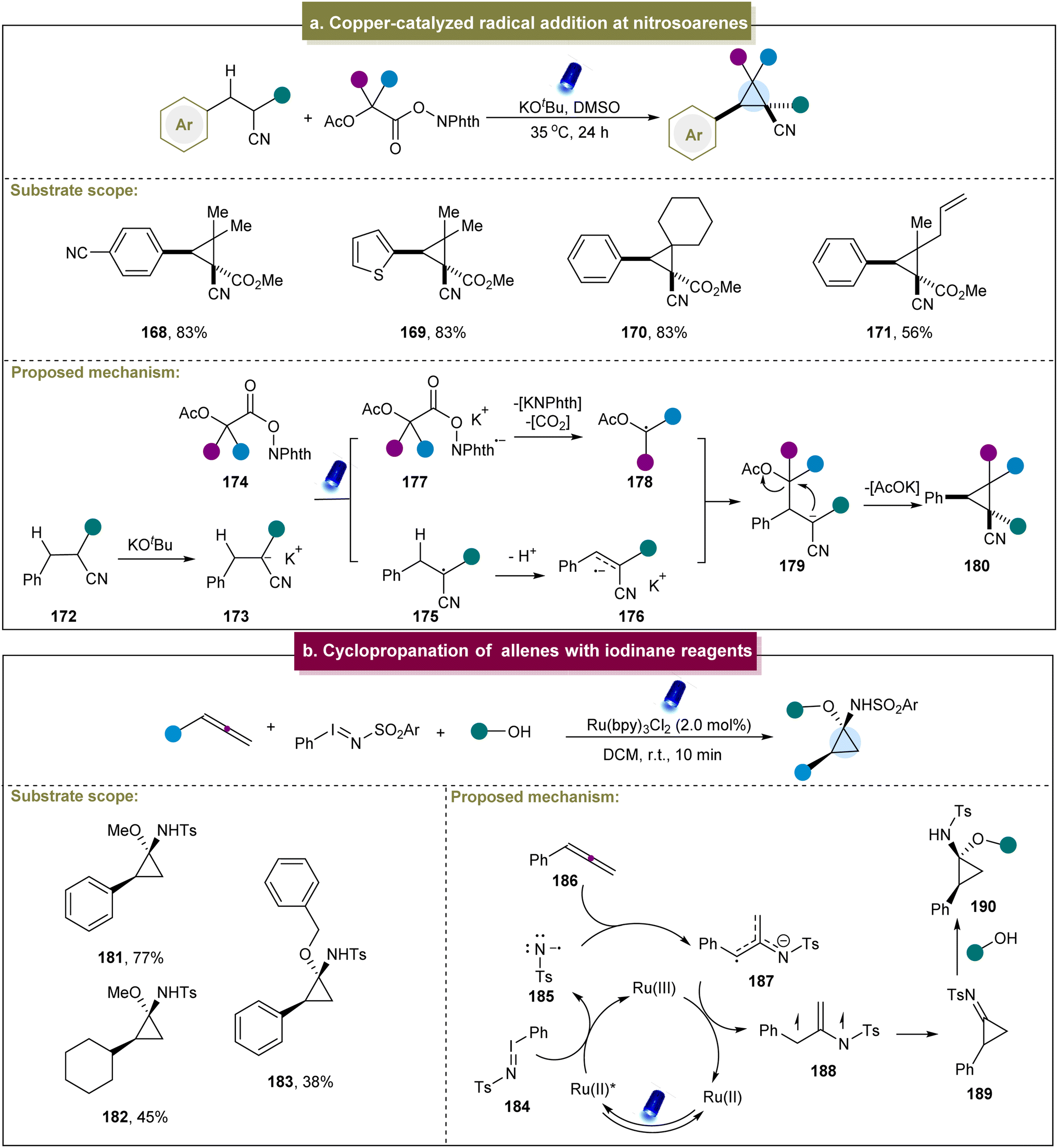 | ||
| Scheme 8 (a) Iridium-catalyzed intramolecular cyclopropanation of alkenes. (b) Cyclopropanation of allenes with iodinane reagents. | ||
Furthermore, Koenigs et al. reported a photocatalyzed cyclopropanation of allenes using iodinane reagents.94 Exploration of the substrate scope revealed that various aromatic allenes with different functional groups efficiently produced amino cyclopropanes in high yields, while aliphatic allenes showed decreased reactivities (181–183). Finally, a plausible mechanism for this reaction was proposed (Scheme 8b). Initially, iodinane 184 underwent the SET with the photo-excited catalyst, generating the nitrene radical anion 185, which was added to the allenic carbon atom to form 187. Subsequently, 187 underwent further oxidation with the ruthenium catalyst, followed by cyclization, leading to the formation of the highly reactive cyclopropyl imine intermediate 189. The experimental results indicated that the cyclopropyl imine intermediate could react with methanol to form the cyclopropyl amine product. Jubault et al. reported a method for electrochemically generating cyclopropylamines from the corresponding cyclopropyl amides, primarily using the Hofmann rearrangement strategy.147
In summary, distinct from the radical, carbene, and alkene initiation pathways for cyclopropane construction, there were still intriguing and promising strategies under photo/electrocatalytic conditions, such as EDA complex formation or direct photolysis. These approaches could facilitate the design of simpler and greener cyclopropanation methods.
4 Synthesis of aziridines by photo/electrochemical approaches
In recent years, significant advancements have been achieved in the synthesis of aziridines using photo/electrochemical methods.79 These include donor-initiated strategies that utilize nitrenes, nitrene radical anions, N-radicals, N-iodoamines, and formaldimines to selectively produce aziridines from various alkenes.80,81 Additionally, acceptor-initiated approaches involving alkene radical cations and dicationic alkene–thianthrene adducts have been developed.82 These methodologies have been applied diversely, including the aziridination of styrene derivatives, coumarins, and aliphatic amines.83–854.1 Aziridination through donor-initiated methods
In 2016, Yoon et al. documented a visible-light-mediated aziridination method that selectively formed triplet nitrenes from azidoformates.79 Mechanistic studies have confirmed that the aziridination was mediated through the [3 + 2] cycloaddition of the carbamoyl azide, followed by the photocatalytic decomposition of the 1,2,3-triazoline intermediate. Particularly, diverse alkenes including styrenes, aliphatic alkenes, tetrasubstituted alkenes, etc. were accommodated in this system (Scheme 9a, 191–194). Furthermore, the reaction could be conducted on a preparative scale by using a flow reactor. However, there have been no reports of aromatic alkenes containing electron-donating groups, and no explicit mechanism has been reported. Inspired by this elegant work, the group of Lu developed a selective intramolecular o-allylphenyl azidoformate aziridination through an energy transfer mechanism.148 This method involved the generation of triplet nitrene through the release of N2 by triplet azides. The triplet nitrene then undergoes radical addition with an alkene, followed by low-energy barrier radical coupling to yield the desired aziridine product. A limitation of the manuscript was the lack of in-depth exploration regarding the introduction of allyl groups. Similarly, Zhu et al. developed catalyst-free visible-light-induced alkene aziridination.149 This reaction proved effective for the terminal alkenes, intramolecular alkenes, electron-deficient alkenes, and dienes. However, the system suggested the intolerance towards heteroatom-containing alkenes and electron-donating groups.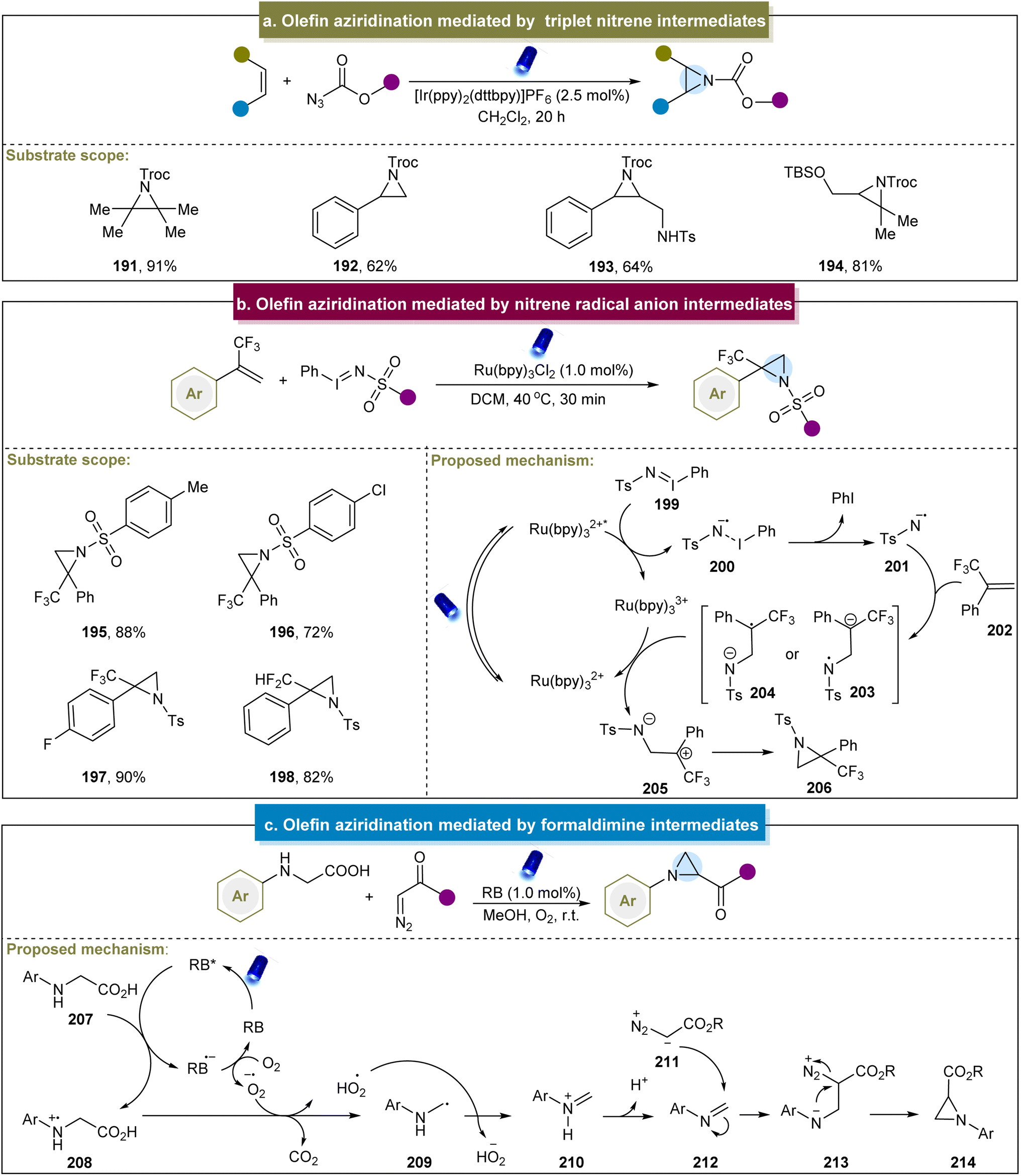 | ||
| Scheme 9 (a) Alkene aziridination mediated by a triplet nitrene intermediate. (b) Alkene aziridination mediated by a nitrene radical anion intermediate. (c) Alkene aziridination mediated by formaldimine intermediates. | ||
Furthermore, Koenigs et al. reported the direct aziridination of iodinane with α-trifluoromethyl styrenes under photocatalytic conditions.80 This strategy utilized iminoiodinanes to generate nitrene radical anions (200) via the oxidative quenching of the excited state of Ru(bpy)32+ (Scheme 9b). The nitrene radical anion proved to be a versatile intermediate, enabling the synthesis of fluorinated aziridines, including perfluoroalkyl and difluoromethyl aziridines (195–198). However, alkylsulfonyl groups were incompatible under these conditions, and limitations were observed with ortho-substituted aromatic rings, pyridine, and aliphatic alkenes. Building on Königs's work, Rastogi et al. reported a photoredox aziridination strategy for synthesizing chalcones.150
