
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal π-Lewis base activation in palladium(0)-catalyzed trans-alkylative cyclization of alkynals†
Lei
Zhu‡
 ab,
Bo
Zhao‡
a,
Ke
Xie
c,
Wu-Tao
Gui
c,
Sheng-Li
Niu
a,
Peng-Fei
Zheng
a,
Ying-chun
Chen
ac,
Xiao-Wei
Qi
*b and
Qin
Ouyang
*a
ab,
Bo
Zhao‡
a,
Ke
Xie
c,
Wu-Tao
Gui
c,
Sheng-Li
Niu
a,
Peng-Fei
Zheng
a,
Ying-chun
Chen
ac,
Xiao-Wei
Qi
*b and
Qin
Ouyang
*a
aCollege of Pharmacy, Third Military Medical University, Shapingba, Chongqing 400038, China. E-mail: ouyangq@tmmu.edu.cn
bBreast and Thyroid Surgery, Southwest Hospital, Third Military Medical University, Shapingba, Chongqing 400038, China. E-mail: qxw9908@foxmail.com
cKey Laboratory of Drug-Targeting and Drug Delivery System of the Ministry of Education and Sichuan Research Center for Drug Precision Industrial Technology, West China School of Pharmacy, Sichuan University, Chengdu 610041, China
First published on 18th July 2024
Abstract
The Pd(0)-mediated umpolung reaction of an alkyne to achieve trans-difunctionalization is a potential synthetic methodology, but its insightful activation mechanism of Pd(0)–alkyne interaction has yet to be established. Here, a Pd(0)–π-Lewis base activation mode is proposed and investigated by combining theoretical and experimental studies. In this activation mode, the Pd(0) coordinates to the alkyne group and enhances its nucleophilicity through π-back-donation, facilitating the nucleophilic attack on the aldehyde to generate a trans-Pd(II)–vinyl complex. Ligand-effect studies reveal that the more electron-donating one would accelerate the reaction, and the cyclization of the challenging flexible C- or O-tethered substrates has been realized. The origin of regioselectivities is also explicated by the newly proposed metal π-Lewis base activation mode.
Introduction
Transition metal-catalyzed transformations of alkynes have been widely used in the preparation of organic intermediates, bioactive molecules, and optical materials, owing to the unique nucleophilic and electrophilic potentials of the C–C triple bond.1 Among them, palladium catalysis always occupies a leading position and has been applied to the construction of various C–X bonds.2 In the current stage of this art, the activation of alkynes by palladium occurs via two major modes: (1) migratory insertion of organopalladium reagents formed via oxidative addition, C–H bond activation or transmetallation to the alkynes to give vinyl–Pd species (Scheme 1a);1e,3 (2) metal π-Lewis acid activation, in which π-coordination of an electrophilic Pd(II) complex removes electron density from the alkynes and renders an outer-sphere nucleophilic attack (Scheme 1b).4 In general, alkynes act as electrophilic partners in both modes, which might significantly restrict the potential of Pd-catalyzed transformations. In fact, the Pd-mediated nucleophilic addition reaction of alkynes has been significantly underdeveloped. | ||
| Scheme 1 Diverse activation modes for Pd-catalyzed transformations involving alkynes. (a) Migratory insertion; (b) π-Lewis acid activation; (c) Pd(0)-catalyzed intramolecular alkylative cyclization; (d) vinylogous π-Lewis base activation; (e) direct π-Lewis base activation. | ||
Tsukamoto and co-workers uncovered an intriguing Pd(0)-catalyzed intramolecular alkylative cyclization of alkynal substrates followed by a cascade Suzuki reaction with organoboronic reagents.5 Unexpectedly, this transformation proceeded trans-selectively against the Ni or Rh-mediated cis-oxidative cyclometallation process,6 and a formal anti-Wacker-type activation mode, in which the Pd(0) catalyst coordinates with the alkyne guiding the nucleophilic attack on the carbonyl functionality, was proposed (Scheme 1c).7 However, the catalytic mechanism, especially the intrinsic Pd(0)–alkyne interaction in this activation mode remains unclear. Due to the lack of theoretical awareness of such an activation mode, the development of Pd(0)-mediated nucleophilic reactions of alkynes was yet at a standstill in recent years.
In a Chatt–Dewar–Duncanson coordination mode, besides the Pd acting as a π-Lewis acid by accepting π-electrons of alkynes, the empty antibonding molecular orbital of the alkyne (π*) is able to accept electrons from the d-orbitals of Pd via π-back-donation.8 Thus it can provide a possible catalytic mode via π-Lewis base activation.9 Our recent studies indicate that Pd(0) can activate 1,3-dienes10 and 1,3-enynes11 to react with electrophilic partners via η2-coordination in a vinylogous manner, in which the generation of π-allylpalladium complexes supplies the essential driving force (Scheme 1d). These emphasize the potential of Pd(0) as a π-Lewis base catalyst to directly activate alkynes. Consequently, we speculated that a metal π-Lewis base activation mode would be reasonable for Pd(0)-catalyzed alkylative transformation of alkynals. The electron flowing to the alkyne from the electron-rich Pd(0) center through π-back-donation would enhance the nucleophilic reactivity of the η2-coordinated Pd(0)–alkyne complex, which would allow a Friedel–Crafts type reaction with electrophilic partners to generate a vinyl–Pd(II) complex (Scheme 1e).
Herein, we carried out a mechanistic study on Pd(0)-catalyzed alkyne transformation, using Tsukamoto's report as a model reaction to elucidate our newly proposed metal π-Lewis base activation mode (Scheme 2).5a We performed a combined theoretical and experimental investigation on the activation mechanism, ligand effects, and origin of selectivity. Importantly, inspired by the theoretical understandings, this Pd(0)-catalyzed transformation was consequently extended to adapt the more flexible C-tethered or O-tethered alkynal substrates.
