
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity of metal hydrides with CO2: going beyond formate with a high-valent cationic pentahydride Mo(VI) complex†
Nicolas
Queyriaux
 *a,
Jorge J.
Cabrera-Trujillo
b,
Nina
Durvin
a,
Laure
Vendier
a,
Karinne
Miqueu
*b and
Antoine
Simonneau
*a
*a,
Jorge J.
Cabrera-Trujillo
b,
Nina
Durvin
a,
Laure
Vendier
a,
Karinne
Miqueu
*b and
Antoine
Simonneau
*a
aLCC-CNRS, Université de Toulouse, CNRS, UPS, 205 Route de Narbonne, BP44099, F-31077 Toulouse Cedex 4, France. E-mail: antoine.simonneau@lcc-toulouse.fr; nicolas.queyriaux@lcc-toulouse.fr
bCNRS/UPPA, IPREM UMR 5254, E2S-UPPA, Hélioparc, 2 Avenue du Président Angot, 64053 Pau Cedex 09, France. E-mail: karinne.miqueu@univ-pau.fr
First published on 18th November 2024
Abstract
The cationic molybdenum pentahydride complex [MoH5(depe)2]+ (depe = 1,2-bis(diethylphosphino)ethane) is shown to undergo two consecutive reactions with carbon dioxide. In the initial, room-temperature process, classical insertion of CO2 into a metal–hydride bond is observed, resulting in the formation of the expected formate complex, [MoH2(HCOO)(depe)2]+. Further reactivity is triggered at temperature above 100 °C. Complete conversion into two new complexes is indeed observed, resulting from the formal cleavage of a C–O bond of carbon dioxide, [MoH(CO)2(depe)2]+ and [MoO(HCOO)(depe)2]+. Unprecedented in the absence of ligand assistance, such metal hydride reactivity has been comprehensively studied by a combination of experimental and computational means with the aim of elucidating the underlying mechanism that governs this process.
Introduction
Carbon dioxide (CO2) is an appealing primary source of carbon atoms for the development of new sustainable synthetic procedures of both carbon-based commodity chemicals and fuels.1–6 Although inexpensive, relatively abundant and non-toxic, CO2 suffers from severe thermodynamic and kinetic limitations that significantly restrict its use as a routine reagent. Transition metal-based complexes have commonly been employed to overcome this inherent kinetic reluctance.7–9 They have been demonstrated to be highly effective catalysts for the conversion of CO2 into a range of hydrogenated C1 products (HCOOH, CH3OH and CH4), either through electro-assisted approaches or via traditional catalytic hydrogenation. In the majority of such processes, the formation of a metal formate species through CO2 insertion into metal hydride bonds represents a fundamental step in catalytic cycles.10 Consequently, a substantial focus has been directed toward elucidating thermodynamic11–13 and kinetic14–18 aspects of this reaction.Although rare, alternative reactivities of metal hydrides have also been reported (Fig. 1). The group of Parkin described C–O bond cleavage upon exposure of [Cp*Mo(PMe3)3H] to a CO2 atmosphere, resulting in the formation of a carbonyl complex together with trimethyl phosphine oxide.19 In the same vein, the group of Milstein investigated the reductive cleavage of CO2 by hydride complexes of group 9 metals (Rh, Ir) supported by PNP pincer ligands.20,21 This reactivity, triggered by metal–ligand cooperation, again gave access to carbonyl derivatives – the second oxygen atom being released as a water molecule. Interestingly, a diisocyanato/dioxo dititanium complex was obtained from the stepwise, double reductive cleavage of CO2 by a dihydride dititanium complex, through the concomitant activation of a dinitrogen ligand.22 On-metal C–O bond activation by metal hydride complexes can also be facilitated by the assistance of exogenous main-group Lewis acids (LA).23 Ultimately, such assistance is capable to promote a cooperative reductive cleavage of CO2, as recently reported by the group of Camp with a strongly polarized heterobimetallic Ir–Al complex.24 In most of those cases, the transfer of an oxo group to a particularly oxophilic exogenous – or remote – partner thus appears to be an important thermodynamic lever that drives the C–O cleavage reaction.
 | ||
| Fig. 1 Examples of C–O bond cleavages in the CO2 molecule mediated by transition metal hydride complexes. | ||
Recently, our group reported on the isolation of a cationic molybdenum(VI) pentahydride complex supported with a bis(phosphine) ligand,25 [MoH5(depe)2]+ (1+, depe = 1,2-bis(diethylphosphino)ethane). Related group 6 pentahydride derivatives were previously postulated, observed or isolated by the groups of Wilkinson,26 Semenenko,27,28 Ito,29 Henderson,30 Cundari/Yousufuddin31 and Chirik.32 Their reactivity towards carbon dioxide remains, however, largely uncharted. In this article, we explore the stoichiometric reactivity of pentahydride complex 1+ towards CO2 and its nitrogen- and sulphur-based analogues (N,N′-diisopropylcarbodiimide – DIC – and CS2). We demonstrate that complex 1+ readily loses H2 in those processes to generate unsaturated derivatives capable of quick reaction with the considered heterocumulenes. In all cases, insertion of the substrates into a metal-hydride bond occurs. Interestingly, further reactivity is observed for CO2 upon thermal activation. A rare example of unassisted, on-metal C–O bond cleavage is evidenced, with the formation of both Mo(II)–carbonyl and Mo(IV)–oxo cationic complexes. Comprehensive computational investigations have been conducted to assess the overall mechanism of this intriguing process.
Results and discussion
We have recently reported the protonation behaviour of the neutral Mo(IV) tetrahydride complex 2 towards a variety of proton sources, highlighting the importance of the nature of the conjugate base of the acid used to warrant protonation-only processes.25 Based on these previous findings, we selected triethylammonium tetraphenylborate [HNEt3][BPh4] – that combines both weakly coordinating counter-anion and conjugate base – to serve as the proton purveyor.Treating a solution of 2 with 1 equiv. of [HNEt3][BPh4] at room temperature resulted in the immediate formation of the expected pentahydride complex [MoH5(depe)2][BPh4], 1·BPh4 (Scheme 1). A diagnostic signal appears as a quintet at −5.0 ppm in the hydride region of the 1H NMR spectrum. This complex can be isolated in good yields (80–90%) as a pale-yellow powder.
 | ||
| Scheme 1 Protonation of 2 employing [HNEt3][BPh4] as the proton source affords 1·BPh4. | ||
Single crystals suitable for X-ray diffraction analysis were obtained upon low temperature storage (−40 °C) of THF–pentane solution of the complex. The resulting structure is almost identical to the one we previously reported, should the nature of the counterion be excluded (see ESI†).25
When a solution of 1·BPh4 in THF was exposed to CO2 (3 bar) at room temperature, the solution rapidly turned red. The newly formed complex 3·BPh4 shows a new signal in the hydride region of the 1H NMR spectrum (in C6D4Cl2), emerging as a pseudo-quintet at −7.94 ppm with an apparent 2J(P–H) coupling of 42.0 Hz. Another notable, new resonance is observed at 7.77 ppm in the form of a singlet that splits into a doublet (1J(C–H) = 209.3 Hz) when 13C-labelled CO2 was used. We assigned this new signal to a formato ligand generated by CO2 insertion into a Mo–H bond of 1·BPh4. This NMR data is in good agreement with previously reported formato ligands.33–37 This was further supported by the appearance of a quintet (3J(C–P) = 3.5 Hz) at 176.1 ppm in the 13C{1H} NMR spectrum of 3·BPh4. The 31P{1H} NMR spectrum features two singlets at 43.9 and 78.3 ppm that expanded into triplets (2J(P–H) = 36.0 and 41.9 Hz, respectively) upon selective 1H-decoupling centred at 1.5 ppm. Collectively, these results point towards the formulation of 3·BPh4 as [MoH2(HCOO-κ2O)(depe)2][BPh4] (Scheme 2).38
 | ||
| Scheme 2 Formation of Mo(IV)–formato complex 3·BPh4 from 1·BPh4 under CO2 atmosphere. | ||
This ion pair crystallised from a THF–pentane solution, and the single-crystal X-ray diffraction (sc-XRD) analysis confirmed the κ2O bidentate coordination of the formate anion, with Mo–O bond lengths of ca. 2.26 Å (Fig. 4). If we restrict to the first coordination sphere, the Mo centre is found in a C2v-symmetric environment. The phosphorus nuclei lie in twos above or under the symmetry plane, in good agreement with the 31P{1H} NMR spectrum showing two resonances. Although the hydrido ligands could not be unambiguously located in the final Fourier difference map, their most likely positions can be estimated from the presence of a large open site located below a mean plane formed by the phosphorus atoms, trans to the formato ligand. It is reasonable to propose that the two hydrides should be staggered relative to the formato ligand, so that d orbital destabilization is minimized in an overall dodecahedral geometry, common for 8-coordination.39
To gain more insight into the reactivity of complex 1+ toward CO2, we carried out Density Functional Theory (DFT) calculations. We initially performed geometry optimizations at B3LYP-D3(BJ)/SDD+f(Mo), 6-31G** (other atoms) level of theory. Further energy refinement through single point calculations was carried out at PCM(THF)-M06-L-D3/def2-TZVPP level of theory considering solvent effects by means of the Polarizable Continuum Model (PCM) method (see ESI†). Optimization of 3+ was first performed in order to validate the reliability of our selected computational level of theory. We found an excellent agreement between the X-ray and DFT geometries, with a relative error between experimental and calculated structures consistently below 1.5% (Table S1†). We then turned our attention to the mechanistic pathway followed by the reaction of 1+ with CO2. As illustrated in the reaction profile (Fig. 2), a reductive elimination of H2 was found to initiate the overall process with a low energy transition state (TS1, ΔG# = 4.0 kcal mol−1) yielding the σ-complex INT1. This is followed by a barrierless dissociation of H2 to afford INT1′ (Fig. S1†). Further κO1 coordination of CO2 to the metal centre then occurs through TS2 (ΔG# = 22.0 kcal mol−1). Formation of the resulting INT2 is slightly endergonic from INT1′ (ΔG = 5.5 kcal mol−1). Interestingly, kinetic measurements performed under two different pressures of CO2 (1.5 vs. 3 bar) showed no influence of the latter (see ESI†). This agrees with the computed energy profile where H2 release is energetically more demanding than CO2 addition. Then, a rapid hydride transfer from the Mo centre to the coordinated CO2 was found to occur (TS3, ΔG# = 0.9 kcal mol−1 from INT2), granting further stabilization. We also considered that the hydride transfer process could occur via an outer-sphere mechanism, but the corresponding TS was found to be energetically prohibitive at 44.9 kcal mol−1 (Scheme S1†). Interestingly, the resulting intermediate INT3, featuring a formato ligand, is nearly isoenergetic with INT1′, suggesting that CO2 insertion-deinsertion processes are easily accessible. Rotation around the C–O bond, allowing the expected κ2O coordination of the formato ligand, finally affords complex 3+ in a global exergonic process (ΔG: −3.0 kcal mol−1). Overall, this mechanism is in good agreement with the concerted CO2 insertion pathway, previously reported by Hazari for group 9 and 10 metal hydride complexes.10