What's more, Zhou and co-workers developed a decarboxylative aza-Darzens reaction to synthesize monosubstituted aziridines via formaldimine intermediates.83 The reaction mechanism, depicted in Scheme 9c, began with the quenching of the excited state RB* by N-aryl glycine (207) to form cationic radical 208. Subsequent decarboxylation of 208 generated the α-amino alkyl radical (229), which underwent oxidation by superoxide radicals to form iminium ion 210. The deprotonation of the iminium ion produces the active imine 212, and aziridine formation occurred through the nucleophilic addition of diazo compounds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, followed by intramolecular nucleophilic attack of the nitrogen atom with N2 as the leaving group. More recently, Xuan et al. introduced a solvent-controlled divergent cycloaddition reaction for the construction of aziridines using α-diazo esters with hexahydro-1,3,5-triazines.151 Mechanistic studies have identified formaldimine as the key intermediate in this strategy. Generally, the activation of nitrogen atoms in donor compounds and their reactions with alkenes represent straightforward and efficient approaches for obtaining aziridines. Current aziridination strategies using photo/electrocatalysis primarily involve nitrenes, nitrene radical anions, N-radicals, N-iodoamines, and formaldimines.
N bond, followed by intramolecular nucleophilic attack of the nitrogen atom with N2 as the leaving group. More recently, Xuan et al. introduced a solvent-controlled divergent cycloaddition reaction for the construction of aziridines using α-diazo esters with hexahydro-1,3,5-triazines.151 Mechanistic studies have identified formaldimine as the key intermediate in this strategy. Generally, the activation of nitrogen atoms in donor compounds and their reactions with alkenes represent straightforward and efficient approaches for obtaining aziridines. Current aziridination strategies using photo/electrocatalysis primarily involve nitrenes, nitrene radical anions, N-radicals, N-iodoamines, and formaldimines.
In addition, Siu and Yudin outlined an electrochemical aziridination method employing N-amino phthalimide as the nitrogen source.152 This approach offers the notable advantage of eliminating the need for stoichiometric amounts of toxic oxidants and metal additives. Moreover, with platinum as the electrode and under constant voltage conditions, both electron-rich and electron-poor aliphatic alkenes were efficiently converted to aziridines (Scheme 10a, 215–217). However, this study could not offer a detailed explanation of this mechanism.
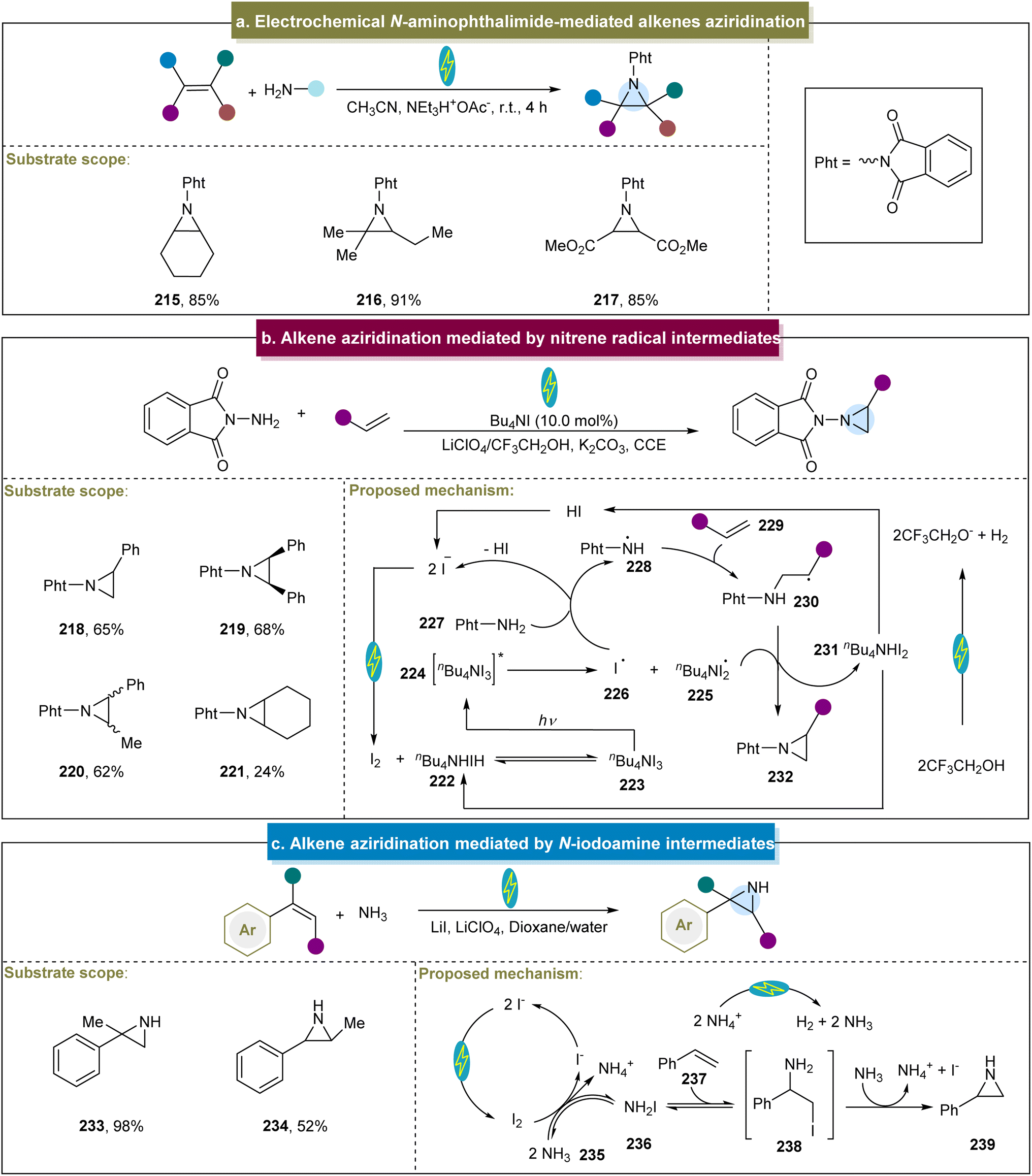 | ||
| Scheme 10 (a) Electrochemical N-aminophthalimide-mediated alkene aziridination. (b) Alkene aziridination mediated by a nitrene radical intermediate. (c) Alkene aziridination mediated by an N-iodoamine intermediate. | ||
In addition to the nitrene and nitrene radical anion methods, another group developed an efficient electrochemical aziridination strategy utilizing N-radicals as key intermediates.81 In this case, with GC (graphitic carbon) and iron as electrodes and under constant current conditions, a wide range of aromatic or aliphatic alkenes had been proved to be compatible with the catalytic system (218–221). However, the reactivity of aliphatic cyclic aromatic hydrocarbons under these reaction conditions was poor, indicating room for further improvement. Initially, the anodic oxidation of iodide generated molecular iodine, which reacted with n-Bu4NI to form ammonium triiodide (n-Bu4NI3, 223, Scheme 10b). Under visible-light irradiation, I˙ (226) was produced and abstracted a hydrogen atom from Pht-NH2 (227), forming the aminyl radical 228. This radical then reacted with the alkene to form radical (230). The homolytic cleavage of the N–H bond in 230, facilitated by I2˙− through hydrogen atom abstraction, was followed by intramolecular cyclization, yielding the aziridine products. Subsequently, Itoh et al. reported a practical aziridination method using the styrene derivatives and p-toluenesulfonamide as the nitrogen source.153 Various functionalized styrenes were adopted in this system, affording the aziridines in excellent yields, whereas the aliphatic alkenes did not react. The proposed mechanism involved TsNH2 reacting with molecular iodine to form the intermediate TsNHI. The photolysis of the N–I bond then generated a nitrogen-centered radical that reacted with the styrene to form the target aziridine. Similarly, the group of Xu developed a method for achieving the stereospecific aziridination of alkenes through photoredox catalysis.154 Aziridines were synthesized using various alkenes and N-protected aminopryridinium salts as the nitrogen radical sources, achieving excellent stereocontrol. However, the reaction conditions were not suitable for alkenes containing vinyl groups with electron-donating substituents.