 | ||
| Scheme 2 Pd(0)-catalyzed alkyne functionalization with electrophiles. | ||
Computational methods
In this work, all of the density functional theory (DFT) calculations were performed using Gaussian 09![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 software packages. The B3LYP13 function together with the Def2-SVP basis set was used for the geometry optimization and frequency analysis. Vibrational frequency calculations were performed for all the stationary points to confirm if each optimized structure is a local minimum or a transition state structure, as well as derive the thermochemical corrections for the enthalpies and free energies. The intrinsic reaction coordinate (IRC) path was performed to check the energy profiles connecting each transition state to two associated minima of the proposed intermediates. After optimization, the M0614 function with the Def2-TZVP basis set was used to calculate the single-point energies to give more accurate energy information. The integration grids defined by the Int=Ultrafine keyword were used for all calculations. Besides, the solvent effect was considered by single-point calculations at the gas-phase stationary points with the SMD solvation model.15 When obtaining the relative Gibbs energy at 298 K, a correction of −2.6 (or 2.6) kcal mol−1 was performed for the transformation involving two molecules into one molecule (or one molecule into two molecules), to reduce the overestimation of entropy contribution.16 Optimized structures are illustrated by using CYLview.17
12 software packages. The B3LYP13 function together with the Def2-SVP basis set was used for the geometry optimization and frequency analysis. Vibrational frequency calculations were performed for all the stationary points to confirm if each optimized structure is a local minimum or a transition state structure, as well as derive the thermochemical corrections for the enthalpies and free energies. The intrinsic reaction coordinate (IRC) path was performed to check the energy profiles connecting each transition state to two associated minima of the proposed intermediates. After optimization, the M0614 function with the Def2-TZVP basis set was used to calculate the single-point energies to give more accurate energy information. The integration grids defined by the Int=Ultrafine keyword were used for all calculations. Besides, the solvent effect was considered by single-point calculations at the gas-phase stationary points with the SMD solvation model.15 When obtaining the relative Gibbs energy at 298 K, a correction of −2.6 (or 2.6) kcal mol−1 was performed for the transformation involving two molecules into one molecule (or one molecule into two molecules), to reduce the overestimation of entropy contribution.16 Optimized structures are illustrated by using CYLview.17
Results and discussion
Explication of Pd(0)–π-Lewis base activation mode
At the outset of this study, we devoted ourselves to investigating the nature of Pd(0)-mediated π-Lewis base activation of alkynes. As depicted in our aforementioned metal π-Lewis base activation mode, when an alkyne coordinates to the Pd(0) center, the electron flows from the electron-rich Pd(0) center to the alkyne through back donation. It would result in the increase of the HOMO-energy of the η2-coordinated Pd(0)–alkyne complex as well as the enhancement of electron density for the alkyne moiety. To validate this, we conducted the molecular orbitals (MO) and natural population analysis (NPA) for the Pd(0)–alkyne complex.As shown in Fig. 1a, the complex of acetylene and (PMe3)2Pd(0) was employed as a model for the molecular orbital interaction analysis. The d-orbitals are considered to have π-symmetry, which could interact with π and π* of the alkyne. For the d/π interaction, the bonding combination of dxz and π gives rise to 1# orbital, and the antibonding combination results in 2# orbital. Meanwhile, the 3# orbital is derived by the bonding combination of dxy and π* echoing the d/π* interaction, which indicates a significant back donation. The 3# and 2# orbitals are the HOMO and HOMO-1 for the (Me3)P2Pd(0)–acetylene complex, which is much higher than the HOMO of acetylene (−8.06 eV). It suggests that the nucleophilicity of the (Me3)P2Pd(0)–acetylene complex is essentially enhanced and qualified for a Friedel–Crafts type process, due to the activation by the Pd(0) catalyst. Moreover, the computed Mulliken charge of the PdL2 fragment in the (Ph3)P2Pd(0)–alkynal complex int-1 is +0.173, indicating a considerable amount of charge transfer from PdL2 to alkynal 1a (Fig. 1b).18 It further reveals the increase of electron density for the alkyne moiety in the π-Lewis base activation mode.
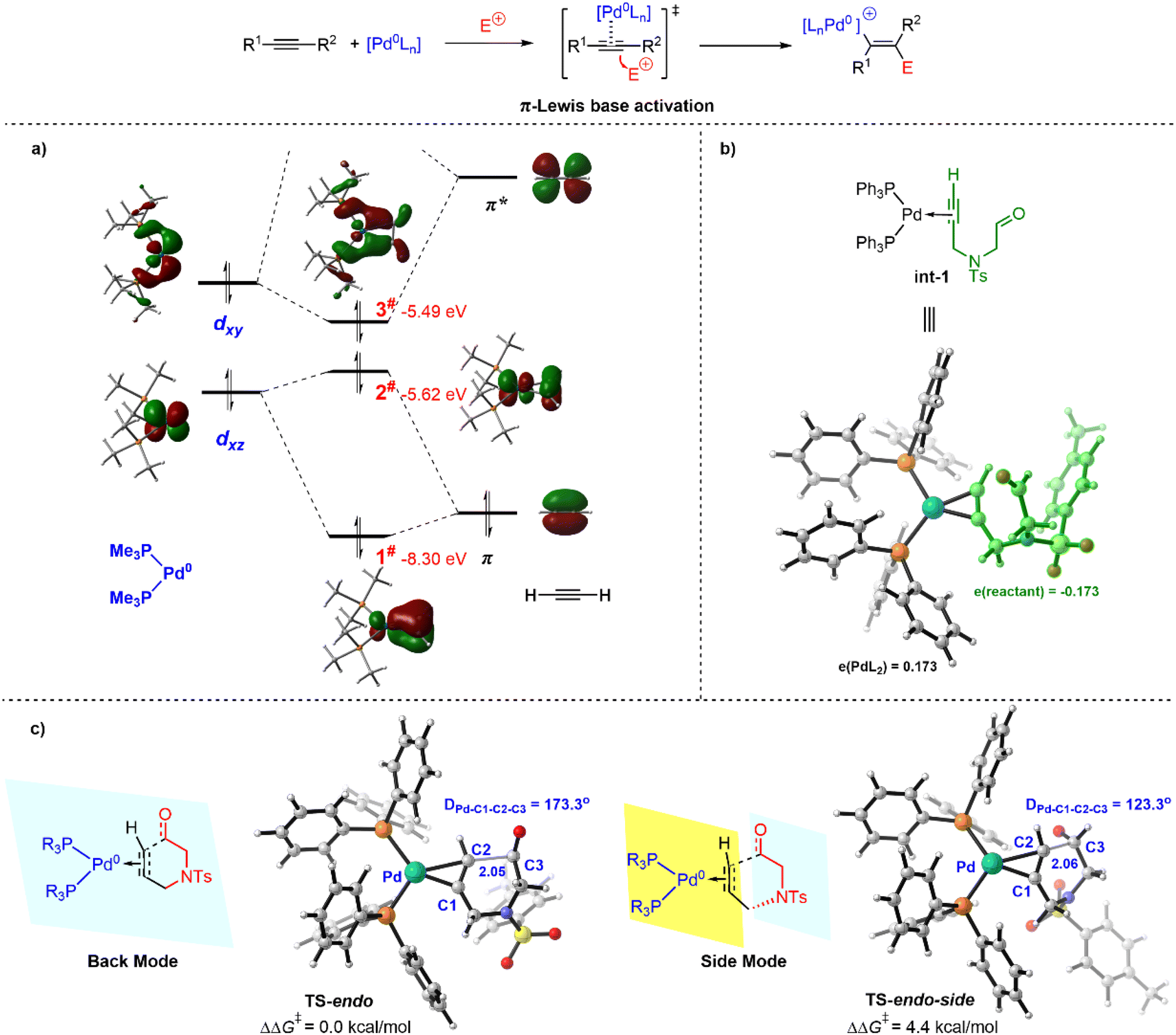 | ||
| Fig. 1 Pd(0)–π-Lewis base activation of alkynes. (a) Molecular orbital analysis for Pd(0) and alkyne. (Me3)P2Pd(0) and acetylene were employed as a simplified model. (b) Mulliken charge distribution of η2-coordinated Pd(0)–alkynal complex. (c) Comparison of back and side modes for π-Lewis base activation. | ||
The nodal planes of orbital 2# and 3# are perpendicular and their orbital energy levels are close, so both of them may exhibit related reactivity. It means the electrophilic partners could react in the back (referring to 3#) or side direction (referring to 2#) of the alkyne. Therefore, we then evaluated the reactivity of the back and side modes for π-Lewis base activation. As depicted in Fig. 1c, the dihedral angle DPd-C1-C2-C3 is 173.3° in the transition state TS-endo, and the corresponding dihedral angle is 123.3° in TS-endo-side, relating to the back and side modes, respectively. Calculated results show that the relative free energy of TS-endo is 4.4 kcal mol−1 lower than that of TS-endo-side, which is correlated with the molecular orbital energy of 2# and 3#. It suggests that the back mode is more favorable in this Pd(0)-catalyzed π-Lewis base activation of alkynal 1a.