 | ||
| Fig. 2 Computed profile for the reaction of complex 1+ with CO2. Free energy values (ΔG) and distances are given in kcal mol−1 and Å, respectively. All data have been computed at the PCM(THF)-M06-L-D3/def2-TZVPP//B3LYP-D3(BJ)/SDD+f(Mo), 6-31G** (other atoms) level of theory. | ||
An o-dichlorobenzene solution of the 3·BPh4 complex was heated at 100 °C for 24 hours under an atmosphere of CO2, resulting in a complex mixture of compounds. Large, unresolved signals were notably observed in the 1H NMR spectrum, suggesting the formation of a paramagnetic molybdenum species identified as [Mo(depe)2Cl2] (see ESI†). Simultaneously, the appearance of new resonances in the aromatic domain is also noted. We tentatively assigned those new signals to the formation of biphenyl (see ESI†), arising from the radical-driven reductive rearrangements of the tetraphenylborate anion.40,41 Slight chemical shift deviations are, however, noted in comparison with an authentic biphenyl sample, which we attribute to the influence of [Mo(depe)2Cl2] paramagnetism. We thus decided to opt for the HB(C6F5)3− anion, that displays increased redox stability. 3·HB(C6F5)3 could easily be prepared by the reaction of 2 with [HPtBu3][HB(C6F5)3],25 followed by room temperature reaction with CO2. Under similar conditions (reaction time increased to 36 h), 3·HB(C6F5)3 indeed granted access to a much cleaner reactivity. More specifically, two new complexes 4·HB(C6F5)3 and 5·HB(C6F5)3 are formed in an approximate 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :64 ratio (Scheme 3, top), together with H2 evolution. The positively charged part of the minor species was identified as [MoH(CO)2(depe)2]+, 4+. This monohydride cationic complex bearing two carbonyl ligands in a trans configuration was independently prepared from the reaction of 1+ with CO (Scheme 3, bottom, and Fig. 4). The second complex that formed, 5+, features a singlet at 46.0 ppm in the 31P{1H} NMR spectrum. The latter unresolvedly broadens upon selective decoupling of the aliphatic protons, suggesting the absence of hydrido ligands. This is further confirmed by the 1H NMR spectrum, which is devoid of any new hydride signals. Interestingly, a new proton signal resonating at 7.31 ppm was observed, which correlates with a singlet at 163.8 ppm in the 13C{1H} NMR spectrum. This signal splits upon 13C-labeling (1J(C–H) = 204.3 Hz), indicating the formation of a new formato derivative. High-resolution mass spectrometry was used to gain further insight into the nature of compound 5+. The isotopic pattern of the resulting signal strongly suggests the presence of an additional oxo ligand. Taken together, those results point towards 5+ being formulated as [MoO(HCOO)(depe)2]+, a cationic Mo(IV) complex. Despite several attempts, independent synthesis of 5+ was unsuccessful: it is, however, interesting to note that 31P NMR features are in line with similar cationic d2 molybdenum–oxo complexes.42 Along these two main products, small amounts of free formate was also noticed, accounting for 8% of the non-gaseous compounds ultimately present in solution.
:64 ratio (Scheme 3, top), together with H2 evolution. The positively charged part of the minor species was identified as [MoH(CO)2(depe)2]+, 4+. This monohydride cationic complex bearing two carbonyl ligands in a trans configuration was independently prepared from the reaction of 1+ with CO (Scheme 3, bottom, and Fig. 4). The second complex that formed, 5+, features a singlet at 46.0 ppm in the 31P{1H} NMR spectrum. The latter unresolvedly broadens upon selective decoupling of the aliphatic protons, suggesting the absence of hydrido ligands. This is further confirmed by the 1H NMR spectrum, which is devoid of any new hydride signals. Interestingly, a new proton signal resonating at 7.31 ppm was observed, which correlates with a singlet at 163.8 ppm in the 13C{1H} NMR spectrum. This signal splits upon 13C-labeling (1J(C–H) = 204.3 Hz), indicating the formation of a new formato derivative. High-resolution mass spectrometry was used to gain further insight into the nature of compound 5+. The isotopic pattern of the resulting signal strongly suggests the presence of an additional oxo ligand. Taken together, those results point towards 5+ being formulated as [MoO(HCOO)(depe)2]+, a cationic Mo(IV) complex. Despite several attempts, independent synthesis of 5+ was unsuccessful: it is, however, interesting to note that 31P NMR features are in line with similar cationic d2 molybdenum–oxo complexes.42 Along these two main products, small amounts of free formate was also noticed, accounting for 8% of the non-gaseous compounds ultimately present in solution.
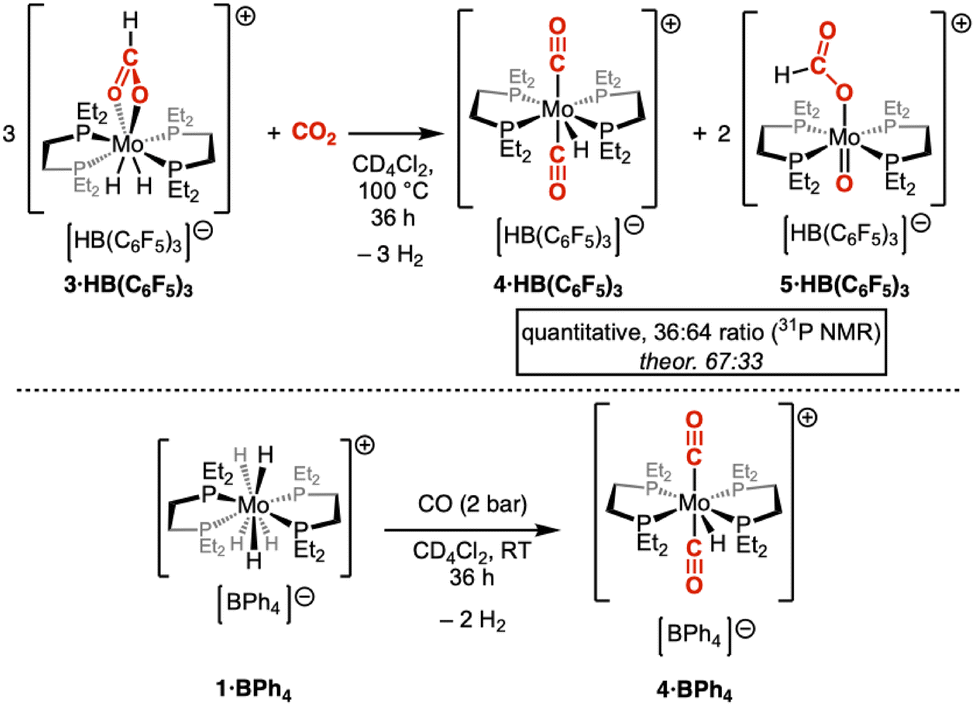 | ||
| Scheme 3 CO2 cleavage induced by thermal activation of the formato complex 3·HB(C6F5)3 (top) and independent synthesis of the monohydrido bis(carbonyl) complex 4·BPh4 (bottom). | ||
Interested in this unusual process – formally resulting from the cleavage of a CO2 molecule C–O bond43 – we again decided to investigate the mechanism of this transformation by the use of DFT calculations. We employed the PCM(o-dichlorobenzene)-M06-L-D3/def2-TZVPP//B3LYP-D3(BJ)/SDD+f(Mo), 6-31G** (other atoms) level of theory and corrected the free energies at 373.15 K using GoodVibes package.44 Two different pathways have been considered for the thermal evolution from complex 3+ to complex 5+. As illustrated in Scheme 4, the first pathway (Pathway I) begins with the reductive elimination of H2, followed by the coordination of CO2. Then, the breaking of the C–O bond in the CO2 ligand occurs, followed by the release of CO leading to the final compound 5+. The second pathway (Pathway II) involves as a first step a hydride transfer from the formato moiety to Mo centre—formally a de-insertion of CO2, the reverse of the final elementary step in the mechanism leading from 1+ to 3+. H2 is subsequently released, followed by the C–O bond cleavage in the CO2 ligand. Finally, two routes were envisaged to achieve the formation of the final product 5+: either CO2 can be added followed by the release of CO, or CO can be released first, followed by the addition of CO2. According to our DFT calculations, Pathway I can be safely ruled out due to several high energy barriers in the process (see the complete reaction profile in Fig. S2†). First, the release of H2 from 3+ is highly endergonic, with the complex INT4-I being 25.3 kcal mol−1 higher in energy than 3+. Additionally, subsequent addition of CO2 requires an overall activation barrier of 45.1 kcal mol−1, which cannot be reached under the reaction conditions. The transition state for the next step (C–O bond cleavage in the CO2 molecule) also has a prohibitive high energy at 43.9 kcal mol−1. In the reaction profile corresponding to Pathway II (Fig. 3), the first step formally corresponds to the hydride transfer from the formato moiety to the Mo centre, which is the back-side reaction from 3+ to INT2, previously described in Fig. 2. The reverse activation barrier of 21.9 kcal mol−1, corrected with GoodVibes software to take the temperature into account, was found accessible at 100 °C. Then, the release of H2 proceeds very easily through TS4, with a rather low barrier of 1.7 kcal mol−1 from INT2, affording the η2-CO2–Mo complex INT4.45 The subsequent C–O bond cleavage is energetically more costly, with a transition state, TS5, located at 31.3 kcal mol−1 above 3+. Finally, DFT calculations suggest that CO is released (TS6) before addition of CO2 (TS7) to form the final product 5+. In this process, CO2 plays the role of a hydride abstractor, generating a formato moiety, which finally coordinates on Mo in 5+. In Pathway II, the rate-determining step (RDS) corresponds to the addition of CO2 (TS7), with an overall activation barrier of 33.2 kcal mol−1 from 3+, which is consistent with a high temperature for the reaction to occur. It is important to note that the released CO (TS6 in Pathway II) is necessary for the thermal evolution of complex 3+ into complex 4+ (Fig. S3†). Additionally, the CO2 needed for the final step in Pathway II (TS7) is derived from the reaction profile described in Fig. S3† (thermal evolution from 3+ into 4+). Consequently, formation of 4+ and 5+ are intimately connected.
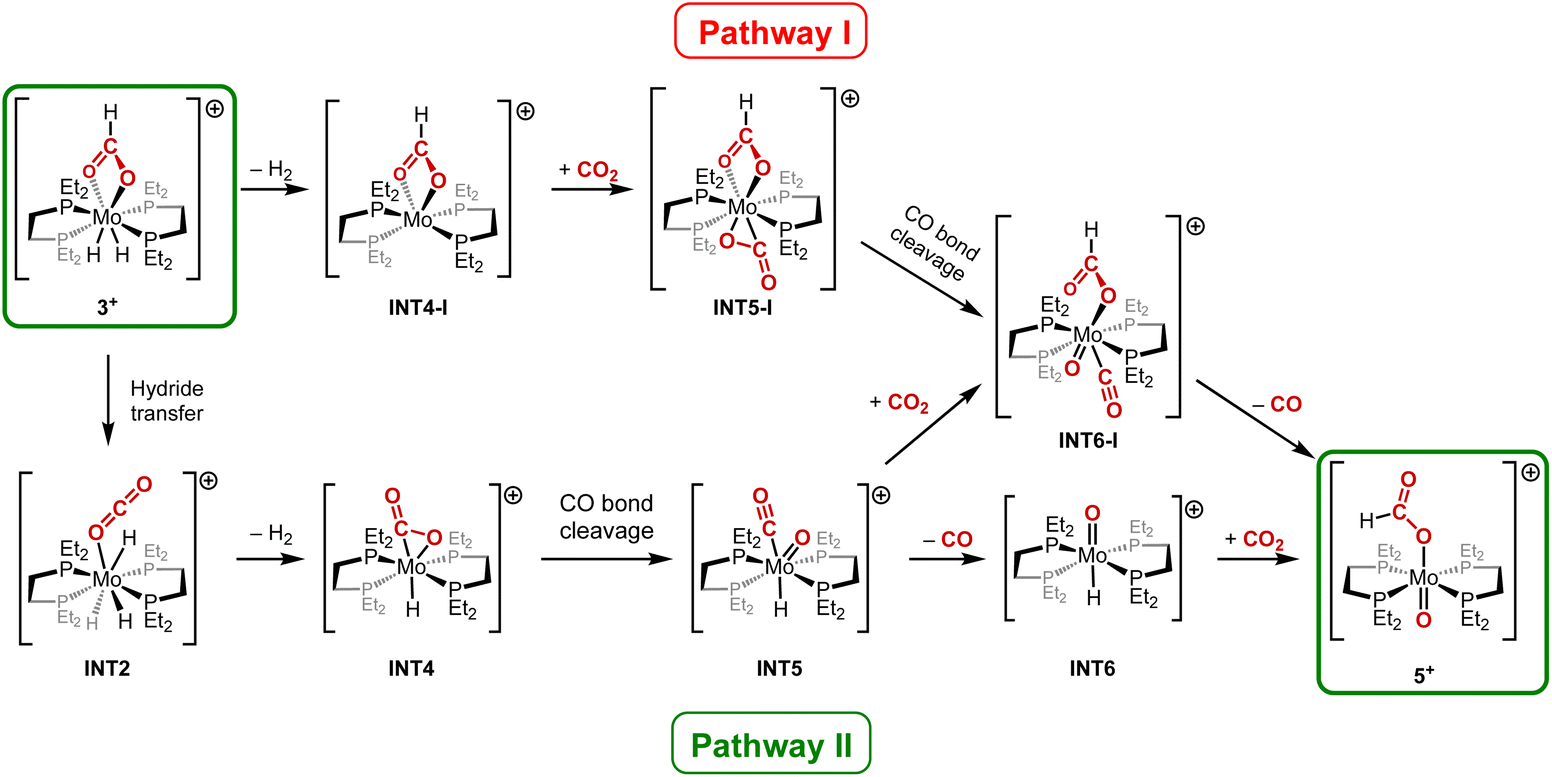 | ||
| Scheme 4 Plausible reaction pathways for the thermal evolution from 3+ to 5+. | ||
 | ||
| Fig. 3 Energy profile for the thermal evolution of complex 3+ into complex 5+ according to Pathway II (Scheme 4). All data have been computed at the PCM(o-dichlorobenzene)-M06-L-D3/def2-TZVPP//B3LYP-D3(BJ)/SDD+f(Mo), 6-31G** (other atoms) level of theory. Free energies (ΔG) have been corrected at 373.15 K. All activation barriers are referred to 3+. Distances and energies are given in Å and kcal mol−1, respectively. | ||
As an extension of our investigation on the activation of CO2, we studied the reactivity of 1·BPh4 with CS2 and DIC (Scheme 5). Upon treatment of complex 1·BPh4 with 1 equiv. of CS2, an immediate darkening of the THF solution is observed. The formation of complex 6·BPh4 was characterized by the appearance of a new quintet at −6.09 ppm (2J(P–H) = 44.0 Hz) and a singlet at 5.76 ppm in the 1H NMR spectrum, as well as a 31P{1H} resonance at 69.1 ppm. The latter splits into a doublet (2J(P–H) = 44.0 Hz) upon selective decoupling of the aliphatic protons. Unlike the behaviour observed in the presence of CO2, these data support a 4e−-reduction of CS2 through double insertion into Mo–H bonds,46–49 resulting in a monohydride methanedithiolate derivative, [MoH(S2CH2)(depe)2][B(C6H5)4] (6·BPh4, Scheme 5). Despite several attempts, 6+ remained reluctant to crystallization and isolation of an analytically pure complex could not be reached (92% spectroscopic purity, see ESI†). Addition of 1 equiv. of DIC to 1·BPh4 in THF led to the formation of the expected M–H insertion product 7·BPh4 (Scheme 5). In the 1H NMR spectrum, loss of the hydride resonance of 1·BPh4 was observed along with the formation of two new diagnostic signals: a multi-lined hydride signal at −7.97 ppm and a singlet at 7.95 ppm assigned to the formamidinate methine proton. Typical of the methine carbon,50–53 a signal at 159.4 ppm is observed in the 13C{1H} NMR spectrum. The 31P{1H} features two triplets at 71.0 and 41.5 ppm. Contrary to 6·BPh4, complex 7·BPh4 easily crystallized from a THF–pentane solution, as orange prisms (η = 54%). In the solid-state structure of 7·BPh4 (Fig. 4), the formamidinate ligand is found in a κ2N coordination, with a N–Mo–N bite angle of 59.0°. As expected, this value and other relevant metrics are very similar to those determined in the molecular structure of 3·BPh4. Again, the exact location of the hydrido ligands could not be determined, although their most probable positions will similarly be found trans to the formamidinate ligand to fulfil the expected distorted dodecahedral coordination sphere.
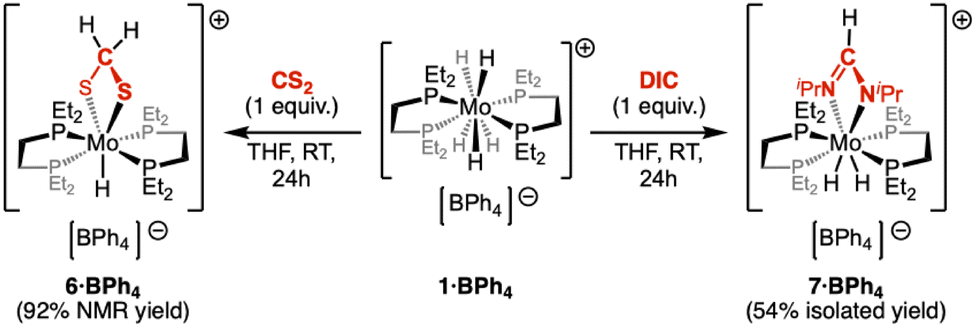 | ||
| Scheme 5 Formation of 6·BPh4 and 7·BPh4 from 1·BPh4. | ||
 | ||
| Fig. 4 From left to right, molecular structures of compounds 3·BPh4, 4·BPh4 and 7·BPh4 in the solid state. The BPh4− counter ions and the hydrogen atoms (except those on the reduced formato or amidinato ligand or bound to the metal centre) were omitted for clarity. Thermal ellipsoids are shown at the 25% level of probability. Compounds 4·BPh4 and 7·BPh4 crystallized with two molecules in the asymmetric unit (Z′ = 2), only one is depicted for clarity. | ||
Conclusions
In summary, we have thoroughly investigated the reactivity of a high-valent cationic pentahydride Mo(VI) complex 1·BPh4 towards CO2 and related heteroallenes (CS2, DIC). The expected insertion of substrate molecules into Mo–H bonds was typically observed, resulting either in two-electron (CO2, DIC) or four-electron reduction of the substrate (CS2). Overall, and despite its high-valent character, complex 1·BPh4 behaves as a potent reductant. Most of those reactivities is triggered by facile hydrogen release, suggesting 1·BPh4 could be better described as a masked Mo(IV) complex. Beyond this expected insertion chemistry, we have been able to evidence a rare example of C–O bond cleavage mediated by a metal hydride complex, that occurs without assistance of a ligand or another proximal reactive centre. Indeed, upon thermal activation, the formato complex 5+ was shown to further evolve quantitatively towards a duet of complexes, namely Mo(II)–carbonyl and Mo(IV)–oxo derivatives. This reaction was particularly scrutinized via a combination of experimental and theoretical tools. The intimate mechanism responsible for the formation of both carbonyl and oxo derivatives has been carefully deciphered and the importance of high temperature reversible CO2 hydrogenation–dehydrogenation process was evidenced. Ultimately, this reversibility – together with the high thermodynamic drivers associated with Mo–O bond formation and transient CO liberation – allows on-metal C–O bond cleavage to be accessed.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
N. Q. and N. D. run the experiments and analysed the data. J. J. C.-T. performed DFT calculations. L. V. recorded single-crystal X-ray diffraction data and solved the structures. K. M. and A. S. obtained funding for the project, managed it and took part in the analysis of the data. N. Q. wrote the first draft of the manuscript. K. M. and J. J. C.-T. analysed the theoretical results and wrote the computational part of the manuscript. The manuscript was then further revised through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. Q. and A. S. are indebted to the European Research Council for funding (ERC Starting Grant No. 757501). Mass Spectrometry Service (N. Martins-Froment) of the Institute of Chemistry of Toulouse-UAR 2599 (Université de Toulouse, CNRS, Toulouse, France – https://www.ict.ups-tlse.fr) is acknowledged for technical assistance with ESI-HRMS experiments. J. J. C.-T. and K. M. would like to thank the Carnot ISIFoR institute for its support and funding. The “Direction du Numérique” of the Université de Pau et des Pays de l'Adour and the Mésocentre de Calcul Intensif Aquitain (MCIA) are acknowledged for the support of computational facilities. This work was also granted access to the HPC resources of IDRIS under the allocation 2023-AD010800045R2 made by GENCI. All authors thank their respective institution for providing access to facilities.Notes and references
- S. De, A. Dokania, A. Ramirez and J. Gascon, ACS Catal., 2020, 10, 14147–14185 CrossRef CAS
.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. Surya Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS
- H.-Q. Liang, T. Beweries, R. Francke and M. Beller, Angew. Chem., Int. Ed., 2022, 61, e202200723 CrossRef CAS PubMed
- N. W. Kinzel, C. Werlé and W. Leitner, Angew. Chem., Int. Ed., 2021, 60, 11628–11686 CrossRef CAS PubMed
- A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948–15982 CrossRef CAS PubMed
- N. Hazari and J. E. Heimann, Inorg. Chem., 2017, 56, 13655–13678 CrossRef CAS PubMed
- K. R. Brereton, N. E. Smith, N. Hazari and A. J. M. Miller, Chem. Soc. Rev., 2020, 49, 7929–7948 RSC
- K. M. Waldie, A. L. Ostericher, M. H. Reineke, A. F. Sasayama and C. P. Kubiak, ACS Catal., 2018, 8, 1313–1324 CrossRef CAS
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS
- J. E. Heimann, W. H. Bernskoetter, N. Hazari and J. M. Mayer, Chem. Sci., 2018, 9, 6629–6638 RSC
- D. W. Cunningham and J. Y. Yang, Chem. Commun., 2020, 56, 12965–12968 RSC
- M. R. Espinosa, M. Z. Ertem, M. Barakat, Q. J. Bruch, A. P. Deziel, M. R. Elsby, F. Hasanayn, N. Hazari, A. J. M. Miller, M. V. Pecoraro, A. M. Smith and N. E. Smith, J. Am. Chem. Soc., 2022, 144, 17939–17954 CrossRef CAS PubMed
- M. R. Elsby, M. R. Espinosa, M. Z. Ertem, A. P. Deziel, N. Hazari, A. J. M. Miller, A. H. Paulus and M. V. Pecoraro, Organometallics, 2023, 42, 3005–3012 CrossRef CAS
- J. E. Heimann, W. H. Bernskoetter and N. Hazari, J. Am. Chem. Soc., 2019, 141, 10520–10529 CrossRef CAS
- J. H. Shin, D. G. Churchill and G. Parkin, J. Organomet. Chem., 2002, 642, 9–15 CrossRef CAS
- A. Anaby, M. Feller, Y. Ben-David, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2016, 138, 9941–9950 CrossRef CAS PubMed
- M. Feller, U. Gellrich, A. Anaby, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6445–6454 CrossRef CAS PubMed
- Q. Zhuo, J. Yang, Z. Mo, X. Zhou, T. Shima, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2022, 144, 6972–6980 CrossRef CAS PubMed
- Y. Jiang, O. Blacque, T. Fox and H. Berke, J. Am. Chem. Soc., 2013, 135, 7751–7760 CrossRef CAS PubMed
- L. Escomel, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2021, 143, 4844–4856 CrossRef CAS PubMed
- N. Queyriaux, N. Durvin, D. Leon, M.-C. Boegli, L. Vendier and A. Simonneau, Eur. J. Inorg. Chem., 2023, e202300426 CrossRef CAS
- E. Carmona-Guzman and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1977, 1716–1721 RSC
- A. P. Borisov, V. D. Makhaev, G. N. Boiko and K. N. Semenenko, Koord. Khim., 1978, 4, 1274 CAS
- V. D. Makhaev, A. P. Borisov, G. N. Boiko and K. N. Semenenko, Koord. Khim., 1982, 8, 963–969 CAS
- T. Ito, A. Takahashi and S. Tamura, Bull. Chem. Soc. Jpn., 1986, 59, 3489–3494 CrossRef CAS
- R. A. Henderson, J. Chem. Soc. Chem. Commun., 1987, 1670–1672 RSC
- C. C. Carter, B. R. Allen, D. V. Bui, D. A. Daley, J. M. Goncalves, C. M. Holinej, S. Lira, D. E. Muniz, A. I. Santiago, N. Sukran, T. R. Cundari and M. Yousufuddin, Inorganica Chim. Acta, 2020, 508, 119638 CrossRef CAS
- S. Kim, Y. Park, J. Kim, T. P. Pabst and P. J. Chirik, Nat. Synth., 2022, 1, 297–303 CrossRef
- S. Burling, G. Kociok-Köhn, M. F. Mahon, M. K. Whittlesey and J. M. J. Williams, Organometallics, 2005, 24, 5868–5878 CrossRef CAS
- M. L. Christ, S. Sabo-Etienne, G. Chung and B. Chaudret, Inorg. Chem., 1994, 33, 5316–5319 CrossRef CAS
- A. Caise, A. E. Crumpton, P. Vasko, J. Hicks, C. McManus, N. H. Rees and S. Aldridge, Angew. Chem., Int. Ed., 2022, 61, e202114926 CrossRef CAS PubMed
- Q. Zhu, J. C. Fettinger and P. P. Power, Dalton Trans., 2021, 50, 12555–12562 RSC
- S. Bontemps, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 1671–1674 CrossRef CAS PubMed
- For examples of CO2 insertion into an Mo–H bond, see:
(a) D. Lyons, G. Wilkinson, M. Thornton-Pett and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1984, 695–700 RSC
- J. K. Burdett, R. Hoffmann and R. C. Fay, Inorg. Chem., 1978, 17, 2553–2568 CrossRef CAS
- J. J. Eisch, J. H. Shah and M. P. Boleslawski, J. Organomet. Chem., 1994, 464, 11–21 CrossRef CAS
- P. K. Pal, S. Chowdhury, M. G. B. Drew and D. Datta, New J. Chem., 2002, 26, 367–371 RSC
- H. B. Vibbert, A. S. Filatov and M. D. Hopkins, Angew. Chem., Int. Ed., 2020, 59, 10581–10586 CrossRef CAS PubMed
- For other examples of C–O cleavage of carbon dioxide with Mo complexes see ref. 19 and:
(a) R. Alvarez, J. L. Atwood, E. Carmona, P. J. Perez, M. L. Poveda and R. D. Rogers, Inorg. Chem., 1991, 30, 1493–1499 CrossRef CAS
- G. Luchini, J. V. Alegre-Requena, I. Funes-Ardoiz and R. S. Paton, F1000Research, 2020, 9, 291 Search PubMed
- For examples of structurally characterized η2-CO2–Mo complexes, see ref. 43c and:
(a) R. Alvarez, E. Carmona, J. M. Marin, M. L. Poveda, E. Gutierrez-Puebla and A. Monge, J. Am. Chem. Soc., 1986, 108, 2286–2294 CrossRef CAS PubMed
- J. Okuda, Z. Für Naturforschung, 1990, 45, 753–754 CrossRef CAS
- W. D. Jones and A. D. Selmeczy, Organometallics, 1992, 11, 889–893 CrossRef CAS
- T. Gandhi, M. Nethaji and B. R. Jagirdar, Inorg. Chem., 2003, 42, 667–669 CrossRef CAS PubMed
- Q. X. Dai, H. Seino and Y. Mizobe, Eur. J. Inorg. Chem., 2011, 2011, 141–149 CrossRef
- E. Lu, Y. Chen and X. Leng, Organometallics, 2011, 30, 5433–5441 CrossRef CAS
- J. Cheng and Z. Hou, Chem. Commun., 2011, 48, 814–816 RSC
- S. Schulz, T. Eisenmann, S. Schmidt, D. Bläser, U. Westphal and R. Boese, Chem. Commun., 2010, 46, 7226–7228 RSC
- Y. Yu, A. R. Sadique, J. M. Smith, T. R. Dugan, R. E. Cowley, W. W. Brennessel, C. J. Flaschenriem, E. Bill, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2008, 130, 6624–6638 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, computational details, NMR and FTIR spectra and crystallographic data. CCDC 2365476–2365478 and 2365557. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04345f |
| This journal is © The Royal Society of Chemistry 2024 |