Additionally, other methods have been developed for the construction of aziridines using N-iodoamine intermediates. He et al. initially reported the electrochemical aziridination of chalcones using simple amines as the nitrogen sources.155 Unfortunately, anilines and non-chalcone-type alkenes were not suitable for this approach. Furthermore, Cheng et al. described the ArI-mediated electrochemical aziridination of electron-deficient alkenes in an undivided cell.82 The reaction proceeded through a stepwise pathway involving in situ-generated iodine nitrene and acetyl hydrazine as the aziridination reagents. This method enabled a scale-up to 10 g, and the product served as a precursor for α-amino acids. Recently, De Vos et al. utilized inexpensive ammonia as the nitrogen source and iodide as a redox mediator for N–H aziridination (Scheme 10c).156 Using carbon and Ni as electrodes under constant current conditions, this work generated a series of unprotected N–H aziridines without the need for pre-oxidized nitrogen sources or subsequent organic de-protection steps, demonstrating the excellent atomic economy. Moreover, mechanistic studies have indicated that straightforward N–I species, such as NH2I, are effective tools for aziridination.
4.2 Aziridination through the activation of alkenes
Considerable efforts have been devoted to exploring pathways for accessing aziridines through donor-initiated methods. The activation of alkenes to facilitate aziridination has emerged as a crucial approach. In 2013, Cho et al. developed a visible-light-induced trifluoromethylation of allylamines to synthesize CF3-containing aziridines, emphasizing selective nucleophilic reactions over intermediate elimination reactions.87 Moreover, perfluoroalkylations with other perfluoroalkyl iodides such as C3F7I and C4F9I proceeded efficiently under standard conditions. Subsequently, Zhao et al. introduced a strategy for direct fluorinated aziridination of alkenes, leveraging the non-covalent interactions between N-allylanilines and fluoroalkyl iodides.85 However, the use of heteroaromatic substrates was not suitable for this reaction, limiting its application in practical synthesis. The potential mechanism of this reaction is shown in Scheme 11a. Initially, a non-covalent interaction occurred between amine (240) and the C–I bond. Upon irradiation with visible light, the carbon radical (243) was generated. The subsequent cyclization proceeded via two possible pathways: (1) the resulting radical (244) abstracted an iodine atom from RFI, forming an intermediate (245). Finally, the desired aziridine was obtained in the presence of a base via additional cyclization reactions. (2) Intermediate (245) was formed via the SET process between 244 and RFI, leading to the generation of the desired product (247) through subsequent cyclization reactions.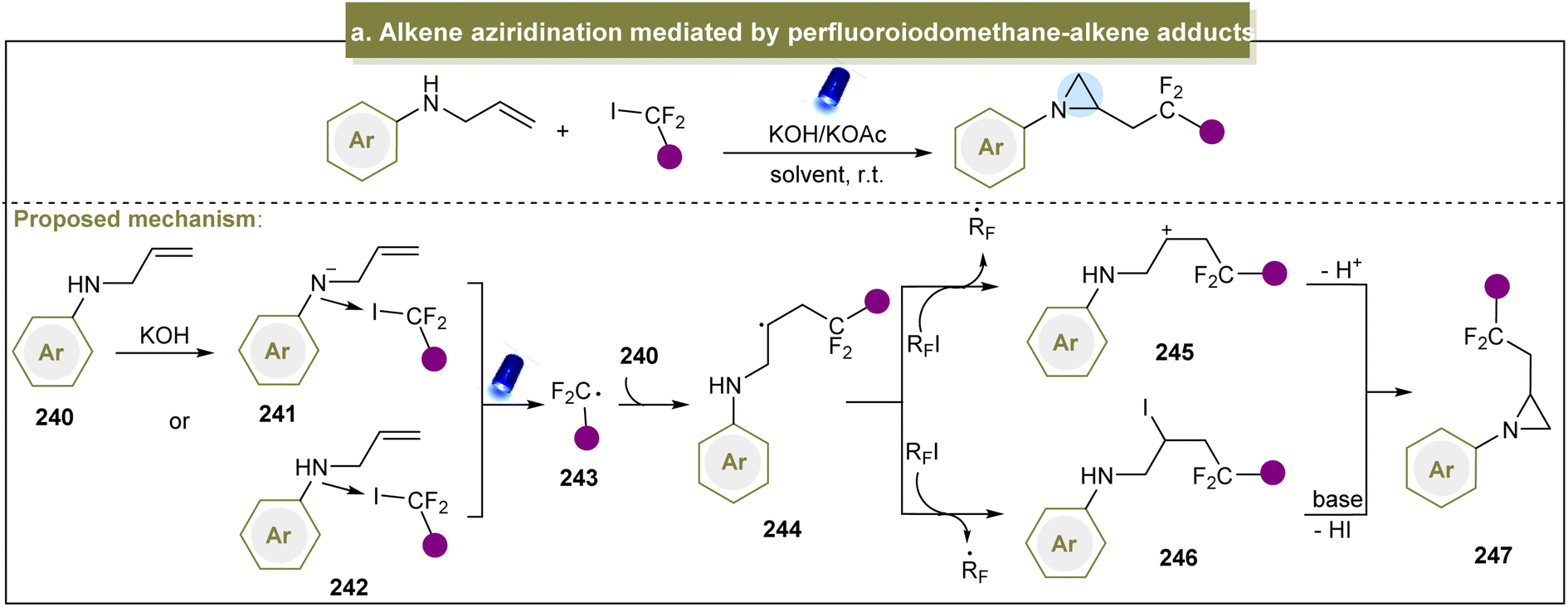 | ||
| Scheme 11 (a) Alkene aziridination mediated by a perfluoroiodomethane–alkene adduct. | ||
Recently, Wickens et al. identified an electrochemical strategy to obtain a metastable dicationic intermediate from aliphatic alkenes.84 With RVC as the electrode and under constant current conditions, the experiments demonstrated that the aziridine products were obtained from a broad range of amine nucleophiles, significantly broadening the limited scope of feasible N-substituents in aziridines synthesized from alkenes using traditional electrophilic nitrogen reagents (Scheme 12a, 248 and 249). Unfortunately, intramolecular and aryl alkenes were not suitable in this case, and the method still did not address the challenge of unreactive aromatic amines. The proposed reaction mechanism indicated that the mono- and bis-TT dicationic adducts (254 and 256) could be formed in high yields by reacting terminal alkenes and thianthrene (250) under the optimized electrolytic conditions. By simultaneously adding a primary alkylamine and an inorganic base without further separation, both cationic species were consumed to yield the desired aziridine products. In 2018, Li et al. developed an electrochemical strategy for generating aziridines using sulfamates as the nitrogen source.157 With the use of graphite felt electrodes and the condition of constant voltage, the direct aziridination of triaryl substituted alkenes was realized for the first time. Even though this electrochemical reaction was extended to use other multisubstituted alkenes, aliphatic alkenes were not feasible in this system (Scheme 12b).
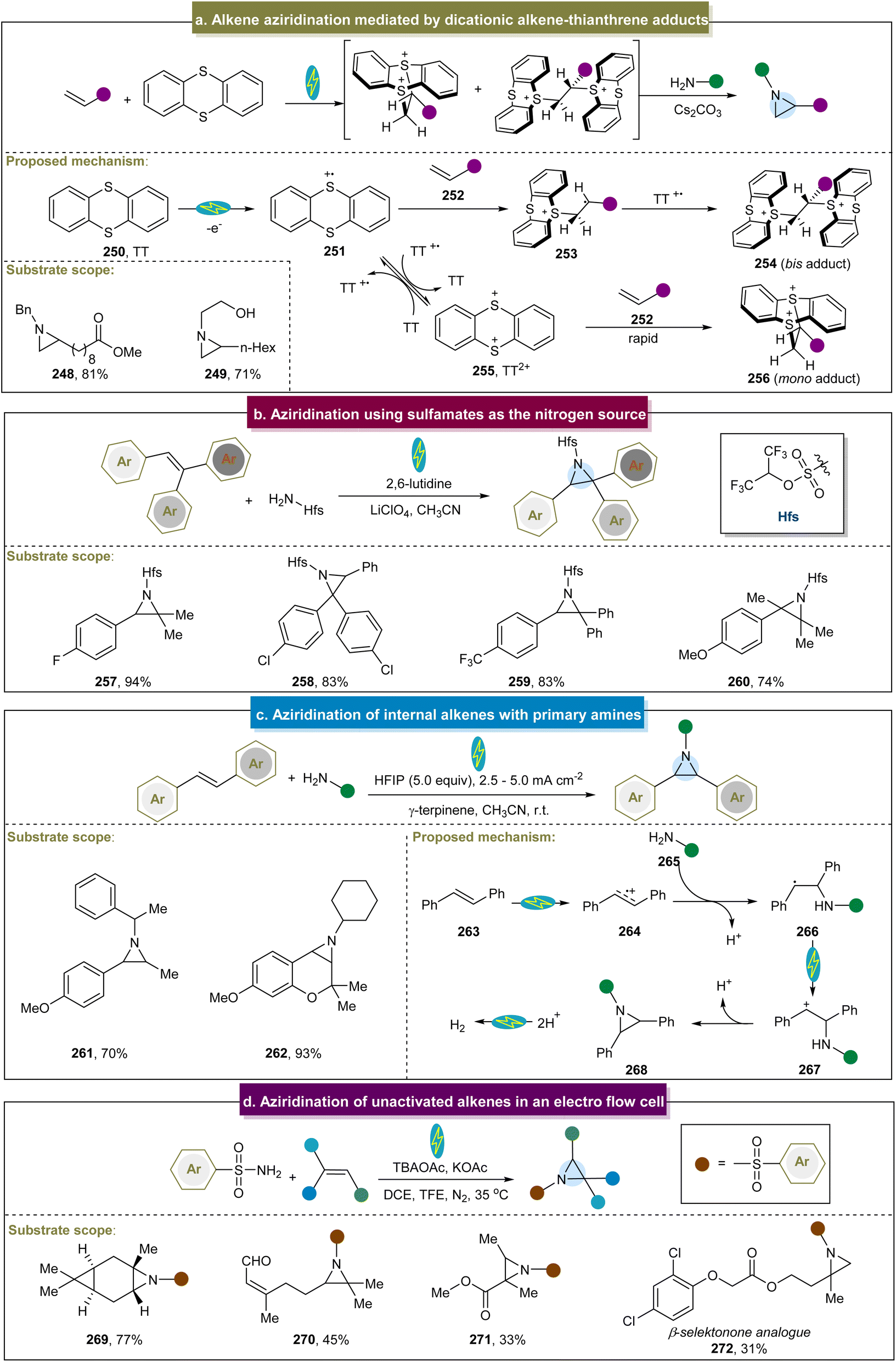 | ||
| Scheme 12 (a) Alkene aziridination mediated by a dicationic alkene–thianthrene adduct. (b) Aziridination using sulfamates as the nitrogen source. (c) Aziridination of internal alkenes with primary amines. (d) Aziridination of unactivated alkenes in an electro flow cell. | ||
Noël et al. proposed an electrochemical method for preparing aziridines through oxidative coupling between alkenes and primary alkylamines (Scheme 12c).158 With the use of carbon and Fe as electrodes, and under constant current conditions, this conversion was effectively conducted using primary alkylamines and alkenes with various structures and electronic properties (261 and 262). These findings indicated that the anodic oxidation of the alkene initiated the electrochemical aziridination process. Upon formation of the radical cation (274), it readily reacted with the amine (265) coupling partner. Following the deprotonation and subsequent SET, a carbocation (267) formed and underwent rapid, barrier-free intramolecular ring closure and deprotonation to yield the desired aziridine product. Unfortunately, electron-withdrawing functional groups and heterocyclic aromatics were not amenable, and this strategy didn't involve terminal alkenes and aromatic amines. Recently, Lei et al. introduced an electro-oxidative flow method that successfully achieved aziridination of natural products without requiring additional oxidants (Scheme 12d), and the flow cell was equipped with carbon paper, a platinum plate was used as the cathode and electrolyzed at a constant current.159 Two possible mechanisms were proposed for this reaction. The first pathway involved the initial oxidation of alkenes to form alkene radical cations, which then react with sulfonamides. The second pathway involved the simultaneous oxidation of sulfonamides and alkenes, followed by polar cross-coupling to produce the desired product. This strategy had a wide range of aliphatic substrates (269–271), and a series of aziridine derivatives from bioactive molecules were synthesized efficiently in good yields including 272.