Comparison of different activation modes
With the activation mode constructed, we then evaluated the reactivity difference for the π-Lewis base activation and others. As shown in Fig. 2, three activation modes including π-Lewis base activation (Path-A), oxidative cyclization (Path-B), and π-Lewis acid activation (Path-C) were taken into consideration. The active (Ph3)P2Pd(0)–alkynal complex int-1 could be generated after the ligand exchange between Pd(PPh3)4 and reactant alkynal 1a, which is endergonic by 10.9 kcal mol−1. Starting from int-1, the π-Lewis base activation (Path-A) proceeds viaTS-endo with a moderate activation free energy of 28.0 kcal mol−1. As a result, the zwitterionic trans-Pd(II)–vinyl complex int-2 was formed, which allows a follow-up Suzuki reaction to access the trans-difunctionalized product. Meanwhile, MeOH and arylboronic acid are considered to stabilize the alkoxide in the π-Lewis base activation mode but these processes possess high energy barriers (Fig. S2†). The vinyl five-membered palladacycle complex int-oxcyc could be generated through oxidative cyclization (Path-B).19 Although this process is less endothermic than Path-A, the activation free energy is 36.9 kcal mol−1 (referring to TS-oxcyc), indicating it is kinetically unfavorable. This is consistent with the observation of the absence of cis-difunctionalized products. In addition, the Pd(0) catalyst could be considered as a Lewis acid to activate the aldehyde moiety (Path-C). However, the activation free energy of this pathway is extremely high, about 54.1 kcal mol−1 (referring to TS-LA), which reveals the insufficient Lewis acidity of the Pd(0) complex. Therefore, these results suggest that this Pd(0)-catalyzed trans-difunctionalization of alkynal 1a would preferentially undergo the π-Lewis base activation pathway. | ||
| Fig. 2 Comparison of the π-Lewis base activation, oxidative cyclization, and π-Lewis acid activation modes. ΔG in this figure is in kcal mol−1 with respect to the Pd(PPh3)4 and reactant alkynal 1a. | ||
Catalytic cycle
As shown in Fig. 3, starting from the zwitterionic Pd(II)–vinyl complex int-2, the protonation by methanol affords the neutral trans-Pd(II)–vinyl–OMe complex int-3, and the generated counter ion MeO− could be considered as an activator of phenylboronic acid to realize the following Suzuki reaction.20 It proves that MeOH was a critical solvent in the experiment.5a