
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting exciton dissociation in anion and cation co-doped polymeric semiconductor for selective oxidation reaction†
Junkang
Ge
,
Jun
Zhao
,
Lei
Li
 ,
Zhihao
Li
,
Hui
Wang
*,
Xiaodong
Zhang
* and
Yi
Xie
,
Zhihao
Li
,
Hui
Wang
*,
Xiaodong
Zhang
* and
Yi
Xie
Hefei National Research Center for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: wanghuig@ustc.edu.cn; zhxid@ustc.edu.cn
First published on 4th October 2024
Abstract
The inherently low dielectric properties and weak shielding effect of polymeric semiconductors cause excitons to dominate their photoexcitation process, which greatly restricts the photocatalytic performances mediated by charge carriers. Here, an anion and cation co-doping strategy was proposed to weaken the binding energy of excitons by forming distinct positive and negative charge regions, where the charge asymmetry produced an external potential to drive exciton dissociation. Using polymeric carbon nitride as a typical model framework, we show that the incorporation of anions (Cl−, Br−, I−) and cations (Na+, K+) could create a significant spatial separation of electrons and holes, thereby promoting exciton dissociation. Specifically, K+ and Cl− co-doped polymeric carbon nitride could effectively promote the dissociation of excitons into hot carriers, contributing to the outstanding efficiency in hot-electron-involved photocatalytic processes, such as the generation of superoxide radicals (O2˙−) and the oxidation of phenylboric acid under visible light. This work presents a practical approach for promoting excitons dissociation through the introduction of charge asymmetry.
Introduction
Nowadays, with the rapid development of social productivity, human demand for energy has far exceeded historical levels, and the utilization of clean energy is in great demand.1–3 Photocatalysis is considered to be a sustainable way to convert solar energy into chemical energy, whose efficiency is closely related to the concentration of photoinduced charge carriers in catalysts.4–6 As one of the most important types of photocatalysts, polymeric semiconductors have attracted great attention for their properties such as low biotoxicity, tunable band structure, and ease of structural modification.7,8 However, the strong Coulomb interaction and the weak dielectric shielding effect of polymeric semiconductors make excitons the dominant photogenerated species, where only a few of them can dissociate into free carriers to participate in the artificial photocatalytic process. Meanwhile, a high concentration of excitons in the system leads to the occurrence of exciton–exciton annihilation, resulting in the consumption of photoinduced species.9,10 Thus, the dissociation of excitons into free electrons and holes would be a practical strategy to enhance the quantum efficiency of photocatalysts, and thus, it is in demand for oxidation–reduction reactions involving charge carriers.11,12As excitons are electrically neutral quasiparticles that are formed by Coulomb interactions between the electrons and holes, the introduction of charge asymmetry generates an external potential to drive their dissociation.13–15 Among the various strategies, elemental doping is an efficient way to break the local charge symmetry of photocatalysts by introducing external ions with various levels of electronegativity.16 In addition, defective structures can serve as reactive sites to improve the separation efficiency of photo-generated charge carriers.17 Both the defective structures in the construction and elemental doping methods could enhance the concentration of photo-generated charge carriers by creating different structural units. Although single-element doping may partially promote exciton dissociation into hot carriers, the possible formation of a steady charge-transfer state prevents further separation of carriers.18,19 Herein, using typical polymeric carbon nitride (PCN) as an example, we proposed an anion and cation co-doping method to promote the exciton dissociation by introducing strong charge asymmetry. As depicted in Scheme 1, the pristine carbon nitride with uniform distribution of the surface electrostatic potential indicates the symmetry distribution of charges, further confirming its potentially strong excitonic effect. In addition, doping with either individual anions or cations in polymeric carbon nitride results in the collection of negative or positive charges in the PCN framework, partially weakening the binding of electrons and holes. As for co-doping, anions (Cl−, Br−, I−) and cations (Na+, K+) located at different sites of the PCN framework exhibit a distinct distribution of negative and positive charges, where the as-formed electrostatic potential facilitates the dissociation of excitons.