The synthesis of aziridines using photo/electrocatalytic methods has attracted considerable attention, particularly for electrocatalytic applications. Numerous strategies have been proposed for providing diverse methodological options. However, certain methods are constrained by specific substrates or functional groups, limiting their applicability. Furthermore, there are relatively few reports on the photoinduced activation of alkenes for aziridination. Therefore, there is a pressing need to develop more universal and efficient methods to overcome substrate limitations and meet the broader demands for aziridine syntheses.
5 Synthesis of epoxides by photo/electrochemical approaches
The formation of epoxides often involves the participation of O2, which can be converted into 1O2 or O2˙− to facilitate epoxide formation. Additionally, epoxides can be generated by reacting alkenes with perfluoroiodomethane–alkene adducts, followed by the reactions with hydroxyl groups.87 Furthermore, the advancements in epoxide production have significantly expanded renewable energy applications,160–162 as summarized by Banerjee et al. in the electrocatalytic synthesis.163 Therefore, our focus was on the synthesis of small molecules containing epoxides, with a discussion of both the classic methods and recent advancements.Itoh et al. (2009) developed a convenient method for the epoxidation of various alkenes under fluorescent lamp irradiation.164 This method proved effective for aromatic-substituted alkenes, alkyl-substituted internal alkenes, and terminal alkenes but was unsuitable for alkyl-substituted alkenes. Subsequently, Katsuki et al. suggested that the Ru(NO)-salen complex could epoxidize aromatic alkenes under visible-light irradiation.165 However, this approach did not work well with substrates containing electron-withdrawing functional groups, and the mechanism remained unclear. Similarly, Luo et al. reported the visible-light-induced epoxidation of α,β-unsaturated ketones.166 The experimental and DFT calculations supported a mechanism involving the single linear oxygen. This method yielded a range of aromatic/aliphatic α,β-epoxy ketones but required long reaction times, which could be mitigated by employing the flow reaction. The reaction required precise temperature control, and compounds containing amide substituents were incompatible under these conditions. Later, Jin et al. discovered a visible-light-induced aerobic epoxidation method for the synthesis of spiro-epoxyoxindole derivatives (Scheme 13a).167 They extensively explored the substrate scope of the two aromatic rings, achieving smooth reactions and synthesizing target compounds in moderate to excellent yields. However, the presence of various substituents at different positions on 3-benzalindolin-2-ketone affected the efficiency of the reaction, and there has been limited exploration of aryl heterocycles as substrates under these conditions. In 2022, the Coeffard group described a photocatalytic method for epoxide synthesis.168 The reaction likely involved the singlet oxygen in oxidative dearomatization to form a hydroperoxide intermediate, followed by intramolecular oxygen atom transfer to the alkene side chain to yield epoxides.
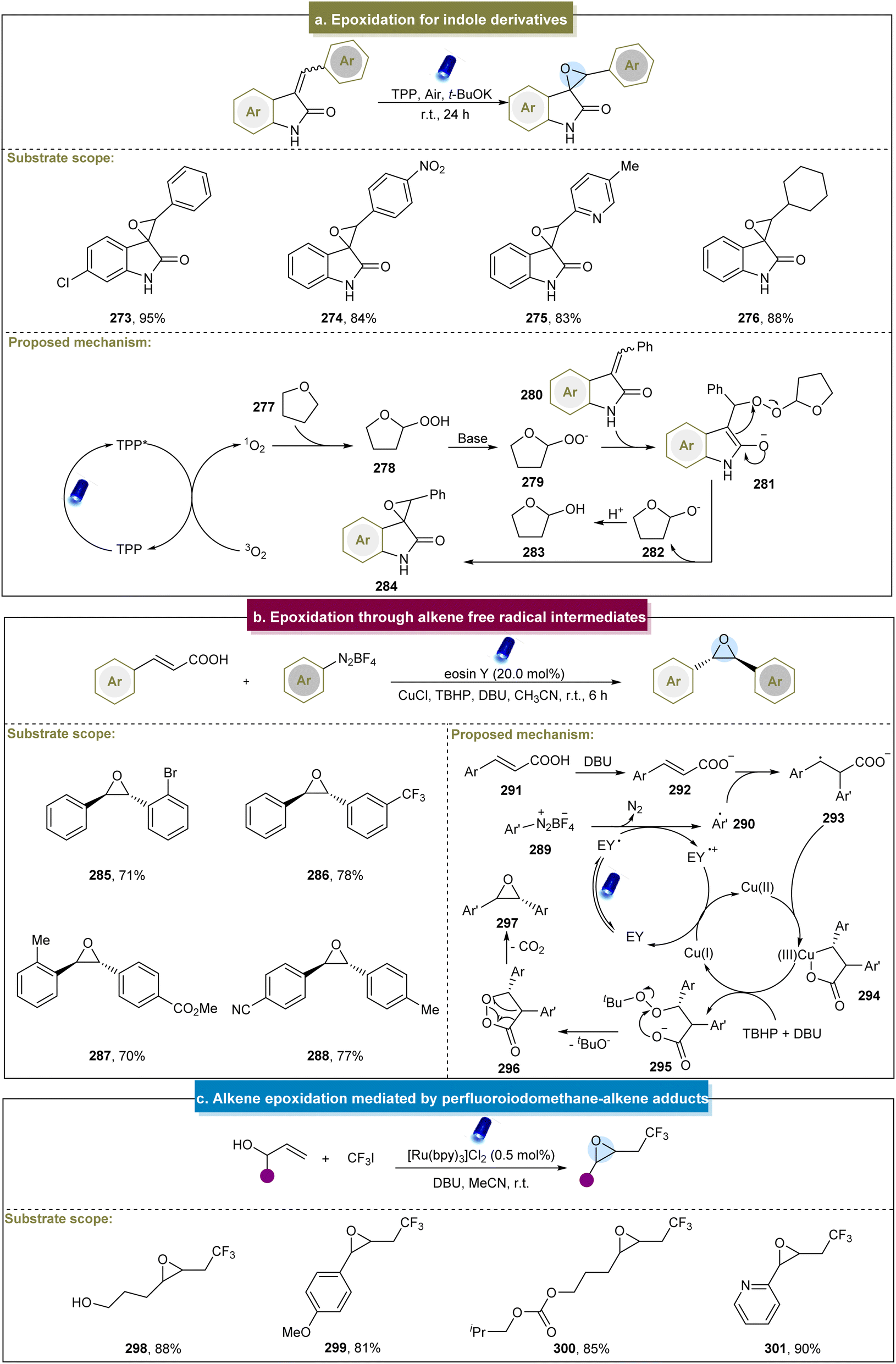 | ||
| Scheme 13 (a) Epoxidation through alkene free radical intermediates, TPP (meso-tetraphenylporphyrin). (b) Pd/ZnO nanoparticles catalyze epoxidation of alkenes. (c) Alkene epoxidation mediated by a perfluoroiodomethane–alkene adduct. | ||
In 2020, Huang et al. reported an epoxidation strategy using pyrenediones as the photocatalysts.169 This system proved effective for various aromatic and aliphatic alkenes. However, a thorough exploration of the reaction mechanism is lacking. Subsequently, Chao et al. described an epoxidation catalyzed by acetylated vitamin B2, targeting α,β-enones formed in situ via major SET and minor energy transfer pathways.170 This catalytic system successfully epoxidized a range of electron-deficient alkenes. However, it was incompatible with heterocyclic alkenes, and the reaction of α,β-enones containing sequences of aryl and aliphatic groups was unsatisfactory.