Subsequent transmetallation proceeds via the four-centered cyclic transition state TS-TM with an energy barrier of 16.7 kcal mol−1, in which one of the PPh3 was dissociated to afford unsaturation.21 This transmetalation process is driven by the formation of a stable B–O bond in MeO–B(OH)2, and gives the Pd(II)–vinyl–phenyl complex int-4 irreversibly. Then the C–C reductive elimination occurs rapidly to release the final trans-difunctionalized product 3a, and the active (Ph3)P2Pd(0)–alkynal complex int-1 is also regenerated.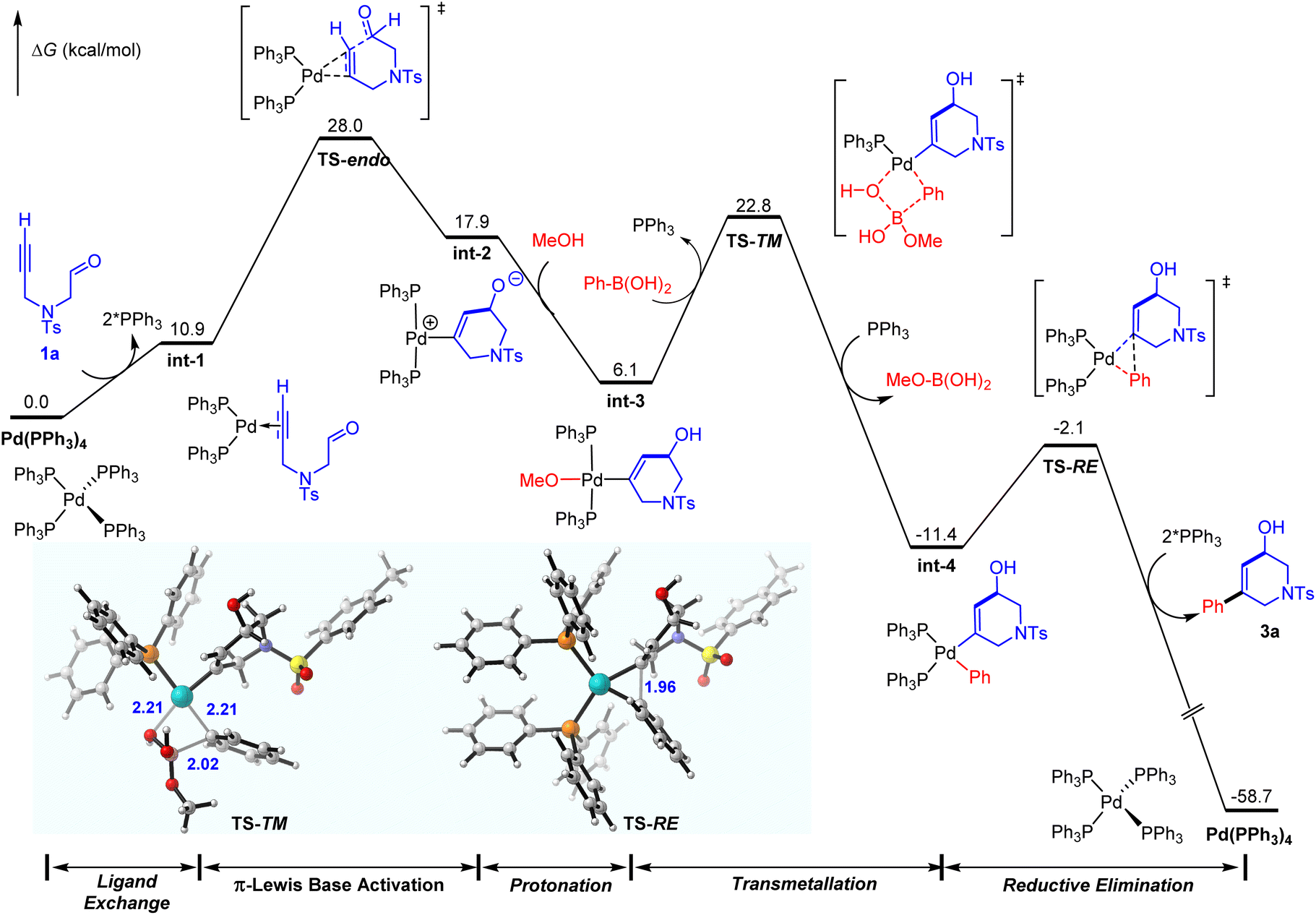 | ||
| Fig. 3 Whole free energy profile for the Pd(0)-catalyzed trans-alkylative cyclization of the alkynal. | ||
Moreover, to exclude the possibility of the Pd(II)-catalyzed mechanism involving sequential transmetalation, alkyne 1,2-carbopalladation, Z/E-isomerization, intramolecular aldehyde 1,2-addition, and protonation, further calculations and control experiments were also conducted. The calculated results indicate that Z/E-isomerization is the rate-determining step of the Pd(II)-mediated reaction pathway. Its activation energy is 28.6 kcal mol−1 (TS-3 in Fig. S4†), which is close to the activation energy of the Pd(0)-mediated reaction pathway in Fig. 3 (28.0 kcal mol−1, viaTS-endo). However, the reduction of Pd(II) with the assistance of phenylboronic acid to generate a Pd(0) catalyst is more favorable than the Pd(II)-mediated reaction pathway (Fig. S4†). Moreover, the corresponding biphenyl by-product was detected in the further control experiment, which corroborates the calculated results (Fig. S5†). Further kinetic studies of the comparison of Pd(0) and Pd(II) catalysts show a low reaction rate as well as an obvious induction period in the Pd(II)-catalyzed system (Fig. 4). These results validate our mechanistic speculation of the reduction of Pd(II) with the assistance of phenylboronic acid to generate the active Pd(0) catalyst in situ. (See the ESI† for more details.)
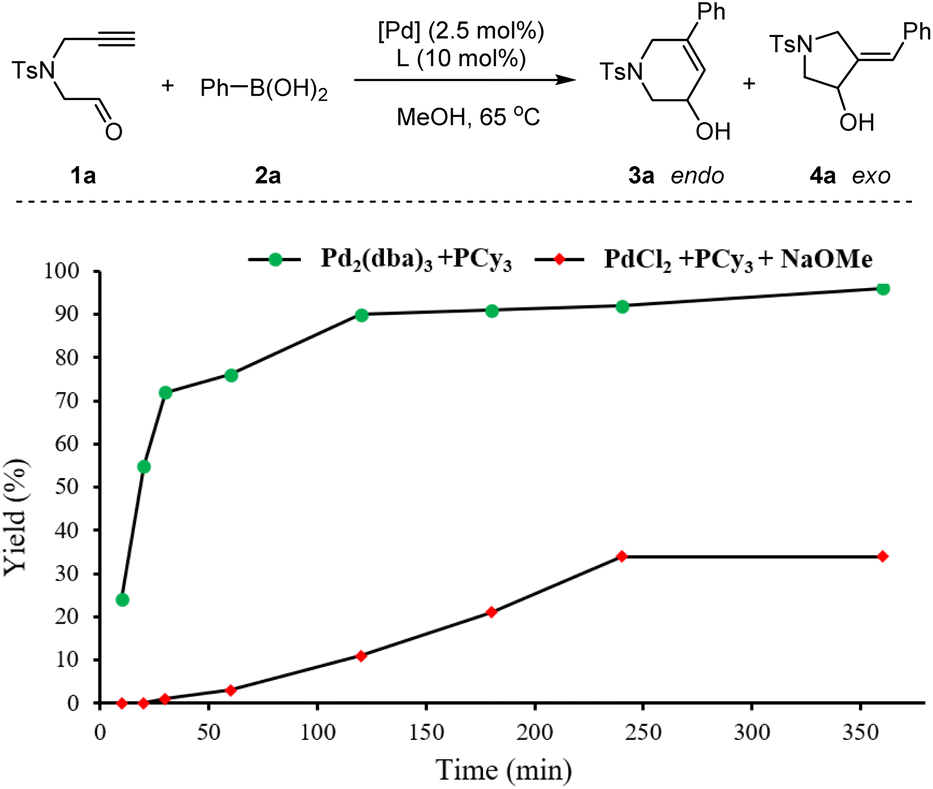 | ||
| Fig. 4 Kinetic studies. The yield indicates the total yield of 3a and 4a. | ||
Thus far, the whole catalytic cycle of the Pd(0)-catalyzed trans-alkylative cyclization of alkynals was constructed, which undergoes a sequential π-Lewis base activation, protonation, transmetallation, and reductive elimination (Fig. 5). Among them, the π-Lewis base activation step is identified as the rate-determining step.
 | ||
| Fig. 5 Proposed catalytic cycle. | ||
Ligand effects
Ligands were recognized to regulate the reactivity and selectivity in this catalytic system. We then turned to explore the ligand effects for this Pd(0)-catalyzed π-Lewis base activation. As shown in Fig. 6a, the transition state TS-endo (marked as a Y-Shape) presents a trigonal planar geometry, in which the bite angle AP–Pd–P is 110°. The reacting alkyne is parallel to the P–Pd–P plane. Meanwhile, another transition state was also found [TS-endo (T-Shape)], in which the conformation of the transition state rearranged to a T-shape.3c,22 The relative free energy of TS-endo (T-Shape) is 4.2 kcal mol−1 higher than that of TS-endo (Y-Shape). This energy discrepancy could be attributed to the reduced orbital overlap between Pd and reactant, owing to the reacting alkyne being perpendicular to the P–Pd–P plane in TS-endo (T-shape) (Fig. S7†). In a sense, the perpendicular conformation would release the steric hindrance to the reactant–ligand and ligand–ligand. We thus speculated that when bulky ligands were employed, the T-shape conformation could be preferred. To clarify this, a series of ligands PPh2Cy (L2), PPhCy2 (L3), and PCy3 (L4), with steric hindrance increased, were then evaluated. Expectedly, the corresponding energy discrepancy was reduced when one cyclohexyl was introduced (Fig. 6b), and it was inverted when two-cyclohexyl substituted L3 was used (Fig. 6c). Furthermore, the T-shape conformation (TS-L4-endo (T-Shape)) becomes plenarily preferred in the PCy3 (L4) involved case (Fig. 6d).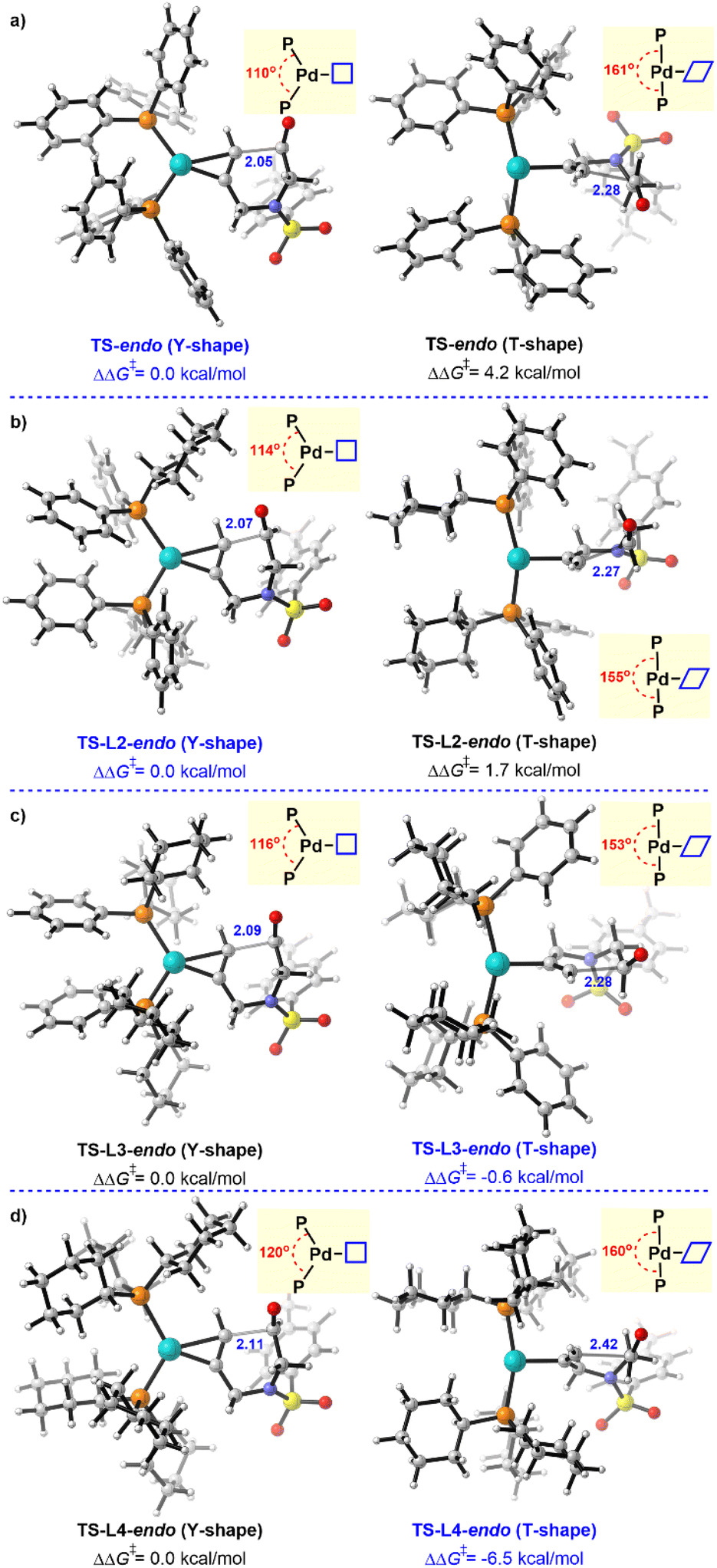 | ||
| Fig. 6 Ligand effects for Pd(0)–π-Lewis base activation of alkynals. | ||
These results suggest that the Pd(0)-catalyzed π-Lewis base activation could undergo different transition state conformations for PPh3 (L1) and PCy3 (L4), which would result in different reactivities.
The crucial feature of the Pd(0)–π-Lewis base activation mode is the electron donation of Pd(0), so the electron effect of the ligand should also matter. Calculated results show that the amounts of charge transfer from PdL2 to alkynal 1a are gradually increased along with the enhanced electron-donating ability of ligands (L1 → L4) (Fig. 7). Moreover, the free energy barrier of the L4-involved case is 8.7 kcal mol−1 lower than that of L1 (Fig. S8†), correlating with the electron-donating effects. It indicates that ligands with greater electron-donating ability would accelerate the Pd(0)-catalyzed π-Lewis base activation reaction. To verify this concept, we then conducted the kinetic experiments. As shown in Fig. 8, the reaction could achieve over 70% yield within 30 minutes in the presence of PCy3 (L4), and it was almost complete within 2 hours.23 In contrast, the corresponding reaction with PPh3 (L1) takes 6 hours to complete, which demonstrates the significant acceleration of the electron-donating ligand PCy3 (L4).
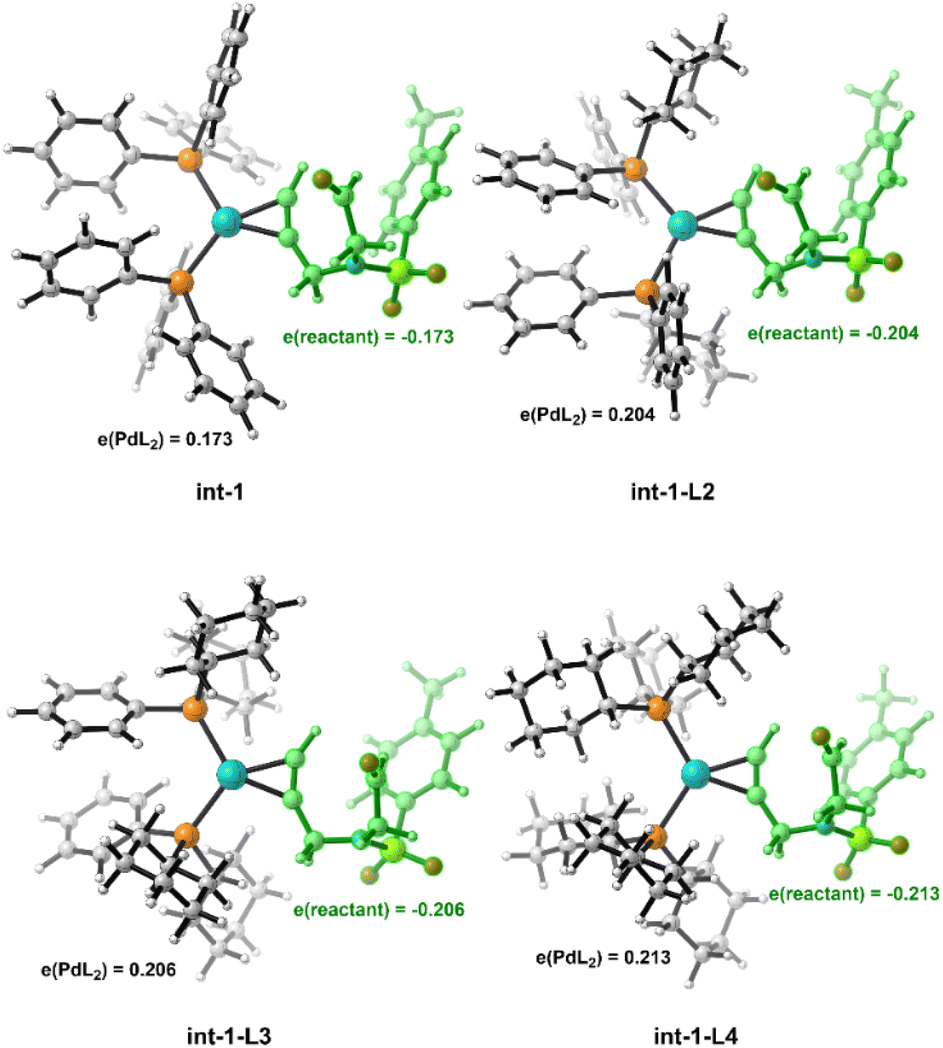 | ||
| Fig. 7 Mulliken charge distribution of the η2-coordinated Pd(0)–alkynal complex with PPh3 (L1), PPh2Cy (L2), PPhCy2 (L3), and PCy3 (L4). | ||
 | ||
| Fig. 8 Kinetic studies. The yield indicates the total yield of 3a and 4a. | ||
Based on the above results, we speculate if the more electron-donating ligand PCy3 (L4) could realize the Pd(0)-catalyzed π-Lewis base activation reaction of challenging C- or O-tethered 1,5-alkynals. We initiated the reaction between O-tethered 1,5-alkynal 1b and p-tolylboronic acid 2b under the catalysis of Pd2(dba)3 and PPh3 (L1). Unsurprisingly, there are no desired products 3b or 4b detected from 60 to 100 °C (Table 1, entry 1), which distinguishes the inertness of the O-tethered substrate 1b. However, when PCy3 (L4) was used, 3b was obtained in a moderate yield with over 19:1 regioselectivity at 80 °C. The yield could be improved to 97% (85% isolated yield) at 100 °C. Moreover, the reaction of C-tethered substrate 1c could also give an acceptable yield for product 3c. These results further emphasize the crucial role of electron-donating effects in the Pd(0)–π-Lewis base activation of alkynes.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | L | Temp. (°C) | Yieldb (%) | endo/exo |
| a Conditions: 1 (0.2 mmol), 2 (0.3 mmol), Pd2(dba)3 (5 mol%), L (30 mol%), MeOH (0.1 M), for 24 h under a nitrogen atmosphere. b Determined by 1H NMR analysis. c Isolated yield. The Ar of Ar–B(OH)2 represents p-tolyl. | |||||
| 1 | 1b | L1(PPh3) | 60–100 | N.D. | — |
| 2 | 1b | L4(PCy3) | 80 | 3b, 64 | >19:1 |
| 3 | 1b | L4(PCy3) | 100 | 3b, 97 (85)c | >19:1 |
| 4 | 1c | L4(PCy3) | 100 | 3c, 43 | >19:1 |

Origin of regioselectivity
The reaction only gave endo-products no matter whether alkyl (1a) or aryl (1d) substituted reactants were used in the presence of PPh3 (L1), while the corresponding regioselectivities were changed when PCy3 (L4) was employed (Fig. 9).5a We then turned to reveal the origin of regioselectivity for this Pd(0)–π-Lewis base activation, based on the understanding of ligand effects. The calculated results show that the relative free energy of TS-endo is 4.0 kcal mol−1 lower than that of TS-exo, indicating the priority of endo-selectivity, which corroborates with the experimental observation. The steric hindrance between the proximal methylene and ligand makes TS-exo present a heterogenic trigonal planar geometry (referring to the change of the P–Pd–C angle from 115° (TS-endo) to 95° (TS-exo)), which results in its high energy. For aryl-tethered 1d, the electron-withdrawing aryl substituent within the tether may enhance the reactivity of the internal position of the alkyne to access the exo-product 4d. But this steric hindrance caused by the aryl group makes the reacting alkyne deviate from the P–Pd–P plane in TS-exo-1d (the corresponding dihedral angle DP-Pd-C1-C2 is 155°), which results in its relative free energy still being 1.7 kcal mol−1 higher than that of TS-endo-1d. Thus, the endo-product 3d was majorly obtained in the presence of PPh3 (L1). These results suggest that the steric effect dominates the regioselectivity in the PPh3 (L1) involved case.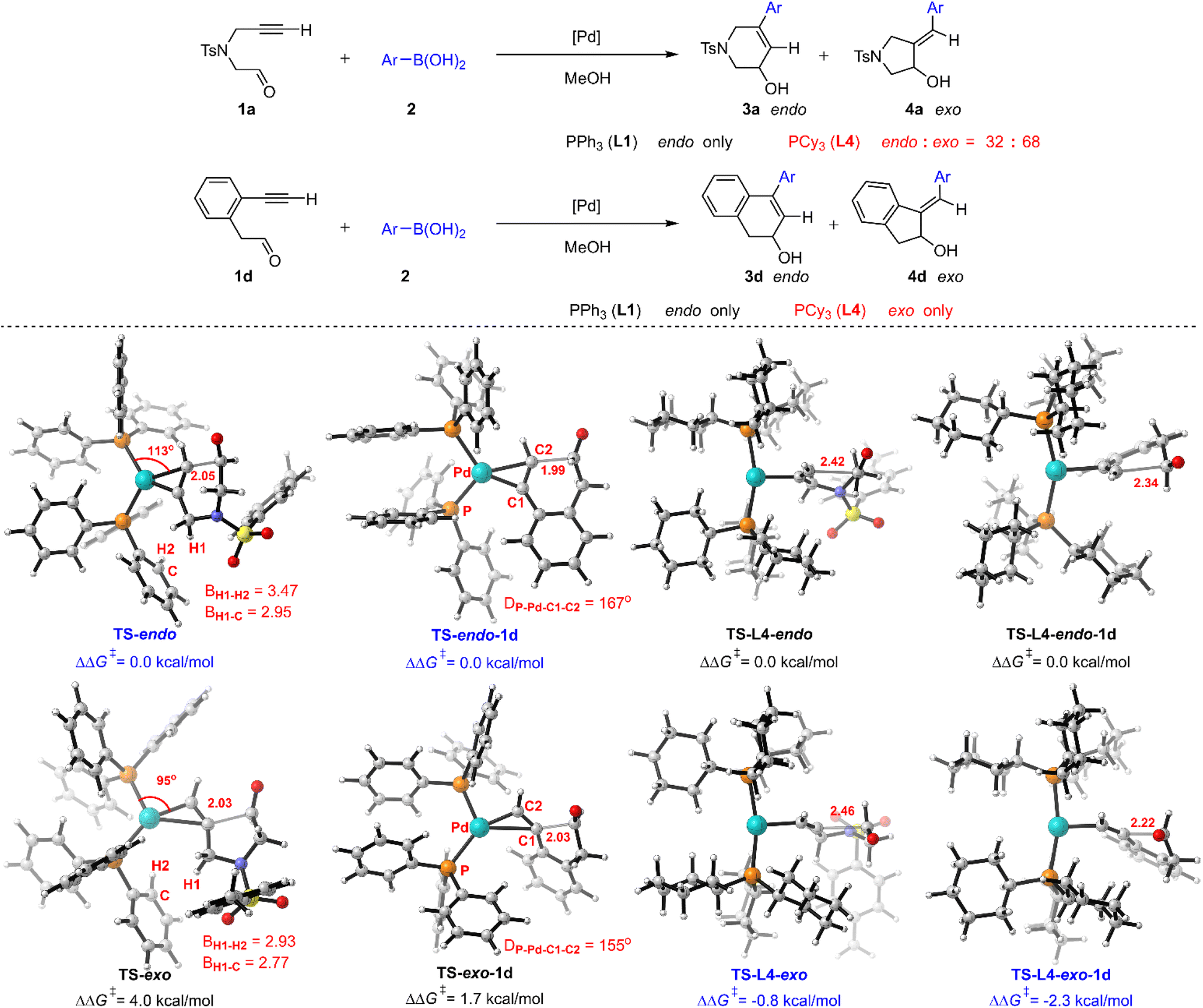 | ||
| Fig. 9 Origin of regioselectivities. The Ar of Ar–B(OH)2 represents p-tolyl. | ||
Moreover, the T-shape conformation is preferred in the PCy3 (L4) involved case, in which the steric hindrance is partially released. Thus, the relative free energy of TS-L4-exo is 0.8 kcal mol−1 lower than that of TS-L4-endo due to the weak electron-donating effects of the alkyl moiety at 1a, which is consistent with the observation of the mixture (endo/exo = 32:68) in the experiment. Meanwhile, the electron-withdrawing aryl group at 1d guides the nucleophilic attack, in which the relative free energy of TS-L4-exo-1d is 2.3 kcal mol−1 lower than that of TS-L4-endo-1d, leading to the exo-selectivity. Furthermore, large substituents at the terminal position of NT-tethered reactants 1e and 1f lead to endo-selectivity in the presence of L4, and electron-withdrawing aryl substituent 1g is more endo-selective (Fig. S9†). These results indicate that the regioselectivity depends on both steric and electronic effects in the PCy3 (L4) involved case.
Conclusions
In summary, the metal π-Lewis base activation mode for the Pd(0)-catalyzed trans-alkylative cyclization of alkynals has been disclosed by a combined theoretical and experimental study. In this activation mode, the electrons flow to the alkyne from the electron-rich Pd(0) center through back-donation to enhance the nucleophilic reactivity of the alkyne moiety. Thus the Pd(0)-coordinated alkyne could proceed with a nucleophilic addition to the aldehyde generating the trans-Pd(II)–vinyl complex. The computational results show that this metal π-Lewis base activation mode is more favorable than the oxidative cyclization and π-Lewis acid activation. Ligand effect investigations indicate that the more electron-donating ligand would accelerate this reaction. As a result, the reaction for the challenging flexible C- or O-tethered substrates has been realized by using PCy3. The origin of regioselectivities is also explicated by the newly proposed metal π-Lewis base activation mode. Our studies provide a comprehensive mechanistic understanding of the Pd(0)-mediated umpolung reaction of alkynes, which could be notably extended to other related systems. We anticipate that it will provide a practical theoretical guide for further experimental development of Pd(0)-catalyzed transformations of alkynes.Data availability
All experimental procedures, details of the calculations, and additional data can be found in the ESI†.Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge the National Key R&D program of China (No. 2022YFC2502501), the National Natural Science Foundation of China (No. 22001264 and 22201302), and the Outstanding Youth Fund of Chongqing (2023NSCQ-JQX0081) for financial support.Notes and references
- (a) J. Corpas, P. Mauleón, R. G. Arrayás and J. C. Carretero, ACS Catal., 2021, 11, 7513–7551 CrossRef CAS; (b) M. M. Haley, Angew. Chem., Int. Ed., 2015, 54, 8332 CrossRef CAS; (c) A. L. Shi Shun and R. R. Tykwinski, Angew. Chem., Int. Ed., 2006, 45, 1034–1057 CrossRef; (d) T. T. Talele, J. Med. Chem., 2020, 63, 5625–5663 CrossRef CAS; (e) Y. Yamamoto, Chem. Soc. Rev., 2014, 43, 1575–1600 RSC.
- (a) R. Chinchilla and C. Najera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS; (b) X. Huang, S. Liao, L. Xin, F. Zhang and Y. Yu, Synthesis, 2020, 53, 238–254 Search PubMed; (c) J. Li, D. He, Z. Lin, W. Wu and H. Jiang, Org. Chem. Front., 2021, 8, 3502–3524 RSC; (d) W. Wu and H. Jiang, Acc. Chem. Res., 2012, 45, 1736–1748 CrossRef CAS PubMed.
- (a) V. P. Boyarskiy, D. S. Ryabukhin, N. A. Bokach and A. V. Vasilyev, Chem. Rev., 2016, 116, 5894–5986 CrossRef CAS PubMed; (b) J. Escorihuela, A. Lledos and G. Ujaque, Chem. Rev., 2023, 123, 9139–9203 CrossRef CAS PubMed; (c) L. Xue and Z. Lin, Chem. Soc. Rev., 2010, 39, 1692–1705 RSC.
- (a) P. Davies and H. Adcock, Synthesis, 2012, 44, 3401–3420 CrossRef; (b) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285–2309 CrossRef CAS PubMed.
