
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAluminium porphyrins catalyse the hydrogenation of CO2 with H2†
Nitin
Kumar
 a,
Gabriela
Gastelu
b,
Martin
Zábranský
a,
Jaroslav
Kukla
c,
Jorge G.
Uranga
*b and
Martin
Hulla
*a
a,
Gabriela
Gastelu
b,
Martin
Zábranský
a,
Jaroslav
Kukla
c,
Jorge G.
Uranga
*b and
Martin
Hulla
*a
aDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Albertov 6, 128 00 Praha 2, Czech Republic. E-mail: martin.hulla@natur.cuni.cz
bInstituto de Investigaciones en Físico-Química Córdoba Universidad Nacional de Córdoba (INFIQC-CONICET), Córdoba, 5000, Argentina. E-mail: jorge.uranga@unc.edu.ar
cInstitute for Environmental Studies, Faculty of Science, Charles University, Albertov 6, 128 00 Praha 2, Czech Republic
First published on 20th November 2024
Abstract
Boron-based frustrated Lewis pairs (FLPs) have become well-established catalysts for the hydrogenation of a wide range of functional groups. Conversely, aluminium-based FLP hydrogenation catalysts are less common, especially for CO2 reduction. They are mostly confined to the hydrogenation of imines, alkenes, and alkynes even though aluminium is much more abundant than boron and forms structurally related compounds. Moreover, aluminium forms penta- and hexa-coordinated complexes, which remain untested in FLP hydrogenation catalysis. Herein, we demonstrate that cationic, hexa-coordinated diaqua-meso-tetraphenylporphyrin aluminium complexes [Al(TPP)(OH2)2]X and [Al(tBuTPP)(OH2)2]X (X = Cl−, OTf−, ClO4−) form FLPs with nitrogen bases, activate H2, and reductively couple CO2 to amines, yielding N-formylamines and water. Our experimental results and DFT analysis indicate that H2 activation involves the formation of an FLP, base-promoted CO2 reduction and formate salt elimination from the FLP, as proposed for transition metal-catalyzed N-formylations. These similarities in the reaction mechanism and structure of aluminium complexes brings Al-based FLPs closer to transition metal catalysis and may enable us to apply this knowledge to ligand design to enhance main group metal-promoted hydrogenations.
Introduction
Frustrated Lewis pairs (FLPs) are combinations of Lewis acids (LA) and bases (LB) sterically hindered from generating classical LA-LB adducts. FLPs have been used to activate small molecules,1 especially H2,2,3 and to catalyse hydrogenations.4 As a case in point, boron-based FLPs catalytically hydrogenate ketones, esters, imines, enamines, N-heterocyclic compounds, silyl ethers, oxime ethers, alkenes, alkynes, aromatics and to some extent even CO2.5–8 In contrast, functional aluminium-based FLP hydrogenation catalysts remain rare, even though aluminium is chemically related to and, by far, much more abundant than boron.In fact, aluminium is the most abundant metal in the Earth's crust and, like boron, forms predominantly trivalent compounds, which are LAs and, as such, potentially suitable for FLP chemistry. Aluminium-based FLPs are well established9,10 and known to facilitate hydroborations,11–13 to react with ammonia boranes,14 C–H,15,16 C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C,17,18 C
C,17,18 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O19–21 bonds and CO2 (ref. 22–25) and even to activate H2,12,26 in some cases.
O19–21 bonds and CO2 (ref. 22–25) and even to activate H2,12,26 in some cases.
Produced in the same reaction, Al-hydrides are, however, less stable than B-hydrides (c.f. NaBH4 and LiAlH4),27 and Al–C bonds of parent LAs are less thermally and hydrolytically stable than their boron equivalents.28,29 For example, B(C6F5)3 can be used as the LA component of FLP hydrogenation catalysts at temperatures up to 100 °C, even in wet solvents,30,31 while Al(C6F5)3 decomposes at 60 °C, facilitating only the stoichiometric reduction of alkenes.32
Alkenes have been catalytically reduced by incorporating three coordinate aluminium catechol LAs into porous organic polymers (Scheme 1a), but this approach failed to hydrogenate other substrates, such as ketones and imines.33 By then, though, imine hydrogenation had already been catalysed by Al-based FLPs, with di-isobutylaluminium hydride and tri-isobutylaluminium (DIBAL, TIBAL) LAs at 103 bar of H2.34 More recently, this system has also proved effective with MAlH4 (M = Li, Mg, Ca, Sr) at 1 bar of H2 (Scheme 1b).35,36 Under these reaction conditions, aluminium likely binds to the imine substrate, thereby forming an FLP suitable for H2 activation.37,38 Imine hydrogenation has also been catalysed by [(DippBIAN)Al-(H)2–Li(OEt2)2]39 at 50 bar of H2 and 100 °C and by cationic (β-diketiminate)AlMe+ (ref. 40) at 1.5 bar of H2 and 60 °C, but attempts at expanding the scope of hydrogenation activity beyond imines, alkenes and alkynes have not been successful thus far.40 In other words, Al-catalysed hydrogenations continue to fail when applied to other substrates, such as aldehydes, ketones and CO2, and succeed only with catechol-borane when using Al-based FLPs.29,41,42
 | ||
| Scheme 1 (a) Hydrogenation of alkenes and alkynes; (b) hydrogenation of imines by Al-based catalysts; (c) N-formylation of amines with CO2 and H2 catalysed by Al-porphyrin. | ||
In this study, we demonstrate that cationic, hexa-coordinate diaqua Al-porphyrin complexes form FLPs with nitrogen bases, activate H2, and act as CO2 hydrogenation catalysts in the presence of amines, yielding N-formylamines and water (Scheme 1c). Our experimental results and DFT analysis indicate that weaker bases, such as 2,4,6-collidine (pKa of conjugate acid 7.43 in H2O), activate H2 more readily than morpholine (pKa of conjugate acid 8.36 in H2O) or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU pKa of conjugate acid 23.9 in MeCN), which bind to CO2 and the LAs and hinder H2 activation. Furthermore, the N-formylation reaction proceeds as proposed for well-established transition metal catalysts,43–59 suggesting that the hexa-coordinate aluminium complexes behave as transition metals in hydrogenation chemistry. Combined, our findings may stimulate ligand and catalyst design knowledge transfer from the field of transition metal catalysis to FLPs.
Result and discussion
Diaqua-meso-tetraphenylporphyrin aluminium complexes [Al(TPP)(OH2)2]X, where X = Cl−, OTf−, ClO4− and diaqua-meso-tetra(4-tert-butylphenyl)porphyrin aluminium chloride [Al(tBuTPP)(OH2)2]Cl, were prepared as reported in the literature (ESI†).60N-formylation of morpholine with CO2 and H2 was selected as the initial model hydrogenation reaction because CO2 reduction is particularly challenging with FLPs5,7,61 but well established with main group hydrides.62 For the reaction to proceed, these main group hydrides must be formed during FLP-catalysed hydrogenation from H2. The preliminary reaction conditions (1 mmol of morpholine, 10 mol% of [[Al(TPP)(OH2)2]Cl:2,4,6-collidine], 4 bar of CO2, 96 bar of H2, 180 °C, 4 mL of sulfolane) were selected based on the only FLP hydrogenation catalyst [R3SnX:2,4,6-collidine] known for this reaction.63 Under these conditions, the desired product, N-formylmorpholine, was synthesized in 33% yield, a value that is below average as the yields reported for [R3SnX:2,4,6-collidine] FLPs range from 39 to 95%, depending on the R and X groups.63Moreover, [[Al(TPP)(OH2)2]Cl:2,4,6-collidine] and [[Al(TPP)(OH2)2]Cl:morpholine] can be formed in the system (vide infra) and act as FLP in H2 activation and CO2 reduction, as confirmed by the results. At RT, the Lewis bases deprotonate the aqua ligands, but at high temperatures, the same ligands are reversibly displaced (ESI†). Minor adjustments were then made to the reaction conditions to suit the Al-porphyrin system (Scheme 2) before introducing structural modifications to the LA and LB used as the FLP catalyst (Table 1).
 | ||
| Scheme 2 Standard reaction conditions for the study of Al-based FLPs in the N-formylation reaction of morpholine. | ||
| Entry | LA | Added LB | Yield (%) | Errorb (σ) |
|---|---|---|---|---|
| a Reaction conditions: morpholine (1 mmol), sulfolane (4 mL), LA and LB (10 mol%), CO2 (7 bar), H2 (100 bar), 200 °C, 24 h, average yield after three runs. b The error indicated is one standard deviation rounded to a whole number. Yield was determined by 1H NMR with an internal standard. | ||||
| 1 | [Al(TPP)(OH2)2]Cl | 2,4,6-col. | 60 | 4 |
| 2 | [Al(TPP)(OH2)2]OTf | 2,4,6-col. | 44 | 2 |
| 3 | [Al(TPP)(OH2)2]ClO4 | 2,4,6-col. | 24 | 2 |
| 4 | [Al(tBuTPP)(OH2)2]Cl | 2,4,6-col. | 53 | 1 |
| 5 | [Al(Pht)Cl] | 2,4,6-col. | 19 | 1 |
| 6 | [Al(TPP)(OH2)2]Cl | — | 26 | 2 |
| 7 | [Al(TPP)(OH2)2]Cl | DBU | 30 | 5 |
| 8 | [Al(TPP)(OH2)2]Cl | NMM | 15 | 4 |
| 9 | [Al(TPP)(OH2)2]Cl | 2,6-Lut | 20 | 6 |
| 10 | TPP | 2,4,6-col. | 4 | — |
| 11 | — | 2,4,6-col. | 4 | — |
| 12 | [Al(TPP)(OH)] | 2,4,6-col. | 4 | — |
| 13 | [Al(TPP)(OH)] + [morpholineH]Cl | 2,4,6-col. | 4 | — |
Under these modified conditions and with [Al(TPP)(OH2)2]Cl, N-formylmorpholine was formed in 60% yield (Table 1, entry 1). Substituting the counter anion Cl− for OTf− or ClO4− decreased the yield to 44 and 24%, respectively (Table 1, entries 2 and 3). By contrast, the ions OTf− and ClO4− interact less in the tetra-substituted R3SnX (X = Cl−, OTf−, ClO4−), accelerating the catalytic turnovers.63 During catalysis, the counter anion likely interacts with the cationic complex [Al(TPP)(OH2)2]+ and promotes the reaction, as shown by adding tetrabutylammonium chloride (10 mol%) to [Al(TPP)(OH2)2]OTf, which increases the reaction yield from 44 to 50% (ESI†).
In addition, the axial ligand of the porphyrin complex must be labile because the stable [Al(TPP)(OH)] complex is inactive (Table 1, entry 12). Together with morpholinium or collidinium chloride, [Al(TPP)(OH)] may be a reaction intermediate (vide infra). But adding morpholinium chloride to the N-formylation reaction with [Al(TPP)(OH)] and 2,4,6-collidine does not promote formamide formation (Table 1, entry 13). Therefore, [Al(TPP)(OH)] should not be formed during the reaction.
Substituting the meso-tetraphenylporphyrin ligand for meso-tetra(4-tert-butylphenyl)porphyrin [Al(tBuTPP)(OH2)2]Cl slightly decreased the catalytic activity, leading to 53% N-formyl morpholine yield (Table 1, entry 4). The electron-donating tert-butyl group on the porphyrin ligand likely diminishes the Lewis acidity of the complex, decreasing the activity. The commercial phthalocyanine complex [Al(Pht)Cl] further decreased the yield of N-formylmorpholine to only 19% (Table 1, entry 5).
Considering that porphyrins and phthalocyanines are planar, steric hindrance should not account for the decrease in reactivity. This difference more likely derives from electronic effects associated with changes in the LA of the metal centre and, above all, from the cationic charge of [Al(TPP)(OH2)2]+, which enthalpically promotes the reaction (vide infra).
FLP catalysts can also be electronically controlled by LB, so we assessed the LB effect on the reaction. Two possible FLPs can be formed in the N-formylation of morpholine with [Al(TPP)(OH2)2]X and 2,4,6-collidine, namely [[Al(TPP)(OH2)]Cl:2,4,6-collidine] and [[Al(TPP)(OH2)]Cl:morpholine], since the reaction substrate is also a LB. Moreover, morpholine is a stronger base (pKa 8.36) than 2,4,6-collidine (pKa 7.43) and, accordingly, should more efficiently activate H2 in combination with LA. Yet, when 2,4,6-collidine was removed from the reaction mixture, the product yield decreased from 60 to 26% (Table 1, entries 1 and 6), indicating that 2,4,6-collidine is crucial for the reaction.
Morpholine may react with CO2, forming a carbamic acid, which removes the base from the reaction, thereby preventing FLP formation.63 This hypothesis was partly supported by the lack of changes in yield when using stronger bases, such as DBU (pKa 23.9 in MeCN). DBU also reacts with CO2, forming carbamates and the desired product in 30% yield, which was as high as that obtained when running the reaction without DBU (Table 1, entry 7). In turn, N-methylmorpholine (NMM, pKa 7.38) and 2,6-lutidine (pKa 6.6) decreased the yield to 15 and 20%, respectively (Table 1, entries 8 and 9). Although aliphatic NMM and 2,4,6-collidine have similar basicities (pKa of conjugate acid in H2O of 7.38 and 7.43, respectively), NMM is presumably not sterically hindered enough for FLP formation. By contrast, 2,6-lutidine has the same steric profile as 2,4,6-collidine but is too weak of a LB.
When optimizing the reaction conditions, we observed that the reaction only proceeded at temperatures above 160 °C, suggesting a high energy barrier (Fig. 1A). In line with the ease of CO2 reduction by main group hydrides,62,63 the reaction readily proceeded at low CO2 partial pressures between 2 and 10 bar (Fig. 1B). Conversely, transition metal-catalysed reactions usually require a high CO2 partial pressure, typically between 25 and 30 bar.43–59 Nevertheless, high partial pressures of H2 are required to improve the N-formylmorpholine yield from 4% at 20 bar to 60% at 100 bar (Fig. 1C), further indicating that H2 activation or its binding is, most likely, limiting the reaction.
 | ||
| Fig. 1 Effect of H2 (C) and CO2 (B) partial pressure and temperature (A) on the N-formylation reaction of morpholine with CO2, H2 and [Al(TPP)(OH2)2]Cl catalyst. All yields were determined by 1H NMR with an internal standard as the averages of three runs, and with error bars representing one standard deviation. | ||
Despite extensive efforts to optimize the conditions, the reaction yields did not surpass 60% (Fig. 1). Neither prolonging the reaction nor adding more catalyst, whether [Al(TPP)(OH2)2]Cl or 2,4,6-collidine, increased the yield. Thus, reaction equilibria, not insufficient reaction time or catalyst instability, accounted for the maximum reaction yield.
Knowing that the reaction is thermodynamically favourable, we studied its mechanism in detail using both experimental approaches and DFT calculations (Fig. 2). Numerous reaction pathways have been hypothesized for N-formylations catalysed by FLPs, which differ in CO2 reduction and N-formylation steps.63 The first, main group-like hydride reduction of CO2, presumably involves CO2 insertion into the Al–H bond and [Al(TPP)(OCHO)] and [baseH]Cl formation followed by a reaction with morpholine, yielding the desired N-formylmorpholine, [Al(TPP)(OH)] and [baseH]Cl. The catalyst is regenerated by water elimination from [Al(TPP)(OH)] through protonation with [baseH]Cl. Nevertheless, [Al(TPP)(OH)] is catalytically inactive in the presence of an equimolar quantity of [baseH]Cl (Table 1, entry 13). Even with a 10-fold excess morpholinium chloride, required for [Al(TPP)(OH)] protonation, the reaction is slower than with [Al(TPP)(OH2)2]Cl, so [Al(TPP)(OH)] formation is unlikely to significantly contribute to the reaction. Similarly, the second reaction pathway via direct carbamate reduction leads to inactive [Al(TPP)(OH)]. Both reaction pathways contradict the experimental results and are, therefore, unlikely to be the working catalytic cycles.
 | ||
| Fig. 2 DFT study of the reaction mechanism of N-formylation of morpholine with CO2 and H2 catalyzed by Al-based FLPs at the M06-2X/6-311++G** level of theory, using IEFPCM model for solvation corrections; the reaction pathway with [Al(TPP)Cl:morpholine] FLP is indicated in blue (left-hand side), the one with [Al(TPP)Cl:2,4,6-collidine] FLP is indicated in orange and the one with [[Al(TPP)(OH2)]Cl:2,4,6-collidine] FLP is indicated in green (right-hand side). Relative enthalpies are indicated in kcal mol−1, and E0 represents the sum of the enthalpies of the initial compounds without any interaction between them. | ||
The third catalytic cycle includes H2 splitting, subsequent CO2 reduction to formate, formate release and regeneration of the initial FLP, which closes the catalytic cycle. To gain further insights into the third reaction mechanism, we performed solvation-corrected DFT calculations at the M06-2X/6-311++G** level of theory so as to describe the enthalpy of the reaction coordinate at 473 K and 100 bar (ESI†). At first, the effect of 2,4,6-collidine on the reaction was studied using [Al(TPP)Cl:morpholine] i.e., without 2,4,6-collidine (Fig. 2, blue pathway), and with [Al(TPP)Cl:2,4,6-collidine] FLP when this base was present (Fig. 2, orange pathway). The effect of the cationic complex [Al(TPP)(OH2)2]Cl, replacing the neutral [Al(TPP)Cl], was then assessed on the more favourable pathway in the presence of 2,4,6-collidine (Fig. 2, green pathway).
In the presence of a LB, morpholine (Ba) or 2,4,6-collidine (Bb), [Al(TPP)Cl] immediately completes its coordination sphere, forming a hexa-coordinate octahedral complex (Fig. 3, Ra and Rb), as observed in transition metal catalysts during the N-formylation reaction.64 In the presence of morpholine, this complex is stabilized by −24.8 kcal mol−1 (Fig. 2, Ra), with a bond length between morpholine and the aluminium centre of 2.17 Å and an almost straight angle of 177.3° between TPP and the aluminium centre (Fig. 3, Ra). Similar energetics are found for morpholinium carbamate binding (−25.4 kcal mol−1), suggesting reversible CO2 insertion into the Al-morpholine bond (ESI†). In contrast, the octahedral complex with 2,4,6-collidine is stabilized by only −14.3 kcal mol−1 (Fig. 2, Rb). Since Rb can be converted into Ra, Ra acts as an off-cycle intermediate for the catalytic cycle with 2,4,6-collidine. In Rb, the distance between 2,4,6-collidine and aluminium is 3.35 Å, and the distortion from octahedral geometry is larger, with an angle of 161.0° between TPP and aluminium (Fig. 3, Rb). As previously proposed,65,66 the distance between 3 and 5 Å is optimal for H2 activation by FLP and, accordingly, favoured by Rb.
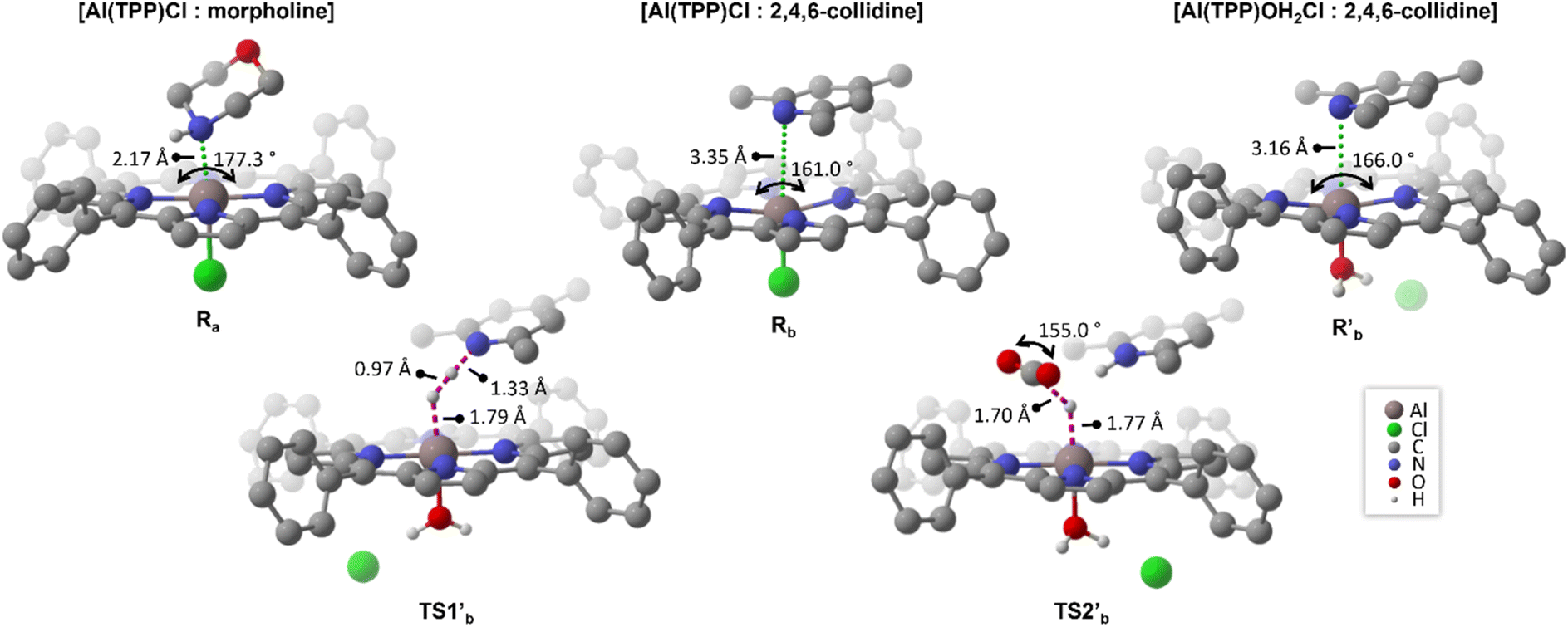 | ||
| Fig. 3 Optimized geometries of [Al(TPP)Cl:morpholine] (Ra), [Al(TPP)Cl:2,4,6-collidine] (Rb) and [[Al(TPP)(OH2)]Cl:2,4,6-collidine] (R′b) (top), and the corresponding transition states for H2 splitting and CO2 hydrogenation for [[Al(TPP)(OH2)]Cl:2,4,6-collidine] catalysts (TS1′b and TS2′b) (bottom). C–H hydrogens are omitted for a better visualization, at the M06-2X/6-311++G** level of theory, using IEFPCM model for solvation corrections. | ||
H2 activation proceeds in a heterolytic fashion.66,67 H2 activation from Ra has a higher energy transition state (Fig. 2, TS1a) than H2 activation from Rb (Fig. 2, TS1b). The local activation barrier for direct H2 activation from Ra is 31.7 kcal mol−1, and only 20.3 kcal mol−1 from Rb. Moreover, considering the equilibrium between Ra and Rb, the energy barrier for H2 activation from Ra is favoured by its transformation to Rb, followed by H2 activation viaTS1b, with the energy barrier (30.8 kcal mol−1) mildly favouring this process. The Al-hydrides, I1a and I1b, are formed in endothermic reactions with relative enthalpies of 5.5 and 2.9 kcal mol−1, respectively. Based on these positive energies, their formation is enthalpically unfavourable, so they are likely rapidly protonated by [baseH]+ or by the ever present carbamic acid, which is produced in the reaction of morpholine with CO2.
Al-hydride protonation is a standard decomposition pathway of such compounds.27 Hydrides of comparably planar calix[4]pyrrole aluminium complexes exist only in an unfavourable equilibrium and are rapidly displaced by even weakly basic ligands, such as THF.27,68 An unfavourable pre-equilibrium between H2 and Al-hydrides, I1a and I1b, is thus expected, where I1b is slightly favoured over I1a and should persist longer or in larger quantities in the reaction. Together with the high local activation energy for H2 activation, the unfavourable pre-equilibrium is likely responsible for the high H2 partial pressure(s) required for the reaction. Experimentally, dihydrogen activation is confirmed when using D2, which results in complete deuteration of the formamide moiety of N-formylmorpholine. Thus, dihydrogen is the sole source of reducing agent of the reaction (ESI†).
The high reactivity of Al-hydrides produced in the reaction is also clearly shown by the low local activation barrier for CO2 reduction (Fig. 2, TS2a and TS2b). Although most Al-hydrides formed in this reaction are likely protonated back to H2 in the reverse reaction, this reaction can proceed thanks to the comparably low local energy barriers of CO2 reduction (6.5 and 4.3 kcal mol−1, respectively). Attempts at isolating [Al(H)(TPP)] failed, but adding CO2 to the solution led to its immediate reduction to formate, as detected by 1H NMR (ESI†).
Based on the enthalpic energetic span of the catalytic cycle, the transition states TS2a and TS2b for CO2 reduction are also the turnover frequency (TOF)-determining transition states (TDTS), with a global energy barrier of 36.8 kcal mol−1 starting from the TOF-determining intermediate (TDI) RaviaTS1a and I1a. For the 2,4,6-collidine pathway (orange), the global energy barrier is only 21.5 kcal mol−1 between Rb and TS2bviaTS1b and I1b. Alternatively, the energy span from Ra to TS2bviaRb, TS1b and I1b is 32.0 kcal mol−1 considering the equilibrium between Rb and Ra and the ability of Ra to act as an off-cycle thermodynamic well for the 2,4,6-collidine pathway. In effect, the reaction with 2,4,6-collidine is favoured in all cases, in line with the experimental results (Table 1, entries 1 and 6).
After TS2a and TS2b, stable, intermediate complexes between [Al(TPP)Cl], formic acid and morpholine (I2a) or 2,4,6-collidine (I2b) are formed. With morpholine, I2a has a stabilization energy similar to that of Ra (−23.2 vs. −24.8 kcal mol−1, respectively), potentially allowing the reverse reaction. The reverse reaction, HCOOH decomposition to CO2 and H2, is also well established in the literature69 and was confirmed here by evolution of H2 gas, characterized by 1H NMR, from a solution of formic acid and [Al(TPP)(OH2)2]Cl (ESI†). Considering these results, a thermodynamic equilibrium may be established in the reaction, thereby limiting the reaction yield.
Thermodynamically, the reaction is driven by [morpholinium][formate] elimination from either I2a or I2b, which spontaneously condenses into N-formylmorpholine and water under these reaction conditions.47,64 This condensation also regenerates the FLP catalyst and allows the reaction to proceed. Formate salts are eliminated in transition metal-catalysed reactions,47,64 whereas the formate remains attached to the central element70–72 in reactions of main group hydrides, including LiAlH4,73 so the [Al(TPP)Cl] catalyst behaves like a transition metal rather than a main group reductant. In the complex I2, the protonated base continuously interacts with the LA and the generated formate. Therefore, another base, such as morpholine, is required for the reaction to proceed (Fig. 2, TS3a and b) prior to catalyst regeneration.
If the reaction is limited by an equilibrium established between R(a or b) and I2(a or b) and driven by morpholine, higher concentrations of morpholine should promote the reaction. Indeed, increasing the quantity of morpholine increased the turnover number (TON) in the 24 hours reaction (Fig. 4). More specifically, increasing the substrate concentration from 1 to 22.5 mmol increased the TON from 7 to 80. This increase confirmed that the 60% yield obtained with 1 mmol of morpholine (Table 1, entry 1) is limited by the quantity of the substrate, not by the efficiency of the catalyst.
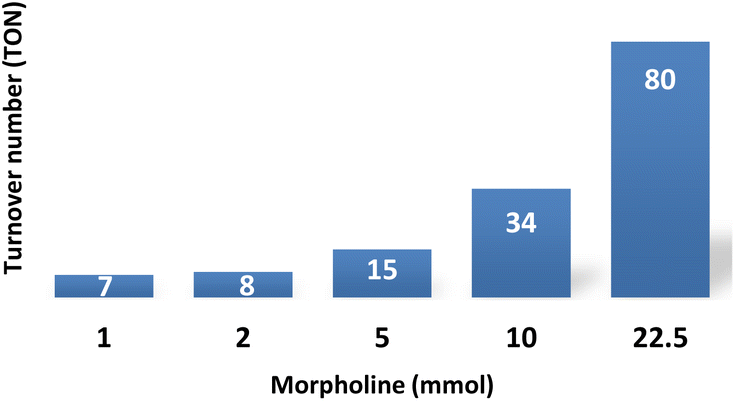 | ||
| Fig. 4 Morpholine effect on the TON of the catalytic system; TON calculated based on the quantity of [Al(TPP)(OH2)2]Cl under the following reaction conditions: morpholine (1 to 22.5 mmol), sulfolane (4 mL), [[Al(TPP)(OH2)]Cl:2,4,6-collidine] (0.1 mmol), CO2 (7 bar), H2 (100 bar), 200 °C, 24 h. The reaction yield was determined by 1H NMR with an internal standard. | ||
In the catalytic cycle with the [Al(TPP)(OH2)2]Cl pre-catalyst (Fig. 5), dissociation of one aqua ligand leads to the [[Al(TPP)(OH2)]Cl:2,4,6-collidine] FLP (R′b), whose geometry is similar to, but more stable than, that of [Al(TPP)Cl:2,4,6-collidine] (Rb) (Fig. 3). Nevertheless, the subsequent transition states, TS1′b and TS2′b (Fig. 3), are more stabilized (Fig. 2, TS1′b and TS2′b), lowering the overall activation barrier of the catalytic cycle. The H2 splitting step proceeds via a late transition state TS1′b, wherein the H–H bond is 0.2 Å more stretched than in H2. Despite having the largest local activation barrier (16.9 kcal mol−1), this transition viaTS1′b is still much more energetically favourable than the transition from the neutral complex Rb to TS1b (20.3 kcal mol−1). In comparison with the neutral TS2b, TS2′b stabilization decreases the energetic span of the catalytic cycle from 21.5 (Rb → TS2b) to 17.5 (R′b → TS2′b) kcal mol−1. When including potential off-cycle intermediates, Ra and R′a, where morpholine binds to the catalyst, the energy span decreases from 32.0 (Ra → Rb → TS2b) to 29.4 (R′a → R′b → TS2′b) kcal mol−1 (ESI†). This difference supports our hypothesis that the cationic aqua complexes of aluminium and the associated FLP with collidine [[Al(TPP)(OH2)]Cl:2,4,6-collidine] is the most favourable catalyst in the system.
 | ||
| Fig. 5 View of the molecular structure of [Al(TPP)(OH2)2]Cl as observed in the crystal structure of [Al(TPP)(OH2)2]Cl·CH2Cl2·2 Et2O. C–H hydrogen atoms were omitted for clarity. Thermal displacement ellipsoids are plotted at the 30% probability level. Applied colours: C – black, H – black contour, Cl – green, N – blue, O – red, Al – yellow. | ||
While the DFT calculations fit the experimental data, the pathways involve, at most, tri-molecular transition states and the formation of an Al-hydride. [Al(TPP)(OH2)]Cl is a hard LA, whose interactions with the relatively soft hydride donor are potentially less favourable than with the considerably harder CO2. Alternative pathway(s) may involve H2 activation by a tetra-molecular transition state involving CO2 interactions with the Al-catalyst and H2 activation by the C-atom of CO2 and the LB, resulting in the direct CO2 reduction to formate. Such a possibility is currently under investigation in our laboratory.
After studying the catalytic system, we assessed the substrate effect on the N-formylation of amines with CO2 and H2 (Scheme 3). In the N-formylation reaction, all secondary amines tested yielded the corresponding N-formylated products between 40 and 60% yield, in line with the expected reaction equilibrium. The alcohol, tertiary amine and amide functional groups were well tolerated. Diisopropyl amine was almost unreactive in the only other FLP system reported thus far, namely [R3SnX:2,4,6-collidine].63 But in our system, due to its low steric hindrance around the LA centre, diisopropyl amine provided an average yield of 54%. Primary aliphatic amines were also N-formylated for the first time with FLP catalysts, albeit in yields ≤20%. Aromatic and benzylic amines were unreactive.
 | ||
| Scheme 3 Substrate scope (a) N-formylation of amines with CO2 and H2. Reaction conditions: amine (1 mmol), sulfolane (4 mL), [[Al(TPP)(OH2)]Cl:2,4,6-collidine] (10 mol%), CO2 (7 bars), H2 (100 bars), 200 °C, 24 h. All yields were determined by 1H NMR, and the structures were confirmed by GC-MS. | ||
Conclusion
Hexa-coordinated complexes of aluminium incorporating a labile ligand such as water can be used as LAs in FLP chemistry, H2 activation, and hydrogenation chemistry. Catalytic hydrogenation of CO2 in the presence of amines yields formamides with [Al(TTP)(OH2)2]Cl as the Lewis acid and the sterically demanding 2,4,6-collidine as the Lewis base. Energetically, the reaction is favoured when using the cationic complex [Al(TTP)(OH2)2]Cl over the neutral [Al(TPP)Cl], which is also unstable and hydrolyses to the cationic form. The advantages of this FLP system are the absence of labile and hydrolytically unstable Al–C bonds, which allows hydrogenation reactions that produce water. Structural and reactivity similarities of the LAs to transition metal complexes may facilitate knowledge transfer from transition metal catalysis to FLP in ligand and catalyst design. Ligand modifications may enable us to tailor Lewis acidity and steric hindrance around the metal centre to improve the reactivity of FLPs and to tune their reactivity to reaction requirements.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Czech Science foundation (GAČR 21-27431M) for funding this study, the National Scientific and Technical Research Council (Consejo Nacional de Investigaciones Científicas y Técnicas – CONICET) for Gabriela Gastelu's fellowship and Charles University Research Centre program No. UNCE/24/SCI/010 for supporting Martin Zábranský. We also thank Carlos V. Melo for editing the manuscript and Alexandros Paparakis for preparing 4-piperidinemethanol, which was used in the substrate scope. This research relied on computational resources of the High-Performance Computing Center (Centro de Computación de Alto Desempeño – CCAD) of the National University of Córdoba (Universidad Nacional de Cordoba – UNC) (https://ccad.unc.edu.ar/), part of the National System of High-Performance Computing (Sistema Nacional de Computación de Alto Desempeño – SNCAD) of the Ministry of Science, Technology and Innovation (Ministerio de Ciencia, Tecnología e Innovación – MinCyT), Argentina.References
- D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625–2641 RSC.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2021, 143, 20002–20014 CrossRef CAS PubMed.
- J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592–3612 RSC.
- A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839–9843 CrossRef CAS PubMed.
- M. A. Courtemanche, A. P. Pulis, É. Rochette, M. A. Légaré, D. W. Stephan and F. G. Fontaine, Chem. Commun., 2015, 51, 9797–9800 RSC.
- T. Wang, M. Xu, A. R. Jupp, Z. W. Qu, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2021, 60, 25771–25775 CrossRef CAS PubMed.
- J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592–3612 RSC.
- M. J. Ingleson, Annu. Rep. Prog. Chem., Sect. A. Inorg. and Phys. Chem., 2013, 109, 28–52 RSC.
- S. A. Weicker and D. W. Stephan, Bull. Chem. Soc. Jpn., 2015, 88, 1003–1016 CrossRef CAS.
- D. M. C. Ould, J. L. Carden, R. Page and R. L. Melen, Inorg. Chem., 2020, 59, 14891–14898 CrossRef CAS PubMed.
- G. Ménard, L. Tran and D. W. Stephan, Dalton Trans., 2013, 42, 13685–13691 RSC.
- N. Sarkar, R. Kumar Sahoo and S. Nembenna, Chem.–Eur. J., 2023, 29, e202203023 CrossRef CAS PubMed.
- C. Appelt, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2013, 52, 4256–4259 CrossRef CAS PubMed.
- G. Ménard, J. A. Hatnean, H. J. Cowley, A. J. Lough, J. M. Rawson and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 6446–6449 CrossRef PubMed.
- G. Ménard and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 4409–4412 CrossRef PubMed.
- Y. Chen, W. Jiang, B. Li, G. Fu, S. Chen and H. Zhu, Dalton Trans., 2019, 48, 9152–9160 RSC.
- C. Appelt, H. Westenberg, F. Bertini, A. W. Ehlers, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2011, 50, 3925–3928 CrossRef CAS.
- M. Lange, J. C. Tendyck, P. Wegener, A. Hepp, E.-U. Würthwein and W. Uhl, Chem.–Eur. J., 2018, 24, 12856–12868 CrossRef CAS.
- D. Pleschka, M. Layh, F. Rogel and W. Uhl, Philos. Trans. R. Soc., A, 2017, 375, 20170011 CrossRef PubMed.
- W. Uhl, P. Wegener, M. Layh, A. Hepp and E. U. Würthwein, Z. Naturforsch., B: Chem. Sci., 2016, 71, 1043–1050 CrossRef CAS.
- G. Ménard, T. M. Gilbert, J. A. Hatnean, A. Kraft, I. Krossing and D. W. Stephan, Organometallics, 2013, 32, 4416–4422 CrossRef.
- P. Federmann, T. Bosse, S. Wolff, B. Cula, C. Herwig and C. Limberg, Chem. Commun., 2022, 58, 13451–13454 RSC.
- P. Federmann, R. Müller, F. Beckmann, C. Lau, B. Cula, M. Kaupp and C. Limberg, Chem.–Eur. J., 2022, 28, e202200404 CrossRef CAS.
- S. Roters, C. Appelt, H. Westenberg, A. Hepp, J. C. Slootweg, K. Lammertsma and W. Uhl, Dalton Trans., 2012, 41, 9033–9045 RSC.
- S. Styra, M. Radius, E. Moos, A. Bihlmeier and F. Breher, Chem.–Eur. J., 2016, 22, 9508–9512 CrossRef CAS PubMed.
- M. M. D. Roy, A. A. Omaña, A. S. S. Wilson, M. S. Hill, S. Aldrich and E. Rivard, Chem. Soc. Rev., 2021, 121, 12784–12965 CrossRef CAS.
- N. A. Shcherbina, A. V. Pomogaeva, A. S. Lisovenko, I. V. Kazakov, N. Yu. Gugin, O. V. Khoroshilova, Y. V. Kondrat'ev and A. Y. Timoshkin, Z. Anorg. Allg. Chem., 2020, 646, 873–881 CrossRef CAS.
- M. A. Courtemanche, J. Larouche, M. A. Légaré, W. Bi, L. Maron and F. G. Fontaine, Organometallics, 2013, 32, 6804–6811 CrossRef CAS.
- D. J. Scott, T. R. Simmons, E. J. Lawrence, G. G. Wildgoose, M. J. Fuchter and A. E. Ashley, ACS Catal., 2015, 5, 5540–5544 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed.
- G. Ménard and D. W. Stephan, Angew. Chem., 2012, 124, 8397–8400 CrossRef.
- J. Camacho-Bunquin, M. Ferrandon, U. Das, F. Dogan, C. Liu, C. Larsen, A. E. Platero-Prats, L. A. Curtiss, A. S. Hock, J. T. Miller, S. T. Nguyen, C. L. Marshall, M. Delferro and P. C. Stair, ACS Catal., 2017, 7, 689–694 CrossRef CAS.
- J. A. Hatnean, J. W. Thomson, P. A. Chase and D. W. Stephan, Chem. Commun., 2014, 50, 301–303 RSC.
- H. Elsen, C. Färber, G. Ballmann and S. Harder, Angew. Chem., Int. Ed., 2018, 57, 7156–7160 CrossRef CAS.
- H. Elsen, J. Langer, M. Wiesinger and S. Harder, Organometallics, 2020, 39, 4238–4246 CrossRef CAS.
- H. Elsen, J. Langer, G. Ballmann, M. Wiesinger and S. Harder, Chem.–Eur. J., 2021, 27, 401–411 CrossRef CAS PubMed.
- Z. Qu, H. Zhu and S. Grimme, ChemCatChem, 2021, 13, 3401–3404 CrossRef CAS.
- J. Pölker, D. Schaarschmidt, J. Bernauer, M. Villa and A. Jacobi von Wangelin, ChemCatChem, 2022, 14, e202200144 CrossRef PubMed.
- A. Friedrich, J. Eyselein, H. Elsen, J. Langer, J. Pahl, M. Wiesinger and S. Harder, Chem.–Eur. J., 2021, 27, 7756–7763 CrossRef CAS PubMed.
- F. Krämer, Angew. Chem., Int. Ed., 2024, 63, e202405207 CrossRef PubMed.
- F. Krämer, J. Paradies, I. Fernández and F. Breher, Chem.–Eur. J., 2024, 30, e202303380 CrossRef PubMed.
- P. Ju, J. Chen, A. Chen, L. Chen and Y. Yu, ACS Sustain. Chem. Eng., 2017, 5, 2516–2528 CrossRef CAS.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 Search PubMed.
- C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef CAS PubMed.
- C. Ziebart, C. Federsel, P. Anbarasan, R. Jackstell, W. Baumann, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2012, 134, 20701–20704 CrossRef CAS PubMed.
- P. Daw, S. Chakraborty, G. Leitus, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2017, 7, 2500–2504 CrossRef CAS.
- Z. Liu, Z. Yang, Z. Ke, X. Yu, H. Zhang, B. Yu, Y. Zhao and Z. Liu, New J. Chem., 2018, 42, 13933–13937 Search PubMed.
- Z. He, H. Liu, H. Liu, Q. Qian, Q. Meng, Q. Mei and B. Han, ChemCatChem, 2017, 9, 1947–1952 Search PubMed.
- H. Liu, Q. Mei, Q. Xu, J. Song, H. Liu and B. Han, Green Chem., 2017, 19, 196–201 RSC.
- M. A. Affan and P. G. Jessop, Inorg. Chem., 2017, 56, 7301–7305 Search PubMed.
- L. Zhang, Z. Han, X. Zhao, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2015, 54, 6186–6189 CrossRef CAS PubMed.
- K. Zhang, L. Zong and X. Jia, Adv. Synth. Catal., 2021, 363, 1335–1340 CrossRef CAS.
- A. A. Dabbawala, N. Sudheesh and H. C. Bajaj, Indian J. Chem., Sect. A:Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2015, 54A, 753–756 Search PubMed.
- K. Kudo, H. Phala, N. Sugita and Y. Takezaki, Chem. Lett., 1977, 6, 1495–1496 CrossRef.
- Y. Zhang, J. Wang, H. Zhu and T. Tu, Chem.–Asian J., 2018, 13, 3018–3021 CrossRef CAS PubMed.
- S. Schreiner, J. Y. Yu and L. Vaska, J. Chem. Soc., Chem. Commun., 1988, 602–603 RSC.
- S. Schreiner, J. Y. Yu and L. Vaska, Inorg. Chim. Acta, 1988, 147, 139–141 Search PubMed.
- M. Minato, D. Y. Zhou, K. I. Sumiura, R. Hirabayashi, Y. Yamaguchi and T. Ito, Chem. Commun., 2001, 1, 2654–2655 Search PubMed.
- Y. Qin, H. Guo, X. Sheng, X. Wang and F. Wang, Green Chem., 2015, 17, 2853–2858 RSC.
- M. A. Courtemanche, A. P. Pulis, É. Rochette, M. A. Légaré, D. W. Stephan and F. G. Fontaine, Chem. Commun., 2015, 51, 9797–9800 Search PubMed.
- M. Hulla and P. J. Dyson, Angew. Chem., Int. Ed., 2020, 59, 1002–1017 CrossRef CAS PubMed.
- A. Paparakis, R. C. Turnell-Ritson, J. S. Sapsford, A. E. Ashley and M. Hulla, Catal. Sci. Technol., 2023, 13, 637–644 Search PubMed.
- U. Jayarathne, N. Hazari and W. H. Bernskoetter, ACS Catal., 2018, 8, 1338–1345 CrossRef CAS.
- L. L. Zeonjuk, N. Vankova, A. Mavrandonakis, T. Heine, G. Röschenthaler and J. Eicher, Chem.–Eur. J., 2013, 19, 17413–17424 CrossRef CAS PubMed.
- L. Liu, B. Lukose, P. Jaque and B. Ensing, Green Energy Environ., 2019, 4, 20–28 CrossRef.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- F. Ebner, H. Wadepohl and L. Greb, J. Am. Chem. Soc., 2019, 141, 18009–18012 CrossRef CAS PubMed.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS.
- S. Ishida, T. Hatakeyama, T. Nomura, M. Matsumoto, K. Yoshimura, S. Kyushin and T. Iwamoto, Chem.–Eur. J., 2020, 26, 15811–15815 CrossRef CAS PubMed.
- M. Hulla, G. Laurenczy and P. J. Dyson, ACS Catal., 2018, 8, 10619–10630 CrossRef CAS.
- G. Gastelu, D. Savary, M. Hulla, D. Ortiz, J. G. Uranga and P. J. Dyson, ACS Catal., 2023, 13, 2403–2409 CrossRef CAS.
- A. E. Finholt and E. C. Jacobson, J. Am. Chem. Soc., 1952, 74, 3943–3944 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2357273, 2357274 and 2386191. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03665d |
| This journal is © The Royal Society of Chemistry 2024 |