Recently, Singh et al. synthesized trans-oxirane oxide using TBHP as an oxidant.86 Various cinnamic acids and aryl diazonium salts reacted effectively (285–288). However, some aliphatic/heteroaromatic α,β-unsaturated carboxylic acids did not yield the desired products. The proposed mechanism is illustrated in Scheme 13b. Initially, the aryl diazonium salt (289) and excited eosin Y (EY*) underwent the SET process to form the aryl radical (290). The resulting interaction between the aryl radical and cinnamate (292) formed an intermediate (293), which chelated with Cu(II) to form a five-membered ring (294). In the presence of TBHP and DBU, this chelated ring proceeded through intermediate (295) to generate another species, 1,2-dioxolan-3-one (296), which ultimately underwent decarboxylation to produce the desired epoxides.
In addition to the 1O2 or O2˙− pathways, Cho et al. developed a method in 2013 for the visible-light-induced trifluoromethylation of allylic alcohols. This method involved a selective nucleophilic reaction via elimination of an intermediate to yield CF3-containing epoxides.87 Mild reaction conditions made this possible for a series of inactivated alkenes with different functional groups (Scheme 13c, 298–301). Unfortunately, this strategy has not yet been applied to the epoxidation of internal alkenes. Itoh et al. described the one-pot synthesis of α,β-epoxy ketones using benzyl alcohols and styrenes.171 This method involved the simultaneous oxidation of benzyl alcohols and styrenes to form benzaldehydes and phenacyl iodides, followed by the Darzens reaction to produce epoxides. Additionally, there was room for improvement, particularly because the styrenes with electron-donating groups exhibited poor reactivity. Furthermore, De Vos et al. introduced an electrocatalytic method for epoxidation.172 This approach achieved high yields with various aromatic- and aliphatic-substituted alkenes. Mechanistic studies have revealed that Br2 first formed Br+ species with alkenes under electric influence. Subsequently, under alkaline conditions, 2-bromoalkanol compounds were generated and the final products were obtained by eliminating bromide.
Various strategies have been developed for synthesizing epoxides, primarily involving the generation of superoxide intermediates to attack alkenes for epoxidation or to cyclize through the attack of neighboring leaving groups by hydroxyl groups. However, the existing literature often limits the substrate scope, highlighting significant opportunities for further exploration in this area. Given the importance of epoxides, there remains a substantial unresolved need to design novel, straightforward, and widely applicable epoxidation methods. Incorporating these important motifs into complex molecules via such strategies requires further investigation.
6 Synthesis of other three-membered rings by photo/electrochemical approaches
In addition to the syntheses of cyclopropanes, aziridines, and epoxides, methods for synthesizing cyclopropenes, vinyloxaziridines, and azirines have also been reported.91,92,95,96 Although some photo/electrochemical methods have been developed, there remains a demand for alternative pathways to construct these compounds, compared to cyclopropanes or aziridines.In 2018, Königs et al. reported visible-light-induced cyclopropenation between diazo esters and aryl alkynes.91 Various electron-donating and electron-withdrawing arylacetylenes efficiently produced the desired cyclopropenes in high yields, such as 302. Moreover, 1,2-diaryl alkynes or alkyl-aryl-substituted alkynes also yielded the target products (Scheme 14a, 303–305). Notably, this reaction was conducted rapidly in continuous-flow devices, which significantly enhanced the reaction yield. This approach offered several advantages, including the absence of metal catalysts, no need for air exclusion, and straightforward operation. However, the reaction exhibited limited reactivity towards heterocyclic aryl acetylenes. Subsequently, they reported photochemical carbene transfer reactions using diazoesters as the carbene precursors, which enabled cyclopropenation with unprotected propargyl alcohols.173 Unfortunately, the diazoesters reacted effectively only with the propargyl alcohols bearing electron-donating groups, restricting the scope of the reaction.
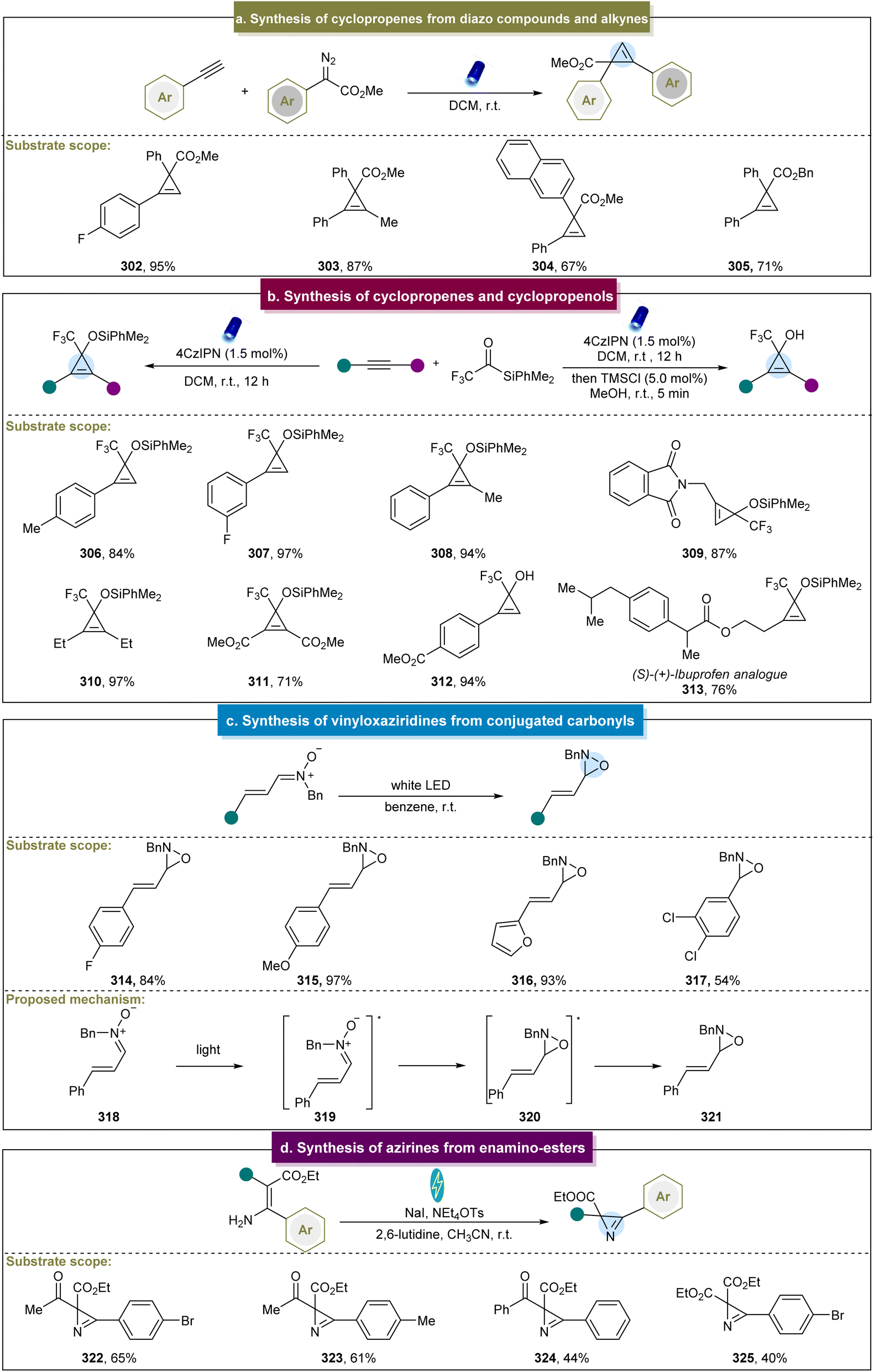 | ||
| Scheme 14 (a) Cyclopropenation of diazo compounds with alkynes. (b) Synthesis of cyclopropenes and cyclopropenols. (c) Synthesis of vinyloxaziridines from conjugated carbonyls. (d) Synthesis of azirines from enamino-esters. | ||
In 2021, researchers continued their investigations in this field and reported the photochemical cyclopropenation of aryl/aryl diazoalkanes with alkynes.174 Depending on the electronic nature of different substituents, selective generation of different products was achieved. Detailed calculations and experimental studies have indicated that the introduction of electron-donating and electron-withdrawing groups can significantly affect the singlet–triplet energy level splitting of diaryl carbene intermediates and the activation energy of consecutive carbene transfer reactions. This strategy can overcome the classical limitations of diaryl carbene reactions, enabling the aryl/aryl diazoalkanes to participate in photochemical carbene transfer reactions controlled by the singlet and triplet spin states. In addition to the diazo pathway, Shen et al. first reported a visible light-induced [2 + 1] cycloaddition reaction for the synthesis of cyclopropenes and cyclopropenols.92 The key intermediate in this reaction, α-CF3 siloxycarbene, served as the donor–acceptor-type carbene. Various terminal and internal aryl alkynes were compatible with this reaction system (Scheme 14b, 306–312), demonstrating good adaptability even in complex biologically active molecules such as (S)–(+)-ibuprofen derivative 313. Moreover, this strategy efficiently transformed the obtained cyclopropenes into cyclopropenols in high yields in a one-pot manner.
In addition to the synthesis of cyclopropenes, Moura-Letts et al. reported a method for synthesizing vinyloxaziridines via visible-light-induced energy transfer.95 The main mechanism involved the condensation reaction between the conjugated carbonyls and hydroxylamines (Scheme 14c). The generated vinyl nitrones underwent photocatalytic isomerization to yield the final products. This reaction utilized simple vinyl nitrones as the starting materials and achieved high yields and stereoselectivity in the formation of vinyloxaziridines (314–317). However, the substrate scope of this method was limited. Furthermore, Hilt et al. introduced an electrochemical method for the intramolecular synthesis of azirines from enamino esters (Scheme 14d, 322–325), and the divided cell was equipped with graphite, and platinum was used as the cathode and electrolyzed at a constant current.96 The success of this strategy relied on the generation of intermediate N-indoamines, offering a novel approach for synthesizing azirines. Furthermore, these azirines can serve as the building blocks to efficiently produce valuable 4-carboxy-oxazoles in high yields, thereby demonstrating the practicality of this method.
Overall, the synthesis of cyclopropenes, vinyloxaziridines, and azirines using photo/chemical methods remained underexplored. Current approaches for the cyclopropene synthesis typically involved the generation of specific carbene intermediates to react with alkynes, yielding the desired products. However, these carbene precursors often required electron-withdrawing groups, highlighting the need for broader exploration of more versatile carbene sources. Additionally, electrocatalytic methods for the synthesis of cyclopropene have not yet been reported. Given the potential pharmaceutical relevance of these structures, further efforts are required to enhance and diversify their synthetic pathways.
7 Conclusions
In conclusion, this review comprehensively outlines the synthesis of three-membered rings using photo/electrochemical approaches. These updates demonstrated the importance of photo/electrochemical methods as viable alternatives for synthesizing these rings, eliminating the need for traditional thermal- and transition-metal-catalyzed conditions. Furthermore, these sustainable methods have diversified the repertoire of three-membered rings, thereby expanding the chemical space relevant to drug discovery. Photo/electrochemical strategies not only enhanced the synthetic efficiency and structural diversity, but also promoted the high atom economy in obtaining these molecules. However, several opportunities and challenges have persisted in this field. For instance, a few studies have demonstrated practical applications, such as the gram-scale synthesis, late-stage modifications, and total synthesis of pharmaceuticals or natural products. Furthermore, tailored substrates were crucial for effective activation. However, these innovative methods have not been widely adopted in the industry. Despite the advancements in structural diversity, synthesizing highly congested three-membered rings has remained challenging owing to the steric hindrance, necessitating continued efforts to overcome this limitation. The enantioselective synthesis of three-membered rings using photo/electrochemical strategies remains a relatively unexplored area. Given the significance of chiral molecules in drug discovery, this challenge is expected to attract increased attention in the future. The advances in synthetic biology and enzyme catalysis have suggested that the enzyme-photo-coupled catalytic systems can facilitate the efficient and rapid synthesis of chiral cyclopropanes. Overall, this review could provide insights into the recent advancements in three-membered ring synthesis via photo/electrochemical methods and inspire further progress in this field in the near future.Data availability
All data are available from the authors on request.Author contributions
Y. Z. and S. D. conceived the project. Y. Z., N. Z. and H. X. conducted the literature survey. H. X., N. Z. and Q. L. wrote the manuscript. Y. Z. and S. D. guided and reviewed the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Chenguang Program of the Shanghai Education Development Foundation and Shanghai Municipal Education Commission (22CGA51, Y. Z.), Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (2024Y08, Y. Z.).References
- C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051–3060 RSC.
- A. de Meijere, S. I. Kozhushkov and H. Schill, Chem. Rev., 2006, 106, 4926–4996 CrossRef CAS PubMed.
- A. Gagnon, M. Duplessis and L. Fader, Org. Prep. Proced. Int., 2010, 42, 1–69 CrossRef.
- A. R. Gomes, C. L. Varela, E. J. Tavares-da-Silva and F. M. Roleira, Eur. J. Med. Chem., 2020, 201, 112327 CrossRef CAS.
- G. S. Singh, Mini-Rev. Med. Chem., 2016, 16, 892–904 CrossRef CAS.
- D. L. Dahlman and P. K. Kadaba, Pestic. Sci., 1988, 22, 71–81 CrossRef CAS.
- P. Xiao, T. Mori, I. Kamei and R. Kondo, FEMS Microbiol. Lett., 2011, 314, 140–146 CrossRef CAS PubMed.
- M. Luo, X. H. Zhang and D. J. Darensbourg, Acc. Chem. Res., 2016, 49, 2209–2219 CrossRef CAS.
- M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- M. M. Littleson, C. M. Baker, A. J. Dalençon, E. C. Frye, C. Jamieson, A. R. Kennedy, K. B. Ling, M. M. McLachlan, M. G. Montgomery, C. J. Russell and A. J. Watson, Nat. Commun., 2018, 9, 1105–1114 CrossRef.
- K. Yamada, M. Ojika and H. Kigoshi, Nat. Prod. Rep., 2007, 24, 798–813 RSC.
- K. E. Hernandez, H. Renata, R. D. Lewis, S. J. Kan, C. Zhang, J. Forte, D. Rozzell, J. A. McIntosh and F. H. Arnold, ACS Catal., 2016, 6, 7810–7813 CrossRef CAS PubMed.
- D. Y. K. Chen, R. H. Pouwer and J. A. Richard, Chem. Soc. Rev., 2012, 41, 4631–4642 RSC.
- C. J. Thibodeaux, W. Chang and H. Liu, Chem. Rev., 2012, 112, 1681–1709 CrossRef CAS.
- S. Ma, D. Mandalapu, S. Wang and Q. Zhang, Nat. Prod. Rep., 2022, 39, 926–945 RSC.
- S. R. Rajski and R. M. Williams, Chem. Rev., 1998, 98, 2723–2796 CrossRef CAS PubMed.
- R. W. Armstrong, M. E. Salvati and M. Nguyen, J. Am. Chem. Soc., 1992, 114, 3144–3145 CrossRef CAS.
- T. Tsuchida, R. Sawa, Y. Takahashi, H. Iinuma, T. Sawa, H. Naganawa and T. Takeuchi, J. Antibiot., 1995, 48, 217–221 CrossRef CAS.
- J. Salaiin and M. Bairtr, Curr. Med. Chem., 1995, 2, 511–542 CrossRef.
- C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651–11679 CrossRef CAS.
- A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213–1270 CrossRef CAS PubMed.
- H. U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151–1196 CrossRef CAS.
- M. Nagarajan, A. Morrell, A. Ioanoviciu, S. Antony, G. Kohlhagen, K. Agama, M. Hollingshead, Y. Pommier and M. Cushman, J. Med. Chem., 2006, 49, 6283–6289 CrossRef CAS PubMed.
- H. M. I. Osborn and J. Sweeney, Tetrahedron: Asymmetry, 1997, 8, 1693–1715 CrossRef CAS.
- L. Huang and W. D. Wulff, J. Am. Chem. Soc., 2011, 133, 8892–8895 CrossRef CAS.
- H. E. Simmons and R. D. Smith, J. Am. Chem. Soc., 1958, 80, 5323–5324 CrossRef CAS.
- L. A. Wessjohann, W. Brandt and T. Thiemann, Chem. Rev., 2003, 103, 1625–1647 CrossRef CAS.
- H. E. Simmons, T. L. Cairns, S. A. Vladuchick and C. M. Hoiness, Org. React., 2004, 20, 1–131 Search PubMed.
- H. M. Davies and A. M. Walji, Org. lett., 2005, 7, 2941–2944 CrossRef CAS.
- R. J. Crawford and M. Ohno, Can. J. Chem., 1974, 52, 3134–3139 CrossRef CAS.
- O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597–2632 CrossRef CAS PubMed.
- A. G. Herraiz and M. G. Suero, Synthesis, 2019, 51, 2821–2828 CrossRef CAS.
- S. D. Burke and P. A. Grieco, Org. React., 2004, 26, 361–475 Search PubMed.
- X. Li, N. Chen and J. Xu, Synthesis, 2010, 20, 3423–3428 Search PubMed.
- G. Callebaut, T. Meiresonne, N. De Kimpe and S. Mangelinckx, Chem. Rev., 2014, 114, 7954–8015 CrossRef CAS.
- L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881–7929 CrossRef CAS.
- G. Dequirez, V. Pons and P. Dauban, Angew. Chem., Int. Ed., 2012, 51, 7384–7395 CrossRef CAS.
- H. Jiang, K. Lang, H. Lu, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2017, 139, 9164–9167 CrossRef CAS PubMed.
- V. V. Thakur and A. Sudalai, Tetrahedron Lett., 2003, 44, 989–992 CrossRef CAS.
- Q. Q. Cheng, Z. Zhou, H. Jiang, J. H. Siitonen, D. H. Ess, X. Zhang and L. Kürti, Nat. Catal., 2020, 3, 386–392 CrossRef CAS.
- B. Bieszczad, X. Chen and S. Z. Zard, Org. Lett., 2022, 24, 9370–9374 CrossRef CAS.
- R. Mello, A. Alcalde-Aragonés, M. E. González Núñez and G. Asensio, J. Org. Chem., 2012, 77, 6409–6413 CrossRef CAS.
- D. G. Nam, S. Y. Shim, H. M. Jeong and D. H. Ryu, Angew. Chem., Int. Ed., 2021, 60, 22236–22240 CrossRef CAS.
- A. W. Johnson and R. B. LaCount, J. Am. Chem. Soc., 1961, 83, 417–423 CrossRef CAS.
- R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134, 12462–12465 CrossRef CAS.
- Q. He, M. P. Pu, Z. Jiang, H. Wang, X. Feng and X. Liu, J. Am. Chem. Soc., 2023, 145, 15611–15618 CrossRef CAS.
- P. Binger and H. M. Buch, Top. Curr. Chem., 1987, 135, 77–151 CrossRef CAS.
- F. L. Carter and V. L. Frampton, Chem. Rev., 1964, 64, 497–525 CrossRef CAS.
- M. Nakamura, H. Isobe and E. Nakamura, Chem. Rev., 2003, 103, 1295–1326 CrossRef CAS.
- J. M. Fox and N. Yan, Curr. Org. Chem., 2005, 9, 719–732 CrossRef CAS.
- I. Marek, S. Simaan and A. Masarwa, Angew. Chem., Int. Ed., 2007, 46, 7364–7376 CrossRef CAS.
- J. Aubé, Chem. Soc. Rev., 1997, 26, 269 RSC.
- C. P. Allen, T. Benkovics, A. K. Turek and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 12560 CrossRef CAS PubMed.
- D. J. Michaelis, C. J. Shaffer and T. P. Yoon, J. Am. Chem. Soc., 2007, 129, 1866 CrossRef CAS.
- T. Patonay, J. Jeko and É. Juhμsz-Tóth, Eur. J. Org Chem., 2008, 1441–1448 CrossRef CAS.
- M. N. Zhao, W. Zhang, X. C. Wang, Y. Zhang, D. S. Yang and Z. H. Guan, Org. Biomol. Chem., 2018, 16, 4333–4433 RSC.
- K. Okamoto, A. Nanya, A. Eguchi and K. Ohe, Angew. Chem., Int. Ed., 2018, 57, 1039–1043 CrossRef CAS.
- D. Y. K. Chen, R. H. Pouwer and J. A. Richard, Chem. Soc. Rev., 2012, 41, 4631–4642 RSC.
- J. M. R. Narayanam and C. R. Stephenson, J. Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS.
- R. Cauwenbergh and S. Das, Synlett, 2022, 33, 129–149 CrossRef CAS.
- V. Jaro and S. Das, Synthesis, 2022, 54, 3383–3398 CrossRef.
- T. Feng, S. Wang, Y. Liu, S. Liu and Y. Qiu, Angew. Chem., Int. Ed., 2022, 61, e202115178 CrossRef CAS PubMed.
- L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem, 2021, 5, 522–545 CrossRef CAS PubMed.
- N. E. Tay, D. Lehnherr and T. Rovis, Chem. Rev., 2021, 122, 2487–2649 CrossRef PubMed.
- J. Q. Liu, A. Shatskiy and M. D. Kärkäs, Science, 2020, 368, 1312–1313 CrossRef CAS PubMed.
- A. M. Del Hoyo, A. G. Herraiz and M. G. Suero, Angew. Chem., Int. Ed., 2017, 56, 1610–1613 CrossRef CAS PubMed.
- C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 130, 15656–15660 CrossRef.
- J. P. Phelan, S. B. Lang, J. S. Compton, C. B. Kelly, R. Dykstra, O. Gutierrez and G. A. Molander, J. Am. Chem. Soc., 2018, 140, 8037–8047 CrossRef CAS PubMed.
- T. Chidley, I. Jameel, S. Rizwan, P. A. Peixoto, L. Pouységu, S. Quideau and G. K. Murphy, Angew. Chem., Int. Ed., 2019, 58, 16959–16965 CrossRef CAS PubMed.
- D. P. Poudel, A. Pokhrel, R. K. Tak, M. Shankar and R. Giri, Science, 2023, 381, 545–553 CrossRef CAS PubMed.
- Z. Wang, A. G. Herraiz, A. M. Del Hoyo and M. G. Suero, Nature, 2018, 554, 86–91 CrossRef CAS PubMed.
- Y. Zhang, G. Zhou, X. Gong, Z. Guo, X. Qi and X. Shen, Angew. Chem., Int. Ed., 2022, 61, e202202175 CrossRef CAS.
- D. Xia, R. Wu, J. Wang, X. Han, Y. Li, Q. Li, X. Luan, X. Hong, Y. Zhang and W. D. Zhang, ACS Catal., 2023, 13, 9806–9816 CrossRef CAS.
- F. J. Sarabia and E. M. Ferreira, Org. Lett., 2017, 19, 2865–2868 CrossRef CAS PubMed.
- B. Xu, L. Troian-Gautier, R. Dykstra, R. T. Martin, O. Gutierrez and U. K. Tambar, J. Am. Chem. Soc., 2020, 142, 6206–6215 CrossRef CAS PubMed.
- M. J. Kim, D. J. Wang, K. Targos, U. A. Garcia, A. F. Harris, I. A. Guzei and Z. K. Wickens, Angew. Chem., Int. Ed., 2023, 62, e202303032 CrossRef CAS PubMed.
- S. O. Scholz, E. P. Farney, S. Kim, D. M. Bates and T. P. Yoon, Angew. Chem., Int. Ed., 2016, 55, 2239–2242 CrossRef CAS PubMed.
- Y. Guo, C. Pei, S. Jana and R. M. Koenigs, ACS Catal., 2021, 11, 337–342 CrossRef CAS.
- J. Chen, W. Q. Yan, C. M. Lam, C. C. Zeng, L. M. Hu and R. D. Little, Org. Lett., 2015, 17, 986–989 CrossRef CAS PubMed.
- F. Liu, J. Dai and X. Cheng, Chin. J. Org. Chem., 2021, 41, 4014–4020 CrossRef CAS.
- Y. Liu, X. Dong, G. L. Deng and L. Zhou, Sci. China. Chem., 2016, 59, 199–202 CrossRef CAS.
- D. E. Holst, D. J. Wang, M. J. Kim, I. A. Guzei and Z. K. Wickens, Nature, 2021, 596, 74–79 CrossRef CAS PubMed.
- X. X. Liu, J. Jia, Z. Wang, Y. T. Zhang, J. Chen, K. Yang, C. Y. He and L. Zhao, Adv. Synth. Catal., 2020, 36, 2604–2608 CrossRef.
- S. Chand, A. K. Sharma, A. K. Pandey and K. N. Singh, Org. Lett., 2022, 24, 6423–6427 CrossRef CAS PubMed.
- E. Kim, S. Choi, H. Kim and E. J. Cho, Chem.–Eur. J., 2013, 19, 6209–6212 CrossRef CAS PubMed.
- M. Ines, A. J. Mendonca, A. P. Esteves, D. I. Mendonca and M. J. Medeiros, C. R. Chim., 2009, 12, 841–849 CrossRef CAS.
- K. Jin, J. H. Maalouf, N. Lazouski, N. Corbin, D. Yang and K. Manthiram, J. Am. Chem. Soc., 2019, 141, 6413–6418 CrossRef CAS.
- W. R. Leow, Y. Lum, A. Ozden, Y. Wang, D.-H. Nam, B. Chen, J. Wicks, T. T. Zhuang, F. Li, D. Sinton and E. H. Sargen, Science, 2020, 368, 1228–1233 CrossRef CAS PubMed.
- R. Hommelsheim, Y. Guo, Z. Yang, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2019, 58, 1203–1207 CrossRef CAS PubMed.
- G. Zhou and X. Shen, Angew. Chem., Int. Ed., 2022, 134, e202115334 CrossRef.
- J. Li, M. J. Lear and Y. Hayashi, Chem.–Eur. J., 2021, 27, 5901–5905 CrossRef CAS.
- Y. Guo, C. Empel, C. Pei, H. Fang, S. Jana and R. M. Koenigs, Chem Catal., 2022, 2, 2012–2023 CrossRef CAS.
- B. E. Austin, R. P. Palner, E. M. Tobias, R. Madiu, E. L. Doran, J. M. Doran, J. M. Doran, A. M. Howard, J. L. Stroud, M. E. Rossi, D. A. Moskovitz, D. A. Rivera, M. D. Mullen, A. H. Zinsky, R. A. Rosario and G. Moura-Letts, Synlett, 2024, 35, 325–329 CrossRef CAS.
- E. Babaoglu and G. Hilt, Chem.–Eur. J., 2020, 26, 8879–8884 CrossRef CAS.
- S. Sengmany, E. Léonel, J. P. Paugam and J. Y. Nédélec, Synthesis, 2002, 4, 0533–0537 CrossRef.
- R. T. Ribeiro, I. L. De Mattos, S. Sengmany, R. Barhdadi, E. Léonel, C. Cachet-Vivier and M. Navarro, Electrochim. Acta, 2011, 56, 7352–7360 CrossRef CAS.
- Y. Zhang, R. Qian, X. Zheng, Y. Zeng, J. Sun, Y. Chen, A. Ding and H. Guo, Chem. Commun., 2015, 51, 54–57 RSC.
- A. M. del Hoyo and M. G. Suero, Eur. J. Org Chem., 2017, 2017, 2122–2125 CrossRef CAS.
- P. Li, J. Zhao, L. Shi, J. Wang, X. Shi and F. Li, Nat. Commun., 2018, 9, 1972–1980 CrossRef PubMed.
- T. Ohtani, Y. Tsuchiya, D. Uraguchi and T. Ooi, Org. Chem. Front., 2019, 6, 1734–1737 RSC.
- A. G. Herraiz and M. G. Suero, Chem. Sci., 2019, 10, 9374–9379 RSC.
- K. Ide, M. Furuta and H. Tokuyama, Org. Biomol. Chem., 2021, 19, 9172–9176 RSC.
- S. K. Jana, M. Maiti, P. Dey and B. Maji, Org. Lett., 2022, 24, 1298–1302 CrossRef CAS PubMed.
- S. S. Luo, H. Shen, S. J. Li, T. Cao, Y. P. Luo, S. Zhang, T. Zhou and X. W. Liu, Org. Chem. Front., 2022, 9, 2627–2633 RSC.
- D. M. Fischer, H. Lindner, W. M. Amberg and E. M. Carreira, J. Am. Chem. Soc., 2023, 145, 774–780 CrossRef CAS.
- C. L. Ji, J. Han, T. Li, C. G. Zhao, C. Zhu and J. Xie, Nat. Catal., 2022, 5, 1098–1109 CrossRef CAS.
- L. Thai-Savard, M. Sayes, J. Perreault-Dufour, G. Hong, L. A. Wells, M. C. Kozlowski and A. B. Charette, J. Org. Chem., 2023, 88, 1515–1521 CrossRef CAS PubMed.
- A. Bosveli, N. Griboura, I. Kampouropoulos, D. Kalaitzakis, T. Montagnon and G. Vassilikogiannakis, Chem.–Eur. J., 2023, 29, e202301713 CrossRef CAS.
- S. Biswas, P. Chandu, S. Garai and D. Sureshkumar, Org. Lett., 2023, 25, 7863–7867 CrossRef CAS.
- T. Guo, L. Zhang, X. Liu, Y. Fang, X. Jin, Y. Yang, Y. Li, B. Chen and M. Ouyang, Adv. Synth. Catal., 2018, 360, 4459–4463 CrossRef CAS.
- J. A. Milligan, J. P. Phelan, V. C. Polites, C. B. Kelly and G. A. Molander, Org. Lett., 2018, 20, 6840–6844 CrossRef CAS PubMed.
- W. Luo, Y. Yang, Y. Fang, X. Zhang, X. Jin, G. Zhao, L. Zhang, Y. Li, W. Zhou, T. Xia and B. Chen, Adv. Synth. Catal., 2019, 361, 4215–4221 CrossRef CAS.
- W. Luo, Y. Fang, L. Zhang, T. Xu, Y. Liu, Y. Li, X. Jin, J. Bao, X. Wu and Z. Zhang, Eur. J. Org Chem., 2020, 11, 1778–1781 CrossRef.
- Y. Liu, W. Luo, J. Wu, Y. Fang, Y. Li, X. Jin, L. Zhang, Z. Zhang, F. Xu and C. Du, Org. Chem. Front., 2020, 7, 1588–1592 RSC.
- R. Nakamura, Y. Sumida and H. Ohmiya, Bull. Chem. Soc. Jpn., 2022, 95, 1001–1005 CrossRef CAS.
- X. L. Huang, Y. Z. Cheng and S. L. You, Org. Chem. Front., 2022, 9, 5463 RSC.
- L. Zhang, J. Shi and Y. Fang, Synthesis, 2023, 55, 907–918 CrossRef CAS.
- L. Huai, L. Zhang, Z. Wang, H. Wu and Y. Fang, Org. Chem. Front., 2023, 10, 1245–1251 RSC.
- J. Hu, M. Tang, J. Wang, Z. Wu, A. Friedrich and T. B. Marder, Angew. Chem., Int. Ed., 2023, 62, e202305175 CrossRef CAS PubMed.
- L. H. Jie, B. Guo, J. Song and H. C. Xu, J. Am. Chem. Soc., 2022, 144, 2343–2350 CAS.
- B. G. Cai, C. Empel, S. Jana, J. Xuan and R. M. Koenigs, ACS Catal., 2023, 13, 11851–11856 CrossRef CAS.
- J. H. G. Teye-Kau, M. J. Ayodele and S. Pitre, Angew. Chem., Int. Ed., 2023, e202316064 Search PubMed.
- X. Zhang, C. Du, H. Zhang, X. C. Li, Y. L. Wang, J. L. Niu and M. P. Song, Synthesis, 2019, 51, 889–898 CrossRef CAS.
- I. D. Jurberg and H. M. Davies, Chem. Sci., 2018, 9, 5112–5118 RSC.
- Y. Guo, T. V. Nguyen and R. M. Koenigs, Org. Lett., 2019, 21, 8814–8818 CrossRef CAS PubMed.
- S. Jana, F. Li, C. Empel, D. Verspeek, P. Aseeva and R. M. Koenigs, Chem.–Eur. J., 2020, 26, 2586–2591 CrossRef CAS PubMed.
- J. Chauhan, M. K. Ravva, L. Gremaud and S. Sen, Org. Lett., 2020, 22, 4537–4541 CrossRef CAS PubMed.
- Y. Guo, C. Empel, C. Pei, I. Atodiresei, T. Fallon and R. M. Koenigs, Org. Lett., 2020, 22, 5126–5130 CrossRef CAS PubMed.
- C. Empel and R. M. Koenigs, J. Flow Chem., 2020, 10, 157–160 CrossRef CAS.
- V. Klöpfer, R. Eckl, J. Floß, P. M. Roth, O. Reiser and J. P. Barham, Green Chem., 2021, 23, 6366–6372 RSC.
- H. Qiu, L. Wen and J. Lv, Synthesis, 2022, 54, 3739–3752 CrossRef CAS.
- Z. L. Chen, C. Empel, K. Wang, P. P. Wu, B. G. Cai, L. Li and J. Xuan, Org. Lett., 2022, 24, 2232–2237 CrossRef CAS PubMed.
- Y. Xu, G. Lv, K. Yan, H. He, J. Li, Y. Luo, R. Lai, H. Li and Y. Wu, Chem.–Asian J., 2020, 15, 1945–1947 CrossRef CAS PubMed.
- V. George and B. König, Chem. Commun., 2023, 59, 11835–11838 RSC.
- F. He, Z. Sun, C. Li, Z. Jiang, H. Miao, Q. Li and C. Wu, Org. Biomol. Chem., 2023, 21, 8273–8278 RSC.
- E. Yamaguchi, H. Inagawa and A. Itoh, Photochem. Photobiol. Sci., 2022, 21, 813–818 CrossRef CAS PubMed.
- A. Bunyamin, C. Hua, A. Polyzos and D. L. Priebbenow, Chem. Sci., 2022, 13, 3273–3280 RSC.
- S. Chen, Y. Zhang, S. Liu and X. Shen, Sci. China: Chem., 2023, 66, 1–7 Search PubMed.
- K. Orłowska, J. V. Santiago, P. Krajewski, K. Kisiel, I. Deperasińska, K. Zawada, W. Chaładaj and D. Gryko, ACS Catal., 2023, 13, 1964–1973 CrossRef.
- Y. Deng, J. Zhang, B. Bankhead, J. P. Markham and M. Zeller, Chem. Commun., 2021, 57, 5254–5257 RSC.
- A. D. Richardson, T. R. Vogel, E. F. Traficante, K. J. Glover and C. S. Schindler, Angew. Chem., Int. Ed., 2022, 61, e202201213 CrossRef CAS.
- J. Ma, S. Chen, P. Bellotti, T. Wagener, C. Daniliuc, K. N. Houk and F. Glorius, Nat. Catal., 2022, 5, 405–413 CrossRef CAS.
- A. N. Vereshchagin, E. O. Dorofeeva, M. N. Elinson and M. P. Egorov, Arkivoc, 2019, vi, 325–335 Search PubMed.
- K. Matsumoto, Y. Hayashi, K. Hamasaki, M. Matsuse, H. Suzuki, K. Nishiwaki and N. Kawashita, Beilstein J. Org. Chem., 2022, 18, 1116–1122 Search PubMed.
- T. Cantin, A. B. Charette, T. Poisson and P. Jubault, Synthesis, 2023, 55, 2943–2950 CrossRef CAS.
- Y. Zhang, X. Dong, Y. Wu, G. L and H. Lu, Org. Lett., 2018, 20, 4838–4842 CrossRef CAS PubMed.
- Q. Xing, D. Jiang, J. Zhang, L. Guan, T. Li, Y. Zhao, M. Di, H. Chen, C. Che and Z. Zhu, Commun. Chem., 2022, 5, 79–87 CrossRef CAS PubMed.
- O. Shikhar Srivastava, V. Anand and N. Rastogi, Asian J. Org. Chem., 2023, e202300415 CrossRef CAS.
- X. Cheng, B. G. Cai, H. Mao, J. Lu, L. Li, K. Wang and J. Xuan, Org. Lett., 2021, 23, 4109–4114 CrossRef CAS PubMed.
- T. Siu and A. K. Yudin, J. Am. Chem. Soc., 2002, 124, 530–531 CrossRef CAS PubMed.
- K. Matsuzawa, Y. Nagasawa, E. Yamaguchi, N. Tada and A. Itoh, Synthesis, 2016, 48, 2845–2850 CrossRef CAS.
- W. L. Yu, J. Q. Chen, Y. L. Wei, Z. Y. Wang and P. F. Xu, Chem. Commun., 2018, 54, 1948–1951 RSC.
- D. Zeng, L. Gu, L. Zhang, G. Li and Y. He, Tetrahedron Lett., 2021, 87, 153413 CrossRef CAS.
- J. R. Vanhoof, P. J. De Smedt, B. Krasniqi, R. Ameloot, D. Sakellariou and D. E. De Vos, ACS Sustainable Chem. Eng., 2021, 9, 11596–11603 CrossRef CAS.
- J. Li, W. Huang, J. Chen, L. He, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2018, 57, 5695–5698 CrossRef CAS PubMed.
- M. Ošeka, G. Laudadio, N. P. van Leest, M. Dyga, A. D. A. Bartolomeu, L. J. Gooßen, B. de Bruin, K. T. de Oliveira and T. Noël, Chem, 2021, 7, 255–266 Search PubMed.
- S. Wang, P. Wang, S. J. Li, Y. H. Chen, Z. J. Sun and A. Lei, Nati. Sci. Rev., 2023, 10, 187–196 CrossRef.
- A. Mohsenzadeh, A. Zamani and M. J. Taherzadeh, ChemBioEng Rev., 2017, 4, 75–91 CrossRef.
- C. Lucky, T. Wang and M. Schreier, ACS Energy Lett., 2022, 7, 1316–1321 CrossRef CAS.
- D. Tang, K. Dang, J. Wang, C. Chen, J. Zhao and Y. Zhang, Sci. China: Chem., 2023, 66, 3415–3425 CrossRef CAS.
- R. Kumar, N. Banerjee, P. Kumar and P. Banerjee, Chem.–Eur. J., 2023, 29, e202301594 CrossRef CAS PubMed.
- N. Tada, H. Okubo, T. Miura and A. Itoh, Synlett, 2009, 18, 3024–3026 Search PubMed.
- H. Tanaka, H. Nishikawa, T. Uchida and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 12034–12041 CrossRef CAS PubMed.
- Y. Wu, G. Zhou, Q. Meng, X. Tang, G. Liu, H. Yin, J. Zhao, F. Yang, Z. Yu and Y. Luo, J. Org. Chem., 2018, 83, 13051–13062 CrossRef CAS PubMed.
- K. Luo, X. Yu, P. Chen, K. He, J. Lin and Y. Jin, Tetrahedron Lett., 2020, 61, 151578–151581 CrossRef CAS.
- L. Peault, S. Rochelle, A. Planchat, P. Nun, E. Le Grognec and V. Coeffard, Adv. Synth. Catal., 2023, 365, 194–200 CrossRef CAS.
- Y. Zhang, X. Yang, H. Tang, D. Liang, J. Wu and D. Huang, Green Chem., 2020, 22, 22–27 RSC.
- D. Shen, T. Ren, Z. Luo, F. Sun, Y. Han, K. Chen, X. Zhang, M. Zhou, P. Gong and M. Chao, Org. Biomol. Chem., 2023, 21, 4955–4961 RSC.
- R. Omura, A. Fujiya, E. Yamaguchi, N. Tada, T. Miura and A. Itoh, Synthesis, 2016, 48, 3971–3975 CrossRef CAS.
- H. Tang, J. R. Vanhoof and D. De Vos, Green Chem., 2022, 24, 9565–9569 RSC.
- F. He and R. M. Koenigs, Chem. Commun., 2019, 55, 4881–4884 RSC.
- S. Jana, C. Pei, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2021, 60, 13271–13279 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |