
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA photoactivated chiral molecular clamp rotated by selective anion binding†
Yiping
Liu
,
Aiyou
Hao
 and
Pengyao
Xing
*
and
Pengyao
Xing
*
Key Laboratory of Colloid and Interface Chemistry of Ministry of Education, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, People's Republic of China. E-mail: xingpengyao@sdu.edu.cn
First published on 28th August 2024
Abstract
Developing chiral molecular platforms that respond to external fields provides opportunities for designing smart chiroptical materials. Herein, we introduce a molecular clamp whose chiral properties can be turned on by photoactivation. Selective anion binding achieves rational tuning of the conformations and chiroptical properties of the clamp, including circular dichroism and circularly polarized luminescence. Cyanostilbene segments were conjugated to chiral amines with a rotatable axis. Negligible chiroptical signals were significantly enhanced through a light illumination-induced isomerization. Binding with halide ions (F−, Cl− and Br−) enables chiroptical inversion and subsequent amplification of the resulting opposite handedness state by photo treatment. In contrast, the larger I− and NO3− ions failed to achieve chiroptical inversion. Also the handedness inversion was hampered in conformationally locked amines. Density-functional theory-based computational studies and experimental results reveal a structural transformation that proceeds from a butterfly-like open geometry to a closed V-shaped state initiated by four hydrogen bonds and the rotatable axis. This work illustrates design protocols for use in smart chiroptical molecular platforms mediated by photo treatment and anion binding.
Introduction
Chirality endows molecules with systematic complexity, giving them fascinating properties and functionalities.1–6 Chiral compounds have found numerous applications in chiroptical related fields such as recognition, sensing, display technologies and asymmetric synthesis.7–15 Noncovalent interactions provide opportunities to support and maintain the asymmetric molecular topology, which facilitates intramolecular chiral folding or intermolecular self-aggregation into chiral arrays.16–21 The dynamic nature of noncovalent forces allows for the flexible manipulation of chiral topology as well as abundant chiral expression.22–26 Precise and rational control over chirality expression, including emergence, conversion, inversion and other chiroptical behaviors, has attracted considerable attention.27–33 The understanding of structure-chiroptical property correlations also accelerates the design of chiral switches and responsive molecular platforms. Utilizing host–guest chemistry or noninvasive photo illumination can effectively alter the geometry and conformations of chiral entities, whereby chiroptical evolution or rational control can be realized.34–38 In addition to the above examples, the use of solvent media as an external field with alterable dielectric constants, polarities and viscosities is vital in directing noncovalent interactions and the evolution of chiral geometries.39–41Anion recognition chemistry has been flourishing for decades.42–45 Molecules with electrically positive pocket regions accommodate anions via electrostatic force-based hydrogen bonds, halogen bonds and chalcogen bonds.46–48 Anion-based host–guest chemistry is widely applied in colorimetric sensing, organocatalysis, extraction, separation, architectonics as well as transmembrane anion transport.49–54 Some inherently folded structures undergo anion binding which alters their geometry to give well-defined chiral architectures, as has been nicely demonstrated by Huc and other groups.55–59 The formation of hydrogen bonds with anions to anchor the chiral geometry, and the enantioselectivity can be controlled by chiral pendants or additional ligands. For instance, Fukuhara et al. reported that halide ion-binding induced the transformation of pyrrole oligomers from random distributions to helices, of which the handedness was controlled by external binap-based ligands.60 The crucial role of anion binding in controlling the chirality of the folded structures has been well established by many other reports,61,62 and the key role of geometry changes in expressing chiroptical signals has been emphasized in related works.63–65 However, the design of chiral platforms and systematic investigations into how the chemical structure (and light, anions, solvent, and temperature) impacts chiroptical properties remain challenging but are important to develop advanced chiral materials.
In this work, we aim to rationally manipulate chiral expression using photoisomerization and anion binding chemistry. Photosensitive cyanostilbene was conjugated to chiral diamines with a rotatable axis. Compound 1 (Scheme 1) has rotatable bonds and shows negligible chirality transfer from the amine skeleton to the cyanostilbene in solution, adopting a butterfly-like open geometry, as demonstrated by experimental and density-functional theory calculations. Photoirradiation caused an E/Z conformational transition that dramatically enhanced the chiroptical response as well as the luminescence by locking the stilbene domain. Binding to F−, Cl− and Br− inverts the initial handedness of 1, the chiroptical responses of which were enhanced in the opposite handedness state. In contrast, larger sized anions, such as I− and NO3−, failed to induce a handedness inversion. The handedness inversion was caused by the topological transition from an open to a closed state facilitated by the rotatable axis of the chiral amines as well as quadruplex hydrogen bonds formed with the halide ions. In contrast, after the chiral amines were locked in compound 2, no chirality evolution was observed (Scheme 1b). This work clarifies the structural factors required for the design of chiroptical molecular platforms responsive to photoirradiation and anion binding, which are important in switchable and smart chiral materials.
