
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHost–guest binding between cucurbit[8]uril and amphiphilic peptides achieved tunable supramolecular aggregates for cancer diagnosis†
Jie
Niu
a,
Jie
Yu
a,
Xuan
Wu
c,
Ying-Ming
Zhang
*a,
Yong
Chen
a,
Zhilin
Yu
 b and
Yu
Liu
*a
b and
Yu
Liu
*a
aCollege of Chemistry, State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: yuliu@nankai.edu.cn; ymzhang@nankai.edu.cn
bKey Laboratory of Functional Polymer Materials, Ministry of Education, State Key Laboratory of Medicinal Chemical Biology, Institute of Polymer Chemistry, College of Chemistry, Nankai University, Tianjin 300071, P. R. China
cSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, Jiangsu 225002, P. R. China
First published on 5th August 2024
Abstract
The manipulation of biocompatible supramolecular nanostructures at subcellular and cellular levels has become one of the increasingly significant topics but remains a formidable challenge in chemical and biological science. In this work, a controllable supramolecular aggregate based on host–guest competitive binding is elaborately constructed using cucurbit[8]uril, methionine-containing amphiphilic peptide, and perylene diimide, displaying in situ oxidation-driven macrocycle-confined fluorescence enhancement for cell imaging and morphological reconstruction for cancer cell death. The experimental results demonstrate that cucurbit[8]uril possesses a high binding affinity with the methionine peptide, while this value sharply decreases after the methionine residue is oxidized to sulfoxide or sulfone. Therefore, perylene diimide can be competitively included by cucurbit[8]uril in the co-assemblies, eventually resulting in a 10-fold fluorescence enhancement and the conversion of topological morphology from nano-sized particles to micron-sized sheets. Moreover, the obtained ternary assemblies can be oxidized by endogenous reactive oxygen species in cancer cells, thus not only providing enhanced fluorescence for cell imaging, but also leading to endoplasmic reticulum dysfunction and significant cell death. Therefore, the controllable and oxidation-responsive morphological transformation based on the host–guest competitive binding in biological media can be viewed as a feasible means for efficient disease theragnosis.
Introduction
In recent decades, the construction of in situ supramolecular assemblies at the subcellular and cellular levels has become one of the hot topics in chemical science and biological materials.1 These biocompatible self-assemblies have been extensively investigated not only for their physiological functions in the regulation of some key metabolic processes, but also for their wide application in drug delivery, biological imaging, and disease diagnosis and treatment. Among the commonly used supramolecular systems, the macrocycle-based ones have stimulated an upsurge of interest.2 In particular, due to their unique structures with rigid skeletons and adjustable cavity sizes, cucurbit[n]urils (CBs, n refers to the number of glycoluril units)3 can encapsulate with many neutral molecules and positively charged molecules with high affinity mainly through hydrophobic and ion–dipole interactions. These molecular binding features can endow CB-involved nanoconstructs with elaborate biological functions in bioimaging,4 targeted drug delivery,5 and regulation of biochemical processes.6 For example, Park and Kim synthesized cyanine 3-modified CB[7] as the host and adamantylamine-conjugated cyanine 5 as the guest. Benefitting from the extremely high host–guest binding affinity, efficient energy transfer from cyanine 3 to cyanine 5 could be realized in living cells, which could be used to indicate the dynamic fusion processes of lysosomes and mitochondria.7 In addition, Wang and co-workers reported the in situ multivalent molecular binding behaviors between CB[7]-grafted hyaluronic acid and adamantine-conjugated triphenylphosphine, thus achieving mitochondrial aggregation and fusion for alleviation of the chemical-induced stress in cells and zebrafish.8 More recently, Shen and Liu also reported an in situ protonation-activated supramolecular self-assembly system in lysosomes based on the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio strong binding between CB[8] and pyridyl-functionalized tetraphenylethylene, which could self-assemble as cubic structures to destruct the lysosome membrane for selectively suppressing cancer cell proliferation.9 Inspired by these fascinating results, it can be anticipated that CBs as host molecules hold great potential in the creation of multivalent cascade supramolecular assemblies and biomaterials. However, to the best of our knowledge, tunable complexation with stimuli-responsive biomolecules and the full utilization of host–guest competitive interaction to simultaneously realize photoluminescence enhancement and induce cancer cell death are still rarely reported in CB-based supramolecular systems.
:1 stoichiometric ratio strong binding between CB[8] and pyridyl-functionalized tetraphenylethylene, which could self-assemble as cubic structures to destruct the lysosome membrane for selectively suppressing cancer cell proliferation.9 Inspired by these fascinating results, it can be anticipated that CBs as host molecules hold great potential in the creation of multivalent cascade supramolecular assemblies and biomaterials. However, to the best of our knowledge, tunable complexation with stimuli-responsive biomolecules and the full utilization of host–guest competitive interaction to simultaneously realize photoluminescence enhancement and induce cancer cell death are still rarely reported in CB-based supramolecular systems.
It is known that the elevated reactive oxygen species (ROS) level is a distinctive characteristic of tumor cells and tissues because of their heightened metabolic activity.10 Some oxidation-responsive supramolecular systems have been reported for bioimaging, drug delivery, and cancer therapy.11 Therefore, it would be more advantageous to harness the ROS microenvironment to drive the topological transformation of supramolecular assemblies and realize organelle dysfunction and cell death. In this work, to reveal the impact of in situ oxidation-driven supramolecular assembling and disassembling processes on anticancer activity, methionine-bearing amphiphilic peptides with n-hexyl chain on the N-terminal (MLGG-6C) were designed and synthesized (Scheme 1). The host–guest complexation between CB[8] and MLGG-6C showed high binding affinity in aqueous solution (>105 M−1). This binary inclusion complex can further co-assemble with the perylene diimide (PDI) fluorophore to form a multivalent supramolecular assembly with controllable aggregation morphology. Importantly, when the methionine residue in MLGG-6C was oxidized from hydrophobic thioether to hydrophilic sulfoxide or sulfone,12 a significant decrease occurred in the binding affinity toward CB[8] (≈103 M−1). These changes could result in the competitive binding of PDI into the cavity of CB[8], which could not only display the enhanced fluorescence emission through the macrocyclic confinement effect, but also realize controllable morphological conversion from nanoparticles to micron-sized sheets by co-assembly with oxidized MLGG-6C. The obtained ternary assembly could further achieve endoplasmic reticulum (ER) imaging and reduce related protein expression in cancer cells, thus leading to the ER dysfunction and cell death. Therefore, it can be envisioned that our in situ ROS-driven controllable supramolecular system may provide a general and reliable approach for disease diagnosis and treatment.
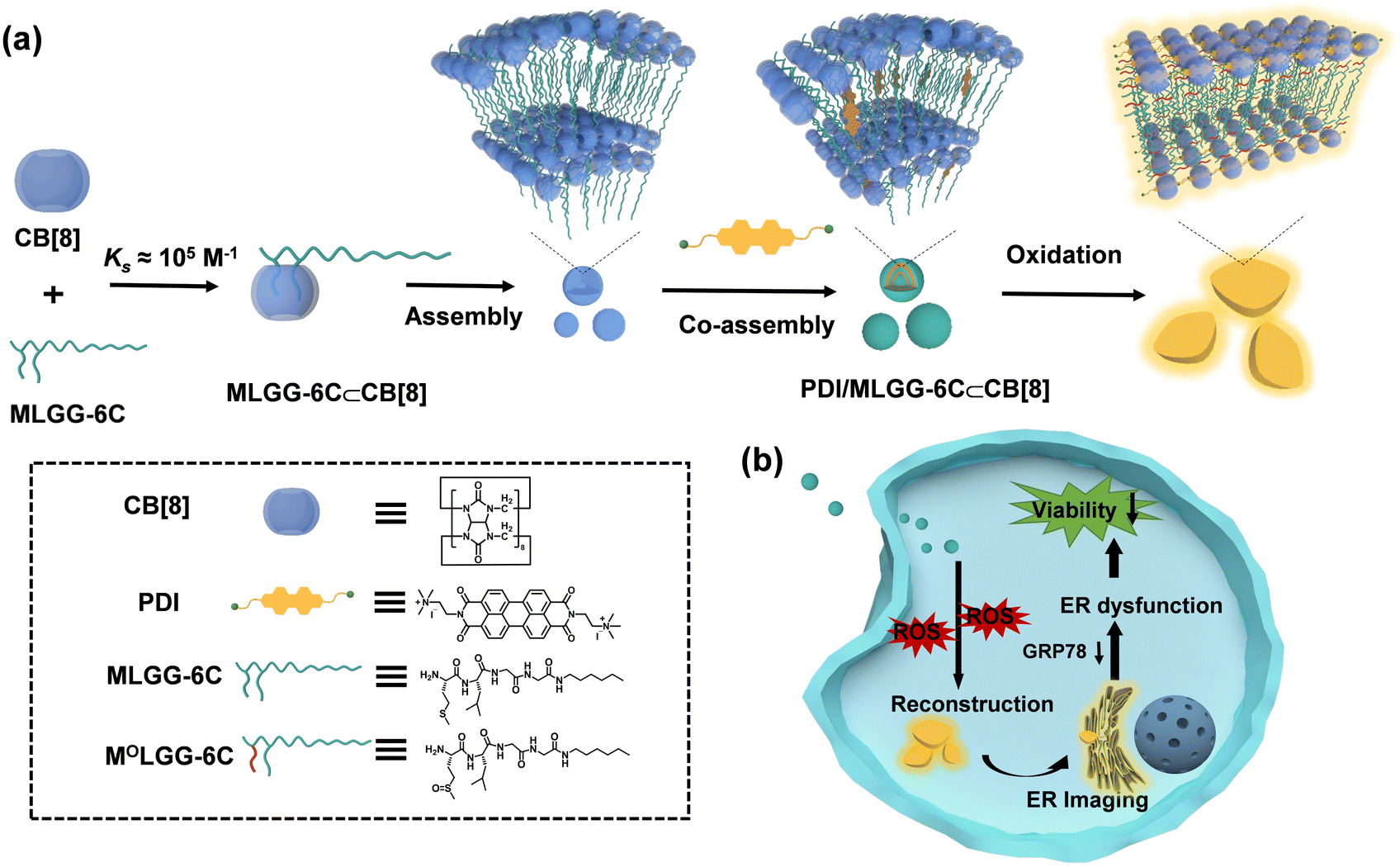 | ||
| Scheme 1 (a) Schematic illustration of the morphological conversion from nanoparticles to micron-sized sheets by the self-assembly of MLGG-6C, CB[8], and PDI; (b) in situ reconstruction of PDI/MLGG-6C⊂CB[8] aggregates for ER imaging and cell death. | ||
Results and discussion
Characterization of compounds
The amphiphilic guest (MLGG-6C) containing a Met-Leu-Gly-Gly sequence and n-hexyl tail was obtained by amide condensation, and the structural characterization is shown in the ESI (Fig. S1–S3, ESI†). Moreover, compounds with different oxidation states and sequences, including MLGGG, MOLGGG, MO2LGGG, LMGGG, LMOGGG, and LMO2GGG, were designed (Fig. S4–S9, ESI†). MLGG-6C and peptides were purchased from Nanjing Genscript Co., Ltd. Meanwhile, quaternary ammonium salt-modified PDI was also synthesized as a potential fluorescent competitive guest (Scheme S1, Fig. S10 and S11, ESI†).13Binding behaviours of MLGG-6C with CB[8]
It has been documented in the previous literature that CB[8] is able to encapsulate specifically sequenced peptides containing methionine (Met) in aqueous solution with a moderate binding affinity in the submicromolar range.12 In our case, given the hydrophilic and electronegative properties of sulfoxide or sulfone groups, it can be expected that the binding affinity between CB[8] and the methionine-containing peptides may remarkably decrease after being oxidized to sulfoxide or sulfone. Therefore, to test our hypothesis, the compounds MLGGG, MOLGGG, MO2LGGG, LMGGG, LMOGGG, LMO2GGG were used to comparatively investigate the selective molecular recognition process.First, 1H NMR spectroscopy was employed to study the host–guest complexation between CB[8] and different peptides. As can be seen from the 1H NMR spectra (Fig. S12, ESI†), the peak resonances of Met and Leu residues in MLGGG underwent a large complexation-induced upfield shift upon addition of CB[8], indicating that the Met residues and Leu residues were encapsulated into the CB[8] cavity. Moreover, the protons of Met and Leu residues in MOLGGG and MO2LGGG also showed upfield shifts with a broadened effect in the presence of CB[8] (Fig. S13 and S14, ESI†). However, these changes in chemical shifts were less significant than that in the MLGGG⊂CB[8] system, demonstrating that the oxidized Met and Leu were still included in the CB[8] cavity but with weaker binding affinity.
Similar phenomena could also be observed in the host–guest complexation of CB[8] with LMGGG. That is, as shown in Fig. S15 (ESI†), the proton signals of Met and Leu residues in LMGGG shifted upfield upon complexation with CB[8], indicating that the Met and Leu residues were encapsulated into the cavity of CB[8]. Comparatively, only the Leu signals in LMOGGG and LMO2GGG underwent complexation-induced upfield shifts, suggesting the exclusive inclusion of Leu residues within the CB[8] cavity (Fig. S16 and S17, ESI†). Taken together, these results demonstrate that CB[8] prefers the encapsulation of the N-terminated Met and Leu residues through hydrophobic and ion–dipole interactions, while the peptides bearing the electronegative oxidized Met residues exhibit weak binding strength with CB[8].
Next, isothermal titration calorimetry (ITC) was employed to quantitatively study the thermodynamic properties between CB[8] and the peptides at different oxidation states, and the corresponding thermodynamic parameters are collected in Table 1 (Fig. S18 and S19, ESI†). The stability constant (KS) between CB[8] and MLGGG was measured to be 3.14 × 105 M−1 in PBS at pH 7.3, which was consistent with the reported results.12 In sharp contrast, the KS value significantly decreased by two orders of magnitude owing to the oxidation of the methionine residue from thioether to sulfoxide (MOLGGG). The binding affinity is further weakened in the case of sulfone-bearing peptides (MO2LGGG). Structurally, the oxidized Met terminus is more electronegative and hydrophilic, which leads to reduced host–guest binding strength by the hydrophobic cavity of CB[8] (Fig. S20, ESI†). Thermodynamically, these binding processes were dominantly governed by the negative enthalpic change, accompanied by the unfavorable entropic loss. The enthalpic gains may be attributed to the combined effect of the hydrophobic and ion–dipole interactions, while the entropic loss may arise from the conformational fixation upon host–guest complexation with CB[8]. In the control experiments, the sequence selectivity of peptide backbones was also examined by reversing the positions of Met and Leu residues. The ITC results demonstrate that the binding stability caused a 10-fold decline in the oxidation states. Moreover, as confirmed by the chemical shift changes and electrostatic potential maps, the inclusion complexation of CB[8] with the N-terminated Leu residue is still maintained after Met oxidation, which may be responsible for the quite similar thermodynamic parameters in the forms of sulfoxide and sulfone. Therefore, given the large disparities in KS values before and after the oxidation process, MLGGG was selected as the optimized peptide chain in the following exploration of the physicochemical and biological performance.
:1 inclusion complexation between CB[8] and peptides at different oxidation states (0.01 M PBS, pH 7.3, 298 K)
| Peptide | K S (M−1) | −ΔG° (kJ mol−1) | −ΔH° (kJ mol−1) | −TΔS° (kJ mol−1) |
|---|---|---|---|---|
| MLGGG | (3.14 ± 0.08) ×105 | 31.4 ± 1.42 | 39.06 ± 0.29 | 7.66 ± 1.71 |
| MOLGGG | (2.30 ± 0.04) ×103 | 19.26 ± 1.24 | 44.96 ± 0.63 | 25.70 ± 1.87 |
| MO2LGGG | (8.92 ± 0.03) ×102 | 11.14 ± 2.78 | 52.99 ± 3.40 | 41.85 ± 0.62 |
| LMGGG | (5.91 ± 0.08) ×105 | 32.98 ± 0.74 | 43.91 ± 0.13 | 10.93 ± 0.87 |
| LMOGGG | (4.16 ± 0.15) ×104 | 26.37 ± 1.20 | 32.23 ± 1.21 | 5.86 ± 0.01 |
| LMO2GGG | (3.40 ± 0.21) ×104 | 25.91 ± 0.96 | 34.37 ± 2.76 | 8.46 ± 1.80 |
The oxidation process was also studied in the presence of 1.0 mM H2O2, suggesting that MLGG-6C could be completely converted to MOLGG-6C form, as jointly evidenced by the reduced retention time from 3.33 to 3.02 min in the high performance liquid chromatography (HPLC) spectra, the clear [M–H]−m/z peak at 474.2751 assigned to MOLGG-6C in the high resolution mass spectra, and the remarkable downfield shifts of the Met residue (Ha–c) in the 1H NMR spectra (Fig. S21–S24, ESI†). Meanwhile, the resonances of Met and Leu residues underwent a large complexation-induced upfield shift upon addition of CB[8], whereas the chemical shifts of the Gly residue and alkyl chain remained unchanged. In addition, the proton signals of Met and Leu residues were broadened and upshifted to some extent after undergoing the oxidation reaction, again indicative of the exclusive host–guest interaction of CB[8] with the oxidized Met and Leu moieties (Fig. 1a). Notably, no obvious change in the chemical shifts of the n-hexyl tail was found before and after oxidation, implying that the introduction of the alkyl chain could not negatively affect the inclusion complexation between CB[8] and MLGG.
 | ||
| Fig. 1 (a) 1H NMR spectra of MLGG-6C, MLGG-6C⊂CB[8] complex, MOLGG-6C, and MOLGG-6C ⊂CB[8] complex in D2O ([MLGG-6C] = 1.0 mM, [CB[8]] = 1.0 mM, and [H2O2] = 3.0 mM). (b) Fluorescence emission spectra of the PDI/MLGG-6C⊂CB[8] assembly in water containing H2O2 at 298 K ([PDI] = 50 μM, [MLGG-6C] = 0.15 mM, [CB[8]] = 0.15 mM, and [H2O2] = 1.0 mM). Inset: photographs of the PDI/MLGG-6C⊂CB[8] assembly before and after oxidation. (c) 1H NMR spectra of PDI, PDI⊂CB[8] complex, and PDI/MLGG-6C⊂CB[8] assembly before and after oxidation in D2O ([PDI] = 0.5 mM, [MLGG-6C] = 1.0 mM, [CB[8]] = 1.0 mM, and [H2O2] = 3.0 mM). (d) Fluorescence quantum yields of PDI, PDI⊂CB[8] complex, and PDI/MLGG-6C⊂CB[8] assembly before and after oxidation. | ||
Competitive binding behaviours of CB[8] with MLGG-6C and PDI
Furthermore, the binding affinity of PDI⊂CB[8] complexation was measured, giving a KS value of (4.78 ± 0.58) × 104 M−1 by fitting the UV-vis absorbance of PDI at 534 nm (Fig. S25, ESI†) according to the non-linear fitting equation.14 The KS value was also obtained as (6.83 ± 0.96) ×104 M−1 from the ITC experiment (Fig. S26 and S27, ESI†), which was consistent with the results of the UV titration experiment. The thermodynamic parameters of ΔH°, TΔS°, and −ΔG° were calculated to be 13.39 ± 5.73, 43.93 ± 4.31, and 30.54 ± 1.42 kJ mol−1, respectively, indicating that the binding process was entropy-driven with a small enthalpy gain in a desolvation process. The KS value is perfectly located in the range between the ones of CB[8] with MLGGG (3.14 × 105 M−1) and MOLGGG (2.30 × 103 M−1). More gratifyingly, the complexation with CB[8] could efficiently prevent PDI from undesired aggregation-caused quenching, thus leading to a 10-fold fluorescence enhancement at 563 and 590 nm (Fig. S28, ESI†). As signaled by a larger downfield shift (Δδ = 0.35 and 0.27 ppm) of the aromatic protons (Fig. S29, ESI†), an effective disaggregation of PDI occurred upon complexation with CB[8]. In addition, 2D DOSY (diffusion-ordered spectroscopy) experiments showed that the diffusion coefficients of free PDI and the PDI⊂CB[8] complex (Fig. S30 and S31, ESI†) were 2.46 × 1010 m2 s−1 and 2.20 × 1010 m2 s−1, respectively, which further excluded the formation of large-sized polymeric aggregates in an end-to-end manner. In comparison, the introduction of CB[7] caused larger upfield shifts (Δδ = −1.34 and −1.12 ppm) and a smaller downfield shift (Δδ = 0.13 ppm) for the nonaromatic end and the aromatic core (Fig. S29, ESI†), respectively, and could not augment the fluorescence emission, indicating encapsulation of CB[7] with the nonaromatic end of PDI (Fig. S32, ESI†).Interestingly, upon addition of MLGG-6C, the fluorescence emission of the PDI⊂CB[8] complex was gradually suppressed and then reached equilibrium in the presence of 3 equiv. of MLGG-6C (Fig. S33, ESI†), indicating that the location of CB[8] was transferred to MLGG-6C. As a control experiment, the fluorescence experiment of PDI with the addition of MLGG-6C was performed, indicating that the PDI fluorescence emission could not be affected by free MLGG-6C (Fig. S34, ESI†). Also, the UV-vis absorbance of PDI⊂CB[8] complexation was recovered to the free PDI state by adding MLGG-6C, indicative of a competitive binding process in this ternary assembly (Fig. S35, ESI†). The ITC data of the PDI⊂CB[8] complex with MLGGG as a competitor also certified the competitive binding process (Fig. S36, ESI†).
Having understood the molecular binding behaviors of CB[8] with MLGG-6C and PDI, their oxidation-responsive assembling abilities were investigated in the presence of H2O2. The control experiment was preliminarily conducted to ensure the structural stability of PDI alone under oxidation conditions (Fig. S37, ESI†). As shown in Fig. 1b, the fluorescence emission of PDI in the PDI/MLGG-6C⊂CB[8] assembly was gradually enhanced by H2O2 in 16 h. Apparently, the oxidation of the Met residue from thioether to sulfoxide could decrease the binding affinity between CB[8] and MLGG-6C and then trigger the inclusion complexation with PDI to dramatically augment its fluorescence intensity. In addition, the 1H NMR spectra also reveal that the aromatic protons of PDI shifted downfield after complexation with CB[8] and then returned back to the free PDI position in the PDI/MLGG-6C⊂CB[8] assembly, indicating that the oxidized MLGG-6C was expelled from the CB[8] cavity and replaced by PDI (Fig. 1c). Accordingly, the calculation of fluorescence quantum yields and lifetimes further certifies that the photoluminescence efficiency of PDI could be almost restored to the binary PDI⊂CB[8] complex after the oxidation of Met in the PDI/MLGG-6C⊂CB[8] assembly (Fig. 1d and S38–S44, ESI†). These results demonstrate the translocation of CB[8] from the Met–Leu moiety to PDI in the oxidation process, which can provide the molecular basis for the subsequent oxidation-responsive modulation in the biological environment.
Additionally, the other peptides containing tyrosine (YLGGG, Fig. S45, ESI†) or cysteine (CLGGG, Fig. S46, ESI†) were designed and applied in the redox experiments. Under the same experimental conditions, the tyrosine unit of YLGGG could not be oxidized or restore PDI fluorescence through competitive binding (Fig. S47–S51, ESI†). Although the cysteine unit of CLGGG was oxidized to form a disulfide dimer, the fluorescence emission intensity of PDI was slightly enhanced (Fig. S52–S56, ESI†), indicating an incomplete competitive binding process. Therefore, the methionine-possessing MLGGG-6C with exceptional oxidation properties and large disparities in binding strengths with CB[8] at different oxidation states is the ideal candidate for constructing tunable supramolecular systems driven by the competitive binding process.
Morphological reconstruction of supramolecular assemblies
Subsequently, the topological morphologies of the PDI/MLGG-6C⊂CB[8] assembly before and after oxidation were studied by microscopic experiments. The transmission electron microscope (TEM) images showed strikingly distinctive morphologies in the different groups. That is, PDI alone was mainly self-assembled into stacked nanosheets via intermolecular π-stacking interaction; the PDI⊂CB[8] complex existed as uniform lamellar structures; the amphiphilic MLGG-6C exhibited loose-knit nanoparticles with a diameter of ca. 500 nm; and the MLGG-6⊂CB[8] assembly showed close-knit nanoparticles with a diameter of ca. 200 nm, respectively (Fig. S57, ESI†). After loading PDI, the diameter of the PDI/MLGG-6C⊂CB[8] ternary supramolecular assembly increased to 300 nm as jointly confirmed by the TEM, scanning electron microscope (SEM), and atomic force microscope (AFM) images (Fig. 2a–c). After being oxidized, the micron-sized sheets were found to be over 2 μM in length and 500 nm in height in the case of the MOLGG-6C/PDI⊂CB[8] assembly (Fig. 2d–f). Along with the microscopic results in the solid state, the dynamic light scattering (DLS) data gave the hydrated diameters of the MLGG-6C/PDI⊂CB[8] assembly, showing dramatic size changes from about 291 nm to 1.3 μM before and after oxidation by H2O2 (Fig. 2g and h). As can be seen from the amplified AFM images (Fig. 2i), it is clearly observed that the ternary supramolecular assembly presented the micro-sheet morphology after oxidation. These results imply that the oxidation-driven guest competitive binding could alter the molecular aggregation modes and give rise to the reconstruction of supramolecular assemblies. The topological morphology transformation from nanoparticles into micron-sized sheets is mainly attributed to the oxidation-driven competitive binding and the assembly of MOLGG-6C with PDI⊂CB[8] via electrostatic interactions. Additionally, it is also found that oxidized MLGG-6C alone displayed a nanorod structure (Fig. S58, ESI†) and the MLGG-6C⊂CB[8] complex has a tendency to form larger-sized assemblies after oxidation.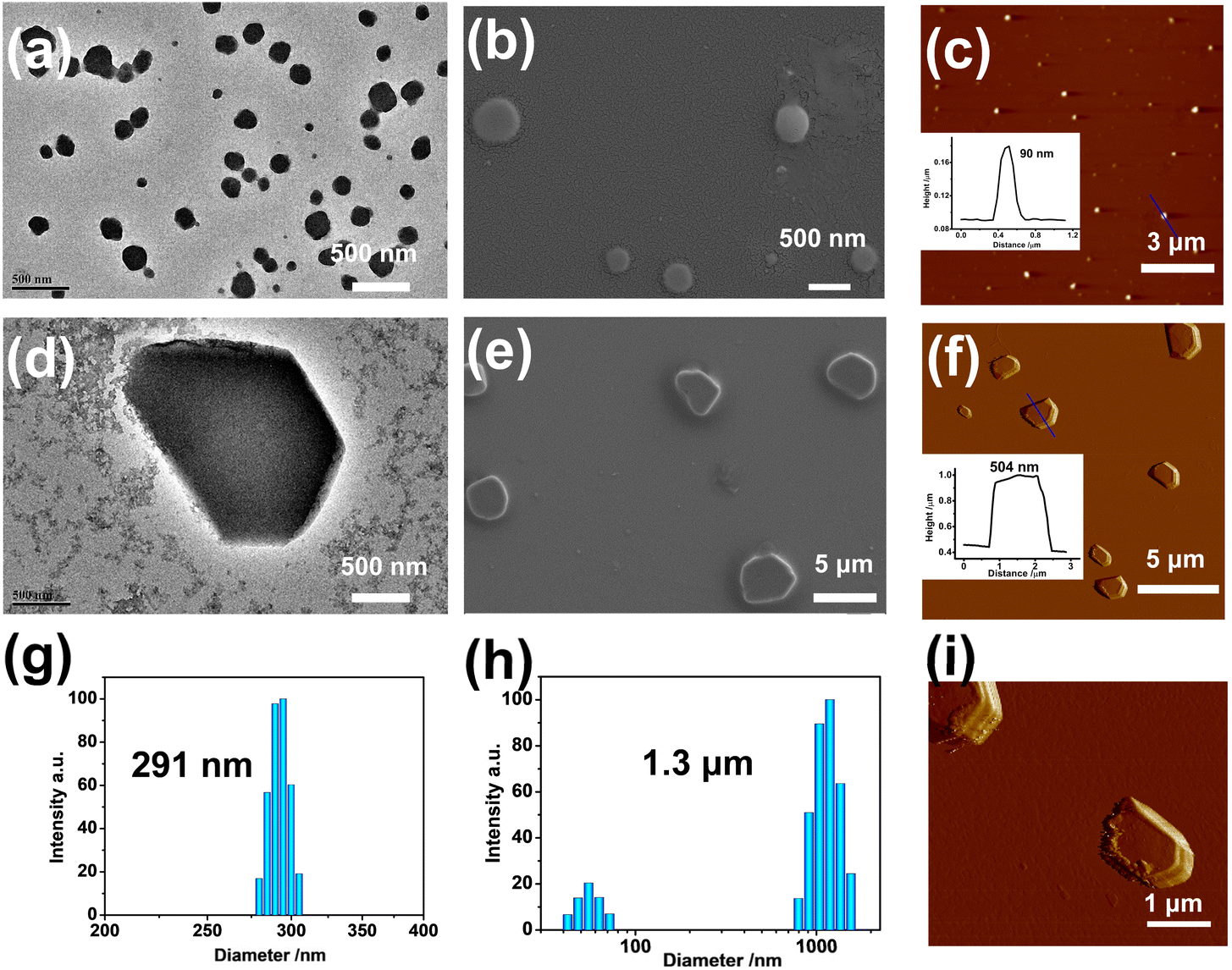 | ||
| Fig. 2 (a) TEM, (b) SEM, (c) AFM images, and (g) DLS results of the PDI/MLGG-6C⊂CB[8] assembly, and (d) TEM, (e) SEM, (f and i) AFM images, and (h) DLS results of the PDI/MLGG-6C⊂CB[8] assembly after oxidation by H2O2. | ||
The intermediate state from nanoparticles to micron-sized sheets has been studied by TEM experiments to further confirm the topological morphology change. It is observed that some small-sized nanoparticles were dispersed around the central large-sized assemblies, which could facilitate the further aggregation of micron-sized sheets (Fig. S59, ESI†). Moreover, the morphology of ternary assemblies at different pH was also studied by TEM experiments. As expected, the conversion of topological morphology from nano-sized particles to micron-sized sheets could be readily achieved in the range of pH 4–7, indicating that the reconstruction may occur in the low pH environment in tumor cells (Fig. S60, ESI†).
In situ oxidation-driven reconstruction of supramolecular assemblies
Given the elevated ROS level in tumor cells and tissues, the endogenous ROS is expected to be used in the ternary PDI/MLGG-6C⊂CB[8] system and realize in situ oxidation-responsive regulation in cancer cells. Therefore, the human cervical cancer cell line (HeLa cells) with overexpressed ROS was selected as the model cells. The molar ratio of the PDI/MLGG-6C/CB[8] assembly was adjusted to 1/2/2 for biological experiments, because such experiments required high-concentration stock solution of the assembly, and the assembly behaviors were further confirmed by fluorescence and TEM experiments (Fig. S61 and S62, ESI†). First, confocal laser scanning microscopy (CLSM) was employed to study the ROS levels in HeLa cancer cells and two normal cells (L929 and RS1 cells) by incubating them with a commercial ROS probe (DCFH-DA). As expected, HeLa cells showed a much higher ROS level, whereas ROS was maintained at a negligible level in L929 and RS1 cells (Fig. S63, ESI†). Then, the reconstruction process of the PDI/MLGG-6C⊂CB[8] assembly in the cellular environment was explored. HeLa cells were incubated in the presence of PDI, PDI⊂CB[8] complex, and PDI/MLGG-6C⊂CB[8] assembly, respectively, to verify their cell imaging performance. As discerned from Fig. 3a, rather weak fluorescence was observed in the cells incubated with PDI, due to its relatively low fluorescence emission ability. Since the inclusion of PDI with CB[8] could greatly enhance its fluorescence emission, the cells treated with the PDI⊂CB[8] complex showed very bright fluorescence, which was consistent with the fluorescence emission in solution. Meanwhile, strong red fluorescence was detected in the group of the PDI/MLGG-6C⊂CB[8] assembly with the dispersion of the micron-sized assemblies, which contributed to the formation of large-sized MOLGG-6C/PDI⊂CB[8] assemblies in the cells. Accordingly, the magnified CLSM images clearly showed the micron-sized structures with strong fluorescence emission (Fig. 3b). In addition, the bio-TEM images of HeLa cells incubated with the PDI/MLGG-6C⊂CB[8] assembly directly visualized the micron-sized assemblies (Fig. 3c). Therefore, combining the CLSM with bio-TEM results, it is concluded that the PDI/MLGG-6C⊂CB[8] assembly could be in situ oxidized in HeLa cells and then reconstructed into large-sized supramolecular assemblies in response to the high ROS level under the cellular conditions. | ||
| Fig. 3 (a) CLSM images of HeLa cells treated with PDI, PDI⊂CB[8] complex, and PDI/MLGG-6C⊂CB[8] assembly. (b) Magnified CLSM images of HeLa cells incubated with the PDI/MLGG-6C⊂CB assembly. The white square region in the merged image is shown in the further magnified column. (c) Bio-TEM images of HeLa cells treated with PDI, PDI⊂CB[8] complex, and PDI/MLGG-6C⊂CB[8] assembly. The white square region highlights the MOLGG-6C/PDI⊂CB[8] assembly. | ||
The PDI/MLGG-6C⊂CB[8] assembly was also treated with the cancer cell line (A549 cells) and normal cell line (L929 and RS1 cells) with elevated and normal ROS levels, respectively. CLSM images of A549 cells showed bright and dotted red fluorescence, which was identical to that in HeLa cancer cells (Fig. S64a, ESI†). However, CLSM images of L929 and RS1 cells displayed negligible fluorescence, suggesting the existence of very few reconstructed assemblies because of the low ROS level (Fig. S64b and c ESI†). Therefore, the obtained PDI/MLGG-6C⊂CB[8] supramolecular system could efficiently distinguish cancer cells from normal cells by visualized fluorescence imaging.
Intracellular ER dysfunction and induced cell death
Then, the co-localization was further assessed to investigate the accumulation of assemblies in subcellular organelles. Satisfactorily, the green fluorescence of ER-Tracker was well overlapped with the red fluorescence of the MOLGG-6C/PDI⊂CB[8] assembly in the HeLa cells (Fig. 4a) and the Pearson's correlation coefficient was 0.86 (Fig. S65, ESI†). Owing to the preferential ER accumulation ability and in situ reconstruction of the PDI/MLGG-6C⊂CB[8] ternary assembly, we wonder if this oxidation-driven supramolecular assembling process may affect the cell activity. Therefore, cell counting kit-8 (CCK8) assays were conducted to investigate the cytotoxicity against cancer cells using this ternary assembly. The HeLa cells were separately incubated with PDI, PDI⊂CB[8], MLGG-6C, MLGG-6C⊂CB[8], PDI/MLGG-6C,and PDI/MLGG-6C⊂CB[8] assembly and then the cell viability was assessed in 24 h. As shown in Fig. 4b and S66, ESI,† the PDI, MLGG-6C and PDI/MLGG-6C group showed ignorable toxicity even at a high concentration. In comparison, the PDI⊂CB[8] complex, MLGG-6C⊂CB[8] complex, and PDI/MLGG-6C⊂CB[8] assembly exhibited concentration dependent cytotoxicity against HeLa cells, and the cell viability was estimated to be 80%, 62%, and 26%, respectively. The significant decrease in cell viability of HeLa cells was probably attributed to the reconstruction of the PDI/MLGG-6C⊂CB[8] assembly and the ER dysfunction in cancer cells. The MLGG-6C⊂CB[8] complex also possessed certain cytotoxicity, which is mainly ascribed to the molecular binding of CB[8] with two side chains (Met and Leu residues) of MLGG-6C and their subsequent oxidation-driven assembly in cells (Fig. S58, ESI†). Furthermore, the ROS-dependent experiments were performed, showing that with the elevated ROS level, the death rate of HeLa cells increased accordingly (Fig. S67 and S68, ESI†). These results demonstrate that the elevated ROS level could lead to high cytotoxicity in HeLa cells. | ||
| Fig. 4 (a) CLSM images of HeLa cells illustrating the co-localization of the PDI/MLGG-6C⊂CB[8] assembly with ER-Tracker Green. (b) Cell viability of HeLa cells treated with PDI, PDI⊂CB[8] complex, and PDI/MLGG-6C⊂CB[8] assembly. The molar ratios of PDI/MLGG-6C/CB[8] were 1:2:2. (c) Western blotting analysis and corresponding quantitative expression of GRP78 in HeLa cells treated with PBS, PDI, PDI⊂CB[8] complex, MLGG-6C⊂CB[8] complex, and PDI/MLGG-6C⊂CB[8] assembly. | ||
In contrast, due to the lack of sufficient ROS in the normal cells, the treatment of L929 and RS1 cells with the PDI/MLGG-6C⊂CB[8] assembly only induced a slight decrease in cell viability (Fig. S69, ESI†). Meanwhile, the ROS levels in L929 and RS1 normal cells could be significantly elevated after being treated with H2O2 and under such elevated ROS conditions, the PDI/MLGG-6C⊂CB[8] assembly showed much higher cytotoxicity (Fig. S70 and S71, ESI†). These results jointly indicate that the oxidation-driven and ER-targeted supramolecular reconstruction may be responsible for the selective cytotoxicity against cancer cells.
Finally, the impact of the PDI/MLGG-6C⊂CB[8] assembly on the ER function was further studied. Glucose-regulated protein 78 (GRP78), which is overexpressed in many tumor cells, is known as the main regulator of the unfolded protein response to maintain ER homeostasis.15 The regulation of GRP78 expression could interfere with ER homeostasis and trigger a cascade of physiological and pathological changes at the cellular level. As shown in Fig. 4c, among all the examined groups, the western blotting (WB) analysis shows that the expression of GRP78 was apparently downregulated in HeLa cells when treated with the PDI/MLGG-6C⊂CB[8] assembly. Therefore, one possible explanation is that in our case, the reconstruction of the PDI/MLGG-6C⊂CB[8] assembly in the ER can efficiently downregulate the GRP78 expression and, consequently, affect ER function and cell proliferation.
Conclusions
In summary, the host–guest complexation between CB[8] and Met-containing amphiphilic peptides was successfully constructed, which could further co-assemble with PDI to form ternary supramolecular nanoparticles. The binding affinity between CB[8] and peptides was greatly weakened by two orders of magnitude after being oxidized from sulfide to sulfoxide of the Met residue. Under such circumstances, PDI could competitively occupy the CB[8] cavity to form the stable inclusion complex, resulting in macrocycle-confined fluorescence enhancement and oxidation-driven morphological transformation from nanoparticles into micron-sized sheets. Significantly, this oxidation-driven reassembling process could also be realized in cancer cells with an elevated ROS level, thus achieving targeted organellar imaging, disturbing ER function, and inducing cell death. Therefore, the present research provides a highly biocompatible and convenient approach for in situ supramolecular reconstruction in the cell microenvironment, which may be beneficial for the development of supramolecular disease diagnosis and treatment.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: JN, YMZ, and YL; methodology: JN, XW, and YMZ; investigation: JN, JY, and XW; visualization: JN, JY, and XW; supervision: YMZ, YC, and ZY; writing—original draft: JN and YMZ; writing—review & editing: JN, ZY, and YL.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22171148, 22371148, 22131008, 22101280, and 22201142), the Natural Science Foundation of Tianjin (21JCZDJC00310), Haihe Laboratory of Sustainable Chemical Transformation, and the Fundamental Research Funds for the Central Universities (Nankai University).Notes and references
- (a) G.-B. Qi, Y.-J. Gao, L. Wang and H. Wang, Adv. Mater., 2018, 30, 1703444 CrossRef PubMed; (b) J. Gao, J. Zhan and Z. Yang, Adv. Mater., 2020, 32, 1805798 CrossRef CAS PubMed; (c) X. Liu, M. Li, J. Liu, Y. Song, B. Hu, C. Wu, A.-A. Liu, H. Zhou, J. Long, L. Shi and Z. Yu, J. Am. Chem. Soc., 2022, 144, 9312–9323 CrossRef CAS PubMed; (d) W. Tan, Q. Zhang, J. Wang, M. Yi, H. He and B. Xu, Angew. Chem., Int. Ed., 2021, 60, 12796–12801 CrossRef CAS PubMed.
- (a) M. Yan, S. Wu, Y. Wang, M. Liang, M. Wang, W. Hu, G. Yu, Z. Mao, F. Huang and J. Zhou, Adv. Mater., 2023, 2304249 Search PubMed; (b) J. Zhou, G. Yu and F. Huang, Chem. Soc. Rev., 2017, 46, 7021–7053 RSC; (c) W.-C. Geng, J. L. Sessler and D.-S. Guo, Chem. Soc. Rev., 2020, 49, 2303–2315 RSC; (d) H. Chen, X. Zeng, H. P. Tham, S. Z. F. Phua, W. Cheng, W. Zeng, H. Shi, L. Mei and Y. Zhao, Angew. Chem., Int. Ed., 2019, 58, 7641–7646 CrossRef CAS PubMed; (e) Y. Cao, X.-Y. Hu, Y. Li, X. Zou, S. Xiong, C. Lin, Y.-Z. Shen and L. Wang, J. Am. Chem. Soc., 2014, 136, 10762–10769 CrossRef CAS PubMed; (f) P. B. Crowley, Acc. Chem. Res., 2022, 55, 2019–2032 CrossRef CAS PubMed.
- (a) S. Garain, B. C. Garain, M. Eswaramoorthy, S. K. Pati and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 19720–19724 CrossRef CAS; (b) Y.-H. Liu, Y.-M. Zhang, H.-J. Yu and Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 3870–3880 CrossRef CAS PubMed; (c) H. Barbero and E. Masson, Chem. Sci., 2021, 12, 9962–9968 RSC.
- (a) J. Yu, J. Niu, J. Yue, L.-H. Wang and Y. Liu, ACS Nano, 2023, 17, 19349–19358 CrossRef CAS PubMed; (b) M. Huo, X.-Y. Dai and Y. Liu, Adv. Sci., 2022, 9, 2201523 CrossRef CAS.
- (a) H. Chen, Y. Chen, H. Wu, J.-F. Xu, Z. Sun and X. Zhang, Biomaterials, 2018, 178, 697–705 CrossRef CAS PubMed; (b) Y. Chen, Z. Huang, H. Zhao, J.-F. Xu, Z. Sun and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 8602–8608 CrossRef CAS PubMed; (c) G. Saravanakumar, J. Kim and W. J. Kim, Adv. Sci., 2017, 4, 1600124 CrossRef PubMed.
- (a) Y.-M. Zhang, J.-H. Liu, Q. Yu, X. Wen and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 10553–10557 CrossRef CAS PubMed; (b) X.-Y. Dai, B. Zhang, Q. Yu and Y. Liu, J. Med. Chem., 2022, 65, 7363–7370 CrossRef CAS PubMed; (c) P. Shi, E. Ju, Z. Yan, N. Gao, J. Wang, J. Hou, Y. Zhang, J. Ren and X. Qu, Nat. Commun., 2016, 7, 13088 CrossRef CAS PubMed; (d) P. Suating, M. B. Ewe, L. B. Kimberly, H. D. Arman, D. J. Wherritt and A. R. Urbach, Chem. Sci., 2024, 15, 5133–5142 RSC.
- M. Li, A. Lee, K. L. Kim, J. Murray, A. Shrinidhi, G. Sung, K. M. Park and K. Kim, Angew. Chem., Int. Ed., 2018, 57, 2120 CrossRef CAS PubMed.
- C. Sun, Z. Wang, L. Yue, Q. Huang, Q. Cheng and R. Wang, J. Am. Chem. Soc., 2020, 142, 16523–16527 CrossRef CAS PubMed.
- X. Wu, M. Liu, J. Niu, Q. Liu, X. Jiang, Y. Zheng, Y. Qian, Y.-M. Zhang, J. Shen and Y. Liu, Chem. Sci., 2023, 14, 1724–1731 RSC.
- (a) S. Ye, J. J. Hu and D. Yang, Angew. Chem., Int. Ed., 2018, 57, 10173–10177 CrossRef CAS PubMed; (b) S. Saikolappan, B. Kumar, G. Shishodia, S. Koul and H. K. Koul, Cancer Lett., 2019, 452, 132–143 CrossRef CAS; (c) S. Prasad, S. C. Gupta and A. K. Tyagi, Cancer Lett., 2017, 387, 95–105 CrossRef CAS PubMed.
- (a) K. Yang, G. Yu, Z. Yang, L. Yue, X. Zhang, C. Sun, J. Wei, L. Rao, X. Chen and R. Wang, Angew. Chem., Int. Ed., 2021, 60, 17570–17578 CrossRef CAS PubMed; (b) H.-J. Yu, Q. Zhou, X. Dai, F.-F. Shen, Y.-M. Zhang, X. Xu and Y. Liu, J. Am. Chem. Soc., 2021, 143, 13887–13894 CrossRef CAS PubMed; (c) G. Saravanakumar, J. Kim and W. J. Kim, Adv. Sci., 2017, 4, 1600124 CrossRef PubMed; (d) M. Pieszka, S. Han, C. Volkmann, R. Graf, I. Lieberwirth, K. Landfester, D. Y. W. Ng and T. Weil, J. Am. Chem. Soc., 2020, 142, 15780–15789 CrossRef CAS PubMed; (e) S. Kim, B. Jana, E. M. Go, J. E. Lee, S. Jin, E.-K. An, J. Hwang, Y. Sim, S. Son, D. Kim, C. Kim, J.-O. Jin, S. K. Kwak and J.-H. Ryu, ACS Nano, 2021, 15, 14492–14508 CrossRef CAS PubMed; (f) S. Mommer, K. Sokołowski, M. Olesińska, Z. Huang and O. A. Scherman, Chem. Sci., 2022, 13, 8791–8796 RSC.
- Z. Hirani, H. F. Taylor, E. F. Babcock, A. T. Bockus, C. D. Varnado, C. W. Bielawski and A. R. Urbach, J. Am. Chem. Soc., 2018, 140, 12263–12269 CrossRef CAS PubMed.
- (a) F. Biedermann, E. Elmalem, I. Ghosh, W. M. Nau and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 7739–7743 CrossRef CAS; (b) S.-W. Tam-Chang, J. Helbley and I. K. Iverson, Langmuir, 2008, 24, 2133–2139 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- D. Dong, M. Ni, J. Li, S. Xiong, W. Ye, J. J. Virrey, C. Mao, R. Ye, M. Wang, L. Pen, L. Dubeau, S. Groshen, F. M. Hofman and A. S. Lee, Cancer Res., 2008, 68, 498–505 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the characterization of target compounds, 1H NMR, UV-vis, and (time-resolved) fluorescence spectra, ITC curves, TEM images, and CCK8 assays. See DOI: https://doi.org/10.1039/d4sc04261a |
| This journal is © The Royal Society of Chemistry 2024 |