
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Germaborene reactivity study – addition of carbon nucleophiles, cycloaddition reactions, coordination chemistry†
Christian
Reik
,
Lukas W.
Jenner
,
Hartmut
Schubert
,
Klaus
Eichele
and
Lars
Wesemann
 *
*
Institut für Anorganische Chemie, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: lars.wesemann@uni-tuebingen.de
First published on 21st June 2024
Abstract
MeNHC substituted germaborenium cation 2 was synthesized directly in reaction of bromo-substituted germaborene 1b with MeNHC. The adamantyl isonitrile substituted germaborenium cation 4 was obtained stepwise: substitution of the chloride atom against adamantyl isonitrile at the B–Cl unit in 1a, simultaneous migration of the chloride to the germanium atom followed by chloride abstraction using Na[BArF4] gives the germaborenium cation 4. Substitution of the bromide atom in 1b against carbon monoxide followed by bromide abstraction using Ag[Al(OtBuF)4] leads to compound 6 exhibiting a B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond substituted at the boron atom by a germylium cation. Treating the germaborene [GeB–Ph] (1c) with selenium, a cycloaddition product 7 was characterised featuring a GeBSe heterocycle. Carbon dioxide reacts with 1b to give a four membered ring molecule 8 as the product of a B–C and Ge–O bond formation. In reaction of 1b with dimethylbutadiene, a product 9 of a [2 + 4] cycloaddition was isolated. Transition metal fragments [Fe(CO)4 (10), CuBr (11), AuCl (12)] show coordination at the germaborene double bond. Molecular structures of the germaborene coordination compounds 10–12 are presented and the ligand properties are discussed. After treating the germaborene [GeB–Br] (1b) with [Cp*Al]4, insertion of a Cp*Al moiety into the B–Br bond was found (13).
C double bond substituted at the boron atom by a germylium cation. Treating the germaborene [GeB–Ph] (1c) with selenium, a cycloaddition product 7 was characterised featuring a GeBSe heterocycle. Carbon dioxide reacts with 1b to give a four membered ring molecule 8 as the product of a B–C and Ge–O bond formation. In reaction of 1b with dimethylbutadiene, a product 9 of a [2 + 4] cycloaddition was isolated. Transition metal fragments [Fe(CO)4 (10), CuBr (11), AuCl (12)] show coordination at the germaborene double bond. Molecular structures of the germaborene coordination compounds 10–12 are presented and the ligand properties are discussed. After treating the germaborene [GeB–Br] (1b) with [Cp*Al]4, insertion of a Cp*Al moiety into the B–Br bond was found (13).
Introduction
In main group element chemistry, studies of boron-element double bonds are an attractive area of research.1–3 In addition to the development of a synthesis strategy, studies on the reactivity of the [BE] unit and the investigation of the electronic structures of the unsaturated molecules are rewarding challenges in molecular chemistry. Research in this area has led to the presentation of many examples of boron-element double bonds: BB,4–8 BC,9–11 BSi,12–16 BGe,17–19 BSn,20 BN,21–29 BP,30–34 BAs,33,35 BO,36–41 BS,41–43 BSe,36,41,43,44 and BTe.41,44 Our research is focused on the chemistry of heavy elements of the group 14. While compounds featuring the BC-double bond have been known for more than forty years, Sekiguchi et al. presented the first example for a BSi double bond in 2006.12 The reactivity of the BC double bond has been of major interest since it was first synthesized. Research groups of Berndt, Nöth and Paetzold studied the chemistry of BC double bonds intensively and presented results of [2 + 2], [2 + 3]-cycloaddition reactions with ketones, alkynes, iminoboranes, and azides.10,45–47 Furthermore, reactions of various reagents like HCl, Br2, MeLi, MeBBr2, tBuNC, HNMe2 with the BC bond were presented. In these reactions, the boron atom reacts as an electrophile, adding, for example, the [CH3]− or [Cl]− anion, and the carbon atom of the BC unit shows the reactivity of a nucleophile. Thus, addition of MeBBr2 leads to formation of B–Br and C–BMeBr units.47 In the current literature, anionic borataalkenes serve as borata-Wittig olefination reagents and borataalkenes were investigated as π-ligands in organometallic chemistry.48–54 The chemistry of Lewis base stabilized alkylideneboranes is also studied intensively with respect to BC bond cleavage, 1,2-dipolar reactivity studies and cycloaddition reactions.48,55–63
Borasilene was reacted with elemental sulphur and selenium to give the three-membered BSiS and BSiSe heterocycles.16 Treating borasilene with oxygen, a splitting of the double bond was observed and 1,3,2,4-dioxasilaboretane was isolated.16 The electrophilicity of the boron atom in the borasilene BSi unit was demonstrated in reaction of the borasilene with lithium trimethylsilylacetylide adding the anionic acetylide at the boron atom.14 The chloride adduct of borasilene exhibits an intramolecular C–H addition reaction under formation of Si–H and B–C bonds.13 This anionic borasilene adduct also shows a reaction with sulphur to give 1,3,2,4-dithiasilaboretane.13 As an interesting addition to the field of B–Si multiple bonds, examples for 2π-aromatic disiladiboretenes exhibiting a planar geometry were published recently.15,64 Kinjo et al. presented the synthesis of an allene type linear [GeBN] molecule exhibiting the first GeB double bond in 2020.17 At the same time, we presented another approach to the synthesis of germaborenes by treating an intramolecular phosphine-germylene Lewis pair with boron trihalides (BCl3, BBr3) followed by Mg reduction.18 The germaborenes 1a, b (Scheme 1) are light sensitive compounds and react at room temperature with light of 530 nm wavelength in a [2 + 2] cycloaddition reaction with a Trip moiety of the terphenyl substituent Ar* (Ar* = 2,6-Trip2C6H3, Trip = 2,4,6-triisopropylphenyl). This reaction, which has been the only example for a cycloaddition reaction of germaborenes so far, is reversible by irradiation with light of 366 nm wavelength, recovering the starting material.18 Germaborene 1a, 1b and also the phenyl substituted derivative 1c were shown to react as a source of borylenes [BX] (X = Cl, Br) and [BPh] in reaction with azides RN3 (R = SiMe3, adamantyl) to yield iminoborane derivatives.65 In analogy to the synthesis of 1, we applied an intramolecular stannylene Lewis pair to this procedure to give the first examples of stannaborenes realized in a stannaborenyl anion and a stannaborenium cation.20 The stannaborenium cation adds ammonia at the SnB double bond to give a B–H and Sn–NH2 unit.20
 | ||
| Scheme 1 Reactions of germaborene (1a X = Cl, 1b X = Br, 1c X = Ph) with carbon-based nucleophiles MeNHC, AdNC and CO (Ar* = 2,6-Trip2C6H3, Trip = 2,4,6-triisopropylphenyl). | ||
We set up the first systematic reactivity study of germaborenes because in terms of orbital overlap and therefore bonding energy, the germaborene consists of a less favourable element combination, which should result in high reactivity. Furthermore, the different substituents on the boron atom in 1 allow to study the influence of the substituents on the reactivity. Since the chemistry of the homologous boraalkenes and borasilenes has been reported, a comparative study with germaborene reactivity is of interest. Finally, the germaborene can be synthesized straightforwardly in a yield of up to 75% from up to 500 mg starting material making a study of germaborene 1 chemistry possible. We present reactions of germaborene with carbon-based nucleophiles leading to unprecedented germaborenium cations. So far unknown cycloaddition reactions with selenium, carbon dioxide and dimethylbutadiene are presented, and results are compared with the chemistry of boraalkenes and borasilenes. The coordination chemistry of the GeB moiety is discussed in iron tetracarbonyl and coinage metal complexes and the ligand properties are compared in view of olefin and boraalkene coordination compounds. An insertion reaction of [Cp*Al]4 into the B–Br unit of the germaborene is shown as an example for an electropositive substituent on boron.
Results and discussion
Halide substituted germaborenes 1 (a: X = Cl, b: X = Br) were treated with MeNHC, AdNC and CO (Scheme 1). Interestingly, the Lewis bases do not attack the germanium atom and instead form a bond with the boron atom although the Ge–B σ- and π-bonds in the germaborene are polarized toward the boron atom.18 At room temperature, the heterocyclic carbene reacts at the boron atom to directly yield a so far unknown MeNHC-substituted germaborenium cation (2) (Scheme 1). The isonitrile and CO show a stepwise reaction with the halide substituted germaborene. In both cases, a carbon–boron bond was formed, and the halide migrates to the germanium atom. Obviously, the isonitrile is less reactive and heating to 60 °C is necessary for product formation. The carbon monoxide reaction product (5) however, also featuring a halide migration to the germanium atom, is formed at room temperature. Both halides 3 and 5, showing a short B–C interaction, were transformed to the cations 4 and 6 by halide abstraction using Na[BArF4] or Ag[Al(OtBuF)4] (Scheme 1).66,67Compounds 2–6 were characterised by NMR spectroscopy and selected signals are listed in Table 1. In the 11B NMR spectrum the adducts 3 and 5 show a signal at lower frequency in comparison to the cationic products of halide abstraction 4 and 6. 13C NMR signals of the CO-substituent at the boron atom in 5 and 6 were only observed with 13CO-gas (220.4 ppm 5, 203.4 ppm 6, see Fig. 1 and SI for spectra).
| 11B δ [ppm] (1J B–P [Hz]) | 31P δ [ppm] (1J11B–P [Hz]) | |
|---|---|---|
| a q: non-binomial quartet, br: unresolved quartet. | ||
| 1a, b, c18,65 | 17.3, 10.3, 16.2 | 5.2, 7.3, 12.6 |
| 2 | 0.1 (d, 127.9 Hz) | 17.9 (br) |
| 3 | −21.4 (d, 115.0 Hz) | 27.8 (br) |
| 4 | −13.4 (d, 149.0 Hz) | 28.5 (q, 152.4 Hz) |
| 5 | −39.7 (d, 121.3 Hz) | 40.1 (br) |
| 6 | −24.8 (d, 168.2 Hz) | 32.3 (q, 166.1 Hz) |
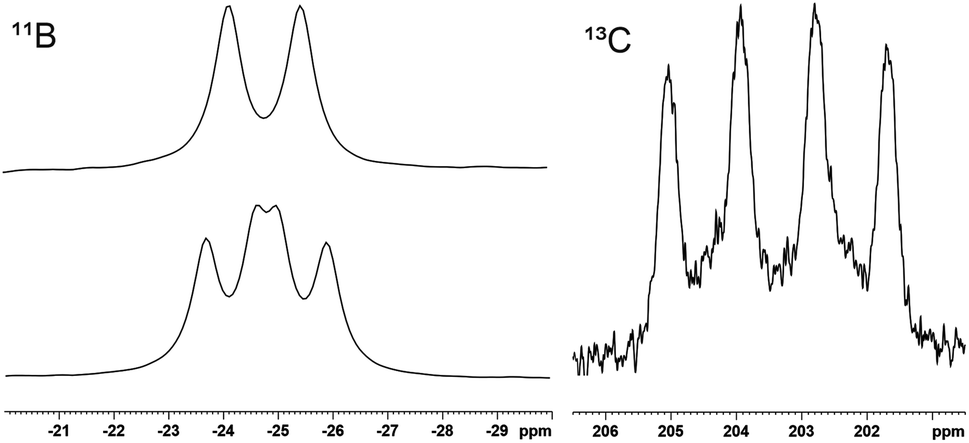 | ||
| Fig. 1 11B NMR (128.37 MHz) (top left, natural abundance 6: d 1J31P–11B = 168 Hz; bottom left, 13CO labelled sample of 6: dd, 1J31P–11B = 160.1 Hz, 1J13C–11B = 119.3 Hz) and 13C NMR (100.62 MHz) carbonyl region (right) of cation 6, 13CO labelled (1J11B–13C = 111.6 Hz). | ||
Adducts 3 and 5 exhibit B–C bond lengths of 1.433(3) and 1.418(3) Å which are comparable with molecules showing BC double bonds [1.401(5)–1.475(8)] and are short distances in comparison with the group of low valent boron isonitrile [1.420(6)–1.569(3) Å]59,68–71 and carbon monoxide [1.445(3)–1.492(4) Å]58,59,70,72–76 adducts (Fig. 2, molecular structure of 3 and 5).77–81 The sum of angles around the boron atom of adduct 3 is with 359.1(1)° close to 360° which can be interpreted as an indicator for delocalisation of the boron electron pair into the BC double bond. In the case of the CO-adduct 5, however, a smaller angle of 353.7(2)° around the boron atom, and therefore a slight pyramidalization, was observed, which indicates a partially localized electron pair on the boron atom.
 | ||
| Fig. 2 ORTEPs of the molecular structures of adducts 3 and 5. Thermal ellipsoids are shown at 50% probability level. Hydrogen atoms and iPr groups have been omitted (Table 2). | ||
| Ge–B | B–C4 | C4–E | Ge–B–P | Ge–B–C4 | P–B–C4 | |
|---|---|---|---|---|---|---|
| 1a 18 | 1.886(2) | 103.1(1) | ||||
| 2 | 1.890(2) | 1.562(3) | 102.6(1) | 134.6(2) | 122.8(2) | |
| 3 | 2.016(1) | 1.433(3) | E = N 1.212(2) | 105.1(1) | 134.1(1) | 119.9(1) |
| 4 | 1.912(3) | 1.483(4) | E = N 1.155(3) | 104.2(1) | 133.3(2) | 122.4(2) |
| 5 | 1.999(2) | 1.418(3) | E = O 1.158(3) | 106.8(1) | 130.7(2) | 116.2(2) |
| 6 | 1.931(2) | 1.437(3) | E = O 1.143(3) | 106.2(1) | 133.6(2) | 120.0(2) |
In the series of cationic Lewis-base adducts 2, 4 and 6 (Fig. 3) the B–C interatomic distances [1.562(3), 1.483(4), 1.437(3) Å] decrease while the Ge–B [1.890(2), 1.912(3), 1.931(2) Å] bond lengths increase. MeNHC-adduct 2 features a short GeB double bond17,18,65,82 and a B–C single bond with the NHC-donor.83–85 Apparently for steric reasons, an angle of 74.6° was found between the [MeNHC-ligand] and [GeBPC2C3] planes in compound 2, which makes π-back donation from the boron atom to the NHC-molecule less favourable. The isonitrile- and CO-donor cations (4, 6) (Fig. 3) show shorter B–C and longer Ge–B interatomic distances in comparison to cation 2. The isonitrile donor in 4 shows less back bonding from the boron atom to the carbon atom C4 upon cationization which goes along with a larger angle at the nitrogen atom C4–N–C5 [3: 127.6(2), 4: 175.0(3)°] (Fig. 3) and a longer B–C bond together with a shorter C–N bond in comparison to 3. The B–C–N bond length found in 4 are comparable with isonitrile adducts of low valent boron compounds [B–C: 1.420(6)–1.569(3); C–N 1.152(3)–1.243(3) Å].59,68–71 The B–C distance in 6 is comparable with a long double bond between these elements.77–81 CO adducts of low valent boron compounds exhibit longer B–C bond lengths [1.445(3)–1.492(4) Å].58,59,70,72–76 The IR stretching frequencies for the CO unit in 5 (1984 cm−1) and 6 (2024 cm−1) reflect a considerable amount of π-back donation by the borylene boron atom. CO adducts of borylenes like [(DippNC)(OC)BTp] (1930, 2094 cm−1) and [(OC)2BTp] (1942, 2060 cm−1) exhibit comparable CO frequencies [Dipp = 2,6-diisopropylphenyl, Tp = 2,6-di(2,4,6-triisopropylphenyl)phenyl].70,72,74,76,86 The direct substitution of a bromide substituent at a low valent boron atom against a carbon monoxide was observed by Xie et al. reacting a bissilylene stabilized bromoborylene with tungsten hexacarbonyl Table 2.76,87
 | ||
| Fig. 3 ORTEPs of the molecular structures of cations 2, 4 and 6. Thermal ellipsoids are shown at 50% probability level. Hydrogen atoms, iPr groups and the anions have been omitted (Table 2). | ||
To evaluate the electronic situations in the molecules 2–6, DFT calculations [BP86/ωB97X-D3BJ, def2-SVP/TZVP(Ge,B,C4)] together with NBO analysis (Table 3) have been carried out on the basis of the solid state molecular structures (see ESI†). The leading Lewis structures of compounds 2–6 are shown in Scheme 2 and the HOMOs of cations 2, 4 and 6 are depicted in Fig. 4.
| 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|
| Ge–B [Å] | 1.89855 | 2.00338 | 1.89128 | 1.99776 | 1.94257 |
| q [e] Ge, B | 1.25, −0.76 | 1.39, −0.83 | 1.54, −1.04 | 1.32, −1.00 | 1.51, −0.99 |
| Wiberg/Löwdin | 1.51/1.60 | 0.87/1.11 | 1.34/1.49 | 0.904/1.14 | 1.10/1.34 |
| σ-bond occ. | 1.9069 | 1.8645 | 1.9180 | 1.8688 | 1.8710 |
| Ge–B% (NBO) | 43, 57 | 41, 59 | 40, 60 | 40, 60 | 40, 60 |
| π-bond occ. | 1.7099 | 1.7489 | 1.6285 | 1.7397 | 1.6547 |
| Ge–B% (NBO) | 37, 63 | 27, 73 | |||
| B–C% (NBO) | 55, 45 | 59, 41 | 60, 40 | ||
| σ-bond occ. | 1.9625 | 1.9661 | 1.9671 | 1.9757 | 1.9754 |
| B–C% (NBO) | 34, 66 | 38, 62 | 37, 63 | 39, 61 | 39, 61 |
| B–C [Å] | 1.54180 | 1.42172 | 1.47599 | 1.42612 | 1.43407 |
| Wiberg/Löwdin | 0.96/1.32 | 1.56/1.71 | 1.19/1.45 | 1.51/1.72 | 1.44/1.63 |
 | ||
| Scheme 2 Leading Lewis structures for adducts 3, 5 and cations 2, 4, 6. | ||
 | ||
| Fig. 4 HOMOs representing the Ge–B π-bond in 2 and 4 and B–C π-bond in 6 (contour value 0.062).88 | ||
In all cases, the Ge–B σ-bond shows a slight polarisation towards the boron atom and the B–C σ-bond is polarized to the carbon atom. In the case of the adducts 3 and 5, a BC π-bond was observed exhibiting a polarisation to the boron atom. To a small extent, the BC π-bond of 5 exhibits hyperconjugation with the Ge–Br σ*-bond. The cations 2 and 4 exhibit a Ge–B π-bond with a polarisation toward the boron atom, which is more distinctive for the isonitrile adduct 4. Thus, 2 and 4 are examples for unprecedented germaborenium cations. The bonding situation in MeNHC-adduct 2 can be compared with the homologous MeNHC-supported stannaborenium cation, featuring a SnB double bond.20 The CO-cation 6 however, features a B–C π-bond, which is polarized to the boron atom. Obviously the π-accepting character of the CO-ligand dominates the delocalisation of the electron pair. The formation of the germyl cation in 6 does not lead to formation of a GeB double bond like in the case of 4. The difference between isonitrile versus CO delocalisation of the electron pair at a low valent boron atom can be compared with the electronic situation found in [Ar*B(CO)CNDipp].86,89
A cycloaddition reaction of germaborenes has been only reported in the case of the intramolecular reversible [2 + 2] addition between GeB double bond and an arene ring of the terphenyl substituent.18 To further investigate the reactivity of the GeB double bond in germaborenes, reactions with selenium, carbon dioxide and dimethylbutadiene were carried out. Selenium reacts at room temperature with the germaborene 1c and formation of a so far unknown GeBSe-heterocycle was characterised by single crystal structure analysis (Scheme 3, Fig. 5) and shows a signal in the 77Se NMR spectrum at −386.8 ppm. This type of addition was reported for the homologues boraalkene and borasilene and also for digermenes and diborenes leading to the corresponding three-membered ring molecules.16,62,90–94 The Ge–B bond in 7 shows an elongation [2.0570(17) Å] compared to the starting material and is close to the value of a Ge–B single bond [2.095(5) Å].18 In 7, a Ge–Se bond [2.3576(2) Å] and a B–Se bond [2.1012(17) Å] were formed. Both bond lengths and the angle at the Se atom of 54.6(1)° are comparable with distances and angles found in the cycles [CBSe: B–Se 2.097(5), C–Se–B 45.5(2); SiBSe: B–Se 1.963(3), Si–Se–B 54.9(1); Ge2Se: Ge–Se 2.3961(4), 2.4017(4) Å, Ge–Se–Ge 59.2(1); B2Se: B–Se 2.115(2), 2.063(2), 2.073(2), 2.102(5), 2.039(6) Å, B–Se–B: 50.03(9)°].90–93 The signal found for 7 in the 77Se NMR spectrum at −386.8 ppm lies in the range of signals found for comparable three membered ring molecules: CBSe −317.0, −453.4, −368.1; SiBSe −400.7; Ge2Se −331.0; B2Se −361.5 ppm.16,62,90,93
 | ||
| Scheme 3 Reactions of germaborene 1b (X = Br)18 and 1c (X = Ph)65 with selenium, carbon dioxide and dimethylbutadiene (Ar* = 2,6-Trip2C6H3, Trip = 2,4,6-triisopropylphenyl). | ||
 | ||
| Fig. 5 ORTEPs of the molecular structures of 7 and 8. Thermal ellipsoids are shown at 50% probability level. Hydrogen atoms and iPr groups have been omitted. Interatomic distances in Å and angles in (°). 7: Ge–B 2.0570(17), Ge–Se 2.3576(2), B–Se 2.1012(17), B–P 1.9601(18), Ge–Se–B 54.6(1), Ge–B–Se 69.1(1), Se–Ge–B 56.4(1); 8: Ge–B 2.085(2), Ge–O1 1.8969(14), B–P 1.971(2), B–C4 1.629(3), C4–O2 1.210(3), O1–Ge–B 74.0(1), C4–B–Ge 81.9(1), O1–C4–O2 121.7(2), O2–C4–B 132.2(2); 9: Ge–B 2.1089(17), B–C7 1.626(2), C6–C7 1.521(2), C5–C6 1.342(2), C4–C5 1.507(2), Ge–C4 1.9842(15). | ||
Treating the germaborene 1b with carbon dioxide, the product of a [2 + 2] cycloaddition, the first example for a BGeOC heterocycle, was obtained (Scheme 3 and Fig. 5). A B–C 1.629(3) and a Ge–O 1.8969(14) Å single bond were formed and the Ge–B bond length [2.085(2) Å] is elongated and comparable with a single bond between these elements.18,83,84 The homologous boraalkene adds CO2 under formation of a C–C and B–O bond.62 In the boraalkene the boron atom reacts as an electrophile and in the germaborene the boron atom exhibits nucleophilic reactivity. Further examples for carbon dioxide [2 + 2] cycloaddition reactions were presented for a variety of unsaturated low valent main group compounds like, e.g., diborenes,93,95–97 dialumenes98 and disilenes.99
Treating the germaborene 1b with dimethylbutadiene, the product (9) of a [2 + 4] cycloaddition was isolated (Scheme 3), and the molecular structure is shown in the ESI.† In Table 411B and 31P NMR data are listed. The shift of the 11B NMR signals to lower frequencies for compounds 7–9 can be explained with the increase of the coordination number in comparison to the starting material 1. The signal for the carbon atom at boron in the CO2-product 8 was observed at 184.2 ppm using 13CO2.
| 11B δ[ppm] | 31P δ[ppm] | |
|---|---|---|
| 7 | −18.1 | 6.5 |
| 8 | −12.9 | 14.5 |
| 9 | −9.7 | 14.6 |
| 10 | −13.5 | 18.6 |
| 11 | 2.5 | 4.2 |
| 12 | 0.4 | 1.0 |
| 13 | 0.1 | 36.6 |
| 14 | 3.6 | 35.2 |
In view of a known coordination chemistry of the homologous boraalkenes the ligand properties of the GeB double bond were tested in reactions with Fe2(CO)9, [Me2S·CuBr] and [Me2S·AuCl] (Scheme 4).49–52 First coordination compounds with the germaborene ligand were isolated and the molecular structures together with selected interatomic distances and angles are depicted in Fig. 6 (molecular structure of 11 is shown in the ESI†). Interatomic distances concerning the Fe-coordination at the Ge–B unit (Table 5) can be compared with the Fe–B distance found in the boraalkene Fe(CO)4 complex of amino-9-fluorenylideneborane [Fe–B: 2.125(5) Å].49 The Ge–Fe bond length lies in the range of germylene–iron coordination compounds [2.4112(3)–2.5970(3) Å].100 Copper and gold coordination at the germaborene (11, 12) can be compared with coordination of the coinage metals at homologous borataalkene which shows a slippage from η2 [Cu: Cu–B 2.12(2)], to η1 [Au: Au–B 2.23(1) Å].53 Cu–B and Au–B bond lengths can also be compared with diborene coordination compounds: [Cu–B 2.149(3), 2.146(3); Au–B 2.271(3), 2.354(2), 2.394(8) Å]54,101,102 The Cu–Ge bond length observed in 11 is slightly smaller than distances found for copper coordination at germanium cluster compounds [Cu–Ge 2.4752(4)–2.5043(4) Å].103,104 In the case of the found Au–Ge interatomic distance in 12 the bond length lies in the range of GeCl3 coordination at gold: Au–Ge 2.4150(6)–2.5351(7) Å.105
 | ||
| Scheme 4 Reactions of germaborene with Fe2(CO)9, [Me2S·CuBr], [Me2S·AuCl] and [Cp*Al]4. | ||
 | ||
| Fig. 6 ORTEPs of the molecular structures of 10 and 12. Thermal ellipsoids are shown at 50% probability level. Hydrogen atoms and iPr groups have been omitted. Interatomic distances in Å and angles in (°). 10: Ge–B 2.017(4), Ge–Fe 2.4718(6), Fe–B 2.257(4), Fe–C5 1.802(4), Fe–C6 1.792(4), Fe–C7 1.800(4), Fe–C4 1.797(4), B–P 1.930(4), B–Br 1.990(4), Ge–Fe–B 50.3(1), Fe–Ge–B 59.3(1), Fe–B–Ge 70.4(1), B–Fe–C5 170.8(2), B–Fe–C7 86.1(1); 11: Ge–B 1.9266(18), Cu–B 2.1049(18), Ge–Cu 2.4627(3), B–P 1.9112(17), Cu–Br1 2.2632(3), B–Br2 1.9421(18), B–Ge–Cu 55.7(1), B–Cu–Ge 49.1(1), Cu–B–Ge 75.2(1), Br2–B–Cu 106.9(1), Br1–Cu–Ge 164.7(1); 12: Ge–B 1.958(5), Ge–Au 2.5057(5), Au–B 2.195(5), B–P 1.945(5), Au–Cl1 2.3330(12), B–Cl2 1.780(5), Ge–Au–B 48.7(1), Au–B–Ge 74.0(2), B–Ge–Au 57.4(1). | ||
| M | Ge–B | Ge–M | B–M |
|---|---|---|---|
| 10 Fe | 2.017(4) | 2.4718(6) | 2.257(4) |
| 11 Cu | 1.927(2) | 2.4627(3) | 2.105(2) |
| 12 Au | 1.958(4) | 2.5057(4) | 2.195(5) |
In comparison to the starting material [1a: 1.886(2); 1b: 1.895(3) Å], an increase of the Ge–B bond length upon coordination of the metal fragments was found (Table 5). Coordination of the Fe(CO)4 fragment gives the largest elongation and for CuBr-coordination only a slight increase of the Ge–B bond length was observed. In the IR spectrum of the Fe(CO)4 complex of amino-9-fluorenylideneborane showing coordination of the iron fragment at a BC bond the CO stretching frequencies were found at ν = 2064, 2011, 1962 cm−1. An olefin Fe(CO)4 complex was found to show CO wavenumbers at 2071, 2005, 1975 cm−1.106 The CO stretching frequencies of the germaborene Fe(CO)4 complex 10 were observed at slightly lower wavenumbers 2051, 1983 and 1960 cm−1 indicating the germaborene as a slightly better donor ligand in comparison to the amino-9-fluorenylideneborane boraalkene and olefin ligand.
Investigated by DFT calculations and NBO analyses, the electronic situation of the coordination compounds can be discussed based on the Dewar–Chatt–Duncanson (DCD)107,108 bonding model (see Fig. 7 and Table SI3 in the ESI†). The HOMO of the Fe(CO)4 complex resembles the σ-donor component of the Fe–(Ge–B) interaction and the HOMO-1 the π-acceptor interaction (Fig. 7). The copper and gold complexes with a d10-valence electron count exhibit a small degree of π-back bonding in the π*-MO of the germaborene. The σ-donor interaction however, which can be described as a donation from the π-MO to the s-orbital of the metal, is more pronounced in the case of the gold coordination compound (Table SI3 in the ESI†).
 | ||
| Fig. 7 HOMO-1 and HOMO of 10 representing the π-acceptor and σ-donor interaction between Ge–B double bond and Fe(CO)4 fragment (contour value 0.03).88 | ||
In the case of the stannaborene, we recently presented a magnesium substituted [SnB–MgBr] stannaborene derivative.20 The change of polarity of the GeB–X bond in germaborene chemistry from halide (1a, 1b) or phenyl (1c) to magnesium or another electropositive substituent would make a new reactivity pattern at the boron atom of the GeB unit possible. Therefore, we studied the synthesis of the homologous magnesium derivative. However, so far, we cannot present a reliable procedure. To connect a less electronegative substituent at the boron atom, we also investigated the incorporation of an aluminium substituent reacting [Cp*Al]4 with germaborene 1b to a give a [GeB–Al] unit.109–112 In the final procedure, 1b was treated with [Cp*Al]4 in benzene at 60 °C for 21 hours to give the aluminium substituted product 13.109–112 The colour of the solution changed from red to orange and after evaporation of the solvent, crystals were obtained from n-pentane (yield 52%). The molecular structure of the insertion product 13 of a Cp*Al molecule into a B–Br bond is shown in Fig. 8. The electronic structure of 13 was analysed by DFT calculations together with NBO analysis. The B–Al σ-bond is polarised towards the boron atom: B 74.4%, Al 25.6% (σ-bond occ. 1.93 e−); to compare with 1b: B–Br: B 33.3%, Br 66.7% (σ-bond occ. 1.98 e−). However, the reactivity of this negatively charged GeB-unit should be checked in further investigations. In the following, a MeNHC adduct (14) of this aluminium compound was synthesized showing a slippage from η5-to η1-coordination of the Cp* moiety at aluminium. The phenyl substituted germaborene 1c however, shows no reaction with [Cp*Al]4 at 60 °C in benzene. Reaction of a transient CAAC-adduct of phenylborylene with Cp3tAl [Cp3t = η5-1,3,4-tri(tert-butyl)-cyclopentadienyl] results in the formation of B–Al bond [2.069(2) Å], which is discussed as an example for a B–Al multiple bond.113,114 Although short Al–B bond lengths were observed in 13 and 14 [13: 2.052(2), 14: 2.1103(13) Å], analyses of the electronic situations give no indications for a partial double bond character between boron and aluminium. Furthermore, the Ge–B distances show only a slight elongation in comparison to the germaborene starting material.18 Cationization by halide abstraction was not successful so far.
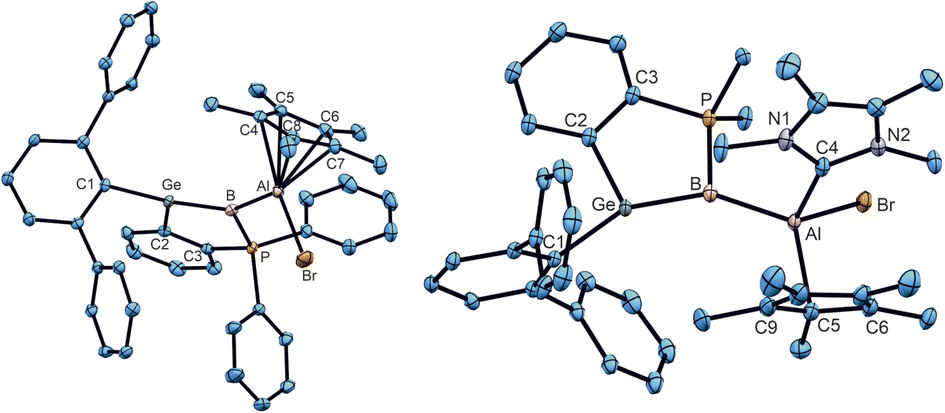 | ||
| Fig. 8 ORTEPs of the molecular structures of 13 and 14. Thermal ellipsoids are shown at 50% probability level. Hydrogen atoms and iPr groups have been omitted. Interatomic distances in Å and angles in (°). 13: Ge–B 1.901(2), B–P 1.8764(19), B–Al 2.052(2), Al–Br 2.3547(6), Al–C4 2.2833(18), Al–C5 2.396(2), Al–C6 2.303(2), Al–C7 2.190(2), Al–C8 2.1626(19), P–B–Ge 99.4(1), P–B–Al 119.2(1), Ge–B–Al 141.3(1), B–Al–Br 107.6(1); 14: Ge–B 1.9163(13), B–Al 2.1103(13), B–P 1.8885(14), Al–C4 2.0609(13), Al–Br 2.4328(4), Al–C5 2.0860(13), Ge–B–P 98.0(1), Ge–B–Al 145.3(1), Al–B–P 116.2(1), B–Al–Br 104.4(1). | ||
Conclusions
Unprecedented germaborenium cations featuring a GeB double bond were isolated substituted by a MeNHC or an adamantyl isonitrile ligand at the boron atom. In the MeNHC case, this cation was obtained directly by reacting bromo substituted germaborene 1b with MeNHC. The formation of the isonitrile substituted cation was realized in two steps: substitution of the chloride atom of the B–Cl unit in germaborene 1a against the adamantyl isonitrile going along with simultaneous transfer of the chloride to the germanium atom followed by chloride abstraction using Na[BArF4]. Due to the strong π-acceptor properties of carbon monoxide, the analogous CO-substitution product at boron exhibits a BC double bond substituted by a germylium cation at the boron atom.
In reaction of the phenyl substituted germaborene 1c with selenium, a so far unknown GeBSe heterocycle was isolated. Carbon dioxide reacts via a [2 + 2] cycloaddition reaction with bromo germaborene 1b. Bond formation between the electrophilic carbon atom and the nucleophilic boron atom gives a four membered GeBCO heterocycle. First coordination compounds with the GeB double bond were observed by coordination of the metal fragments [Fe(CO)4, CuBr, AuCl]. A comparison of IR data of analogue Fe(CO)4 complexes with boraalkene and olefin ligands allows a categorization of the ligands, with the germaborene being the slightly better donor ligand.
By insertion of a Cp*Al fragment into the B–Br bond of germaborene an electropositive substituent was introduced to germaborene chemistry giving the boron atom a putative nucleophilic character, which is to be verified in further germaborene chemistry studies.
Data availability
Full experimental and computational details are provided as part of the ESI.†Author contributions
Investigations, writing, review C. R.; preparation of 2 L. W. J.; special NMR experiments K. E.; discussion and X-ray measurements H. S.; supervision, funding acquisition, DFT calculation, manuscript writing and review L. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support of the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (Justus 2 Cluster).Notes and references
- R. Borthakur and V. Chandrasekhar, Coord. Chem. Rev., 2021, 429, 213647 CrossRef CAS.
- C. Weetman, Chem.–Eur. J., 2021, 27, 1941–1954 CrossRef CAS PubMed.
- A. Agarwal and S. K. Bose, Chem.–Asian J., 2020, 15, 3784–3806 CrossRef CAS PubMed.
- A. Moezzi, R. A. Bartlett and P. P. Power, Angew. Chem., Int. Ed., 1992, 31, 1082–1083 CrossRef.
- Y. Wang, B. Quillian, P. Wei, C. S. Wannere, Y. Xie, R. B. King, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2007, 129, 12412–12413 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420–1422 CrossRef CAS PubMed.
- W. Lu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2018, 140, 1255–1258 CrossRef CAS PubMed.
- L. Zhu and R. Kinjo, Chem. Commun., 2023, 59, 10436–10439 RSC.
- B. Glaser and H. Nöth, Angew. Chem., Int. Ed., 1985, 24, 416–417 CrossRef.
- H. Klusik and A. Berndt, Angew. Chem., Int. Ed., 1983, 22, 877–878 CrossRef.
- R. Boese, P. Paetzold and A. Tapper, Chem. Ber., 1987, 120, 1069–1071 CrossRef CAS.
- N. Nakata and A. Sekiguchi, J. Am. Chem. Soc., 2006, 128, 422–423 CrossRef CAS PubMed.
- Y. Suzuki, S. Ishida, S. Sato, H. Isobe and T. Iwamoto, Angew. Chem., Int. Ed., 2017, 56, 4593–4597 CrossRef CAS PubMed.
- D. Franz, T. Szilvási, A. Pöthig and S. Inoue, Chem.–Eur. J., 2019, 25, 11036–11041 CrossRef CAS PubMed.
- S. K. Kushvaha, P. Kallenbach, S. S. Rohman, M. K. Pandey, Z. Hendi, F. Rüttger, R. Herbst-Irmer, D. Stalke, P. Parameswaran and H. W. Roesky, J. Am. Chem. Soc., 2023, 145, 25523–25527 CrossRef CAS PubMed.
- N. Nakata and A. Sekiguchi, Chem. Lett., 2007, 36, 662–663 CrossRef CAS.
- B. Rao and R. Kinjo, Angew. Chem., Int. Ed., 2020, 59, 3147–3150 CrossRef CAS PubMed.
- D. Raiser, C. P. Sindlinger, H. Schubert and L. Wesemann, Angew. Chem., Int. Ed., 2020, 59, 3151–3155 CrossRef CAS PubMed.
- P. Tholen, Z. Dong, M. Schmidtmann, L. Albers and T. Müller, Angew. Chem., Int. Ed., 2018, 57, 13319–13324 CrossRef CAS PubMed.
- M. Zweigart, K. Eichele, H. Schubert, C. P. Sindlinger and L. Wesemann, J. Am. Chem. Soc., 2023, 145, 12452–12458 CrossRef CAS PubMed.
- G. Alcaraz, L. Vendier, E. Clot and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2010, 49, 918–920 CrossRef CAS PubMed.
- M. C. MacInnis, R. McDonald, M. J. Ferguson, S. Tobisch and L. Turculet, J. Am. Chem. Soc., 2011, 133, 13622–13633 CrossRef CAS PubMed.
- A. Kumar, N. A. Beattie, S. D. Pike, S. A. Macgregor and A. S. Weller, Angew. Chem., Int. Ed., 2016, 55, 6651–6656 CrossRef CAS PubMed.
- R. Guo, C. Hu, Q. Li, L. L. Liu, C.-H. Tung and L. Kong, J. Am. Chem. Soc., 2023, 145, 18767–18772 CrossRef CAS PubMed.
- M. Sugie, H. Takeo and C. Matsumura, J. Mol. Spectrosc., 1987, 123, 286–292 CrossRef CAS.
- P. Paetzold, E. Schröder, G. Schmid and R. Boese, Chem. Ber., 1985, 118, 3205–3216 CrossRef CAS.
- P. Paetzold, in Adv. Inorg. Chem., ed. H. J. Emeléus and A. G. Sharpe, Academic Press, 1987, vol. 31, pp. 123–170 Search PubMed.
- K. Niedenzu, J. W. Dawson, P. Fritz and H. Jenne, Chem. Ber., 1965, 98, 3050–3052 CrossRef CAS.
- K. Niedenzu, H. Beyer, J. W. Dawson and H. Jenne, Chem. Ber., 1963, 96, 2653–2656 CrossRef CAS.
- X. Feng, M. M. Olmstead and P. P. Power, Inorg. Chem., 1986, 25, 4615–4616 CrossRef CAS.
- R. T. Paine and H. Noeth, Chem. Rev., 1995, 95, 343–379 CrossRef.
- A. N. Price and M. J. Cowley, Chem.–Eur. J., 2016, 22, 6248–6252 CrossRef CAS PubMed.
- E. Rivard, W. A. Merrill, J. C. Fettinger and P. P. Power, Chem. Commun., 2006, 3800–3802 RSC.
- E. A. LaPierre, B. O. Patrick and I. Manners, J. Am. Chem. Soc., 2023, 145, 7107–7112 CrossRef CAS PubMed.
- M. A. Petrie, S. C. Shoner, H. V. R. Dias and P. P. Power, Angew. Chem., Int. Ed., 1990, 29, 1033–1035 CrossRef.
- Y. K. Loh, K. Porteous, M. Á. Fuentes, D. C. H. Do, J. Hicks and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 8073–8077 CrossRef CAS PubMed.
- D. Vidovic, J. A. Moore, J. N. Jones and A. H. Cowley, J. Am. Chem. Soc., 2005, 127, 4566–4567 CrossRef CAS PubMed.
- Y. Wang, H. Hu, J. Zhang and C. Cui, Angew. Chem., Int. Ed., 2011, 50, 2816–2819 CrossRef CAS PubMed.
- Y. K. Loh, C. C. Chong, R. Ganguly, Y. Li, D. Vidovic and R. Kinjo, Chem. Commun., 2014, 50, 8561–8564 RSC.
- A. K. Swarnakar, C. Hering-Junghans, M. J. Ferguson, R. McDonald and E. Rivard, Chem. - Eur. J., 2017, 23, 8628–8631 CrossRef CAS PubMed.
- H. Dolati, L. Denker, B. Trzaskowski and R. Frank, Angew. Chem., Int. Ed., 2021, 60, 4633–4639 CrossRef CAS PubMed.
- D. Franz, E. Irran and S. Inoue, Angew. Chem., Int. Ed., 2014, 53, 14264–14268 CrossRef CAS PubMed.
- H. Wang, J. Zhang, H. Hu and C. Cui, J. Am. Chem. Soc., 2010, 132, 10998–10999 CrossRef CAS PubMed.
- S. Liu, M.-A. Légaré, D. Auerhammer, A. Hofmann and H. Braunschweig, Angew. Chem., Int. Ed., 2017, 56, 15760–15763 CrossRef CAS PubMed.
- B. Glaser, E. Hanecker, H. Nöth and H. Wagner, Chem. Ber., 1987, 120, 659–667 CrossRef CAS.
- R. Boese, P. Paetzold, A. Tapper and R. Ziembinski, Chem. Ber., 1989, 122, 1057–1060 CrossRef CAS.
- A. Tapper, T. Schmitz and P. Paetzold, Chem. Ber., 1989, 122, 595–601 CrossRef CAS.
- C. Chen, G. Kehr and G. Erker, Org. Chem. Front., 2023, 10, 4161–4166 RSC.
- S. Helm and H. Nöth, Angew. Chem., Int. Ed., 1988, 27, 1331–1337 CrossRef.
- S. W. Helm, G. Linti, H. Nöth, S. Channareddy and P. Hofmann, Chem. Ber., 1992, 125, 73–86 CrossRef CAS.
- K. S. Cook, W. E. Piers and S. J. Rettig, Organometallics, 1999, 18, 1575–1577 CrossRef CAS.
- K. Watanabe, A. Ueno, X. Tao, K. Škoch, X. Jie, S. Vagin, B. Rieger, C. G. Daniliuc, M. C. Letzel, G. Kehr and G. Erker, Chem. Sci., 2020, 11, 7349–7355 RSC.
- N. A. Phillips, R. Y. Kong, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2021, 60, 12013–12019 CrossRef CAS PubMed.
- F. Krämer, M. Radius, H. Berberich, I. Fernández and F. Breher, Chem. Commun., 2022, 58, 3905–3908 RSC.
- J. Han, C. Hu, Q. Li, L. L. Liu, C.-H. Tung, P. Cui and L. Kong, Inorg. Chem., 2023, 62, 18820–18824 CrossRef CAS PubMed.
- A. Hermann, F. Fantuzzi, M. Arrowsmith, T. Zorn, I. Krummenacher, B. Ritschel, K. Radacki, B. Engels and H. Braunschweig, Angew. Chem., Int. Ed., 2020, 59, 15717–15725 CrossRef CAS PubMed.
- J. Böhnke, H. Braunschweig, T. Dellermann, W. C. Ewing, T. Kramer, I. Krummenacher and A. Vargas, Angew. Chem., Int. Ed., 2015, 54, 4469–4473 CrossRef PubMed.
- J. Böhnke, H. Braunschweig, T. Dellermann, W. C. Ewing, K. Hammond, J. O. C. Jimenez-Halla, T. Kramer and J. Mies, Angew. Chem., Int. Ed., 2015, 54, 13801–13805 CrossRef PubMed.
- H. Asakawa, K.-H. Lee, Z. Lin and M. Yamashita, Nat. Commun., 2014, 5, 4245 CrossRef CAS PubMed.
- L. Kong, W. Lu, Y. Li, R. Ganguly and R. Kinjo, Angew. Chem., Int. Ed., 2016, 55, 14718–14722 CrossRef CAS PubMed.
- X. Jie, C. Chen, C. G. Daniliuc, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2023, 62, e202214700 CrossRef CAS PubMed.
- C. Chen, C. G. Daniliuc, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2021, 60, 19905–19911 CrossRef CAS PubMed.
- C. Chen, C. G. Daniliuc, G. Kehr and G. Erker, J. Am. Chem. Soc., 2021, 143, 21312–21320 CrossRef CAS PubMed.
- J. Fan, L. Yue, C. Liu, B. Rao, G. Zhou, A. Li and B. Su, J. Am. Chem. Soc., 2024, 146, 39–44 CrossRef CAS PubMed.
- D. Raiser, H. Schubert, H. F. Bettinger and L. Wesemann, Chem.–Eur. J., 2021, 27, 1981–1983 CrossRef CAS PubMed.
- I. Krossing, Chem.–Eur. J., 2001, 7, 490–502 CrossRef CAS.
- I. Krossing, H. Brands, R. Feuerhake and S. Koenig, J. Fluorine Chem., 2001, 112, 83–90 CrossRef CAS.
- H. Wang, L. Wu, Z. Lin and Z. Xie, Angew. Chem., Int. Ed., 2018, 57, 8708–8713 CrossRef CAS PubMed.
- H. Braunschweig, M. A. Celik, R. D. Dewhurst, K. Ferkinghoff, A. Hermann, J. O. C. Jimenez-Halla, T. Kramer, K. Radacki, R. Shang, E. Siedler, F. Weißenberger and C. Werner, Chem.–Eur. J., 2016, 22, 11736–11744 CrossRef CAS PubMed.
- M. Nutz, B. Borthakur, C. Pranckevicius, R. D. Dewhurst, M. Schäfer, T. Dellermann, F. Glaab, M. Thaler, A. K. Phukan and H. Braunschweig, Chem.–Eur. J., 2018, 24, 6843–6847 CrossRef CAS PubMed.
- T. J. Hadlington, T. Szilvási and M. Driess, Chem. Sci., 2018, 9, 2595–2600 RSC.
- C. Pranckevicius, C. Herok, F. Fantuzzi, B. Engels and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 12893–12897 CrossRef CAS PubMed.
- M. Arrowsmith, J. Böhnke, H. Braunschweig and M. A. Celik, Angew. Chem., Int. Ed., 2017, 56, 14287–14292 CrossRef CAS PubMed.
- J. Böhnke, M. Arrowsmith and H. Braunschweig, J. Am. Chem. Soc., 2018, 140, 10368–10373 CrossRef PubMed.
- A. Hofmann, M.-A. Légaré, L. Wüst and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 9776–9781 CrossRef CAS PubMed.
- H. Wang, L. Wu, Z. Lin and Z. Xie, J. Am. Chem. Soc., 2017, 139, 13680–13683 CrossRef CAS PubMed.
- M. M. Olmstead, P. P. Power, K. J. Weese and R. J. Doedens, J. Am. Chem. Soc., 1987, 109, 2541–2542 CrossRef CAS.
- M. Pilz, J. Allwohn, P. Willershausen, W. Massa and A. Berndt, Angew. Chem., Int. Ed., 1990, 29, 1030–1032 CrossRef.
- C.-W. Chiu and F. P. Gabbaï, Angew. Chem., Int. Ed., 2007, 46, 6878–6881 CrossRef CAS PubMed.
- J. Möbus, G. Kehr, C. G. Daniliuc, R. Fröhlich and G. Erker, Dalton Trans., 2014, 43, 632–638 RSC.
- F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159–13163 CrossRef CAS PubMed.
- R. H. Kern, M. Schneider, K. Eichele, H. Schubert, H. F. Bettinger and L. Wesemann, Angew. Chem., Int. Ed., 2023, 62, e202301593 CrossRef CAS PubMed.
- M. Arrowsmith, J. I. Schweizer, M. Heinz, M. Härterich, I. Krummenacher, M. C. Holthausen and H. Braunschweig, Chem. Sci., 2019, 10, 5095–5103 RSC.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, 1–19 RSC.
- T. Heitkemper and C. P. Sindlinger, Chem.–Eur. J., 2020, 26, 11684–11689 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 CrossRef CAS PubMed.
- H. Wang, J. Zhang, H. K. Lee and Z. Xie, J. Am. Chem. Soc., 2018, 140, 3888–3891 CrossRef CAS PubMed.
- G. A. Zhurko, CHEMCRAFT, http://www.chemcraftprog.com Search PubMed.
- S. Fujimori and S. Inoue, J. Am. Chem. Soc., 2022, 144, 2034–2050 CrossRef CAS PubMed.
- T. Sasamori, H. Miyamoto, H. Sakai, Y. Furukawa and N. Tokitoh, Organometallics, 2012, 31, 3904–3910 CrossRef CAS.
- H. Braunschweig, P. Constantinidis, T. Dellermann, W. C. Ewing, I. Fischer, M. Hess, F. R. Knight, A. Rempel, C. Schneider, S. Ullrich, A. Vargas and J. D. Woollins, Angew. Chem., Int. Ed., 2016, 55, 5606–5609 CrossRef CAS PubMed.
- S. R. Wang, M. Arrowsmith, J. Böhnke, H. Braunschweig, T. Dellermann, R. D. Dewhurst, H. Kelch, I. Krummenacher, J. D. Mattock, J. H. Müssig, T. Thiess, A. Vargas and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8009–8013 CrossRef CAS PubMed.
- J. Fan, P.-T. Chia, Z.-F. Zhang, M.-C. Yang, M.-D. Su and C.-W. So, Angew. Chem., Int. Ed., 2022, 61, e202212842 CrossRef CAS PubMed.
- Y. Wang, X. Zhang, J. Han, Q. Li, R. Wei, D. A. Ruiz, L. L. Liu, C.-H. Tung and L. Kong, Angew. Chem., Int. Ed., 2022, 61, e202117053 CrossRef CAS PubMed.
- A. Stoy, J. Böhnke, J. O. C. Jiménez-Halla, R. D. Dewhurst, T. Thiess and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 5947–5951 CrossRef CAS PubMed.
- A. Stoy, M. Härterich, R. D. Dewhurst, J. O. C. Jiménez-Halla, P. Endres, M. Eyßelein, T. Kupfer, A. Deissenberger, T. Thiess and H. Braunschweig, J. Am. Chem. Soc., 2022, 144, 3376–3380 CrossRef CAS PubMed.
- J. Fan, J.-Q. Mah, M.-C. Yang, M.-D. Su and C.-W. So, J. Am. Chem. Soc., 2021, 143, 4993–5002 CrossRef PubMed.
- C. Weetman, P. Bag, T. Szilvási, C. Jandl and S. Inoue, Angew. Chem., Int. Ed., 2019, 58, 10961–10965 CrossRef CAS.
- N. Wiberg, W. Niedermayer, K. Polborn and P. Mayer, Chem.–Eur. J., 2002, 8, 2730–2739 CrossRef CAS PubMed.
- A. Jana, V. Huch, H. S. Rzepa and D. Scheschkewitz, Organometallics, 2015, 34, 2130–2133 CrossRef CAS.
- W. Lu, A. Jayaraman, F. Fantuzzi, R. D. Dewhurst, M. Härterich, M. Dietz, S. Hagspiel, I. Krummenacher, K. Hammond, J. Cui and H. Braunschweig, Angew. Chem., Int. Ed., 2022, 61, e202113947 CrossRef CAS PubMed.
- P. Bissinger, A. Steffen, A. Vargas, R. D. Dewhurst, A. Damme and H. Braunschweig, Angew. Chem., Int. Ed., 2015, 54, 4362–4366 CrossRef CAS PubMed.
- F. S. Geitner and T. F. Fässler, Inorg. Chem., 2020, 59, 15218–15227 CrossRef CAS.
- K. Mayer, L. J. Schiegerl and T. F. Fässler, Chem.–Eur. J., 2016, 22, 18794–18800 CrossRef CAS PubMed.
- A. Bauer and H. Schmidbaur, Dalton Trans., 1997, 1115–1116 RSC.
- G. J. Reiß and M. Finze, J. Organomet. Chem., 2011, 696, 512–516 CrossRef.
- M. J. S. Dewar, Bull. Soc. Chim. Fr., 1951, 18, C79 Search PubMed.
- J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939–2947 RSC.
- C. Ganesamoorthy, S. Loerke, C. Gemel, P. Jerabek, M. Winter, G. Frenking and R. A. Fischer, Chem. Commun., 2013, 49, 2858–2860 RSC.
- P. E. Romero, W. E. Piers, S. A. Decker, D. Chau, T. K. Woo and M. Parvez, Organometallics, 2003, 22, 1266–1274 CrossRef CAS.
- A. V. Protchenko, J. Urbano, J. A. B. Abdalla, J. Campos, D. Vidovic, A. D. Schwarz, M. P. Blake, P. Mountford, C. Jones and S. Aldridge, Angew. Chem., Int. Ed., 2017, 56, 15098–15102 CrossRef CAS PubMed.
- M. Bonath, D. Schädle, C. Maichle-Mössmer and R. Anwander, Inorg. Chem., 2021, 60, 14952–14968 CrossRef CAS PubMed.
- A. Hofmann, M.-A. Légaré, L. Wüst and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 9776–9781 CrossRef CAS.
- A. Hofmann, C. Pranckevicius, T. Tröster and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 3625–3629 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2350051–2350063. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03743j |
| This journal is © The Royal Society of Chemistry 2024 |