 | ||
| Scheme 1 (a) Transformation of the structure and properties of compound 1 and reference compound 2 by photoisomerization and anion binding. (b) Schematic representation of the chiroptical molecular movement invoked by anion binding. | ||
Results and discussion
Th syntheses of compounds 1 and 2 are demonstrated in the ESI.† The compounds were synthesized through several condensation steps with high yields and characterized thoroughly using nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry.66 The cyanostilbene domains are sensitive to photoirradiation, which causes an E to Z conformational transition in diluted solutions rather than photocycloaddition. In diluted tetrahydrofuran (THF) solutions of R1, photo irradiation at 365 nm results in gradual spectral variations, showing decreased absorption at around 350 nm, accompanied by an increase in absorbance at around 270 nm (Fig. 1a). The absorption changes correspond to the photoisomerization from the E to the Z form. Further inspection of the emission properties was also conducted (Fig. 1b), and illustrates that the gradual enhancement of the fluorescence peaked at around 400 nm. The fluorescent intensity was enhanced by a factor of 400 after sufficient photoirradiation, with quantum yields ranging from 0.9% to 4.9% for compound 1. The significant emission enhancement is associated with the rigidity of the Z-stilbene conformation, which locks the geometry due to restriction of the rotation between the benzene and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group. During photoisomerization, the terminally linked naphthalene ring transforms close to the benzene region, corresponding to increased intramolecular interactions and steric hindrance. It is reminiscent of the aggregation-induced emission process. Moreover, the rigid structure is beneficial to reduce vibration relaxation and thus enhance emission. Similar trends were observed in the spectrum of R2, a molecule that has a similar skeleton and photosensitive domains to R1 (Fig. 1c and d). The spectrum of R2 shows a decrease in absorption at 345 nm and an increase at around 270 nm, the trend of which after irradiation was roughly the same as that of R1. In addition, an enhanced emission was also seen for R2, suggesting that the similar optical properties are only caused by light absorption that induces the isomerization of the cyanostilbene and that they are almost unaffected by the linked amines. We also conducted illumination experiments on R1 and R2 in dimethyl sulfoxide (DMSO) (Fig. S1†), where a similar trend of spectral variations was recorded. Furthermore, in DMSO-d6, the 1H NMR spectra of R1 indicate a transition from the E to the Z conformation (Fig. 1e and f). The molar ratio of E/Z decreased from 7
C group. During photoisomerization, the terminally linked naphthalene ring transforms close to the benzene region, corresponding to increased intramolecular interactions and steric hindrance. It is reminiscent of the aggregation-induced emission process. Moreover, the rigid structure is beneficial to reduce vibration relaxation and thus enhance emission. Similar trends were observed in the spectrum of R2, a molecule that has a similar skeleton and photosensitive domains to R1 (Fig. 1c and d). The spectrum of R2 shows a decrease in absorption at 345 nm and an increase at around 270 nm, the trend of which after irradiation was roughly the same as that of R1. In addition, an enhanced emission was also seen for R2, suggesting that the similar optical properties are only caused by light absorption that induces the isomerization of the cyanostilbene and that they are almost unaffected by the linked amines. We also conducted illumination experiments on R1 and R2 in dimethyl sulfoxide (DMSO) (Fig. S1†), where a similar trend of spectral variations was recorded. Furthermore, in DMSO-d6, the 1H NMR spectra of R1 indicate a transition from the E to the Z conformation (Fig. 1e and f). The molar ratio of E/Z decreased from 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 to 3:7 after photo illumination for 5 min based on integration. The spectral variations suggested the successful construction of a photosensitive chiral platform.
:3 to 3:7 after photo illumination for 5 min based on integration. The spectral variations suggested the successful construction of a photosensitive chiral platform.
 | ||
| Fig. 1 Photoisomerization process. (a and b) Absorption and emission (λex = 340 nm) spectra along with the photo irradiation of R1 (0.1 mM) at 365 nm. (c and d) Absorption and emission (λex = 340 nm) spectra and the photo irradiation of R2 (0.1 mM) at 365 nm. Absorption and emission spectra were obtained in THF. (e) Partial 1H NMR spectra during the photo irradiation of R1 in DMSO-d6 at 365 nm. (f) The photoisomerization of R1. | ||
Electronic circular dichroism (CD) spectroscopy records the ground state chirality of chromophores. Its intensity or Cotton effect is associated with geometrical rigidity, which also correlates with the dot products of the electric and magnetic transition dipole moments. In diluted solution (0.1 mM in DMSO), negligible CD signals were observed (Fig. 2a and b). Although the skeletons of the amines are chiral, and compounds 1 and 2 ought to prefer a V-shaped chiral geometry, the negligible CD signal implies that the conformations of the cyanostilbene luminophore are diversified with flexibility. The free rotation between the benzene and CC group results in muted chiroptical activity. Photo irradiation arouses the E/Z transition that surprisingly boosts the CD signals due to the restrained geometry of the Z conformer. A strong positive signal was observed for both S1 and S2 (Fig. 2a and b), which suggests a photoactivated chiroptical behavior that is sufficient for intramolecular chirality transfer to occur from the chiral amines to the luminophores. In addition, the chiroptical photoactivity shows an expected enantiomeric effect, whereby mirror CD signals were obtained (Fig. 2c). The enhancement approaches a plateau at around 10 min of photo irradiation, and finally achieved dissymmetry g-factors of 7.0 × 10−4 and 1.6 × 10−3 for 1 and 2, respectively, at 290 nm (Fig. 2d). Obviously, 2 has a higher dissymmetry factor primarily thanks to the locked geometry of the cyclohexanediamine that to a large extent further anchors the flexibility of the E-cyanostilbenes. The chiroptical photoactivation is influenced by the solvent media, so that in THF, slightly smaller CD enhancements are present (Fig. S2†). It is believed that solvation effects are prominent in the folded geometries. Different solvents with distinct polarities would impact on the expression of chirality.
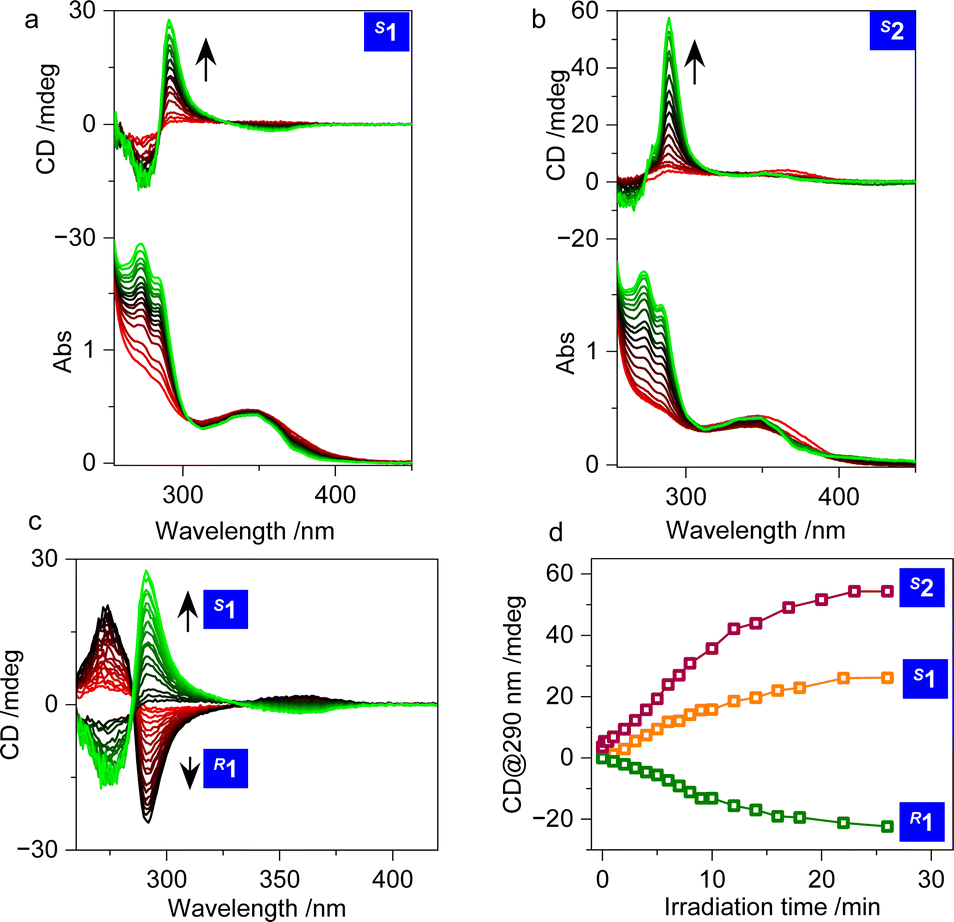 | ||
| Fig. 2 (a–c) CD spectra of compounds 1 and 2 in DMSO under irradiation with 365 nm UV light (0.1 mM). (d) Cotton effect intensity at 290 nm for R1, S1 and S2 as a function of photo irradiation time. | ||
The V-shaped orientation of the double amide groups creates a great opportunity for conducting anion binding chemistry. The supramolecular pocket comprises an abundance of hydrogen bonding donors for anions that facilitate host–guest interactions. In diluted THF solution, the introduction of F−, Cl− and Br− in the form of tetrabutylammonium halides caused absorption changes (Fig. 3a and S3†). The binding fits well to the 1:1 binding model with a small association constant (Ka) at a 104 magnitude (Fig. 3b, c and Table S1†). The F−, Cl− and Br− anions give gradually higher Ka values contrary to their decreasing electronegativities, a phenomenon which might be due to size compatibility. Next, we evaluated whether the high binding affinity could impact on the chiroptical responses. Introducing F−, Cl− and Br− to a THF solution of 1 could induce the chiroptical inversion in the absorption region of cyanostilbene, even though they share very small CD signals (Fig. S4, S5,†3d and e). We also obtained comparative CD signals, as shown in Fig. 3f, including the larger sized I− and NO3− anions in their tetrabutylammonium form (Fig. S6†). It turns out that in the presence of I− and NO3− the handedness of the CD signal was maintained, while in the presence of F−, Cl− and Br− an inversion was observed. In agreement with the absorption titrations, Br− and F− give the highest and smallest Ka, which is consistent with the critical transition molar equiv. of the halides. The critical molar equiv. are 4, 2, and 1 respectively for F−, Cl− and Br−, which means that Br− is more effective at triggering chiroptical inversion than the other halides, probably thanks to its suitable size. In sharp contrast, even the addition of an excess of any of the halides could barely invert the intrinsic CD signal of compound 2 (Fig. S7†). Absorption titration evidenced the fact that 2 still possess a considerable Ka of around 105 with the halides (Fig. S8†), while the changes of chiroptical activities could hardly be expressed, possibly due to the locked geometry of the cyclohexanediamine. It should also be noted that the solvent medium has a critical impact on the chirality expression. In DMSO, all tested anions failed to trigger any changes in the CD spectra (Fig. S9†).
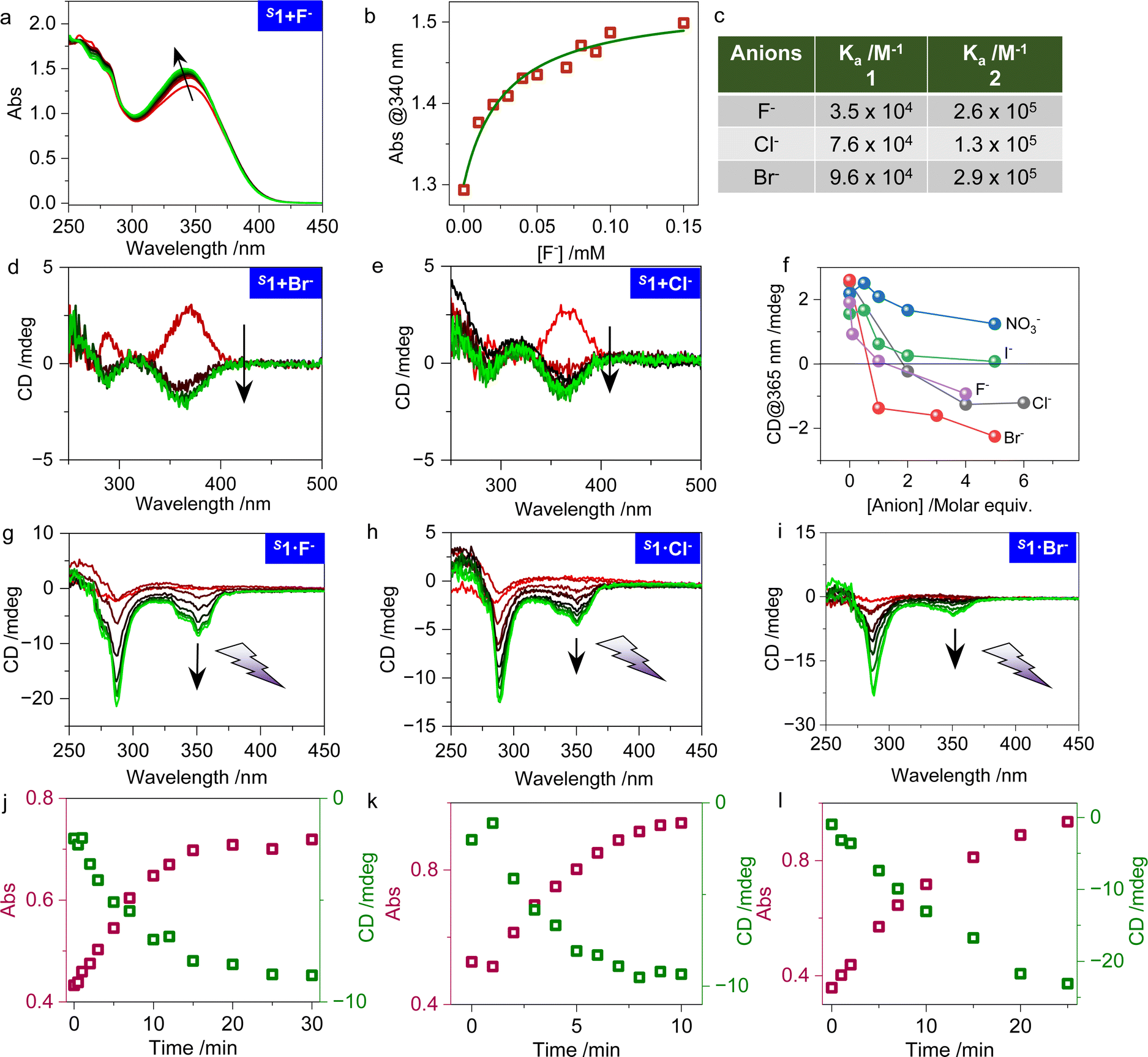 | ||
| Fig. 3 Manipulation of chirality via anion binding chemistry. (a and b) Absorption titration studies and the 1:1 fitted curve of S1 (0.05 mM in THF) and F−. (c) Summary of the binding constants obtained from the absorption titrations. (d and e) CD spectra changes of S1 (0.1 mM in THF) with increasing molar equiv. of Br− and Cl−. (f) CD signal at 365 nm of S1 (0.1 mM in THF) as a function of anion molar equiv. of F−, Cl−, Br−, I− and NO3−. (g–i) CD spectra of S1˙F−, S1˙Cl− and S1˙Br− (0.1 mM, 1:1 in THF) upon photo irradiation. (j–l) The corresponding absorption and CD intensity changes at 290 nm upon photo irradiation. | ||
We wondered whether photo irradiation could boost the small CD signals in the opposite direction. Fig. 3g–i and S10† illustrate the results of the photo irradiation treatment of the anion adducts S1˙F−, S1˙Cl−and S1˙Br−, which expectedly show enhanced Cotton effects with an opposite handedness compared to S1, while the same handedness was enhanced for the adduct S1˙NO3− (Fig. S11†). Compound 1 performs as an ideal molecular photoactivated platform with folded chirality tuned by anion binding chemistry (Fig. 3j and i). Then, we took another route to confirm the role of the anion binding chemistry. As shown in Fig. S12,† the Cotton effects of photoactivated S1 with strong chiroptical activity were entirely inverted in the presence of halides (F−, Cl− and Br− except for NO3−). This means that the binding affinities of 1 and 1′ are similar, while halides could invert the handedness in both the E and Z form. Moreover, a thorough temperature variable test was carried out to evaluate the influence of temperature to the systems. Here, tiny changes were found during the process of temperature change (Fig. S13 and S14†), indicating the presence of thermally stable chiroptical activity, either on the molecules on their own or in complexes with anions. SEM images showed the morphology of the assemblies (Fig. S15†), from which spherical shapes were observed whether anions are added or not. In addition, using the distribution statistics, images at an amplification of ten times the particle size were obtained for the systems combined with anions. In general, the increase of the size illustrated the formation of complexes of the compounds upon the addition of anions, but the chirality inversion was not observed using these macroscopic detection techniques.
Computational studies help to understand the geometrical transitions in photoisomerization as well as anion binding (Fig. 4). R1 was optimized at the CAMB3LYP/6-31G(d) level of theory (Fig. 4a). Its cyanostilbene arms adopts an open geometry with a N–C–C–N dihedral angle of 176.55°. The butterfly geometry is stabilized by intramolecular folding driven by the consistent hydrogen bonds centered by a carbonyl oxygen with two donor hydrogens from the adjacent benzene and diphenylethane groups. The bond distances are determined to be 2.459 and 2.510 Å. After photoisomerization into the Z form, R1′, the conformation adopts a similar open geometry with a slightly altered hydrogen bond length (2.453 and 2.507 Å) as well as a fixed dihedral angle (176.55°). Based on the analysis of the optimized structures before and after the photoisomerization, the E/Z transition has a very limited impact on the skeleton. The distance between the naphthalene units in the two arms is shorter by around 5.1 Å, while the dihedral angle between the benzene and naphthalene groups linked by the CC bond decreased by 3.0°. The increased steric hindrance fixes the twisted chiral structure, indicating enhanced chiroptical properties. In order to understand the influence of isomerization on the chiroptical responses, the CD spectra calculated at the CAMB3LYP/6-31G(d) level of theory based on time-dependent density-functional theory were obtained based on the optimized geometries (Fig. 4c–e).67 The calculated CD spectra of R1 and R1′ are consistent with the experimental results. Molecular orbitals (MOs) were visualized to reveal the electronic transitions (Fig. S16†). The S0 → S1 transition contained the information in the CD spectra, which was analyzed from the distribution of the HOMO and LUMO. The frontier orbitals are located on the cyanostilbene unit, where the HOMO is mainly distributed in the naphthalene region while the LUMO is spread around the benzene ring and CC bond. Compared to R1, R1′ shows a similar HOMO but a larger energy gap, which changes the transitions. Meanwhile, R1˙Cl− displays a similar gap corresponding to a similar absorption. The dissymmetry g-factor at the maximum wavelength was elevated from −2.6 × 10−4 to −8.0 × 10−4 after isomerization, which also agrees well with the experimentally observed enhanced Cotton effects. From the optimized structures, the restricted rotation between the benzene and CC group might play the key role. This change is described as a transition from “free rotating” to “single locking” for R1 and R1′ in Scheme 1a. Due to the fixed geometry, the calculations neglect the rotation and diversified geometries formed in solution, yet still consider the angles and strength of the transition dipole moments at the specific electronic transitions. For the calculated ECD spectra (Fig. 4c), the dominant geometry was used to qualitatively illustrate the changes of the signs and values, but there might be a certain degree of deviation in the numerical value. The calculation results suggest that chiroptical photoactivation is caused by the limited rotation of the stilbene moiety as well as the enhanced dot product values of the electronic dipole transition moments. After coordination with the halide ions, in this case Cl−, the two arms adopt a closed geometry driven by the four hydrogen bonds formed between Cl− and the amide and aryl protons (Fig. 4a). The lengths of the hydrogen bonds range from 2.428 to 2.739 Å, and the dihedral angle declined to 46.70°. The calculated CD spectra shown in Fig. 4d imply that R1 and R1˙Cl− have opposite signs. The slight blue-shift of the absorbance and CD spectra agrees well with the absorption titration studies displayed in Fig. 3a. Apparently, anion binding triggers a conformational transition from an open to a closed state that alters the relative orientation of the electronic transition dipole moments of the cyanostilbene domains (Fig. 4f), thus resulting in sign inversion. Further evidence to support this assumption was given by the calculated E–M angles between the electronic and magnetic transition dipole moments. Based on the equation  ,68 an angle of 96.16° is obtained for R1, greater than 90°, and thus corresponding to a negative signal. Conversely, an angle of 73.83° for R1˙Cl− represents the opposite signal. As a control, R2 with locked amine segments, spontaneously adopts a V-shaped geometry with a dihedral angle of 53.56°. Fig. 4e demonstrated nearly identical ECD signals for R2 and R2˙Cl−, indicating that after coordination of Cl−, the conformation of the basic skeleton undergoes no apparent transition, despite also forming four hydrogen bonds (Fig. 4b). The binding energies of R1˙Cl− and R2˙Cl− were determined to be −52.09 and −52.94 kcal mol−1, respectively. Therefore, the fitted values for the complexation constants of compounds 1 or 2 with the corresponding ions are quite similar. In addition, the slightly larger values of R2˙Cl− might be due to the greater binding energy and a more favourable structure for binding with anions that does not require conformational transformation. From the 1H NMR spectra (Fig. 1e), there are two groups of peaks about the E- and Z-isomers obtained by photoirradiation. Under photoisomerization, the spectra did not show any obviously shifted peaks for any of the (P and M) diastereomers, like reported results.69 Moreover, the 1H NMR spectra (Fig. S17†) in THF-d8 were also obtained to recognize the mechanism of anion binding. Though the peaks in the aromatic region are difficult to ascribe to specific H atoms, almost all the chemical shifts were changed for the systems bound with anions, as compared to S1. In particular, the H atoms connected to the chiral carbon atoms and to the CC bond showed obvious shifts toward lower field, which proved the conformation transition with the addition of anions.
,68 an angle of 96.16° is obtained for R1, greater than 90°, and thus corresponding to a negative signal. Conversely, an angle of 73.83° for R1˙Cl− represents the opposite signal. As a control, R2 with locked amine segments, spontaneously adopts a V-shaped geometry with a dihedral angle of 53.56°. Fig. 4e demonstrated nearly identical ECD signals for R2 and R2˙Cl−, indicating that after coordination of Cl−, the conformation of the basic skeleton undergoes no apparent transition, despite also forming four hydrogen bonds (Fig. 4b). The binding energies of R1˙Cl− and R2˙Cl− were determined to be −52.09 and −52.94 kcal mol−1, respectively. Therefore, the fitted values for the complexation constants of compounds 1 or 2 with the corresponding ions are quite similar. In addition, the slightly larger values of R2˙Cl− might be due to the greater binding energy and a more favourable structure for binding with anions that does not require conformational transformation. From the 1H NMR spectra (Fig. 1e), there are two groups of peaks about the E- and Z-isomers obtained by photoirradiation. Under photoisomerization, the spectra did not show any obviously shifted peaks for any of the (P and M) diastereomers, like reported results.69 Moreover, the 1H NMR spectra (Fig. S17†) in THF-d8 were also obtained to recognize the mechanism of anion binding. Though the peaks in the aromatic region are difficult to ascribe to specific H atoms, almost all the chemical shifts were changed for the systems bound with anions, as compared to S1. In particular, the H atoms connected to the chiral carbon atoms and to the CC bond showed obvious shifts toward lower field, which proved the conformation transition with the addition of anions.
 | ||
| Fig. 4 Computational studies. (a and b) DFT geometry optimized structures of R1, R1′, R1˙Cl−, R2 and R2˙Cl−. (c–e) Calculated electronic CD spectra of different species. (f) A schematic representation of the halide-ion induced geometry transition. | ||
CPL refers to the differential between left and right-handed emissions, giving related chiroptical information about the luminescent materials in the excited state. Improving the dissymmetry factor (glum) and realizing the effective regulation of CPL is a challenge for exploring CPL materials. On account of the adjustable Cotton effects and luminescence, the compounds reported here are potential candidates to explore controllable CPL properties. As shown in Fig. S18 and S19,†R1 and R1˙X− (X− = F−, Cl−, Br−) showed weak emissions and silent CPL signals. The work reported by Mori and co-workers demonstrated the correlation between the g-factors of CD and CPL signals,70 where the CPL signals obtained were smaller than the CD signals. On the one hand, the system without photo-irradiation resulted in small CD signals, and thus chirality expression in the excited state might be very weak. On the other hand, the fluorescence intensity could restrict the detection of the CPL emission. In comparison, an active and positive CPL signal was observed in R1′ after irradiation for 10 min with 365 nm UV light. The emergence of the CPL is consistent with the photoactivated chiroptical responses. Besides, anion-induced chiroptical inversion was also found in the CPL spectra, where all the systems of R1′˙X− (X− = F−, Cl−, Br−) displayed negative CPL peaks at around 420 nm (Fig. 5a). Meanwhile, mirror CPL signals were obtained for enantiomers of the 1′˙X− systems with glum values of ±3.0 × 10−3 (Fig. 5b–d and S19†). From the experimental results, the signs of the first Cotton effect of the CD signal for the system do not agree with the CPL signal in our report. For example, the S1′˙Cl− system showed negative CD signals but a positive CPL emission. In fact, the values and the signs of CD and CPL signals were not in agreement for the chiral systems, considering the structures of the ground and excited states.71,72 However, an effective high energy structure is necessary for the calculations but is hard to obtain. Mirror CPL emissions for the enantiomers and inversed signals for the systems with anions were successfully detected. It was inferred that inversed supramolecular chirality was successfully constructed, even in the excited state, due to the stable geometry anchored by the anions. Therefore, simple activation and regulation of CPL were realized via the combination of the effects of UV light and the halogen anions.
 | ||
| Fig. 5 CPL spectra of the systems after irradiation at 365 nm for 10 min. (a) Comparison of R1, R1˙F−, R1˙Cl− and R1˙Br− (0.1 mM, 1:1 in THF). (b–d) CPL spectra of R1/S1˙X− (X− = F−, Cl−, Br−), (λex = 340 nm). | ||
Conclusions
In conclusion, this work reports the design of a chiral molecular clamp, of which the conformation and chiroptical properties could be precisely manipulated by photochemistry and anion binding. Compound 1 was constructed by covalently appending a cyanostilbene moiety with chiral amines, which, in diluted solution, showed negligible CD signals. After photoisomerization, 1 displays emergent Cotton effects as well as luminescence ascribed to an E/Z transformation that limits molecular rotations. Additionally, binding with halide ions, including F−, Cl− and Br−, selectively initiates chiroptical inversion, which could further be amplified by photo irradiation. In contrast, switching to larger sized anions, such as I− and NO3−, or using a conformationally locked cyclohexanediamine molecule failed to initiate chiroptical inversion. DFT computational studies unveiled a conformational transition from a butterfly-like open state to a closed state after anion binding, thanks to the rotatable axis present in the molecule. This work established a protocol to design functional chiroptical platforms responsive to external fields.Data availability
We state that the original data can be acquired by request to the authors.Author contributions
Y. Liu carried out the main experiments and data analysis. P. Xing and A. Hao proposed the assumptions and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 22171165, 22371170).Notes and references
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS.
- J. Sheng, D. R. S. Pooler and B. L. Feringa, Chem. Soc. Rev., 2023, 52, 5875–5891 RSC.
- M. L. Ślęczkowski, M. F. J. Mabesoone, P. Ślęczkowski, A. R. A. Palmans and E. W. Meijer, Nat. Chem., 2021, 13, 200–207 CrossRef.
- T. Mori, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS.
- M. Zhang, M. Kim, W. Choi, J. Choi, D. H. Kim, Y. Liu and Z. Lin, Prog. Polym. Sci., 2024, 151, 101800 CrossRef CAS.
- W. Ma, L. Xu, A. F. De moura, X. Wu, H. Kuang, C. Xu and N. A. Kotov, Chem. Rev., 2017, 117, 8041–8093 CrossRef CAS PubMed.
- M. Hu, H.-T. Feng, Y.-X. Yuan, Y.-S. Zheng and B. Z. Tang, Coord. Chem. Rev., 2020, 416, 213329 CrossRef CAS.
- J. Y. C. Lim, I. Marques, V. Felix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 12228–12239 CrossRef CAS.
- X. Chen, R. Zhu, B. Zhang, X. Zhang, A. Cheng, H. Liu, R. Gao, X. Zhang, B. Chen, S. Ye, J. Jiang and G. Zhang, Nat. Commun., 2024, 15, 3314 CrossRef CAS PubMed.
- X. Wu, X. Han, Q. Xu, Y. Liu, C. Yuan, S. Yang, Y. Liu, J. Jiang and Y. Cui, J. Am. Chem. Soc., 2019, 141, 7081–7089 CrossRef CAS.
- M.-Y. Zheng, Z.-B. Jin, Z.-Z. Ma, Z.-G. Gu and J. Zhang, Adv. Mater., 2024, 36, 2313749 CrossRef CAS PubMed.
- L. Zhang, H.-X. Wang, S. Li and M. Liu, Chem. Soc. Rev., 2020, 49, 9095–9120 RSC.
- X. Feng, W. Meng and H. Du, Chem. Soc. Rev., 2023, 52, 8580–8598 RSC.
- C.-L. Wang, J. Wang, J.-K. Jin, B. Li, Y. L. Phang, F.-L. Zhang, T. Ye, H.-M. Xia, L.-W. Hui, J.-H. Su, Y. Fu and Y.-F. Wang, Science, 2023, 382, 1056–1065 CrossRef CAS.
- T. Zheng, R. Chen, J. Huang, T. P. Gonçalves, K.-W. Huang and Y.-Y. Yeung, Chem, 2023, 9, 1255–1269 CAS.
- J. M. Ovian, P. Vojackova and E. N. Jacobsen, Nature, 2023, 616, 84–89 CrossRef CAS PubMed.
- S. Tribedi and R. B. Sunoj, Chem. Sci., 2022, 13, 1323–1334 RSC.
- X.-H. Hu, L. Fu, J. Hou, Y.-N. Zhang, Z. Zhang and H.-F. Wang, J. Phys. Chem. Lett., 2020, 11, 1282–1290 CrossRef CAS.
- Z. Wang, Z. Cao, A. Hao and P. Xing, Chem. Sci., 2024, 15, 6924–6933 RSC.
- J. Cao, P. Weng, Y. Qi, K. Lin and X. Yan, Chem. Commun., 2024, 60, 1484–1487 RSC.
- C. Du, Z. Li, X. Zhu, G. Ouyang and M. Liu, Nat. Nanotechnol., 2022, 17, 1294–1302 CrossRef CAS PubMed.
- M. Lago-Silva, M. M. Cid, E. Quiñoá and F. Freire, Angew. Chem., Int. Ed., 2023, 62, e202303329 CrossRef CAS PubMed.
- M. Lago-Silva, M. M. Cid, E. Quiñoá and F. Freire, J. Am. Chem. Soc., 2024, 146, 752–759 CrossRef CAS PubMed.
- L. Gao, C. Xing, X. Dou, Y. Zou, C. Zhao and C. Feng, Angew. Chem., Int. Ed. Engl., 2022, 61, e202211812 CrossRef CAS.
- D. Niu, Y. Jiang, L. Ji, G. Ouyang and M. Liu, Angew. Chem., Int. Ed., 2019, 58, 5946–5950 CrossRef CAS.
- K. Maeda and E. Yashima, Top. Curr. Chem., 2017, 375, 72 Search PubMed.
- T. Pal and D. Chaudhuri, J. Am. Chem. Soc., 2023, 145, 2532–2543 CrossRef CAS.
- B. Ni, D. Vivod, J. Avaro, H. Qi, D. Zahn, X. Wang and H. Cölfen, Nat. Commun., 2024, 15, 2042 CrossRef CAS.
- H. Li, L. Kou, L. Liang, B. Li, W. Zhao, X.-J. Yang and B. Wu, Chem. Sci., 2022, 13, 4915–4921 RSC.
- J. Li, Y. Cui, Y. L. Lu, Y. Zhang, K. Zhang, C. Gu, K. Wang, Y. Liang and C.-S. Liu, Nat. Commun., 2023, 14, 5030 CrossRef CAS PubMed.
- G. Liu, M. G. Humphrey, C. Zhang and Y. Zhao, Chem. Soc. Rev., 2023, 52, 4443–4487 RSC.
- X. Meng, Y. Wang, D. Bukharina, Y. Fei, J. Wang, Y. He, Q. Miao, R. Yu, B. Jin, X. Wang, G. Chen, K. Li, V. V. Tsukruk, K. Wang and C. Ye, Adv. Opt. Mater., 2024, 12, 2303204 CrossRef CAS.
- J. S. S. K. Formen and C. Wolf, Angew. Chem., Int. Ed., 2021, 60, 27031–27038 CrossRef CAS.
- Y. Lv, C. Xiao, J. Ma, D. Zhou, W. Wu and C. Yang, Chin. Chem. Lett., 2024, 35, 108757 CrossRef CAS.
- M. Quan, X.-Y. Pang and W. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202201258 CrossRef CAS PubMed.
- Y. Hashimoto, T. Nakashima, D. Shimizub and T. Kawai, Chem. Commun., 2016, 52, 5171–5174 RSC.
- C. Petermayer and H. Dube, J. Am. Chem. Soc., 2018, 140, 13558–13561 CrossRef CAS PubMed.
- S. Guo, L.-Y. Hu, Q.-Y. Meng, Y.-Y. Zhang, C.-C. Zhang, L.-J. Xing, H. Yu and H.-L. Sun, J. Colloid Interface Sci., 2024, 657, 913–920 CrossRef CAS.
- K. Takaishi, K. Iwachido and T. Ema, J. Am. Chem. Soc., 2020, 142, 1774–1779 CrossRef CAS PubMed.
- T. Ikai, S. Okuda, M. Aizawa and E. Yashima, Macromolecules, 2022, 55, 7023–7031 CrossRef CAS.
- T. Ogoshi, T. Akutsu, D. Yamafuji, T. Aoki and T. Yamagishi, Angew. Chem., Int. Ed., 2013, 52, 8111–8115 CrossRef CAS PubMed.
- N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754 CrossRef CAS PubMed.
- J. Y. C. Lim and P. D. Beer, Chem, 2018, 4, 731–783 CAS.
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS PubMed.
- M. S. Taylor, Coord. Chem. Rev., 2020, 413, 213270 CrossRef CAS.
- S.-Y. Zhuang, Y. Cheng, Q. Zhang, S. Tong and M.-X. Wang, Angew. Chem., Int. Ed., 2020, 59, 23716–23723 CrossRef PubMed.
- J. Y. C. Lim, I. Marques, V. Felix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 12228–12239 CrossRef CAS PubMed.
- G. Sekar, V. V. Naira and J. Zhu, Chem. Soc. Rev., 2024, 53, 586–605 RSC.
- S. Chatterjee, B. Liu and H.-S. Peng, Coord. Chem. Rev., 2024, 508, 215779 CrossRef CAS.
- M. S. Li, S. Zhang, X. Y. Zhang, Y. C. Wang, J. L. Chen, Y. H. Tao and X. H. Wang, Angew. Chem., Int. Ed., 2021, 60, 6003–6012 CrossRef CAS.
- D. Zhang, T. K. Ronson, J. Mosquera, A. Martinez and J. R. Nitschke, Angew. Chem., Int. Ed., 2018, 57, 3717–3721 CrossRef CAS.
- X. Xiao, M. A. Shehzad, A. Yasmin, Z. Ge, X. Liang, F. Sheng, W. Ji, X. Ge, L. Wu and T. Xu, J. Membr. Sci., 2020, 614, 118553 CrossRef CAS.
- L. Zhang, B. Li, R. Li, Y. Wang, S. Ye, P. Zhang and B. Wu, J. Am. Chem. Soc., 2023, 145, 18221–18226 CrossRef CAS PubMed.
- S. Benz, M. Macchione, Q. Verolet, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 9093–9096 CrossRef CAS PubMed.
- Y. Zhong, B. Kauffmann, W. Xu, Z. L. Lu, Y. Ferrand, I. Huc, X. C. Zeng, R. Liu and B. Gong, Org. Lett., 2020, 22, 6938–6942 CrossRef CAS PubMed.
- S. Akine, S. Hotate and T. Nabeshima, J. Am. Chem. Soc., 2011, 133, 13868–13871 CrossRef CAS PubMed.
- G. Y. Shen, F. Gou, J. H. Cheng, X. H. Zhang, X. G. Zhou and H. F. Xiang, RSC Adv., 2017, 7, 40640–40649 RSC.
- K. Fu and G. Liu, ACS Nano, 2024, 18, 2279–2289 CrossRef CAS PubMed.
- E. Lee, H. Ju, I.-H. Park, J. H. Jung, M. Ikeda, S. Kuwahara, Y. Habata and S. S. Lee, J. Am. Chem. Soc., 2018, 140, 9669–9677 CrossRef CAS PubMed.
- T. Kinoshita, Y. Haketa, H. Maeda and G. Fukuhara, Chem. Sci., 2021, 12, 6691–6698 RSC.
- H. Maeda, Y. Bando, K. Shimomura, I. Yamada, M. Naito, K. Nobusawa, H. Tsumatori and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9266–9269 CrossRef CAS PubMed.
- H. Li, L. Kou, L. Liang, B. Li, W. Zhao and B. Wu, Chem. Sci., 2022, 13, 4915–4921 RSC.
- Y. Okayasu, K. Wakabayashi and J. Yuasa, Inorg. Chem., 2022, 61, 15108–15115 CrossRef CAS.
- A. Homberg, E. Brun, F. Zinna, S. Pascal, M. Gorecki, L. Monnier, C. Besnard, G. Pescitelli, L. Di Bari and J. Lacour, Chem. Sci., 2018, 9, 7043–7052 RSC.
- Y. Imai, Y. Nakano, T. Kawai and J. Yuasa, Angew. Chem., Int. Ed., 2018, 57, 8973–8978 CrossRef CAS.
- L. Zhu, C. Y. Ang, X. Li, K. T. Nguyen, S. Y. Tan, H. Ågren and Y. Zhao, Adv. Mater., 2012, 24, 4020–4024 CrossRef CAS PubMed.
- B. Fernandez, R. Rodriguez, A. Rizzo, E. Quinoa, R. Riguera and F. Freire, Angew. Chem., Int. Ed., 2018, 57, 3666–3670 CrossRef CAS PubMed.
- J. L. Greenfield, J. Wade, J. R. Brandt, X. Shi, T. J. Penfoldd and M. J. Fuchter, Chem. Sci., 2021, 12, 8589–8602 RSC.
- I. H. Delgado, S. Pascal, A. Wallabregue, R. Duwald, C. Besnard, L. Guenee, C. Nancoz, E. Vauthey, R. C. Tovar, J. L. Lunkley, G. Muller and J. Lacour, Chem. Sci., 2016, 7, 4685–4693 RSC.
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
- S. Sun, X. Li, C. Xu, Y. Li, Y. Wu, B. L Feringa, H. Tian and X. Ma, Natl. Sci. Rev., 2023, 10, nwad072 CrossRef.
- S. Tong, J.-T. Li, D.-D. Liang, Y.-E. Zhang, Q.-Y. Feng, X. Zhang, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 14432–14436 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, experimental details, and additional CD, 1H NMR, 13C NMR, SEM and MS spectra. See DOI: https://doi.org/10.1039/d4sc04216f |
| This journal is © The Royal Society of Chemistry 2024 |