 | ||
| Scheme 1 Simulated electrostatic potential of polymeric carbon nitride under various doping conditions. | ||
To further investigate this, density functional theory (DFT) calculations were conducted to study the influence of incorporating K+ and Cl− ions for co-doping on the exciton dissociation of the polymeric carbon nitride matrix. Fig. 1a and b depict the spatial distributions of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the polymeric carbon nitride. The uniform distributions and significant overlaps on the tri-s-triazine structure rule out the possibility of the dissociation of excitons within the framework. In contrast, a significant spatial separation between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) was observed in the K+ and Cl− co-doped model (Fig. 1c and d). Specifically, Cl−-containing units with electron-rich features dominate the distribution of the highest occupied molecular orbital (HOMO), while a K+-containing unit dominates the distribution of the lowest unoccupied molecular orbital (LUMO). Such spatial separation of electrons and holes between the two distinct motifs indicates the potential dissociation of excitons in the K+, Cl− co-doped polymeric carbon nitride system.
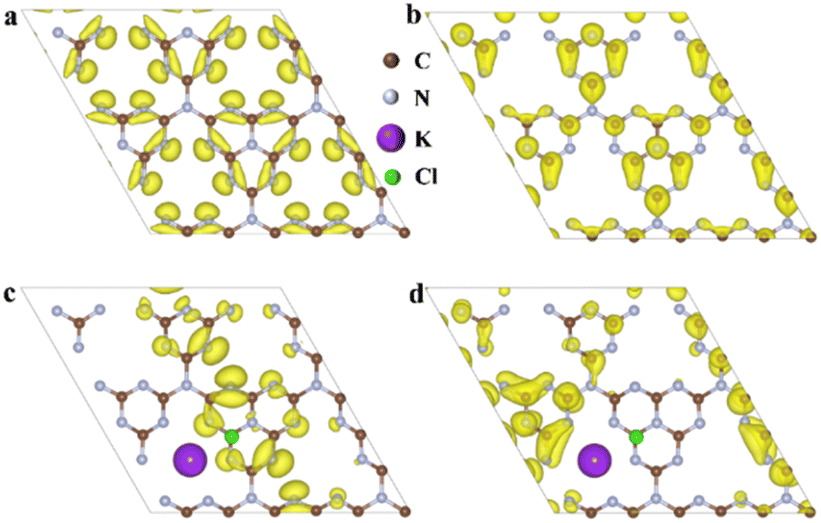 | ||
| Fig. 1 The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) distributions in PCN (a and b), and K+, Cl− co-doped CN (c and d), respectively. | ||
Results and discussion
In this study, PCN co-doped with anions (Cl−, Br−, I−) and cations (Na+, K+) was synthesized through a modified thermal polymerization of thiourea and the corresponding doping element-containing compounds (See detail in ESI†). Using the typical K+, Cl− co-doped polymeric carbon nitride (denoted as KCl–CN) as an example, a range of KCl–CN samples with varying contents of doping element and carbon nitride samples doped with individual K+/Cl− (denoted as K–CN and Cl–CN, respectively) were synthesized for comparison. The powdered X-ray diffraction (PXRD) patterns of KCl–CN (Fig. 2a and S1†) exhibit a weakened (100) peak and (002) peak, indicating a decrease in crystallinity and an increase in structural distortion of PCN after elemental doping.20 As shown in Fig. 2b, the KCl–CN spectrum exhibits a similar Fourier transform infrared (FT-IR) spectrum as that of PCN, indicating that both retained essentially tri-s-triazine ring units. However, compared to the PCN, the FT-IR spectrum of KCl–CN exhibits a prominent peak at 2181 cm−1, which can be attributed to the stretching vibration of cyano groups (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) formed from the amine groups (–NH2) in the heptazine rings.21 Furthermore, KCl–CN shows two new absorption peaks at 992 and 1154 cm−1, which are attributed to the ion–dipole interaction of heptazine with alkali ions (K+) and halide ions (Cl−) over KCl–CN. Solid-state 13C NMR spectra (Fig. 2c) show that KCl–CN presents two new peaks at 122.7 and 171.6 ppm, which are likely associated with the C (3) atom in the cyano group (–CN) and the C (4) atom in the polymeric carbon nitride structure.22,23 In addition, elemental mapping with the energy-dispersive X-ray spectra (EDS) (Fig. S4†) illustrates the uniform distribution of K+ and Cl− in the KCl–CN. X-ray photoelectron measurements (XPS) were performed to further explore the elements and chemical states of samples. In the high-resolution C 1s-K 2p XPS spectra (Fig. 2d), KCl–CN exhibits two distinct new peaks at 295.6 and 292.7 eV, corresponding to the K 2p3/2 and K 2p1/2 peaks of the K element. Meanwhile, KCl–CN exhibits a significant Cl 2p3/2 and Cl 2p1/2 signal in the Cl 2p spectrum (Fig. 2e). As compared to PCN, the peak corresponding to the C–NHx at 288.3 eV in KCl–CN exhibits an obvious shift toward low energy, owing to the formation of cyano groups (–CN) with a strong electron-withdrawing effect during the polymerization process. Similar low-energy shifts were observed in the N 1s spectra (Fig. S5b†).24 Interestingly, the binding energy of K+ and Cl− in KCl–CN is lower and higher than that of the KCl, respectively, implying the K+ and Cl− are incorporated into PCN and bonded with the surrounding C and N atoms.25 In the synchrotron X-ray absorption near-edge structure (XANES) spectra (Fig. 2f), as compared with PCN, KCl–CN presented new peaks at 298.2 and 302.5 eV, corresponding to K L2,3.26,27 Based on these results, it is safe to conclude that K+ and Cl− were successfully introduced into PCN. At the same time, the other anions, i.e., Br− and I−, and cations, i.e., Na+ and K+, were also successfully co-doped into the PCN framework (Fig. S6 and S7†).
N) formed from the amine groups (–NH2) in the heptazine rings.21 Furthermore, KCl–CN shows two new absorption peaks at 992 and 1154 cm−1, which are attributed to the ion–dipole interaction of heptazine with alkali ions (K+) and halide ions (Cl−) over KCl–CN. Solid-state 13C NMR spectra (Fig. 2c) show that KCl–CN presents two new peaks at 122.7 and 171.6 ppm, which are likely associated with the C (3) atom in the cyano group (–CN) and the C (4) atom in the polymeric carbon nitride structure.22,23 In addition, elemental mapping with the energy-dispersive X-ray spectra (EDS) (Fig. S4†) illustrates the uniform distribution of K+ and Cl− in the KCl–CN. X-ray photoelectron measurements (XPS) were performed to further explore the elements and chemical states of samples. In the high-resolution C 1s-K 2p XPS spectra (Fig. 2d), KCl–CN exhibits two distinct new peaks at 295.6 and 292.7 eV, corresponding to the K 2p3/2 and K 2p1/2 peaks of the K element. Meanwhile, KCl–CN exhibits a significant Cl 2p3/2 and Cl 2p1/2 signal in the Cl 2p spectrum (Fig. 2e). As compared to PCN, the peak corresponding to the C–NHx at 288.3 eV in KCl–CN exhibits an obvious shift toward low energy, owing to the formation of cyano groups (–CN) with a strong electron-withdrawing effect during the polymerization process. Similar low-energy shifts were observed in the N 1s spectra (Fig. S5b†).24 Interestingly, the binding energy of K+ and Cl− in KCl–CN is lower and higher than that of the KCl, respectively, implying the K+ and Cl− are incorporated into PCN and bonded with the surrounding C and N atoms.25 In the synchrotron X-ray absorption near-edge structure (XANES) spectra (Fig. 2f), as compared with PCN, KCl–CN presented new peaks at 298.2 and 302.5 eV, corresponding to K L2,3.26,27 Based on these results, it is safe to conclude that K+ and Cl− were successfully introduced into PCN. At the same time, the other anions, i.e., Br− and I−, and cations, i.e., Na+ and K+, were also successfully co-doped into the PCN framework (Fig. S6 and S7†).
 | ||
| Fig. 2 (a) Powder XRD patterns, (b) FT-IR spectra, (c) solid-state 13C NMR spectra (inset: normalization intensity of solid-state 13C NMR spectra) and (d) C 1s XPS spectra of PCN and KCl–CN; (e) Cl 2p spectrum of KCl–CN; (f) C K-edge XANES curves of PCN and KCl–CN. | ||
The introduction of K+ and Cl− into PCN is anticipated to have a significant impact on its excitonic effects, where spectroscopic measurements were carried out to investigate the photoexcitation processes involved. Compared PCN, KCl–CN narrows the band gap and enhances the absorption capacity of visible light, as illustrated in ultraviolet-visible (UV-vis) spectra and the corresponding Tauc plots in Fig. 3a. Based on their light absorption, the excitation wavelength for photoluminescence (PL) measurements was selected at 365 nm. As shown in Fig. 3b, the photoluminescence intensity of KCl–CN is lower than that of PCN, indicating a significant reduction in the population of singlet excitons in KCl–CN. Time-resolved fluorescence spectroscopy was carried out to monitor the corresponding lifetime of emission peaks. The radiative decay results (Fig. 3b inset and Table S1†) could be described as a triple-exponential fitting, yielding lifetimes of 1.96 ns for PCN and 0.41 ns for KCl–CN. The reduction in the radiation emission lifetime can be understood by the reduction in the population of the singlet excitons in KCl–CN.28,29 Phosphorescence spectra (Fig. 3c) were employed to investigate long-lived photoinduced species, revealing that PCN exhibits a more pronounced phosphorescence emission compared to KCl–CN. Furthermore, time-resolved phosphorescence kinetics spectra show that the phosphorescence lifetime of KCl–CN is shorter than that of PCN, indicating a distinct reduction in the population and lifetime of triplet excitons in KCl–CN.18 Considering the competitive relationship between excitons and hot carriers, the reduced number of excitons is beneficial to enhance the generation of charge carriers. Electrochemical measurements were carried out to further verify this conjecture. As shown in Fig. 3d, KCl–CN exhibits a higher surface photovoltage intensity than that of PCN, indicating efficient carrier generation due to co-doping with K+ and Cl−. Furthermore, KCl–CN exhibits a 6.1∼fold higher response on periodic on/off photocurrent to PCN under a 400 nm cut-off filter at a 0.4 V bias voltage. As evidenced by the electrochemical impedance spectroscopy (EIS) results in Fig. 3e, KCl–CN possesses a lower electrochemical impedance than the PCN under illumination, leading to improved electron transport.30,31
 | ||
| Fig. 3 (a) UV-vis spectra (inset: corresponding Tauc plots), (b) photoluminescence spectra (inset: time-resolved fluorescence spectra), (c) phosphorescence spectra (inset: time-resolved phosphorescence spectra), (d) surface photovoltage (inset: periodic on/off photocurrent response curves at a constant potential of approximately 0.4 V vs. Ag/AgCl reference electrode in 0.5 M Na2SO4 under the light on/off condition) and (e) EIS Nyquist plots under visible light conditions of PCN and KCl–CN (EIS was performed over a frequency range of 0.01 Hz to 106 Hz with an AC voltage amplitude of 5 mV); (f) schematic illustration of the exciton dissociation process of anion and cation co-doped samples. | ||
The above results confirm that K+ and Cl− co-doping has significant positive effects on boosting exciton dissociation into hot carriers under light excitation. Similarly, other anion (Cl−, Br−, I−) and cation (Na+, K+) co-doped polymeric carbon nitride samples with enhanced exciton dissociation exhibit greatly enhanced periodic on/off photocurrent compared to that of the corresponding individual anion/cation-doped counterpart (Fig. S12†). Thus, anion (Cl−, Br−, I−) and cation (Na+, K+) co-doped polymeric carbon nitride samples promote the dissociation process of excitons, which can be understood by the processes illustrated in Fig. 3f.
Boosting hot carrier generation and transfer in the anion and cation co-doped samples leads to greatly enhanced photocatalytic activities, which were systematically studied using KCl–CN as an example. The Mott–Schottky curves (Fig. S14†) demonstrate the n-type semiconducting characteristics of the samples, where the flat potentials of PCN and KCl–CN were measured to be −1.14 and −1.04 V vs. NHE, respectively. Consequently, the band structure of PCN and KCl–CN (Fig. S15†) indicates that both samples are capable of driving the reduction in ground-state oxygen (O2) to superoxide radical (O2˙−). Nitrotetrazolium blue chloride (NBT) was used as a feature probe to evaluate the O2˙− generation ability of samples, which can react with O2˙− to produce endoperoxide. As shown in Fig. 4a, the time-dependent NBT oxidation absorption curves demonstrate that KCl–CN can quickly react with NBT, as compared to the PCN, indicating enhanced O2˙− generation of KCl–CN. Furthermore, electron spin resonance (ESR) measurements were used to prove the existence of photogenerated reactive oxygen species (ROS); 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) was used as a trapping agent. As shown in Fig. 4b, compared to PCN, KCl–CN typically exhibits a stronger signal of O2˙−. Given its capabilities in O2˙− generation, the KCl–CN is anticipated to be an efficient catalyst for the aerobic oxidation of organic molecules, such as the selective oxidation of phenylboronic acid to corresponding phenols and derivatives. As is known, phenols and their derivatives are important aromatic chemicals that are widely employed in industrial, medicinal, and cosmetic domains.32,33 As depicted in Fig. 4c, KCl–CN exhibited a robust yield in the conversion of phenylboronic acid to phenol under visible light, while the yields of PCN were negligible. Control experiments conducted under various atmospheric conditions indicated that the oxidation reaction depends on the activation of oxygen (Fig. 4d). The addition of p-BQ (a scavenger for O2˙−) results in a significant reduction of yield during the oxidation process of phenylboronic acid.
 | ||
| Fig. 4 (a) Time-dependent NBT degradation curves, (b) ESR trapping spectra of PCN and KCl–CN in the presence of DMPO in methanol, (c) time-dependent phenylboric acid oxidation curves of PCN and KCl–CN, (d) different reaction conditions for phenylboric acid oxidation spectrum in KCl–CN, (e) illumination wavelength-dependent phenylboric acid oxidation curves of PCN and KCl–CN, and (f) different catalysts for phenylboric acid oxidation spectrum. All of the above tests and experiments were performed under λ ≥ 400 nm. | ||
In addition, Fig. 4e exhibits wavelength-dependent phenylboric acid oxidation, indicating that KCl–CN possesses much higher photocatalytic phenylboric acid oxidation ability than PCN. Fundamentally, Fig. 4f indicates that PCN co-doped with anions and cations has a superior oxidation capacity of phenylboric acid, and thus, it shows greatly enhanced phenylboric acid oxidation yield compared to the corresponding individual anions (Cl−, Br−, I−)/cations (Na+, K+)-doped counterpart (Fig. S18†). At last, the catalytic activity and structural configuration of KCl–CN were maintained during five consecutive cycling experiments, as evidenced using PXRD and FT-IR characterizations (Fig. S20 and S21†).
With the optimal reaction conditions in our hand, we explored another phenylboric acid with different substitutions to evaluate the advantages and scope of photocatalytic selective oxidation reactions over the KCl–CN catalyst. In general, KCl–CN demonstrated exceptional performance, regardless of the presence of electron-withdrawing (2–6, Table 1) or -donating groups (7, Table 1), as well as the positions of the substituents (7–9, Table 1). Overall, KCl–CN demonstrated the ability to catalyze the production of a wide range of phenols under visible light, indicating its potential for application in selective photocatalytic oxidation reactions.

|
Conclusions
An anion (Cl−, Br−, I−) and cation (Na+, K+) co-doping method has been proposed to achieve efficient exciton dissociation in polymeric semiconductors, where boosting the charge carrier generation and photocatalytic efficiency have been demonstrated. Specifically, using the anion and cation co-doped polymeric carbon nitride as an example, we show that the K+ and Cl− co-doped polymeric carbon nitride could create a significant spatial separation of electrons and holes in the formation of charge asymmetry. In addition, all the samples co-doped with anions and cations exhibit enhanced exciton dissociation compared to the corresponding individual anion/cation-doped counterparts. As a result, the anion and cation co-doped samples can readily activate molecular oxygen to form superoxide radicals (O2˙−) under visible light, which subsequently react with phenylboronic acid to produce the corresponding phenol. This work provides a practical approach to efficiently promote exciton dissociation and shows the great advantage of co-doping with anions and cations in the photocatalytic process.Data availability
Additional experimental details and data are provided in the ESI,† including XRD, EDS, XPS, XANES, SEM, TEM, BET, Mott–Schottky plots, EIS Nyquist plots, and ESR.Author contributions
J. G. performed the experiments and collected the data. L. L. and Z. L. conducted and analyzed the DFT calculations. J. Z. assisted in performing the experiments. H. W. and X. Z. cowrote the manuscript. H. W., X. Z. and Y. X. supervised the project. All authors approved the final version of the manuscript.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFA1502903), the National Natural Science Foundation of China (T2122004, 92163105, U2032212, 22275179), the Fundamental Research Funds for the Central Universities (WK2060000039), and the Anhui Provincial Natural Science Foundation (2108085J07). The numerical calculations in this paper were performed on the supercomputing system in the Supercomputing Center of the University of Science and Technology of China. We also thank the Beamlines MCD-A and MCD-B (Soochow Beamline for Energy Materials) at the National Synchrotron Radiation Laboratory (NSRL) for their assistance with characterization. This work was partially carried out at the Instruments Center for Physical Science, University of Science and Technology of China.References
- S. Shafiee and E. Topal, Energy Policy, 2009, 37, 181–189 CrossRef.
- D. G. Vlachos and S. Caratzoulas, Chem. Eng. Sci., 2010, 65, 18–29 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS.
- H. Kisch, Acc. Chem. Res., 2017, 50, 1002–1010 CrossRef CAS PubMed.
- A. Kumar Singh, C. Das and A. Indra, Coord. Chem. Rev., 2022, 465, 214516 CrossRef CAS.
- G. Grando, G. Sportelli, G. Filippini, M. Melchionna and P. Fornasiero, NanoTrends, 2023, 4, 100028 Search PubMed.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- J. Dong, Z. Gong, Y. Chen, G. Hao, W. Zhou, J. Li, M. Yang, R. Deng and L. Wang, Sci. China Mater., 2023, 66, 3176–3188 CrossRef CAS.
- H. Wang, S. Jin, X. Zhang and Y. Xie, Angew. Chem., Int. Ed., 2020, 59, 22828–22839 CrossRef CAS PubMed.
- Y. Li, S. Li, L. Liu, Z. Zhang and W. Ma, Appl. Catal., B, 2023, 321, 122025 CrossRef CAS.
- S. Lin, B. Wu, Q. Li, X. Xiao, M. Zheng, J. Liu, Y. Xie and B. Jiang, Sci. China Mater., 2023, 66, 4680–4688 CrossRef.
- A. Avateev, I. Ghosh, B. König and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 15936–15947 CrossRef.
- G. D. Scholes and G. Rumbles, Nat. Mater., 2006, 5, 683–696 CrossRef CAS.
- M. Dvorak, S. H. Wei and Z. Wu, Phys. Rev. Lett., 2013, 110, 016402 CrossRef PubMed.
- L. Yu, H. Li, H. Shang, P. Xing, B. Zhou, Z. Chen, X. Liu, H. Zhang, Y. Shi and L. Zhang, ACS Nano, 2023, 17, 15077–15084 CrossRef CAS.
- K. H. Eckstein, H. Hartleb, M. M. Achsnich, F. Schöppler and T. Hertel, ACS Nano, 2017, 11, 10401–10408 CrossRef CAS.
- X. N. Yu, S. F. Ng, L. K. Putri, L. L. Tan, A. R. Mohamed and W. J. Ong, Small, 2021, 17, 2006851–2006896 CrossRef CAS.
- P. Zhang, L. Li, J. Zhao, H. Wang, X. Zhang and Y. Xie, Precis. Chem., 2023, 1, 40–48 CrossRef CAS.
- C. K. Lee, L. Shi and A. P. J. Willard, Phys. Chem. Lett., 2016, 7, 2246–2251 CrossRef CAS.
- X. Li, G. Fang, T. Wu, Q. Tian, Q. Yang and Z. Chen, Appl. Surf. Sci., 2023, 635, 157717 CrossRef CAS.
- X. Li, H. Mai, X. Wang, Z. Xie, J. Lu, X. Wen, S. P. Russo, D. Chen and R. A. Caruso, J. Mater. Chem. A, 2024, 12, 5204–5214 RSC.
- B. Jürgens, E. Irran, J. Senker, P. Kroll, H. Müller and W. Schnick, J. Am. Chem. Soc., 2003, 125, 10288–10300 CrossRef.
- H. Ou, P. Yang, L. Lin, M. Anpo and X. Wang, Angew. Chem., Int., Ed., 2017, 56, 10905–10910 CrossRef CAS PubMed.
- Y. Xu, M. Fan, W. Yang, Y. Xiao, L. Zeng, X. Wu, Q. Xu, C. Su and Q. He, Adv. Mater., 2021, 33, 2101455 CrossRef CAS PubMed.
- J. Stoch and M. Ladecka, Appl. Surf. Sci., 1988, 31, 426–436 CrossRef CAS.
- A. W. Gillespie, A. Diochon, B. L. Ma, M. J. Morrison, L. Kellman, F. L. Walley, T. Z. Regier, D. Chevrier, J. J. Dynes and E. G. Gregorich, Biogeochemistry, 2013, 117, 337–350 CrossRef.
- Y. Xu, M. Fan, W. Yang, Y. Xiao, L. Zeng, X. Wu, Q. Xu, C. Su and Q. He, Adv. Mater., 2021, 33, 2101455 CrossRef CAS.
- A. Indra, R. Beltrán-Suito, M. Müller, R. P. Sivasankaran, M. Schwarze, A. Acharjya, B. Pradhan, J. Hofkens, A. Brückner, A. Thomas, P. W. Menezes and M. Driess, ChemSusChem, 2021, 14, 306 CrossRef CAS PubMed.
- H. Wang, X. Sun, D. Li, X. Zhang, S. Chen, W. Shao, Y. Tian and Y. Xie, J. Am. Chem. Soc., 2017, 139, 2468–2473 CrossRef CAS PubMed.
- A. Indra, A. Acharjya, P. W. Menezes, C. Merschjann, D. Hollmann, M. Schwarze, M. Aktas, A. Friedrich, S. Lochbrunner, A. Thomas and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 1653 CrossRef CAS.
- V. Kumar, V. Vyas, D. Kumar, A. Kushwaha and A. Indra, Chem. Sci., 2024, 15, 13218–13226 RSC.
- R. Bal, M. Tada, T. Sasaki and Y. Iwasawa, Angew. Chem., Int. Ed., 2006, 45, 448452 CrossRef.
- K. Dong, Y. Yao, H. Li, H. Li, S. Sun, X. He, Y. Wang, Y. Luo, D. Zheng, Q. Liu, Q. Li, D. Ma, X. Sun and B. Tang, Nat. Synth., 2024, 3, 763–773 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04201h |
| This journal is © The Royal Society of Chemistry 2024 |