
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theory-driven design of cadmium mineralizing layered double hydroxides for environmental remediation†
Zixian
Li
a,
Nuo
Xu
a,
Jing
Ren
a,
Haigang
Hao
c,
Rui
Gao
c,
Xianggui
Kong
a,
Hong
Yan
 a,
Xiao
Hua
d,
Yung-Kang
Peng
e,
Shulan
Ma
f,
Dermot
O'Hare
g and
Yufei
Zhao
*ab
a,
Xiao
Hua
d,
Yung-Kang
Peng
e,
Shulan
Ma
f,
Dermot
O'Hare
g and
Yufei
Zhao
*ab
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: zhaoyufei@mail.buct.edu.cn
bQuzhou Institute for Innovation in Resource Chemical Engineering, Quzhou 324000, Zhejiang, P. R. China
cCollege of Chemistry and Chemical Engineering, Inner Mongolia University, 010021 Hohhot, Inner Mongolia, P. R. China
dDepartment of Chemistry, Lancaster University, Lancaster LA1 4YB, UK
eDepartment of Chemistry, City University of Hong Kong, Hong Kong, Hong Kong SAR 999077, P. R. China
fBeijing Key Laboratory of Energy Conversion and Storage Materials and College of Chemistry, Beijing Normal University, Beijing 100875, P. R. China
gChemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK
First published on 19th July 2024
Abstract
The environmental concern posed by toxic heavy metal pollution in soil and water has grown. Ca-based layered double hydroxides (LDHs) have shown exceptional efficacy in eliminating heavy metal cations through the formation of super-stable mineralization structures (SSMS). Nevertheless, it is still unclear how the intricate coordination environment of Ca2+ in Ca-based LDH materials affects the mineralization performance, which hinders the development and application of Ca-based LDH materials as efficient mineralizers. Herein, we discover that, in comparison to a standard LDH, the mineralization efficiency for Cd2+ ions may be significantly enhanced in the pentacoordinated structure of defect-containing Ca-5-LDH utilizing both density functional theory (DFT) and ab initio molecular dynamics (AIMD) simulations. Furthermore, the calcination-reconstruction technique can be utilized to successfully produce pentacoordinated Ca-5-LDH. Subsequent investigations verified that Ca-5-LDH exhibited double the mineralization performance (421.5 mg g−1) in comparison to the corresponding pristine seven coordinated Ca-7OH/H2O-LDH (191.2 mg g−1). The coordination-relative mineralization mechanism of Ca-based LDH was confirmed by both theoretical calculations and experimental results. The understanding of LDH materials and their possible use in environmental remediation are advanced by this research.
Introduction
Heavy metals including Cd, Ni, Pb, and Cu are being released into our environment at an alarming rate due to the acceleration of global industrialization.1,2 The accumulation of heavy metal ions in organisms presents challenges in terms of metabolism and poses a severe threat to both the ecosystem and human health.3,4 Recently, a range of remediation methods, such as chemical precipitation,5 adsorption,6 ion exchange,7 and electrochemical methods,8 have been explored to remove such severe pollution.9 Due to its simple implementation and quick adsorption properties, the in situ immobilization method—which uses appropriate mineralizers to lower heavy metal concentrations by adsorption and/or precipitation—has attracted the most attention among these techniques.10 Nevertheless, the current capacity and stability of the available mineralizers in “fixing” heavy metals are still deemed unsatisfactory. Consequently, there has been a heightened endeavor to explore smart mineralizers that possess a superior removal capacity and stability.11Anionic clay minerals known as layered double hydroxides (LDHs)12 are made up of layers of divalent and trivalent metal cations with six-octahedral coordination and negatively charged anions acting as interlayer guests.13,14 According to recent studies, LDHs have a powerful potential to mineralize heavy metal contaminants by creating super-stable mineralization structures (SSMS).15,16 Kong et al. used CaAl-LDH as the mineralizer to selectively remove Cd2+ ions from water17 and our group previously reported CaFe-LDH as an effective mineralizer to remove Ni2+ ions from wastewater18 and CaAl-LDH as a rapid stabilizer to mineralize Cu2+, Zn2+, Co2+, and Ni2+ ions.19 Even though the majority of previously published research concentrated on the removal capabilities of Ca-based LDHs, the relationship between the Ca coordination environment and performance has not been fully elucidated. A large amount of research has focused on understanding the structure of amorphous calcium carbonate20 and key outstanding problems include rationalizing its metastable state.21 An appropriate approach is to conduct complex structural evolution simulations.22,23 Similar studies on the structural evolution of calcium in solid minerals are quite rare,24 while previous work has lacked exploration of performance related relationships after structural recognition. Considering that Ca has a substantially higher atomic radius (1 Å) compared to the more typical divalent ions present in LDHs such as Mg (0.72 Å).25 the coordination environments of Ca ions in LDHs exhibit considerable diversity, frequently having coordination numbers of seven or more.26 Such a wide range of coordination environments can significantly influence the catalytic performance, such as in aldol condensations.27–29 Currently, there is a lack of elucidation of a relationship between the coordination environment of Ca cations in the LDH layers and the mineralization performance. Hence, it is imperative to investigate the structural evolution of calcium in solid-state LDHs and how it affects the mineralization properties, to explain this complex chemical kinetic behavior at the atomic level.
Density functional theory (DFT) and ab initio molecular dynamics (AIMD) were used to determine the structure of Ca2Al1-LDH. In the pristine Ca2Al1-LDH structure, Ca ions are six-coordinated by oxygen ions (Ca-6-LDH). The Ca coordination number (CaAl-5-LDH) will decrease with the formation of oxygen vacancies, while additional coordination with molecules in the solution will increase the Ca coordination number (Ca-7x-LDH, x stands for the Cl−, OH− or H2O). This work aims to explore the effect of Ca2+ ion coordination on the mineralization performance (Scheme 1). Our theoretical results indicate that a defective LDH (Ca-5-LDH) will promote the mineralization process towards the removal of Cd2+ ions due to lower steric hindrance and favorable coordination with water molecules, and subsequent experimental results confirmed this conclusion. This work innovatively utilizes theoretical DFT and AIMD methods to quantitatively analyze the coordination environment of Ca ions on LDH surfaces, as well as the thermodynamics and kinetics of Ca-solvent coordination, to provide theoretical guidance for subsequent thermodynamic calculations and dynamic simulations in the study of environmental remediation materials.
 | ||
| Scheme 1 The effect of the Ca2+ coordination environment on the mineralisation of Cd2+ ions by LDHs. | ||
Results and discussion
Structure confirmation
Ca2Al1-Cl-LDH was chosen for theoretical modeling (Fig. S1†) as chloride is a common anion that could be utilized when using Cl-containing salts as the raw source.30 Within the LDH layers, six hydroxyl groups surround each divalent or trivalent cation, forming a hexacoordinated octahedral structure as shown in Fig. S1b† (Ca-6-LDH).30 Furthermore, it is worth noting that hydroxyl defects are commonly present in the layers of LDHs.31 These defects have been found to significantly enhance the catalytic and mineralization capabilities of LDHs.32 The presence of hydroxyl defects leads to a pentacoordinated structure (Ca-5-LDH) as illustrated in Fig. S1a.† Previous literature has reported that due to the large radius of the Ca2+ ions, they form additional coordination with either the intercalated solvent or the interlayer anions.33 Considering the composition of the alkaline environment, it is possible for Cl−, OH−, or H2O to act as the seventh coordination to Ca2+ within the LDH structure, Ca-7Cl-LDH (Fig. S1c†), Ca-7OH-LDH (Fig. S1d†) and Ca-7H2O-LDH (Fig. S1e†), respectively.In order to determine the final stable structure of Ca-7x-LDHs, we initially calculated the energy for three ligands (Cl−, OH− and H2O) combined with Ca2+ (Fig. 1a). It can be seen that the binding energy of the OH− and H2O ligands to Ca2+ is negative (−1.33 eV for the Ca-OH bond and −0.51 eV for the Ca-H2O bond), and that of Cl− is much positive (0.31 eV for the Ca-Cl bond), which suggested that the combination of the OH− and H2O ligands with Ca2+ is thermodynamically spontaneous, while Cl− ligands cannot directly form a bond with Ca2+. We further analyzed the structural parameters of the Ca2+ and the surrounding ligands when the seventh coordination structure was formed (Table S1,† the corresponding structure is displayed in Fig. S2†), and the results show that when Ca2+ forms a bond with the seventh ligand, the bond length with the hydroxyl on the laminate still lies in the Ca–O bonding range (∼2.5 Å).34 This proves that the formation of the seventh coordination did not destroy the original hexacoordinated octahedral structure of the laminate. Crystal orbital Hamilton population (COHP) analysis showed the contribution of bonding and antibonding states to the energy of the band structure and was commonly used to compare the bond strength.35 The COHP analysis of the bond between Ca2+ and the seventh ligand (Cl−, OH− and H2O) is shown in Fig. 1b. IpCOHP was used to quantitatively investigate the bond strength, and the values of Ca-OH and Ca-H2O bonds are 0.04 and 0.17, respectively, and the value of the Ca–Cl bond is −0.04, which proves that Ca2+ tends to form stronger bonds with OH− and H2O, and the orbital repulsion between Ca2+ and Cl− is too strong to form a bond, which is in accordance with Fig. 1a.
 | ||
| Fig. 1 (a) Comparison of binding energies between Ca2+ and the seventh ligand (Ca-Cl/Ca-OH/Ca-H2O bond in Ca-7Cl-LDH/Ca-7OH-LDH/Ca-7H2O-LDH, respectively); (b) COHP analysis of additional coordination bonds, Ca-Cl/Ca-OH/Ca-H2O bonds in the Ca-7Cl-LDH/Ca-7OH-LDH/Ca-7H2O-LDH structure, respectively. IpCOHP is the integral of COHP, the more positive the value, the stronger the bond. The configurations of the AIMD process of the Ca-6-LDH structure at (c) 0 fs; (d) 1500 fs and (e) 3000 fs; (f) RDF analysis of the AIMD process. | ||
In order to further verify the results that the O atom of OH− and H2O is more favorable to bond with Ca2+, we used the AIMD method to simulate the dynamic behavior of the Ca-6-LDH structure in a solution environment. 3000 fs kinetic simulation calculations were performed, and energy and temperature changes are shown in Fig. S3.† The energy and temperature changes prove that our dynamic simulation system has reached equilibrium. Fig. 1c–e are the structural snapshots at 0 fs, 1500 fs, and 3000 fs of the kinetic simulation, respectively. Configurations show that Ca2+ forms an extra bond with O atoms (both from OH− and H2O) in the solution. The radial distribution function (RDF) analysis could be used as an efficient tool to gather statistics of the distribution around the target atoms and therefore to investigate the coordination situation.36 The RDF analysis of the kinetic process is shown in Fig. 1f. The coordination number is about 6.5 when the Ca–O bond length is in the range of about 2.5 Å, and a larger coordination number indicates that Ca2+ forms a new coordination with O (from OH− and/or H2O) in the interlayer region. The distance between Ca and Cl− is mostly concentrated between 5 and 6 Å, which is too far to be in the bonding range, and no obvious coordination structure is observed. DFT calculation combined with AIMD simulation confirmed the possibility of Ca2+ within the LDH structure forming a seven-coordinate structure with OH− and H2O in the interlayer region, while the stability of the constructed pentacoordinated structure in Ca-5-LDH is also explored below.
Isomorphic substitution of Cd2+
After determining the structure of the multi-coordinate environment of Ca2+ ions within the LDH structure, the energy changes upon isomorphic substitution of Ca2+ by Cd2+ ions in Ca-5-LDH, Ca-6-LDH, Ca-7OH-LDH and Ca-7H2O-LDH during the mineralization process have been investigated. According to prior literature studies,19,37,38 dissolution of Ca2+ ions, stabilization of Cd2+ ions, and the formation of CdAl-LDH are proposed for the mineralization process. This is reasonable based on the similar ionic radius of Ca2+ (1.00 Å) and Cd2+ (0.95 Å). From a theoretical perspective, the change in free energy upon gradually replacing Ca2+ ions in CaAl-LDH with Cd2+ ions may be used to probe the isomorphic substitution process.The chemical reaction process is described as follows:
| Ca12Al6(OH)36(Cl)6 + nCd2+ → CdnCa12−nAl6(OH)36(Cl)6 + nCa2+ |
Fig. 2a shows the process of the gradual replacement of Ca2+ in CaAl-LDH by Cd2+ to form CdAl-LDH. The Gibbs free energy change (ΔG) of each step is shown in Fig. 2b. The calculations show that for Ca-6-LDH, Ca-7OH-LDH and Ca-7H2O-LDH, the free energy initially decreases and then increases with each step, and the energy of the final mineralized structure is higher than that of the initial structure. However, in the Ca-5-LDH model, the free energy of each structure during the mineralization process is much lower than that of the initial structure, which indicates that the entire mineralization process in the pentacoordinate structure is energetically favorable. In order to explore the universality of this performance, the mineralization process of Ni2+ ions has also been calculated and shown in Fig. S4.† The performance differences during the mineralization of Ni2+ ions are consistent with the mineralization of Cd2+ ions. The change of Gibbs free energy during isomorphic substitution suggests that the Ca-5-LDH structure with hydroxyl defects is favorable for the mineralization process, and the reasons for this difference will be discussed below.
 | ||
| Fig. 2 (a) Optimized geometries of the corresponding reaction intermediates in the isomorphic substitution of Cd2+ by Ca-6-LDH; (b) relationship between the Gibbs free energy and the ratio of [Cd2+] to [Cd2+ + Ca2+] in the isomorphic substitution process. | ||
Reasons for performance differences
In order to further explore the reasons for the influence of different coordination structures in the LDH structure on the mineralization performance, the weak interaction between Ca2+ and the surrounding environment was estimated using the Independent Gradient Model (IGM)39 approach. The IGM method supports artificially dividing the model into two fragments, and using the electron density gradient value between the fragments to obtain the δg function, so as to analyze the weak interaction region and its characteristics.40,41 Fig. S5a–d† show the color-filled isosurface of the Ca2+ and its surrounding coordination groups. It can be seen intuitively that the weak interaction between the Ca2+ and the surrounding groups in the pentacoordinated Ca-5-LDH model is the smallest, while the seven-coordinated Ca-7OH-LDH and Ca-7H2O-LDH models have additional weak interactions between the Ca2+ and the surrounding seventh groups. The quantitative weak interaction analysis is given in Table S2† in order to further precisely identify the weak interaction. The δg function value is summarized in Fig. S6,† the stronger the interaction between atoms, the larger the δg of the interaction region will be. The δg function values of the Ca-7OH-LDH and Ca-7H2O-LDH models were found to be bigger than those of the Ca-6-LDH and Ca-5-LDH models, and the intensity order represented by δg is consistent with the electron density and potential energy density (Table S2†). However, static weak interaction analysis lacks detailed information on the mineralization process, and provides little understanding of the excellent mineralization performance of Ca-5-LDH. Therefore, AIMD simulation of the Ca-5-LDH structure was conducted to further explore its coordination changes during the mineralization process.In order to simulate the Ca2+/Cd2+ cation exchange process in Ca-5-LDH more accurately, we performed a 20 ps AIMD simulation. Fig. 3a shows the structure of the dynamic simulation, and the energy and temperature changes during the simulation process are shown in Fig. S7.† From Fig. 3a, it can be seen that two Ca2+ ions (Ca1 and Ca2) and the Al3+ ion (Al) all exhibit a pentacoordinated structure due to the absence of hydroxyl groups. Herein, we quantitatively summarized the changes in coordination numbers of Ca1, Ca2, and Al with all surrounding oxygen atoms (Ca1–O, Ca2–O and Al–O) during the simulation process and the results are shown in Fig. 3b. It can be seen that Ca1–O and Ca2–O exhibit a hexacoordination structure during the AIMD process, which is different from the constructed initial model (pentacoordination). However, it should be noted that the Al3+ ion around the defect site always retains pentacoordination with the surrounding O atoms, consistent with the initial structure, which indicates that the laminate has not been totally restored to a hexacoordinated structure during the mineralization process. There are other reasons for the formation of the hexacoordinated structure of Ca2+ ions as discussed below.
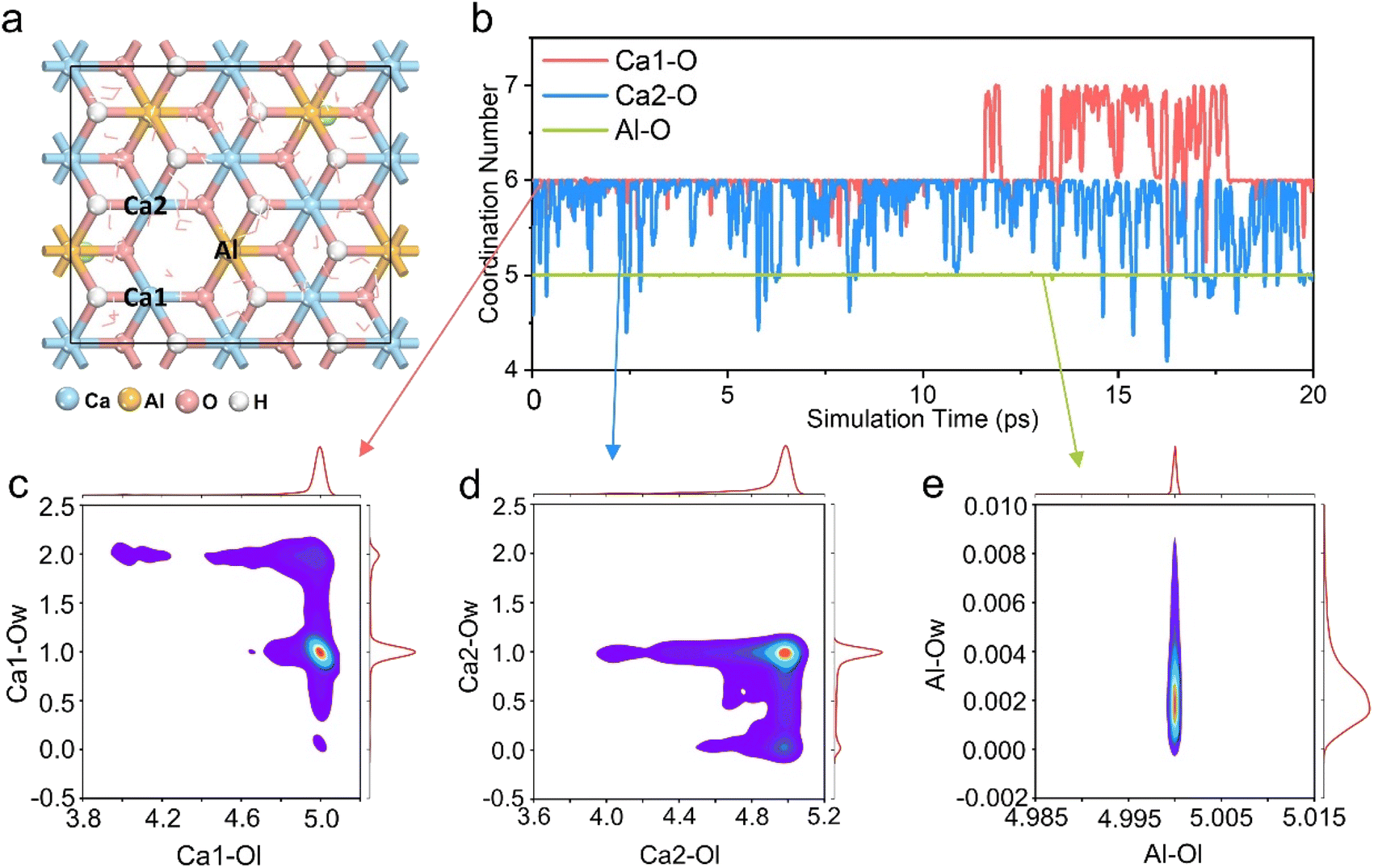 | ||
| Fig. 3 (a) The Ca-5-LDH structure of the 20 ps AIMD simulation, the metal ions around the O vacancy are defined as Ca1, Ca2, and Al, respectively; (b) evolution of coordination number values of Ca1–O, Ca2–O and Al–O during the AIMD simulation of the Ca-5-LDH structure; (c) coordination distribution around the Ca1 ion, the redder the color, the higher the probability; (d) coordination distribution around the Ca2 ion; (e) coordination distribution around the Al ion. | ||
To further explore the hexacoordinated structure composition of Ca1 and Ca2 ions during the mineralization process, the O atoms in the system were divided into two types as shown in Fig. S8,† which are LDH layer O atoms (Ol) and intercalated solvent O atoms (Ow). The distribution of these two kinds of O surrounding the Ca1 ion is shown in Fig. 3c. It can be seen that the Ca1 ion has five-coordination with Ol and one coordination with Ow. The results indicate that during the process of mineralizing, the Ca1 ion forms an additional coordination with the O atom in the solution while maintaining a pentacoordination with the hydroxyl groups in the LDH layers. The analysis of the coordination structure around the Ca2 ion is shown in Fig. 3d. It can be seen that similar to the coordination structure of the Ca1 ion, the Ca2 ion also forms coordination structures with both Ol and Ow. That is to say, while maintaining a five-coordinated structure with O in the layers, Ca2+ forms additional coordination with O in H2O.
The analysis of the coordination structure around Al ions is shown in Fig. 3e; Al forms five-coordination with Ol and basically no coordination with Ow. The coordination distribution around the Ca1, Ca2, and Al ions suggests that Ca-5-LDH can not only maintain the hydroxyl defects, but also that the Ca2+ ions may form additional coordination with O atoms in the solvent environment (Ca-5-LDH-H2O). In order to observe these structures, snapshots taken during the AIMD process at 0 ps, 10 ps and 20 ps are shown in Fig. S9.† The Ca-Ol coordination surrounding the Ca2+ ions can be clearly seen. Following on from these results, it can be concluded that Ca-5-LDH not only exists stably in the solution environment during mineralization, but also forms additional coordination with H2O in solution (Ca-5-LDH-H2O).
To investigate the presence of the aforementioned additional coordination during the mineralization process, we examined the initial three structures. These structures correspond to the stages following the introduction of one Cd2+ ion (CaAlCd1-5-LDH), two Cd2+ ions (CaAlCd2-5-LDH), and three Cd2+ ions (CaAlCd3-5-LDH) into the LDH layers (Fig. S10a–S12a†). We conducted AIMD simulations lasting 10 ps, and the results of the coordination number and metal cation distribution analysis during the simulation are presented in Fig. S10–S12.† The coordination behavior for Ca2+, Cd2+, and Al3+ in CaAlCdx-5 (x = 1, 2 and 3) is found to be similar to that in Ca-5-LDH. That is, Cd2+ and Ca2+ could form additional bonds with H2O during the mineralization process.
Based on the previous analysis of IGM weak interactions and additional coordination phenomena during mineralization, it is reasonable to conjecture that the dissolution of Ca2+ ions is optimal for Ca-5-LDH compared with Ca-6-LDH, Ca-7OH-LDH and Ca-7H2O-LDH, while Ca-5-LDH-H2O further promotes Ca2+ dissolution. Based on this, we calculated the dissolution energy of Ca2+ in different models using DFT (Fig. S13†). The energy required for Ca2+ dissolution from Ca-7OH-LDH and Ca-7H2O-LDH is higher than that for Ca-6-LDH and Ca-5-LDH. Moreover, it can be seen that Ca-5-LDH-H2O facilitates the dissolution of Ca2+ compared with Ca-5-LDH. Additional interactions hinder the dissolution of Ca2+ in Ca-6-LDH and Ca-7x-LDH. Ca-5-LDH forms additional coordination with H2O (Ca-5-LDH-H2O) and thereby promotes the process of isomorphic substitution of Cd2+, which explains the prominent mineralization performance of Ca-5-LDH shown in Fig. 3.
Experimental verification
In the previous theoretical calculation section, we have proposed a reliable strategy to improve the mineralization performance of CaAl-LDH, that is, by changing the coordination number of the Ca2+ ions and constructing an unsaturated coordination LDH structure. To challenge this hypothesis, we set out to prepare low-coordination CaAl-LDH materials containing a large number of hydroxyl defects within the metal hydroxide layers.The memory effect of LDHs refers to the regeneration of the layered structure of LDH materials by placing their calcined products (mixed metal oxide, MMO) in an aqueous solution after calcination. This has been previously shown to be a common method for introducing defects into the metal hydroxide layers.42–44 We first prepared the CaAl-LDH, and then calcined it at different temperatures (t) from 200 °C to 500 °C; the XRD patterns of the synthesized CaAl-LDH and CaAl-t (calcination product) are shown in Fig. S14.† The XRD of the pristine CaAl-LDH is consistent with previous reports. The XRD pattern of the calcination products CaAl-t shows that at higher temperatures, the LDH layers collapse and transform into mixed oxide phases, including Al2O3 and amorphous CaO, as observed by the new Bragg reflections in the XRD.45 The XRD data (Fig. S14†) indicate that the LDH structure begins to collapse above 300 °C, and phase separation occurs at higher temperatures. CaAl-450 was selected as a typical sample to represent the structure after calcination. The SEM images of the initial CaAl-LDH structure and the CaAl-450 structure are shown in Fig. S15a and b.† The initial CaAl-LDH structure shows a layered morphology with a platelet size of a few micrometers, while the CaAl-450 structure shows an agglomerate caused by the calcination. In order to obtain the defective LDH structure, we reconstructed CaAl-450 in water (CaAl-450-r). As shown in Fig. 4a, the XRD shows the typical Bragg reflections (002), (004) and (110) expected for an LDH, indicating that CaAl-450-r has been fully restored to a crystalline LDH. Moreover, the SEM image of CaAl-450-r (Fig. S15c†) reveals a lamellar morphology.
 | ||
| Fig. 4 (a) XRD patterns of CaAl-LDH, CaAl-450-r; (b) K-edge XANES spectra of Ca for Ca(OH)2, CaAl-LDH, CaAl-450-r; (c) magnitude of k3-weighted FT spectra of Ca(OH)2, CaAl-LDH, CaAl-450-r; (d) fitting results of magnitude of k3-weighted FT spectra of Ca(OH)2, CaAl-LDH, CaAl-450-r; (e) coordination model of Ca2+ in CaAl-450-r and CaAl-LDH samples. (f) Mineralization capacities of CaAl-LDH and CaAl-450-r treating 1000 mg L−1 Cd2+ solution. (g) Schematic illustration of comparison between mineralization processes using CaAl-LDH and CaAl-450-r. | ||
We used Ca(OH)2 as a reference material during the XAFS investigation of the coordination environment in both CaAl-LDH and CaAl-450-r. The results are shown in Fig. 4b, c and S16.† From E space analysis (Fig. 4b), comparing Ca(OH)2 which contains normally hexacoordinated Ca2+, a weaker front-edge peak around 4042.5 eV was observed in both CaAl-LDH and CaAl-450-r, which indicates the presence of a geometrically distorted octahedral Ca(OH)6 environment.46–48 This suggests a change in the coordination number of Ca2+ in both CaAl-LDH and CaAl-450-r. More detailed information regarding the coordination number can be obtained by further analysis of the K and R space data. Analysis of the K space (Fig. S16†) shows that the CaAl-450-r forms an LDH structure after reconstruction. Analysis of the R space data (Fig. 4c) shows that there are two main peaks in R space for all samples located at ∼1.83 Å and ∼3.08 Å respectively, corresponding to the first shell Ca–O and the Ca–Ca/Al second shell. Furthermore, the first shell peak intensities for CaAl-LDH are strongest in Fig. 4c, followed by the Ca(OH)2 sample, and finally the CaAl-450-r sample. To further quantify the analysis of Ca–O coordination numbers, R-space data (Fig. 4c) in XAFS characterization results were fitted as shown in Fig. 4d and Table S3.† It should be noted that the coordination number of the first Ca–O shell for CaAl-LDH samples is ∼7.2, much larger than that of the Ca(OH)2 reference (∼6.0).28 The existence of additional Ca-OH coordination in a solid base catalyst, CaAl-LDH has been previously reported,28 while for the CaAl-450-r, the coordination number of the first Ca–O shell is determined to be ∼4.9, lower than that for the Ca(OH)2 reference (∼6.0). These data further substantiate the existence of an oxygen defect-containing CaAl-400-r, which results in a distorted Ca(OH)6 octahedron in E space (Fig. 4b). We have also used electron paramagnetic resonance (EPR) measurements to confirm the existence of the oxygen defects in CaAl-450-r. As shown in Fig. S17,† CaAl-450-r exhibits a strong EPR signal at g = 2.003, indicating the formation of oxygen vacancies in CaAl-450-r.49,50 A schematic representation of the Ca coordination for CaAl-LDH and CaAl-450-r is shown in Fig. 4e. CaAl-LDH contains an additional coordination structure, while CaAl-450-r presents a coordinatively unsaturated structure with oxygen defects. We compared these Ca-LDHs with different coordination environments for super-stable mineralization of Cd2+ ions as discussed below.
The mineralization performance for Cd2+ exhibited by CaAl-LDH and CaAl-400-r is shown in Fig. 4f. The methods used to calculate adsorption capacity have been extensively reported.19,38,44,51 The mineralization performance of CaAl-450-r, which corresponds to a Ca-5-LDH structure, was 421.5 mg g−1, much higher than that of CaAl-LDH (191.2 mg g−1), corresponding to the Ca-7OH-LDH/Ca-7H2O-LDH structure, which is consistent with the theoretical calculations. The XRD characterization of the final mineralized product (named CaAl-LDH-Cd and CaAl-450-r-Cd) is shown in Fig. S18.† From the analysis of the XRD characterization, it can be seen that the mineralized product shows an LDH structure, which suggested that the Cd2+ replaces Ca2+ in the LDH layers, proving that the mineralization mechanism proceeds mainly via cation substitution, similar to our previous report (Fig. 4g).19,38,51 The SEM images of CaAl-LDH-Cd and the CaAl-450-r-Cd sample are shown in Fig. S19.† The apparent lamellar structure suggested that the sample maintains the LDH structure after mineralizing the Cd2+ ions, which is consistent with the XRD data, demonstrating an isomorphic substitution mechanism during the mineralization process. Some previous reports showed that the mechanism of mineralization of heavy metal ions by the LDH material includes isomorphic substitution and surface adsorption, regardless of which, the dissolution of Ca2+ can promote the mineralization process.17 In order to study the effects of calcination and reconstruction on specific surface areas,44 Brunauer–Emmett–Teller (BET) N2 adsorption/desorption experiments were performed. As shown in Fig. S20,† CaAl-LDH exhibited a specific BET surface area of 19.23 m2 g−1, while CaAl-450-r displayed a similar result (24.33 m2 g−1). These similar surface areas indicate that the improvement in the mineralization performance of Cd2+ in the different samples mainly comes from the difference in the coordination structure of Ca2+, not a surface area effect.
Based on the above theoretical and experimental results, the display of the CaAl-LDH micro-coordination structure and the explanation of the mechanism for improved mineralization performance can be summarized in Scheme 2. In the pristine Ca-7OH-LDH/Ca-7H2O-LDH, Ca ions form a seven-coordinate structure surrounded by significant steric hindrance effects that prevent further H2O from coordinating with Ca ions and impede Ca dissolution. The pristine Ca-7OH-LDH/Ca-7H2O-LDH was calcined and transformed into CaAl-MMO, and penta-coordinated Ca-5-LDH was obtained after reconstruction through utilizing the memory effect. Compared with Ca-7OH-LDH/Ca-7H2O-LDH, the steric hindrance effect of Ca-5-LDH was weaker, and subsequent additional H2O coordination promoted the dissolution of Ca ions and thus promoted the mineralization process of Cd ions.
 | ||
| Scheme 2 The display of the LDH micro-coordination structure and the explanation of the mechanism for improving the mineralization performance, based on the combination of calculation and experimental results. | ||
Conclusion
In this work, the cation exchange of Ca2+ by Cd2+ in CaAl-LDH has been investigated using density functional theory (DFT) and ab initio molecular dynamics (AIMD) simulations. A significant enhancement in Cd2+ mineralization performance within the pentacoordinated CaAl-5-LDH structure was predicted. The unsaturated coordination CaAl-LDH was successfully prepared using a calcination-reconstruction process, and its superior mineralization ability for Cd2+ is consistent with our theoretical calculations. This work is of great significance for understanding the mechanism of Ca-LDHs to remove heavy metals and providing guidance for the design of mineralizers with enhanced performance.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Author contributions
Zixian Li: conceptualization, investigation, writing – original draft. Nuo Xu: experiments, investigation. Jing Ren: XAFs investigation. Haigang Hao: experiments and corresponding analysis. Rui Gao: dynamic simulation. Xianggui Kong: investigation. Hong Yan: investigation. Xiao Hua: investigation. Yung-Kang Peng: investigation, supervision. Shulan Ma: investigation, supervision. Dermot O'Hare: investigation, supervision. Yufei Zhao: conceptualization, supervision, project administration, resources, writing – review & editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was supported by the National Natural Science Foundation of China (22278030, 22090032, 22090030, and 22288102). We are thankful for the 1W1B beamline of the Beijing Synchrotron Radiation Facility (BSRF) and BL14W1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF).References
- N. T. Bui, H. Kang, S. J. Teat, G. M. Su, C.-W. Pao, Y.-S. Liu, E. W. Zaia, J. Guo, J.-L. Chen, K. R. Meihaus, C. Dun, T. M. Mattox, J. R. Long, P. Fiske, R. Kostecki and J. J. Urban, A nature-inspired hydrogen-bonded supramolecular complex for selective copper ion removal from water, Nat. Commun., 2020, 11, 3947 CrossRef CAS.
- R. Bruno, M. Mon, P. Escamilla, J. Ferrando-Soria, E. Esposito, A. Fuoco, M. Monteleone, J. C. Jansen, R. Elliani, A. Tagarelli, D. Armentano and E. Pardo, Bioinspired Metal-Organic Frameworks in Mixed Matrix Membranes for Efficient Static/Dynamic Removal of Mercury from Water, Adv. Funct. Mater., 2021, 31, 2008499 CrossRef CAS.
- C. He, A. Hu, F. Wang, P. Zhang, Z. Zhao, Y. Zhao and X. Liu, Effective remediation of cadmium and zinc co-contaminated soil by electrokinetic-permeable reactive barrier with a pretreatment of complexing agent and microorganism, Chem. Eng. J., 2021, 407, 126923 CrossRef CAS.
- W.-M. Yin, Y. Wang, Y.-C. Hou, Y. Sun, J.-G. Zhang, H.-L. Sun, S.-J. Li, Q.-J. Pan and Y.-R. Guo, Petaloid-array hierarchically structured carbon Dots/Mg(OH)2 composite: Design, characterization and removal/recovery of cadmium via slowly releasing, Chem. Eng. J., 2020, 401, 125961 CrossRef CAS.
- T. A. Kurniawan, G. Y. S. Chan, W.-H. Lo and S. Babel, Physico–chemical treatment techniques for wastewater laden with heavy metals, Chem. Eng. J., 2006, 118, 83–98 CrossRef CAS.
- S. Li, M. Dai, Y. Wu, H. Fu, X. Hou, C. Peng and H. Luo, Resource utilization of electroplating wastewater: obstacles and solutions, Environ. Sci.: Water Res. Technol., 2022, 8, 484–509 RSC.
- S. Dong, B. Wang, X. Liu, L. Wang, J. Zhan, A. Qayum, X. Jiao, D. Chen and T. Wang, A reductive ion exchange strategy using NaTi2(PO4)3 for metal removal/recovery from wastewater, J. Mater. Chem. A, 2021, 9, 293–300 RSC.
- S.-Y. Ding, M. Dong, Y.-W. Wang, Y.-T. Chen, H.-Z. Wang, C.-Y. Su and W. Wang, Thioether-Based Fluorescent Covalent Organic Framework for Selective Detection and Facile Removal of Mercury(II), J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS PubMed.
- C. Xu, J. Zeng, Y. Wang, X. Jiang and X. Wang, Graphene and boron nitride foams for smart functional applications, SmartMat, 2023,(4), e1199 CrossRef CAS.
- F. Fu and Q. Wang, Removal of heavy metal ions from wastewaters: A review, J. Environ. Manag., 2011, 92, 407–418 CrossRef CAS PubMed.
- D. Sarma, S. M. Islam, K. S. Subrahmanyam and M. G. Kanatzidis, Efficient and selective heavy metal sequestration from water by using layered sulfide K2xSn4−xS8−x (x = 0.65–1; KTS-3), J. Mater. Chem. A, 2016, 4, 16597–16605 RSC.
- L. Zhang, D. Guo, X. Tantai, B. Jiang, Y. Sun and N. Yang, Synthesis of Three-Dimensional Hierarchical Flower-Like Mg–Al Layered Double Hydroxides with Excellent Adsorption Performance for Organic Anionic Dyes, Trans. Tianjin Univ., 2021, 27, 394–408 CrossRef CAS.
- H. Yan, X.-J. Zhao, Y.-Q. Zhu, M. Wei, D. G. Evans and X. Duan, in The Periodic Table II: Catalytic, Materials, Biological and Medical Applications, ed. D. M. P. Mingos, Springer International Publishing, Cham, 2019, pp. 89–120, DOI:10.1007/430_2019_47.
- P. J. Sideris, U. G. Nielsen, Z. Gan and C. P. Grey, Mg/Al ordering in layered double hydroxides revealed by multinuclear NMR spectroscopy, Science, 2008, 321, 113–117 CrossRef CAS.
- X. Kong, P. Hao and H. Duan, Super-stable mineralization effect of layered double hydroxides for heavy metals: Application in soil remediation and perspective, Explor., 2021, 1, 20210052 Search PubMed.
- X.-J. Zhao, S.-M. Xu, P. Yin, J.-Y. Guo, W. Zhang, Y. Jie and H. Yan, Theoretical study on the mechanism of super-stable mineralization of LDHs in soil remediation, Chem. Eng. J., 2023, 451, 138500 CrossRef CAS.
- X. Kong, R. Ge, T. Liu, S. Xu, P. Hao, X. Zhao, Z. Li, X. Lei and H. Duan, Super-stable mineralization of cadmium by calcium-aluminum layered double hydroxide and its large-scale application in agriculture soil remediation, Chem. Eng. J., 2021, 407, 127178 CrossRef CAS.
- H. Chi, J. Wang, H. Wang, S. Li, M. Yang, S. Bai, C. Li, X. Sun, Y. Zhao and Y.-F. Song, Super-Stable Mineralization of Ni2+ Ions from Wastewater using CaFe Layered Double Hydroxide, Adv. Funct. Mater., 2022, 32, 2106645 CrossRef CAS.
- W. Xiong, J. Wang, X. Kong, G. I. N. Waterhouse, H. Liu, Y. Wang, S. Liu, Y. Wang, S. Li, Y. Zhao and H. Duan, Efficient and Superstable Mineralization of Toxic Cd2+ Ions through Defect Engineering in Layered Double Hydroxide Nanosheets, J. Phys. Chem. C, 2023, 127, 8759–8769 CrossRef CAS.
- D. Gebauer, A. Völkel and H. Cölfen, Stable Prenucleation Calcium Carbonate Clusters, Science, 2008, 322, 1819–1822 CrossRef CAS PubMed.
- T. C. Nicholas, A. E. Stones, A. Patel, F. M. Michel, R. J. Reeder, D. G. A. L. Aarts, V. L. Deringer and A. L. Goodwin, Geometrically frustrated interactions drive structural complexity in amorphous calcium carbonate, Nat. Chem., 2024, 16, 36–41 CrossRef CAS PubMed.
- F. M. Michel, J. MacDonald, J. Feng, B. L. Phillips, L. Ehm, C. Tarabrella, J. B. Parise and R. J. Reeder, Structural Characteristics of Synthetic Amorphous Calcium Carbonate, Chem. Mater., 2008, 20, 4720–4728 CrossRef CAS.
- P. Raiteri, J. D. Gale, D. Quigley and P. M. Rodger, Derivation of an Accurate Force-Field for Simulating the Growth of Calcium Carbonate from Aqueous Solution: A New Model for the Calcite−Water Interface, J. Phys. Chem. C, 2010, 114, 5997–6010 CAS.
- Y. Li, H. Pan, Q. Liu, X. Ming and Z. Li, Ab initio mechanism revealing for tricalcium silicate dissolution, Nat. Commun., 2022, 13, 1253 CAS.
- (a) CRC Handbook of Chemistry and Physics, ed. D. R. Lide, National Institute of Standards and Technology. CRC Press (an imprint of Taylor and Francis Group, Boca Raton, FL, 86th edn, 2005, p. 2544 Search PubMed; (b) D. R. Lide, CRC handbook of chemistry and physics, CRC Press, Boca Raton, FL, 86th edn, 2005, p. 2544 Search PubMed.
- B. Kutus, X. Gaona, A. Pallagi, I. Pálinkó, M. Altmaier and P. Sipos, Recent advances in the aqueous chemistry of the calcium(II)-gluconate system – Equilibria, structure and composition of the complexes forming in neutral and in alkaline solutions, Coord. Chem. Rev., 2020, 417, 213337 CrossRef CAS.
- G. M. Kim, S. M. Park and S. W. Park, Chloride removal of calcium aluminate cements: Reaction and physicochemical characteristics, Case Stud. Constr. Mater., 2023, 18, e01975 Search PubMed.
- W. Bing, L. Zheng, S. He, D. Rao, M. Xu, L. Zheng, B. Wang, Y. Wang and M. Wei, Insights on Active Sites of CaAl-Hydrotalcite as a High-Performance Solid Base Catalyst toward Aldol Condensation, ACS Catal., 2017, 8, 656–664 CrossRef.
- W. Bing, H. Wang, L. Zheng, D. Rao, Y. Yang, L. Zheng, B. Wang, Y. Wang and M. Wei, A CaMnAl-hydrotalcite solid basic catalyst toward the aldol condensation reaction with a comparable level to liquid alkali catalysts, Green Chem., 2018, 20, 3071–3080 RSC.
- F. Cavani, F. Trifirò and A. Vaccari, Hydrotalcite-type anionic clays: Preparation, properties and applications, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- D. Zhou, P. Li, X. Lin, A. McKinley, Y. Kuang, W. Liu, W. F. Lin, X. Sun and X. Duan, Layered double hydroxide-based electrocatalysts for the oxygen evolution reaction: identification and tailoring of active sites, and superaerophobic nanoarray electrode assembly, Chem. Soc. Rev., 2021, 50, 8790–8817 RSC.
- Y. Zhao, L. Zheng, R. Shi, S. Zhang, X. Bian, F. Wu, X. Cao, G. I. N. Waterhouse and T. Zhang, Alkali Etching of Layered Double Hydroxide Nanosheets for Enhanced Photocatalytic N2 Reduction to NH3, Adv. Energy Mater., 2020, 10, 2002199 CrossRef CAS.
- F. Jalilehvand, D. Spångberg, P. Lindqvist-Reis, K. Hermansson, I. Persson and M. Sandström, Hydration of the Calcium Ion. An EXAFS, Large-Angle X-ray Scattering, and Molecular Dynamics Simulation Study, J. Am. Chem. Soc., 2001, 123, 431–441 CrossRef CAS.
- G. Renaudin, J. P. Rapin, E. Elkaim and M. François, Polytypes and polymorphs in the related Friedel's salt [Ca2Al(OH)6]+[X·2H2O]− halide series, Cem. Concr. Res., 2004, 34, 1845–1852 CrossRef CAS.
- M. Ha, A. Hajibabaei, D. Y. Kim, A. N. Singh, J. Yun, C. W. Myung and K. S. Kim, Al-Doping Driven Suppression of Capacity and Voltage Fadings in 4d-Element Containing Li-Ion-Battery Cathode Materials: Machine Learning and Density Functional Theory, Adv. Energy Mater., 2022, 12, 2201497 CrossRef CAS.
- P. Berruyer, C. Cibaka-Ndaya, A. Pinon, C. Sanchez, G. L. Drisko and L. Emsley, Imaging Radial Distribution Functions of Complex Particles by Relayed Dynamic Nuclear Polarization, J. Am. Chem. Soc., 2023, 145, 9700–9707 CrossRef CAS PubMed.
- J. Wang, X. Kong, M. Yang, W. Xiong, Z. Li, H. Zhou, G. I. N. Waterhouse, S.-M. Xu, H. Yan, Y.-F. Song, H. Duan and Y. Zhao, Superstable Mineralization of Heavy Metals Using Low-Cost Layered Double Hydroxide Nanosheets: Toward Water Remediation and Soil Fertility Enhancement, Ind. Eng. Chem. Res., 2022, 62, 365–374 CrossRef.
- J. Wang, Y. Wang, W. Xiong, Z. Li, X. Kong, H. Yan, Y. Lin, H. Duan and Y. Zhao, Super-stable mineralization of multiple heavy metal ions from wastewater for utilization in photocatalytic CO2 reduction and trace precious metal recovery, Chem. Eng. Sci., 2023, 271, 118583 CrossRef CAS.
- C. Lefebvre, G. Rubez, H. Khartabil, J.-C. Boisson, J. Contreras-García and E. Hénon, Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density, Phys. Chem. Chem. Phys., 2017, 19, 17928–17936 RSC.
- T. Lu and Q. Chen, Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS PubMed.
- T. Lu and Q. Chen, in Comprehensive Computational Chemistry, ed. M. Yáñez and R. J. Boyd, Elsevier, Oxford, 1st edn, 2024, pp. 240–264, doi: DOI:10.1016/B978-0-12-821978-2.00076-3.
- J. Han, Y. Dou, M. Wei, D. G. Evans and X. Duan, Erasable Nanoporous Antireflection Coatings Based on the Reconstruction Effect of Layered Double Hydroxides, Angew. Chem., Int. Ed., 2010, 49, 2171–2174 CrossRef CAS PubMed.
- Y. Xu, Z. Wang, L. Tan, Y. Zhao, H. Duan and Y.-F. Song, Fine Tuning the Heterostructured Interfaces by Topological Transformation of Layered Double Hydroxide Nanosheets, Ind. Eng. Chem. Res., 2018, 57, 10411–10420 CrossRef CAS.
- C. Li, Z. Li, J. Wang, W. Xiong, H. Yan, Y. Bai, D. O’Hare and Y. Zhao, Mineralization and electroreduction-recycling of toxic Pb2+ ions using memory-effect of layered double hydroxide structure with ultrahigh capacity, Chem. Eng. J., 2023, 462, 141926 CrossRef CAS.
- W. Xiong, M. Yang, J. Wang, H. Wang, P. Zhao, Z. Li, B. Liu, X. Kong, H. Duan and Y. Zhao, Removal, recycle and reutilization of multiple heavy metal ions from electroplating wastewater using super-stable mineralizer Ca-based layered double hydroxides, Chem. Eng. Sci., 2023, 279, 118928 CrossRef CAS.
- Y. Zhao, G. Chen, T. Bian, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, L. J. Smith, D. O'Hare and T. Zhang, Defect-Rich Ultrathin ZnAl-Layered Double Hydroxide Nanosheets for Efficient Photoreduction of CO2 to CO with Water, Adv. Mater., 2015, 27, 7824–7831 CrossRef CAS.
- A. Shimamura, E. Kanezaki, M. I. Jones and J. B. Metson, Direct observation of grafting interlayer phosphate in Mg/Al layered double hydroxides, J. Solid State Chem., 2012, 186, 116–123 CrossRef CAS.
- D.-Y. Wang, H.-L. Chou, Y.-C. Lin, F.-J. Lai, C.-H. Chen, J.-F. Lee, B.-J. Hwang and C.-C. Chen, Simple Replacement Reaction for the Preparation of Ternary Fe1–xPtRux Nanocrystals with Superior Catalytic Activity in Methanol Oxidation Reaction, J. Am. Chem. Soc., 2012, 134, 10011–10020 CrossRef CAS PubMed.
- H. Sun, W. Zhang, J.-G. Li, Z. Li, X. Ao, K.-H. Xue, K. K. Ostrikov, J. Tang and C. Wang, Rh-engineered ultrathin NiFe-LDH nanosheets enable highly-efficient overall water splitting and urea electrolysis, Appl. Catal., B, 2021, 284, 119740 CrossRef CAS.
- L. Wu, Z. Sun, Y. Zhen, S. Zhu, C. Yang, J. Lu, Y. Tian, D. Zhong and J. Ma, Oxygen Vacancy-Induced Nonradical Degradation of Organics: Critical Trigger of Oxygen (O2) in the Fe–Co LDH/Peroxymonosulfate System, Environ. Sci. Technol., 2021, 55, 15400–15411 CrossRef CAS.
- W. Xiong, M. Yang, J. Wang, H. Wang, P. Zhao, Z. Li, B. Liu, X. Kong, H. Duan and Y. Zhao, Removal, recycle and reutilization of multiple heavy metal ions from electroplating wastewater using super-stable mineralizer Ca-based layered double hydroxides, Chem. Eng. Sci., 2023, 279, 118928 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02860k |
| This journal is © The Royal Society of Chemistry 2024 |