
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic super-reducing photocatalysts generate solvated electrons via two consecutive photon induced processes†
Marco
Villa
 a,
Andrea
Fermi
a,
Francesco
Calogero
a,
Xia
Wu
b,
Andrea
Gualandi
a,
Pier Giorgio
Cozzi
a,
Alessandro
Troisi
*b,
Barbara
Ventura
*c and
Paola
Ceroni
*a
a,
Andrea
Fermi
a,
Francesco
Calogero
a,
Xia
Wu
b,
Andrea
Gualandi
a,
Pier Giorgio
Cozzi
a,
Alessandro
Troisi
*b,
Barbara
Ventura
*c and
Paola
Ceroni
*a
aDepartment of Chemistry Ciamician, Via Selmi 2, 40126 Bologna, Italy. E-mail: paola.ceroni@unibo.it
bDepartment of Chemistry and Materials Innovation Factory, University of Liverpool, Liverpool L69 7ZD, UK. E-mail: a.troisi@liverpool.ac.uk
cInstitute for Organic Synthesis and Photoreactivity (ISOF), National Research Council (CNR), Via P. Gobetti 101, 40129 Bologna, Italy. E-mail: barbara.ventura@isof.cnr.it
First published on 20th August 2024
Abstract
Photocatalysts with extremely strong reducing potential are often thought to operate through a consecutive photoinduced electron transfer (ConPeT) mechanism, where a first photon generates the radical anion of the photocatalyst via electron transfer and a second photon excites the radical anion into a super-reducing agent. Among them, 4CzIPN, (2,4,5,6-tetrakis(9H-carbazol-9-yl) isophthalonitrile) and the analogous 4DPAIPN (2,4,5,6-tetrakis(diphenylamino)isophthalonitrile) are supposed to operate following this principle, but the knowledge of the photophysical properties of the photogenerated radical anions is still very limited. An in-depth spectroscopic and computational study of their radical anions demonstrates that the excited states of 4CzIPN˙− and 4DPAIPN˙− are not behaving as super-reducing agents: they are very short lived (ca. 20 ps), not emissive and not quenched by common organic substrates. Most importantly, longer lived solvated electrons are generated upon excitation of these radical anions in acetonitrile and we propose that it is the solvated electron the species responsible for the exceptional reducing capability of this photocatalytic system.
Introduction
Cyanoarene organic chromophores are emerging as ideal photoreductants in photoredox catalysis.1–3 In search of an expansion of the available cathodic potential window, they were employed in the so-called consecutive photoinduced electron transfer mechanism (ConPeT, Fig. 1a). The ConPeT process was pioneered by König with a perylenebisimide dye4 and it was further expanded to other organic chromophores,5 such as dicyanoanthracene,6 rhodamine7 and eosin.8 Most of the cases are based on neutral photocatalysts and the corresponding radical anions as represented in Fig. 1a, but there are also reports about cationic photocatalysts and the corresponding neutral radicals formed after the first photoinduced electron transfer process.9,10 Recently, cyanoarenes entered the arena and were used for a variety of reactions, including activation of reductively recalcitrant aryl chlorides.11–20 The potent photoreductant activity of reduced cyanoarene was first reported by electrophotocatalysis, where the radical anion is produced electrochemically and is subsequently excited by light absorption.21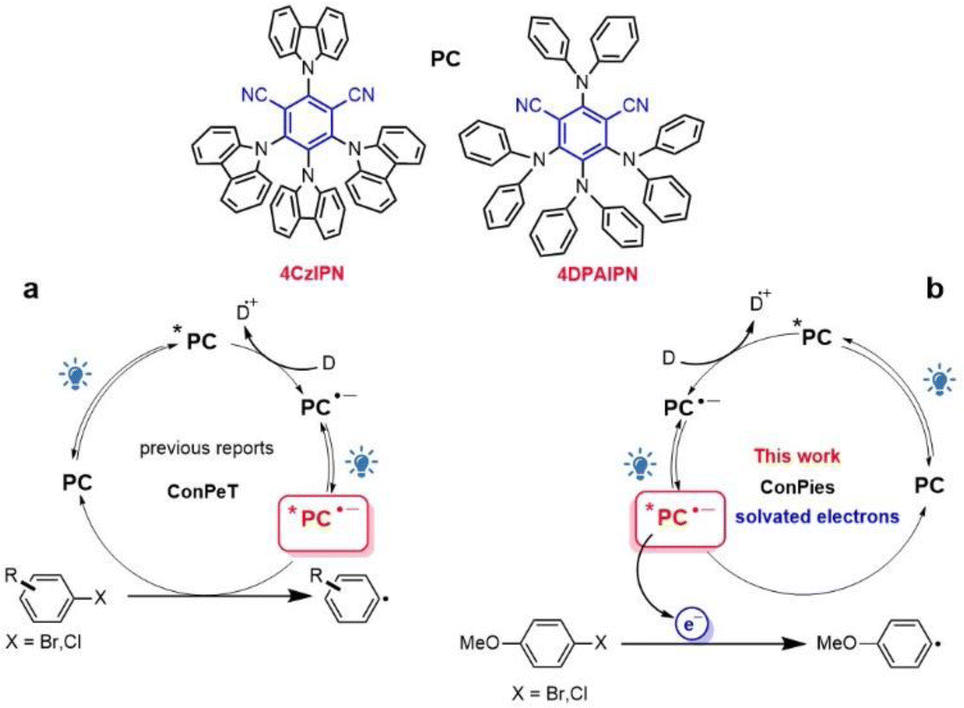 | ||
| Fig. 1 Schematic illustration of the ConPeT mechanism (a) and two consecutive photon induced generation of solvated electrons (ConPies) investigated in the current work (b). | ||
Despite the large body of literature on the topic, direct evidence of the ConPeT mechanism, including the quenching process of the *PC˙− excited state, are elusive. The ConPeT mechanism was supported by:9,14,15 (i) EPR spectrum of the radical anion; (ii) quadratic dependence of the initial photoreaction yield on the power of the excitation light; (iii) presence of a new blue-shifted emission band attributed to the radical anion excited state *PC˙−. However, the EPR spectrum demonstrates the formation of the radical anion, but not the involvement of its excited state in the reaction. The quadratic dependence is expected every time in which two photons are needed to drive the reaction, including the cases in which the chromophore is photodegraded into another photocatalyst that drives the reaction.22,23 The emission band is not unequivocally assigned to the radical anion: for example, the observed changes in the emission spectrum are not reported to be reversible upon air-exposure.9
Recently, it was demonstrated that ConPeT was not the active mechanism in some of the literature reports.24,25 For cyanoarenes, it was reported that reduction of aryl halides can proceed via halogen atom transfer.26,27 A very recent study28 reported that dehalogenation of activated aryl/alkyl halides did not proceed via the ConPeT mechanism, while reduction of highly demanding unactivated alkyl/aryl halides involves a ConPeT mechanism from high-energy excited states of the radical anion. This is an unexpected and quite unlikely process because of the extremely short lifetime of high-energy excited states, which rapidly deactivates by internal conversion to the lower lying excited state, according to Kasha's rule. This limitation can be overcome by preassociation of the radical anion and the substrate, as recently demonstrated.29
It is evident that there is an urgent need to better understand the nature of super-reducing radical photoreagents. Prompted by this complex scenario, we explored the most famous 4CzIPN and 4DPAIPN (Fig. 1). The steady-state and time-resolved (down to 300 fs resolution) spectroscopic investigations, complemented by a computational study, provide the first experimental evidence of solvated electron generation in acetonitrile via sequential two-photon absorption and the reactivity of 4-bromoanisole and 4-chloroanisole with the generated solvated electron.
Results and discussion
4CzIPN displays an absorption band tailing up to 500 nm and an emission band peaked at 545 nm (green lines in Fig. 2a) with a biexponential decay (Table 1) due to prompt (τPF = 13.4 ns) and delayed fluorescence (τTADF = 1.8 μs) in degassed acetonitrile solution. 4DPAIPN presents similar photophysical properties (green lines in Fig. 2b and Table 1) with a red-shift of the lowest energy transition.28 Upon addition of tetrabutylammonium oxalate (TBAOx, 5 mM),30 quenching of the emission of both photocatalysts is observed (dashed black lines in Fig. 2), consistent with the photoinduced electron transfer from the oxalate31 to the TADF chromophore.28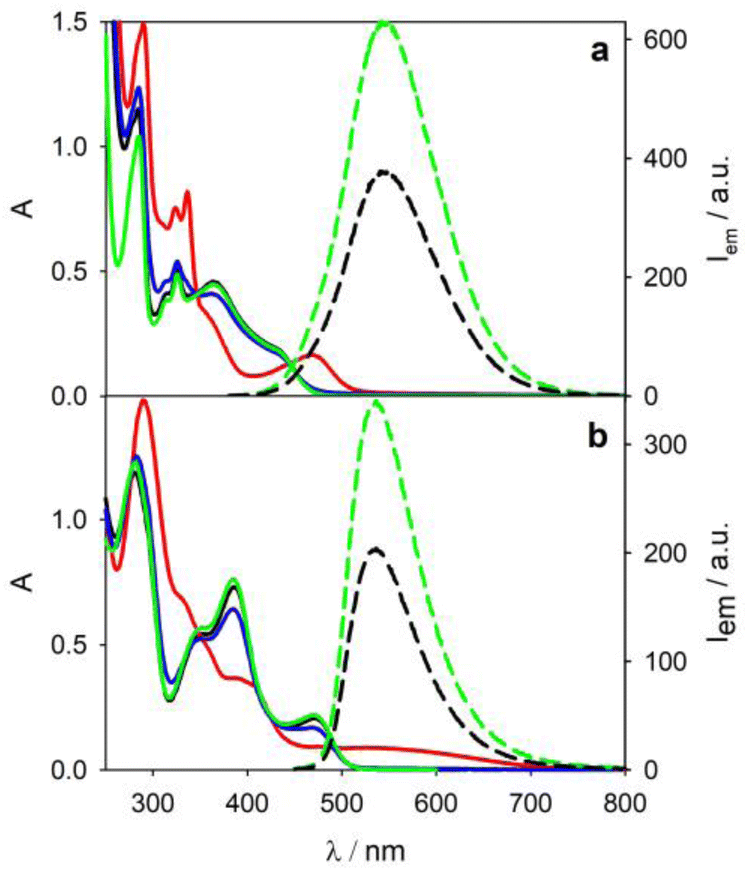 | ||
| Fig. 2 Absorption (solid lines) and emission (dashed lines) spectra in degassed acetonitrile solution of: 4CzIPN 0.03 mM (a) and 4DPAIPN 0.04 mM (b) before (green lines) and after addition of TBAOx (5.2 mM, black lines) and subsequent irradiation at 390 nm (red lines, irradiation time = 30 s). Absorption spectra reported in blue are registered after air bubbling. λex = 405 nm. | ||
| λ abs/nm | λ em/nm | τa/ns | τ /μs | |
|---|---|---|---|---|
| a Lifetime of the lowest in energy excited state. b Lifetime associated to the thermally activated delayed fluorescence, TADF. c Data acquired by transient absorption spectroscopy. | ||||
| 4CzIPN | 430 | 545 | 13.4 | 1.82 |
| 4CzIPN˙− | 460 | — | 0.027c | — |
| 4DPAIPN | 470 | 536 | 3.4 | 67 |
| 4DPAIPN˙− | 550 | — | 0.021c | — |
Upon irradiation at 390 nm of the solution containing 4CzIPN and TBAOx, a new absorption band at lower energy appears (λmax = 468 nm, red line in Fig. 2a), which is consistent with the formation of the radical anion 4CzIPN˙− by comparison with literature.28 By air bubbling, an almost complete (>95%) recovery of the initial spectrum of 4CzIPN is observed (blue line in Fig. 2a).
Similarly, irradiation of a 4DPAIPN solution containing TBAOx leads to the formation of the radical anion (red line in Fig. 2b). The recovery (ca. 90%) of the 4DPAIPN spectrum is obtained upon reoxidation by molecular oxygen. For both 4CzIPN˙− and 4DPAIPN˙− radical anions, we do not observe any new emission by selective excitation of the radical anion at 500 and 600 nm, respectively (or upon excitation at shorter wavelength, e.g., 460 nm), contrary to the previous literature reports.9,14,15 Similar results have been obtained with N,N-diisopropylethylamine (DIPEA) as sacrificial electron donor instead of TBAOx (see Fig. S5 and S6†).
Ultrafast pump–probe experiments have been performed on 4CzIPN˙− and 4DPAIPN˙− in order to explore their excited state dynamics.‡Fig. 3a reports the transient absorption spectra of the radical anion 4CzIPN˙− upon excitation at 480 nm. The signal in the 450–800 nm range decays to zero on a very short timescale, according to a biexponential evolution: τ1ca. 300 fs (of the order of the instrumental time resolution), likely related to internal conversion from upper lying doublet excited state to D1, and τ2 = 27 ps (Fig. 3b and S11†), attributable to the D1 decay back to the ground state (D0, see the computational study reported below). Interestingly, in the NIR spectral region, the featureless absorption signal initially decays with the same fast processes observed in the VIS region, then a broad band peaking at ca. 1440 nm forms and remains stable on longer time scales (Fig. 3a). This band resembles the absorption of the solvated electron observed in acetonitrile upon pulse radiolysis.32,33 The decay of this species is multiexponential with lifetimes ranging from hundreds of ps to few ns (Fig. 4a, the corresponding lifetimes are reported in Table S2†).
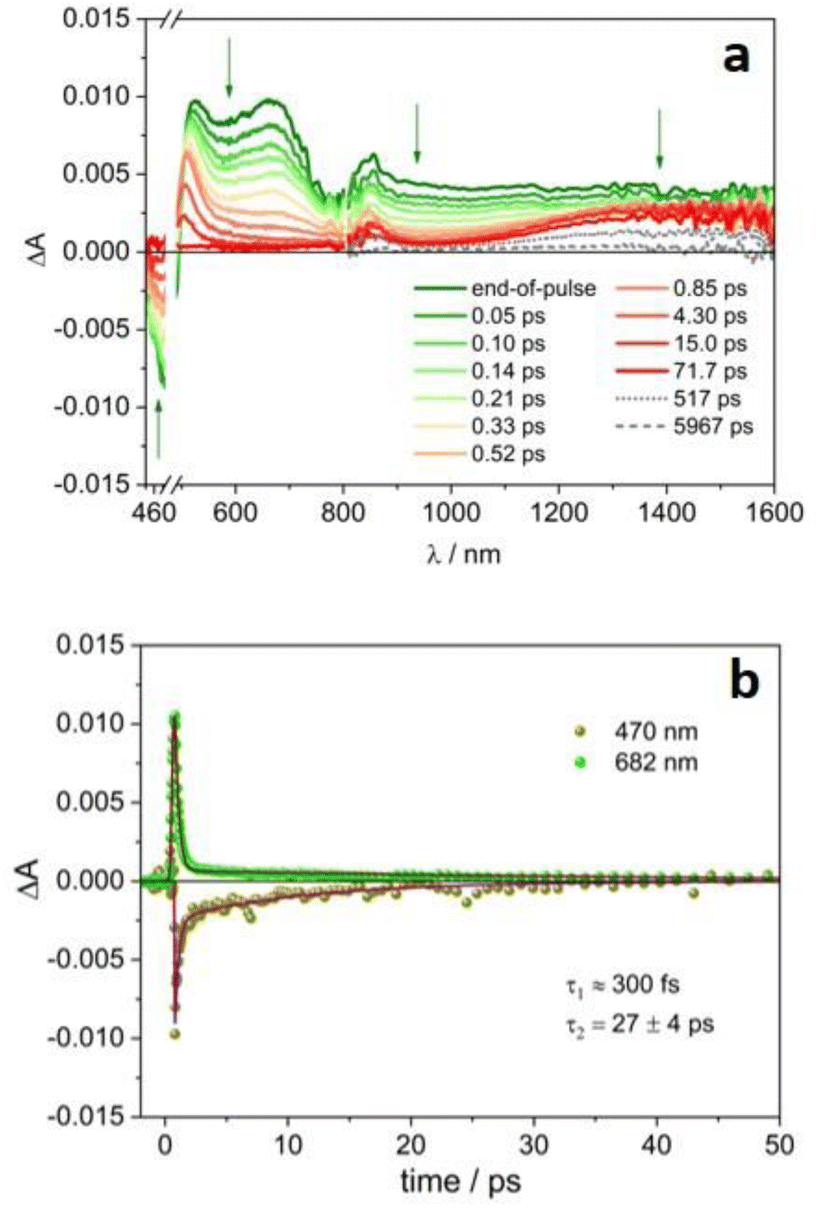 | ||
| Fig. 3 Transient absorption spectra at different delays of a degassed acetonitrile solution of 4CzIPN˙− (a) and the corresponding ΔA temporal evolution at significant wavelengths (b). λex = 480 nm, A480 = 0.28, 0.2 cm optical path, 8 μJ per pulse. | ||
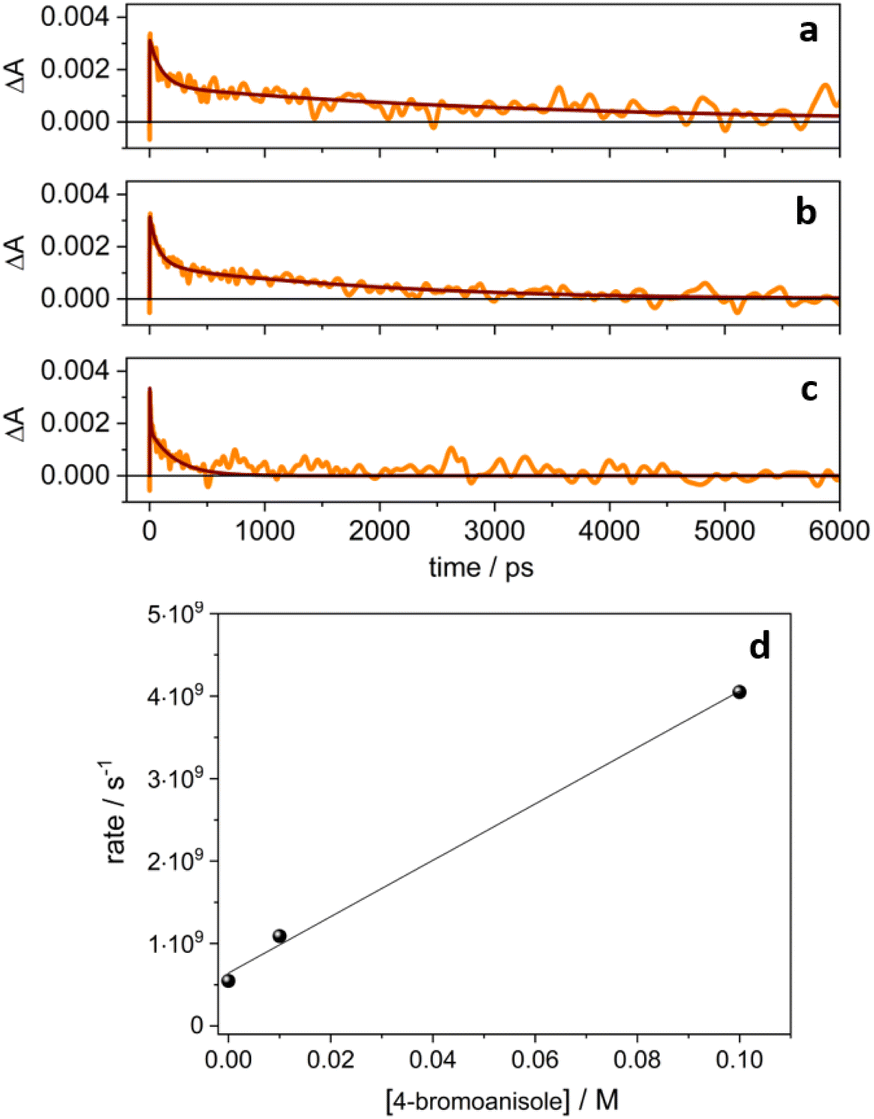 | ||
| Fig. 4 Normalized temporal evolution of ΔA measured at 1440 nm (orange line) for a degassed solution of 4CzIPN˙− (a) and in the presence of 0.01 M (b) and 0.1 M (c) 4-bromoanisole. (d) Rate constant of the ΔA decay at 1440 nm as a function of the concentration of 4-bromoanisole. | ||
The photogeneration of solvated electron is a well-known process, but it usually requires high-energy photons for photoionization of the molecule and aqueous solution to stabilize the generated electron.34–37 For example, the metal complex [Fe(CN)6]4− in water forms hydrated electrons upon irradiation of the metal-to-ligand charge transfer (MLCT) band at 254 nm.38,39 The involvement of solvated electrons in photoredox catalysis has already been reported10,40,41 and most of the examples are in aqueous media,42–45 where the solvated electron is characterized by a blue color with absorption peak at 715 nm (ε715 nm = 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 M−1 cm−1), a redox potential as high as −2.9 V vs. NHE and a lifetime in the microsecond timescale. However, neutral organic chromophores, such as cyanoarenes, are not soluble in aqueous solution and therefore water is not commonly employed in photoredox catalysis, so that it greatly limits the applicability of this approach. As expected, the properties of solvated electrons are strongly dependent on the solvent. Photogeneration of solvated electrons in organic solvents is much less explored in photoredox catalysis10,46 for two main reasons: (i) the shorter lifetime of the solvated electron, which is less stabilized than in water; (ii) the difficult detection of solvated electrons, whose absorption band moves to lower energy in the near-infrared spectral region, e.g. 1450 nm is the band maximum in acetonitrile.32,33,47
700 M−1 cm−1), a redox potential as high as −2.9 V vs. NHE and a lifetime in the microsecond timescale. However, neutral organic chromophores, such as cyanoarenes, are not soluble in aqueous solution and therefore water is not commonly employed in photoredox catalysis, so that it greatly limits the applicability of this approach. As expected, the properties of solvated electrons are strongly dependent on the solvent. Photogeneration of solvated electrons in organic solvents is much less explored in photoredox catalysis10,46 for two main reasons: (i) the shorter lifetime of the solvated electron, which is less stabilized than in water; (ii) the difficult detection of solvated electrons, whose absorption band moves to lower energy in the near-infrared spectral region, e.g. 1450 nm is the band maximum in acetonitrile.32,33,47
Noticeably, in the presence of 4-bromoanisole, that is reported to be reduced by the excited state *4CzIPN˙− according to the ConPeT mechanism,28 no significant change in the lifetime of the D1 excited state is observed (Fig. S14†), but the band at 1440 nm is decreasing with a much faster decay as a function of the concentration of 4-bromoanisole (Fig. 4b and c). These results demonstrate that 4-bromoanisole does not quench the excited states of 4CzIPN˙− and the photogenerated solvated electron is able to reduce 4-bromoanisole (Ered = −2.75 V vs. SCE),5 with a reaction rate constant of 3.4 × 1010 M−1 s−1 (for more details on the data and fitting parameters, see Fig. S15 and Table S2†). This value demonstrates that the reaction is a diffusionally controlled process, as previously reported for other scavengers of solvated electrons in acetonitrile.32
Upon selective excitation of the radical anion 4DPAIPN˙− (Fig. 5) at 600 nm, similar results have been obtained. A broad spectrum in the 600–1600 nm range is detected, as well as ground state bleaching below 600 nm (Fig. 5a). The signal decays very rapidly, with a dominant ultrafast component of the order of 300 fs and a second fast decay of 21 ps (Fig. 5b). The two processes can be attributed to the formation and decay of the lowest excited state D1 of 4DPAIPN˙−. Again, a band at ca. 1440 nm remains on longer time scales: lifetimes of hundreds of ps and ca. 4 ns followed by an infinite component emerge from the fitting (Fig. 5b, inset). The band has the same spectral shape of that recorded for 4CzIPN˙− (Fig. S12†) and can thus be attributed to the generated solvated electron. As a further proof of the presence of a new species in the transient absorption spectra of 4CzIPN˙− and 4DPAIPN˙−, it is worth noting that the kinetic evolutions of the NIR signals observed in Fig. 3–5 are substantially different from those observed in the visible spectral range. On the other hand, the kinetic profiles in the visible and NIR spectral region reported for 4CZIPN or 4DPAIPN (Fig. S7b and S9b†) are identical.
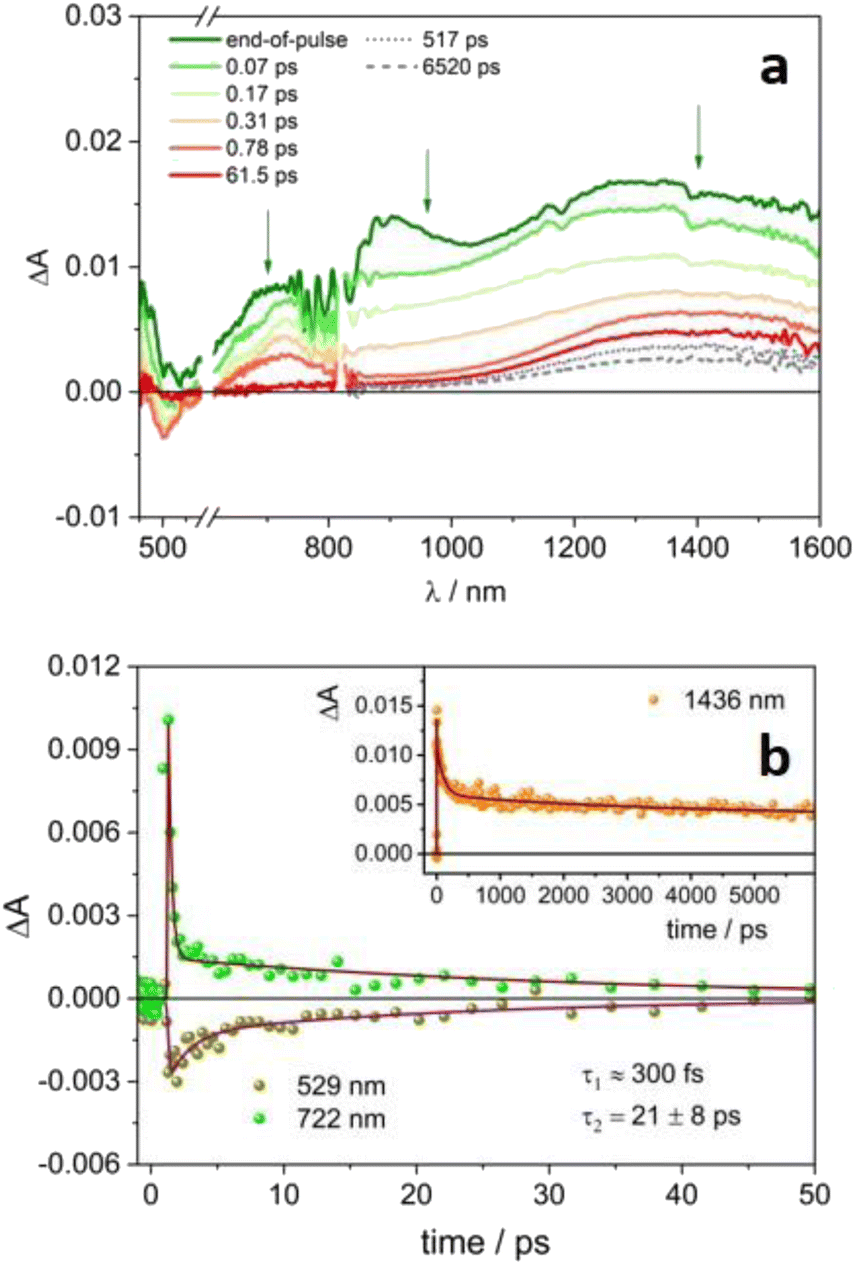 | ||
| Fig. 5 Transient absorption spectra at different delays of a degassed acetonitrile solution of 4DPAIPN˙− (a) and the corresponding ΔA temporal evolution at significant wavelengths (b). λex = 600 nm. A600 = 0.22, 0.2 cm optical path, 8 μJ per pulse. | ||
To better estimate the reduction power of the photogenerated solvated electron, we investigated the reduction of a more challenging substrate, namely 4-chloroanisole. Remarkably, the band at 1440 nm decreases much faster and completely disappears in less than 1 ns upon addition of 4-choloroanisole 0.1 M (Fig. S17†), while no significant change in the lifetime of the D1 excited state is observed. These results demonstrate that the same mechanism is in place with the more energy demanding substrate.
Based on the molar absorption coefficient of the solvated electron at 1450 nm in acetonitrile solution,32 an estimation of the quantum yield of solvated electron production results in 10% for *4CzIPN˙− and 20% for *4DPAIPN˙− (for more details, see ESI†). The typical band of the solvated electron in the NIR spectral region is observed also when the radical anions are produced in the presence of DIPEA instead of TBAOx (Fig. S13†).
Excited states calculations at the TDDFT level (see ESI† for computational details and validation) provide further insight on the proposed mechanism. The computational absorption spectra of 4DPAIPN˙− reveal a quasi-dark state in the infrared region corresponding to the D0 → D1 transition and noted in ref. 28. This state likely accounts for the experimentally observed lack of fluorescence in 4DPAIPN˙−. The first bright peak in the visible is computed to be at 533 nm and attributed to the combined transitions from the D0 state to the D2–D5 states, mirroring the experimental absorption peak at 550 nm (see Fig. 6a). A similar D0 → D1 dark state in the infrared is computed for 4CzIPN˙− (see Fig. S19†) with the lowest bright peak computed to be at 457 nm (in excellent agreement with the experiment), but in this case dominated by the D0 → D6 transition. The electronic configurations determining the excited states of the anions further explain the observed photophysics. As shown in Fig. 6b, frontier orbitals can be clearly separated into orbitals of the phenyl core and orbitals of the four electron accepting peripheral units. The dark D0 → D1 transition is a “core-to-core” transition, involving orbitals in the central phenyl ring. Conversely, the allowed transitions from the D0 state to the D2–D5 states can be characterized as a transfer of electron density from the phenyl core to the 4 peripheral units. This arrangement is likely to facilitate subsequent electron transfer processes from such higher doublet excited states to electron accepting molecules, including the solvent molecules (see Fig. 6c).
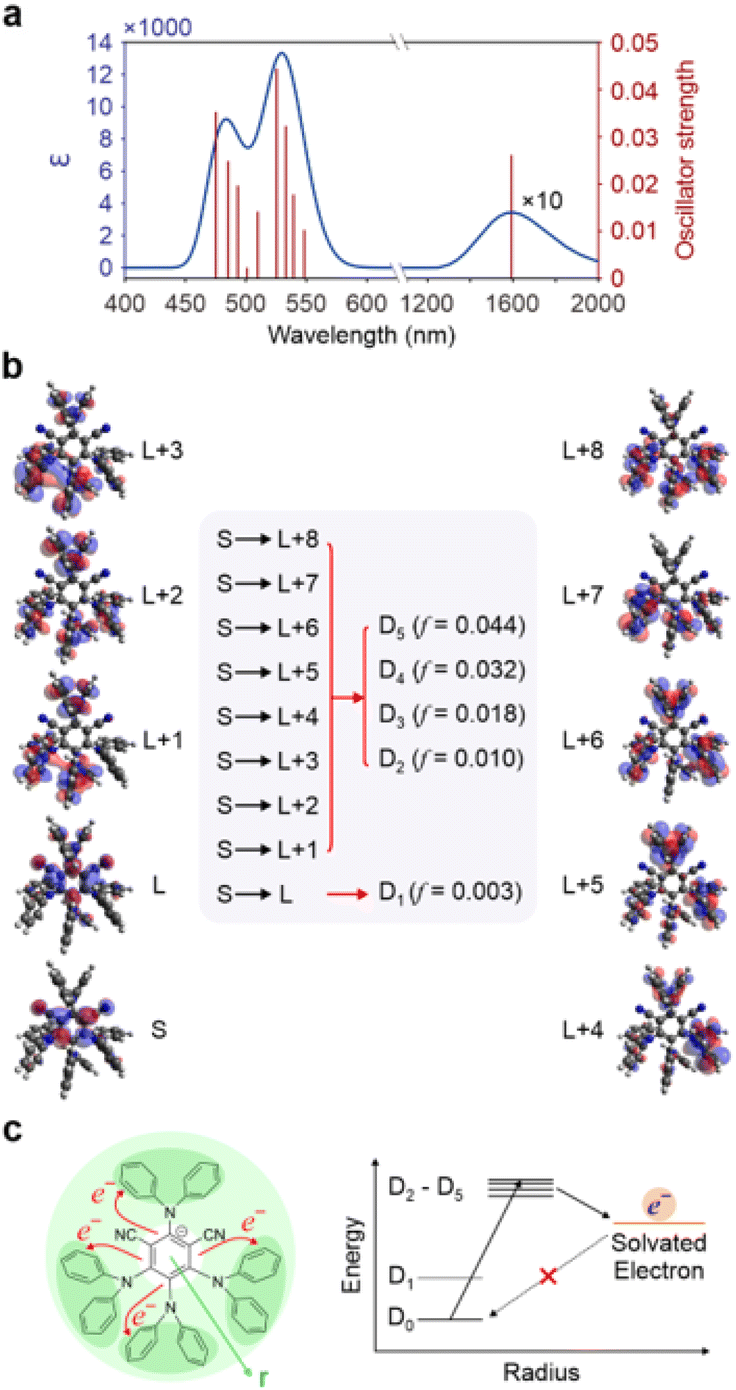 | ||
| Fig. 6 (a) The calculated absorption spectrum of 4DPAIPN˙−, with a different scale use for intensity and ab-sorption profile at long wavelengths. (b) Frontier orbitals in-volved in the main excitations with D2–D5 being electron trans-fer from phenyl core to peripheral units. (c) Schematics illustrating the charge carrier character of the D2–D5 states that fa-cilitates electron transfer to solvent and create a barrier for the back electron transfer. The energy levels are represented to highlight the distance of the negative charge (radius r) from the centre of the molecule in the respective state. | ||
More critically, as the SOMO of the anion is localized on the phenyl core, the back electron transfer to regenerate the anion from an external reducing species, including a solvated electron, is slowed down by the limited overlap between the orbital that should host the back-transferred electron and the surrounding solvent molecules. The exceptional performance of 4DPAIPN˙− as a photoreducing agent is therefore related to the peculiar nature of its geometry and electronic states. As shown in Fig. S20,† the proposed mechanism of photoexcited electron transfer toward the solvent and inhibited back electron transfer is valid also for 4CzIPN˙−, with the corresponding states D2–D5 being also in this case the initial state for the electron transfer to the species to be reduced. In 4CzIPN˙−, however, because of the large oscillator strength (f = 0.137, see Fig. S21†) of the D0 → D6, it is D6 the state that is primarily populated by excitation.
Conclusions
The strong photoreducing abilities of the investigated TADF chromophores is the result of the photogeneration of solvated electrons in a consecutive two-photon induced mechanism (ConPies, Fig. 1b). The generated solvated electron has extremely high diffusivity, which enables photoreactivity to take place beyond the immediate vicinity of the photocatalyst and for a longer time compared to the usual timescale of ConPeT. The elucidation of the mechanism and its relationship with the underlying geometric and electronic structure of the photocatalysts opens completely new avenues for the design of novel photocatalysis.Data availability
Data available on request from the authors.Author contributions
M. V., B. V. and P. C. conceived the study, performed the photophysical analysis, prepared the manuscript and finalized the paper in collaboration with A. F. and F. C., A. G. and P. G. C. provided the organic chromophores employed and prepared the manuscript. X. W. performed the computational study under the supervision of A. T. and together prepared the manuscript. All authors agreed on the submission of the final version of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.3 – Call for tender No. 1561 of 11.10.2022 of Ministero dell’Università e della Ricerca (MUR); funded by the European Union – NextGenerationEU. B. V. acknowledges funding from Italian CNR (PHEEL project). X. W., and A. T. acknowledge funding from Horizon Europe (European Innovation Council, project no. 101057564).Notes and references
- M. M. Bryden and E. Zysman-Colman, Organic thermally ac-tivated delayed fluorescence (TADF) compounds used in pho-tocatalysis, Chem. Soc. Rev., 2021, 50, 7587–7680 RSC.
- E. Speckmeier, T. G. Fischer and K. Zeitler, A toolbox ap-proach to construct broadly applicable metal-free catalysts for photoredox chemistry: deliberate tuning of redox potentials and importance of halogens in donor–acceptor cyanoarenes, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed.
- P. G. Cozzi, P. Ceroni, A. Gualandi, and M. Marchini, Other Nitrogen Heterocycles: Carbazoles, Imides and PDI, mpg-C 3 N 4, Tetrazines, Riboflavin, and BODIPY, in Photoorganocatalysis in Organic Synthesis, ed. M. Fagnoni, S. Protti and D.Ravelli, 2019, pp. 423–469 Search PubMed.
- T. Ghosh, J. Ghosh, I. Bardagi and B. König, Reduction of aryl halides by consecutive visible light-induced electron transfer processes, Science, 2014, 346, 725–728 CrossRef PubMed.
- M. Lepori, S. Schmid and J. P. Barham, Photoredox catalysis harvesting multiple photon or electrochemical energies, Beilstein J. Org. Chem., 2023, 19, 1055–1145 CrossRef CAS PubMed.
- M. Neumeier, D. Sampedro, M. Májek, V. A. de la Peña O'Shea, A. J. von Wangelin and R. Pérez-Ruiz, Dichromatic photo-catalytic substitutions of aryl halides with a small organic dye, Chem.–Eur. J., 2018, 24, 105–108 CrossRef CAS PubMed.
- I. Ghosh and B. König, Chromoselective Photocatalysis: Controlled Bond Activation through Light-Color Regulation of Redox Potentials, Angew. Chem., Int. Ed., 2016, 55, 7676–7679 CrossRef CAS PubMed.
- Y. Huang, J. Hou, L.-W. Zhan, Q. Zhang, W.-Y. Tang and B. D. Li, Photoredox Activation of Formate Salts: Hydrocarboxy-lation of Alkenes via Carboxyl Group Transfer, ACS Catal., 2021, 11, 15004–15012 CrossRef CAS.
- I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- M. M. Hossain, A. C. Shaikh, J. Moutet and T. L. Gianetti, Nat. Synth., 2022, 1, 147–157 CrossRef.
- J. Xu, J. Cao, X. Wu, H. Wang, X. Yang, X. Tang, R. W. Toh, R. Zhou, E. K. L. Yeow and J. Wu, J. Unveiling extreme photore-duction potentials of donor–acceptor cyanoarenes to access aryl radicals from aryl chlorides, J. Am. Chem. Soc., 2021, 143, 13266–13273 CrossRef CAS PubMed.
- A. F. Chmiel, O. P. Williams, C. P. Chernowsky, C. S. Yeung and Z. K. Wickens, Non-innocent radical ion intermediates in photoredox catalysis: parallel reduction modes enable coupling of diverse aryl chlorides, J. Am. Chem. Soc., 2021, 143, 10882–10889 CrossRef CAS PubMed.
- J. Soika, C. McLaughlin, T. Neveselý, C. G. Daniliuc, J. J. Molloy and R. Gilmour, Organophotocatalytic N–O bond cleavage of Weinreb amides: mechanism-guided evolution of a PET to ConPET platform, ACS Catal., 2022, 12, 10047–10056 CrossRef CAS.
- Y. Fang, T. Liu, L. Chen and D. Chao, Exploiting consecutive photoinduced electron transfer (ConPET) in CO2 photoreduction, Chem. Commun., 2022, 58, 7972–7975 RSC.
- Z. Qu, T. Tian, Y. Tan, X. Ji, G.-J. Deng and H. Huang, Redox-neutral ketyl radical coupling/cyclization of carbonyls with N-aryl acrylamides through consecutive photoinduced electron transfer, Green Chem., 2022, 24, 7403–7409 RSC.
- V. J. Mayerhofer, M. Lippolis and C. Teskey, Dual-catalysed intermolecular reductive coupling of dienes and ketones, Angew. Chem., Int. Ed., 2024, 63, e202314870 CrossRef CAS PubMed.
- C. Ma, Y. Tian, Y. Wang, X. He, Y. Jiang and B. Yu, Visible-light-driven transition-metal-free site-selective access to ison-icotinamides, Org. Lett., 2022, 24, 8265–8270 CrossRef CAS PubMed.
- N. Xian, J. Yin, X. Ji, G.-J. Deng and H. Huang, Visible-light-mediated photoredox carbon radical formation from aqueous sulfoxonium ylides, Org. Lett., 2023, 25, 1161–1165 CrossRef CAS PubMed.
- L. Chen, Q. Qu, C.-K. Ran, W. Wang, W. Zhang, Y. He, L.-L. Liao, J.-H. Ye and D.-G. Yu, Photocatalytic carboxylation of C−N bonds in cyclic amines with CO2 by consecutive visible-light-induced electron transfer, Angew. Chem., Int. Ed., 2023, 62, e202217918 CrossRef CAS PubMed.
- C. Huang, C. Huang, P. Xiao, Z.-M. Ye, C.-L. Wang, C. Kang, S. Tang, Z. Wei and H. Cai, Direct C(sp3)–H arylation of unprotected benzyl anilines and alkylarenes by arganocatalysis under visible light, Org. Lett., 2024, 26, 304–309 CrossRef CAS PubMed.
- C. P. Chernowsky, A. F. Chmiel and Z. K. Wickens, Elec-trochemical activation of diverse conventional photoredox catalysts induces potent photoreductant activity, Angew. Chem., Int. Ed., 2021, 60, 21418–21425 CrossRef CAS PubMed.
- S. Grotjahn and B. König, Photosubstitution in dicyano-benzene-based photocatalysts, Org. Lett., 2021, 23, 3146–3150 CrossRef CAS PubMed.
- E. Pinosa, E. Bassan, S. Cetin, M. Villa, S. Potenti, F. Calogero, A. Gualandi, A. Fermi, P. Ceroni and P. G. Cozzi, Light-induced access to carbazole-1,3-dicarbonitrile: a thermally activated delayed fluorescent (TADF) photocatalyst for cobalt-mediated allylations, J. Org. Chem., 2023, 88, 6390–6400 CrossRef CAS PubMed.
- A. J. Rieth, M. I. Gonzalez, B. Kudisch, M. Nava and D. G. Nocera, How radical are “radical” photocatalysts? A closed-shell Meisenheimer complex is identified as a super-reducing photoreagent, J. Am. Chem. Soc., 2021, 143, 14352–14359 CrossRef CAS PubMed.
- M. Marchini, A. Gualandi, M. Mengozzi, P. Franchi, M. Lu-carini, P. G. Cozzi, V. Balzani and P. Ceroni, Mechanistic insights into two-photon-driven photocatalysis in organic synthesis, Phys. Chem. Chem. Phys., 2018, 20, 8071–8076 RSC.
- T. Constantin, F. Juliá, N. S. Sheikh and D. Leonori, A case of chain propagation: α-aminoalkyl radicals as initiators for aryl radical chemistry, Chem. Sci., 2020, 11, 12822–12828 RSC.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed.
- Y. Kwon, J. Lee, Y. Noh, D. Kim, Y. Lee, C. Yu, J. C. Roldao, S. Feng, J. Gierschner, R. Wannemacher and M. S. Kwon, Formation and degradation of strongly reducing cyanoarene-based radical anions towards efficient radical anion-mediated photoredox catalysis, Nat. Commun., 2023, 14, 92 CrossRef CAS PubMed.
- B. Pfund, D. Gejsnæs-Schaad, B. Lazarevski and O. S. Wenger, Picosecond reactions of excited radical ion super-reductants, Nat. Commun., 2024, 15, 4738 CrossRef CAS PubMed.
- F. Draper, E. H. Doeven, J. L. Adcock, P. S. Francis and T. U. Connell, Extending photocatalyst activity through choice of electron donor, J. Org. Chem., 2023, 88, 6445–6453 CrossRef CAS PubMed.
- A. A. Isse, A. Gennaro and F. Maran, Mechanism of the dis-sociative electro-oxidation of oxalate in aprotic solvents, Acta Chem. Scand., 1999, 53, 1013–1022 CrossRef CAS.
- D. C. Grills and S. V. Lymar, Solvated electron in acetonitrile: radiation yield, absorption spectrum, and equilibrium between cavity- and solvent-localized states, J. Phys. Chem. B, 2022, 126, 262–269 CrossRef CAS PubMed.
- S. C. Doan and B. J. Schwartz, Nature of excess electrons in polar fluids: anion-solvated electron equilibrium and polarized hole-burning in liquid acetonitrile, J. Phys. Chem. Lett., 2013, 4, 1471–1476 CrossRef CAS PubMed.
- J. Bonin, I. Lampre and M. Mostafavi, Absorption spectrum of the hydrated electron paired with nonreactive metal cations, Radiat. Phys. Chem., 2005, 74, 288–296 CrossRef CAS.
- V. Svoboda, R. Michiels, A. C. Laforge, J. Med, F. S. P. Slavíček and H. J. Wörner, Real-time observation of water radiolysis and hydrated electron formation induced by extreme-ultraviolet pulses, Sci. Adv., 2020, 6, eaaz0385 CrossRef CAS PubMed.
- L. Turi, W.-S. Sheu and P. J. Rossky, Characterization of excess electrons in water-cluster anions by quantum simulations, Science, 2005, 309, 914–917 CrossRef CAS PubMed.
- T. E. Gartmann, L. Ban, B. L. Yoder, S. Hartweg, E. Chasovskikh and R. Signorell, Relaxation dynamics and genuine properties of the solvated electron in neutral water clusters, J. Phys. Chem. Lett., 2019, 10, 4777–4782 CrossRef CAS PubMed.
- M. Shirom and G. Stein, Excited state chemistry of the ferrocyanide ion in aqueous solution. I. Formation of the hydrated electron, J. Chem. Phys., 1971, 55, 3372–3378 CrossRef CAS.
- M. S. Matheson, W. A. Mulac and J. Rabani, Formation of the hydrated electron in the flash photolysis of aqueous solutions, J. Phys. Chem., 1963, 67, 2613–2617 CrossRef CAS.
- F. Glaser, C. Kerzig and O. S. Wenger, Multi-photon excitation in photoredox catalysis: concepts, applications, methods, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS PubMed.
- M. Schmalzbauer, M. Marcon and B. König, Excited state anions in organic transformations, Angew. Chem., Int. Ed., 2021, 60, 6270–6292 CrossRef CAS PubMed.
- C. Kerzig and M. Goez, Combining energy and electron transfer in a supramolecular environment for the “green” generation and utilization of hydrated electrons through photoredox catalysis, Chem. Sci., 2016, 7, 3862–3868 RSC.
- C. Kerzig, X. Guo and O. S. Wenger, Unexpected hydrated electron source for preparative visible-light driven photoredox catalysis, J. Am. Chem. Soc., 2019, 141, 2122–2127 CrossRef CAS PubMed.
- F. Brandl, S. Bergwinkl, C. Allacher and B. Dick, Consecutive photoinduced electron transfer (conPET): the mechanism of the photocatalyst rhodamine 6G, Chem.–Eur. J., 2020, 26, 7946–7954 CrossRef CAS PubMed.
- A. Shaikh, P. Singh and N. Lal, Solvated electrons: dynamic reductant in visible light photoredox catalysis, Adv. Synth. Catal., 2024, 366, 1906–1921 CrossRef.
- R. Obertík, J. Chudoba, J. Šturala, J. Tarábek, L. Ludvíková, T. Slanina, B. König and R. Cibulka, Highly chemoselective catalytic photooxidations by using solvent as a sacrificial electron acceptor, Chem.–Eur. J., 2020, 28, e202202487 CrossRef PubMed.
- I. P. Bell, M. A. J. Rodgers and H. D. Burrows, Kinetic and thermodynamic character of reducing species produced on pulse radiolysis of acetonitrile, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 315 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04518a |
| ‡ To avoid misinterpreting the time dependent spectral features of the radical anions, we have also characterized the neutral 4CzIPN and 4DPAIPN, see Fig. S7–S10. |
| This journal is © The Royal Society of Chemistry 2024 |