- (a) H. Tsukamoto, T. Ueno and Y. Kondo, J. Am. Chem. Soc., 2006, 128, 1406–1407 CrossRef CAS PubMed; (b) H. Tsukamoto and Y. Kondo, Angew. Chem., Int. Ed., 2008, 47, 4851–4854 CrossRef CAS; (c) H. Tsukamoto, K. Ito, T. Ueno, M. Shiraishi, Y. Kondo and T. Doi, Chem.–Eur. J., 2023, 29, e202203068 CrossRef CAS.
- (a) E. A. Standley, S. Z. Tasker, K. L. Jensen and T. F. Jamison, Acc. Chem. Res., 2015, 48, 1503–1514 CrossRef CAS PubMed; (b) Y. Hoshimoto, M. Ohashi and S. Ogoshi, Acc. Chem. Res., 2015, 48, 1746–1755 CrossRef CAS; (c) V. Piccialli, Synthesis, 2007, 2007, 2585–2607 CrossRef; (d) R. Shintani, K. Okamoto, Y. Otomaru, K. Ueyama and T. Hayashi, J. Am. Chem. Soc., 2005, 127, 54–55 CrossRef CAS PubMed; (e) E. Oblinger and J. Montgomery, J. Am. Chem. Soc., 1997, 119, 9065–9066 CrossRef CAS.
- (a) R. A. Fernandes, A. K. Jha and P. Kumar, Catal. Sci. Technol., 2020, 10, 7448–7470 RSC; (b) J. Takacs and X.-t. Jiang, Curr. Org. Chem., 2003, 7, 369–396 CrossRef CAS; (c) D. H. Camacho, S. Saito and Y. Yamamoto, Tetrahedron Lett., 2002, 43, 1085–1088 CrossRef CAS; (d) L. S. Hegedus, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon: Oxford, 1990, vol. 1994, p. 1571 Search PubMed.
- (a) G. Frenking and N. Frohlich, Chem. Rev., 2000, 100, 717–774 CrossRef CAS PubMed; (b) J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939–2947 RSC.
- B. K. Liebov and W. D. Harman, Chem. Rev., 2017, 117, 13721–13755 CrossRef CAS PubMed.
- (a) X. Yan, X. M. Yang, P. Yan, B. Zhao, R. Zeng, B. Pan, Y. C. Chen, L. Zhu and Q. Ouyang, Chem. Sci., 2023, 14, 4597–4604 RSC; (b) S. Z. Tan, P. Chen, L. Zhu, M. Q. Gan, Q. Ouyang, W. Du and Y. C. Chen, J. Am. Chem. Soc., 2022, 144, 22689–22697 CrossRef CAS PubMed; (c) X. X. Yang, R. J. Yan, G. Y. Ran, C. Chen, J. F. Yue, X. Yan, Q. Ouyang, W. Du and Y. C. Chen, Angew. Chem., Int. Ed., 2021, 60, 26762–26768 CrossRef CAS; (d) B. X. Xiao, B. Jiang, R. J. Yan, J. X. Zhu, K. Xie, X. Y. Gao, Q. Ouyang, W. Du and Y. C. Chen, J. Am. Chem. Soc., 2021, 143, 4809–4816 CrossRef CAS PubMed.
- (a) Z. Wang, J. Wu, W. Lamine, B. Li, J. M. Sotiropoulos, A. Chrostowska, K. Miqueu and S. Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 21231–21236 CrossRef CAS; (b) Q. He, L. Zhu, Z. H. Yang, B. Zhu, Q. Ouyang, W. Du and Y. C. Chen, J. Am. Chem. Soc., 2021, 143, 17989–17994 CrossRef CAS PubMed; (c) Y. Yang, J. Jiang, H. Yu and J. Shi, Chem.–Eur. J., 2018, 24, 178–186 CrossRef CAS PubMed; (d) S. Xu, Y. Zhang, B. Li and S. Y. Liu, J. Am. Chem. Soc., 2016, 138, 14566–14569 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Gaussian Inc., Wallingford, CT, 2013.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- (a) Z. Wang, Y. Zhou, W. H. Lam and Z. Y. Lin, Organometallics, 2017, 36, 2354–2363 CrossRef CAS; (b) T. Fan, K. S. Fu and Z. Lin, Organometallics, 2013, 32, 5224–5230 CrossRef CAS; (c) F. Schoenebeck and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 2496–2497 CrossRef CAS; (d) V. S. Bryantsev, M. S. Diallo and W. A. Goddard III, J. Phys. Chem. B, 2008, 112, 9709–9719 CrossRef CAS; (e) C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2005, 1, 1133–1152 CrossRef CAS PubMed.
- C. Y. Legault CYLview, 1.0b, Université de Sherbrooke, Sherbrooke, Canada, 2009, http://www.cylview.org Search PubMed.
- To distinguish the role of Lewis acid or base in the metal-alkyne complex, the electron density of typical Lewis acid metal-alkyne complexes was considered. Calculated results indicate a significant charge transfer from alkyne to metal in the Pd(II) or Au(I) complexes, which is against the Lewis base mode in the (Ph3)P2Pd(0)–alkynal complex. Please see ESI† for more details.
- (a) K. Zhong, S. Liu, X. He, H. Ni, W. Lai, W. Gong, C. Shan, Z. Zhao, Y. Lan and R. Bai, Chin. Chem. Lett., 2023, 34, 108339 CrossRef CAS; (b) M. Liu, J. Sun and K. M. Engle, Tetrahedron, 2022, 103, 132513 CrossRef CAS.
- (a) M. A. Ortuño, A. Lledós, F. Maseras and G. Ujaque, ChemCatChem, 2014, 6, 3132–3138 CrossRef; (b) B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2116–2119 CrossRef CAS PubMed; (c) C. Amatore, A. Jutand and G. Le Duc, Chem.–Eur. J., 2011, 17, 2492–2503 CrossRef CAS PubMed.
- (a) C. Joshi, J. M. Macharia, J. A. Izzo, V. Wambua, S. Kim, J. S. Hirschi and M. J. Vetticatt, ACS Catal., 2022, 12, 2959–2966 CrossRef CAS PubMed; (b) Y. Wang, X. Qi, Q. Ma, P. Liu and G. C. Tsui, ACS Catal., 2021, 11, 4799–4809 CrossRef CAS.
- (a) P. Steinhoff and M. E. Tauchert, Beilstein J. Org. Chem., 2016, 12, 1573–1576 CrossRef CAS PubMed; (b) M. Yamashita, I. Takamiya, K. Jin and K. Nozaki, Organometallics, 2006, 25, 4588–4595 CrossRef CAS; (c) K. Tatsumi, R. Hoffmann, A. Yamamoto and J. K. Stille, Bull. Chem. Soc. Jpn., 1981, 54, 1857–1867 CrossRef CAS.
- It's worth noting that both 3a and 4a were detected with a ratio of close to 1:1.6 in the presence of PCy3. While only 3a was detected in the presence of PPh3.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04190a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |