
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnified ionic and radical C-4 alkylation and arylation of pyridines†
Qiu
Shi
,
Xiaofeng
Huang
,
Ruizhi
Yang
and
Wenbo H.
Liu
 *
*
School of Chemistry, Sun Yat-sen University, Guangzhou, 510006, China. E-mail: liuwb29@mail.sysu.edu.cn
First published on 4th July 2024
Abstract
C–H Functionalization of pyridines is an efficient strategy to access pyridine derivatives occurring in pharmaceuticals, agrochemicals, and materials. Nucleophilic additions to pyridiniums via both ionic and radical species have proven particularly useful. However, these reactions suffer from poor regioselectivity. By identifying an enzyme-mimic pocket-type urea activation reagent, we report a general platform for pyridine C-4 functionalization. Both ionic and radical nucleophiles can be incorporated to achieve the alkylation and arylation. Notably, the highly regioselective C-4 radical arylation is disclosed for the first time. The broad scope of nucleophiles and pyridines renders this platform applicable to the late-stage functionalization of drug-like molecules and the preparation of complex biologically important molecules.
Introduction
The prevalence of pyridine motifs in pharmaceuticals, agrochemicals, and functional materials underscores the importance of their synthesis.1 Among established synthetic methods, C–H functionalization of simple pyridines is arguably the most straightforward to access complex pyridine derivatives.2 To this end, one prominent strategy is to convert pyridines to pyridiniums, which are more electrophilic than the unactivated pyridines. Nucleophilic addition to the pyridiniums by either ionic or radical nucleophiles (the so-called Minisci reaction) efficiently affords the functionalized pyridines.3 Regarding the ionic nucleophilic addition, an oxidative rearomatization step is usually required in order to obtain the pyridine derivatives. In contrast, radical addition can directly afford the functionalized pyridines. However, because multiple reactive sites are available for unbiased pyridines, both the ionic and radical additions suffer from the regioselectivity (C-2 vs. C-4 isomers) and the overreaction issues3 (Fig. 1a). Uncontrollable formation of both regioisomers remains one of the most important yet unsolved problems in the area of pyridinium functionalization. Regioselective protocols for C–H functionalization of non-biased pyridine are thus in high demand. Compared to C-2 (ref. 4) and C-3,5 C-4 functionalization via direct C–C bond formation is special considering that the C-4 position is far away from the N atom, which, as the key directing group for C–H activation and directed ortho-metalation, is less potent to influence the C-4 position.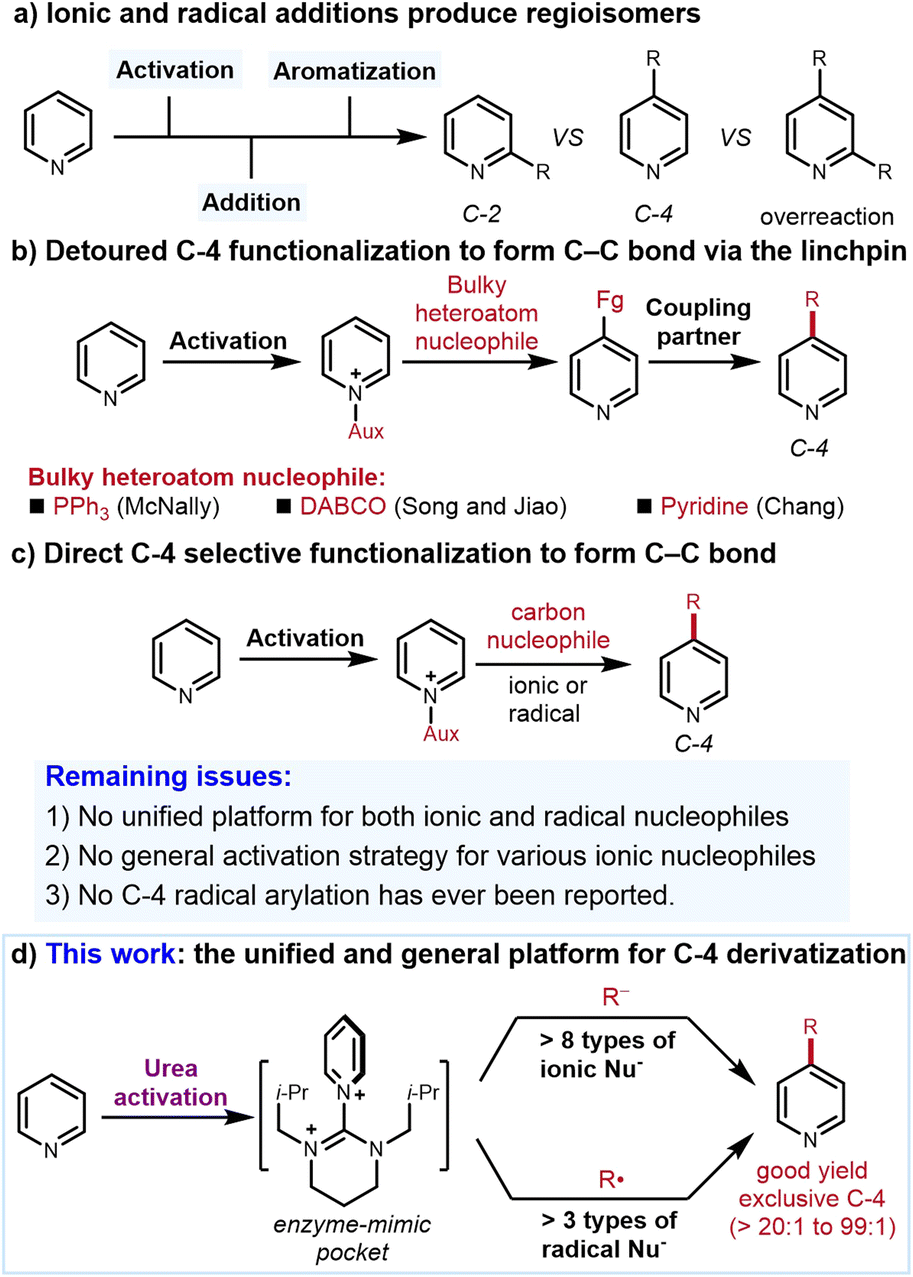 | ||
| Fig. 1 Background and this work. | ||
One powerful C-4 functionalization strategy is to preinstall a transformable heteroatom linchpin selectively, which can then couple with a wide range of C-based nucleophiles. The selectivity of this strategy is enabled by the steric hindrance between bulky heteroatom nucleophiles and N-activation reagents. Notable examples include PPh3 developed by McNally,6a−i DABCO developed by Song and Jiao6j and pyridine developed by Chang6k among others6l−n (Fig. 1b). Despite the high regioselectivity, this strategy requires the formation of heteroatom nucleophile-based intermediates. Directly constructing the C–C bonds between the pyridine and coupling partners is more step-economical but more difficult because unlike PPh3 and DABCO, the structures of the carbon nucleophiles are not fixed and broadly diversified. For instance, the challenges to induce high C-4 regioselectivity for bulky tert-butyl group and the methyl group as the nucleophiles are different. The conditions feasible for the tert-butyl group to afford high selectivity may not be qualified for the methyl. Therefore, establishing a unified protocol for all C-based nucleophiles instead of some specific heteroatom nucleophiles is apparently more difficult (Fig. 1c).
In this context, Knochel et al. reported that the Grignard and organozinc reagents selectively react at the C-4 positions of pyridines via BF3·Et2O activation although only substituted biased pyridines are effective and simple pyridine cannot be derivatized.7a Through silylium catalysis, List et al. found that silyl ketene acetals add onto the silyl-activated pyridinium in high C-4 selectivity.7b Other approaches with alkenes/alkynes/electron-rich arenes as the ionic coupling partners were also known.7c−i Besides the ionic nucleophiles, radicals are also applied to the pyridine C-4 alkylation (but no arylation so far). Hong et al. developed numerous elegant transformations for C-4 functionalization with alkyl bromide, alkane and cyclopropanol as the radical precursors.8a–c Meanwhile, Baran et al. invented a highly selective C-4 radical alkylation, which requires two steps to install the activation reagent. Since these installation steps rely on strong acids and reflux conditions, it seems that only simple pyridines could be alkylated.8d More recently, Studer et al. found that upon acid isomerization, their powerful and versatile C-3-selective protocol can afford C-4 selectivity with both ionic and radical species.8e
These methods represent the state of the art of the field. Nevertheless, some unaddressed issues remain: (1) most examples are specific either to ionic or radical nucleophiles. The protocol that accommodates both ionic and radical nucleophiles is rare. (2) When ionic nucleophiles are concerned, the scope is narrow. One activation strategy is usually only suitable to one specific type of nucleophile. An activation strategy that is appliable to diverse nucleophiles with variable nucleophilicity is elusive. (3) When radical species are used, established protocols are only efficient for C-4 alkylation and no C-4 arylation has ever been reported.3b,c,8 To address these issues, we conceive that the qualified pyridinium candidate should meet several criteria including (1) high stability to tolerate the conditions generating ionic nucleophiles and radicals without causing the activating reagents to fall off; (2) strong electrophilicity to accommodate nucleophiles with variable nucleophilicity and (3) exceptional reliable regiochemical control. With these concerns in mind, herein, we disclose a general and practical C-4 alkylation and arylation of pyridines with both ionic and radical nucleophiles through a unified platform (Fig. 1d). The success of this protocol hinges on the identification of an enzyme-mimic pocket-type urea to convert pyridines to stable, highly electrophilic pyridiniums with both C-2 and C-6 positions perfectly shielded.
Results and discussion
Recently, our laboratory has reported an aromatic C–H oxazolination.9 During this study, modest para-selectivity was observed when substituted oxazolidinone as the oxazolination reagent was employed. Building on this and inspired by the pioneering work of Charette on C-2 functionalization of pyridine with secondary amide as the activation reagent,4a we envision that substituted urea may be feasible to selectively activate the pyridines upon triflic anhydride activation.10 Two substituents of the urea flank the C-2 and C-6 positions of pyridiniums instead of one in the oxazolidinone and amide, thereby creating a more confined enzyme-mimic pocket. Besides, due to the presence of two positive charges, the resulting guanidinium adduct is more electrophilic than normal pyridiniums, thereby implying that a broad range of nucleophiles with variable nucleophilicity can all be accommodated (Fig. 2a).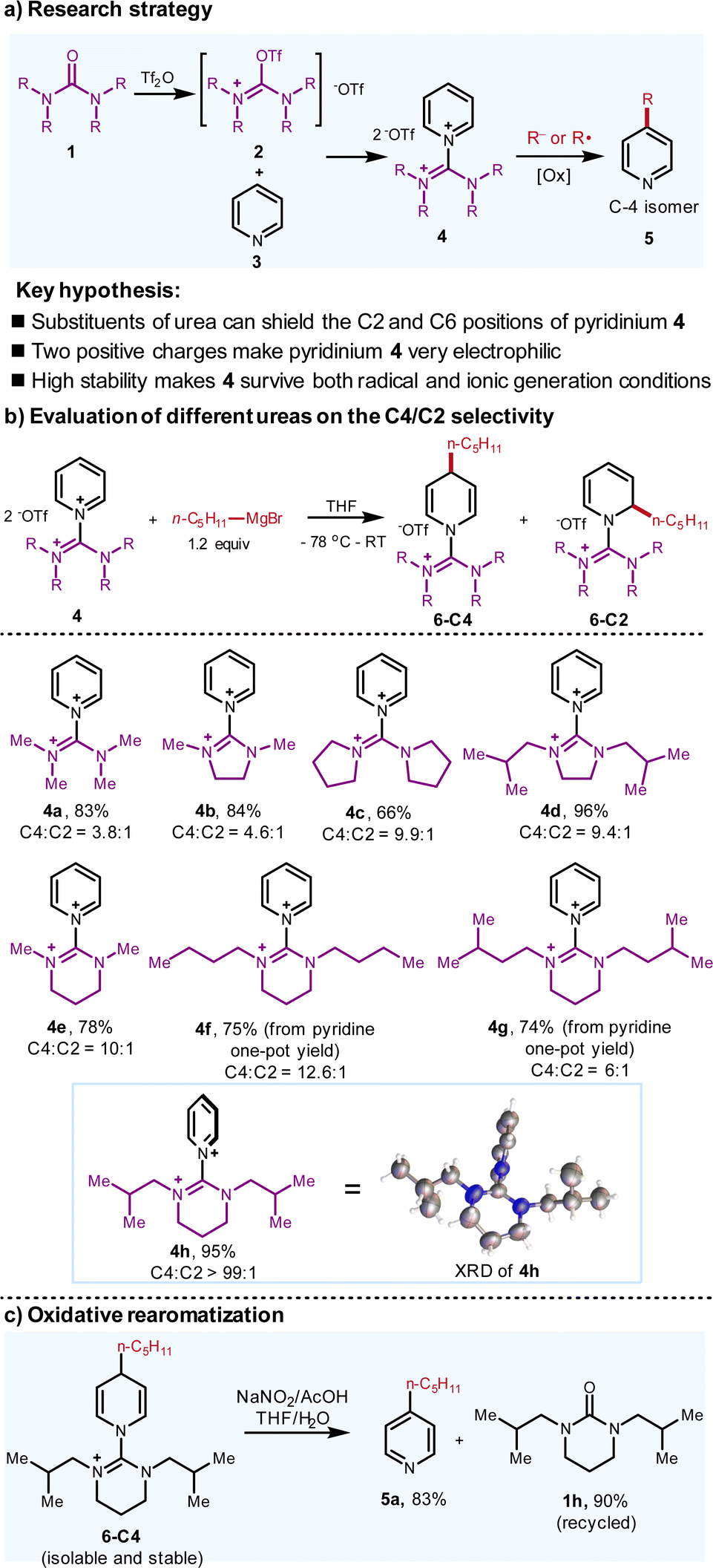 | ||
| Fig. 2 Design strategy and evaluation of diverse ureas with different structures. The yields and ratio of regioisomers were determined by 1H NMR. Please see ESI† for experimental details. | ||
Guided by these propositions, after converting pyridine to the guanidinium-type adduct 4, we have screened various ureas to evaluate the C-4/C-2 selectivity via applying the Grignard reagents as the nucleophile (Fig. 2b). Consistent with our hypothesis, the selectivity is indeed correlated with the steric hindrance of substituents on ureas. By altering the backbones and the substituents, it is concluded that 4h reacts with n-pentyl Grignard reagent to form the dihydropyridine in 95% yield with an excellent C-4/C-2 selectivity (>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by GC-MS). The crystal structure of 4h suggests that the pyridine and the urea rings are twisted with the 90 °C dihedral angle and the two isobutyl groups shelter the left and right faces of C-2 and C-6 of the pyridine. When the LUMO of the pyridinium interacts with the HOMO of the nucleophile, the nucleophilic reaction follows the Bürgi–Dunitz angle, which implies that the attack trajectory of C-2 and C-6 would be hampered by the two isobutyl groups. In contrast, the C-4 is far away from the substituents, which is not affected for the nucleophilic addition.
:1 by GC-MS). The crystal structure of 4h suggests that the pyridine and the urea rings are twisted with the 90 °C dihedral angle and the two isobutyl groups shelter the left and right faces of C-2 and C-6 of the pyridine. When the LUMO of the pyridinium interacts with the HOMO of the nucleophile, the nucleophilic reaction follows the Bürgi–Dunitz angle, which implies that the attack trajectory of C-2 and C-6 would be hampered by the two isobutyl groups. In contrast, the C-4 is far away from the substituents, which is not affected for the nucleophilic addition.
The two isobutyl groups and 6-membered cyclic urea backbone are critical for the twisted structure of 4h. As a comparison, XRD analysis shows almost no shielding effect of C-2 and C-6 in 4a (please see ESI† for the crystal structure of 4a). It should be noticed that the Grignard reagents are classified as hard nucleophiles in terms of HSAB (hard–soft-acid–base) theory, which usually afford C-2 addition unless the copper salt is added to form the soft organocuprate nucleophiles.11 In our case, the selectivity completely switches to C-4, thus demonstrating that this urea activation reagent is robust to shield both C-2 and C-6 positions. Finally, when the addition product 6-C4 was subjected to the oxidative rearomatized conditions, the pyridine derivative 5a was isolated in good yield. Moreover, the urea 1h was isolated in high yield (90%), which can be further recycled (Fig. 2c). Remarkably, because of the electron-withdrawing feature of the guanidinium motifs, this dihydropyridine adduct 6-C4 is isolable and stable to air, which is unique for the dihydropyridine without electron-withdrawing groups on the pyridine rings.
Scope of ionic nucleophiles and pyridines
With the qualified urea activation reagent identified, the scope of both Grignard reagents (Fig. 3) and pyridines (Fig. 4) were investigated. Diversified Grignard reagents including primary (5a, 79%; 5e, 58%), secondary (5b, 91%; 5c, 72%), and tertiary (5d, 45%) smoothly react with the pyridiniums to produce C-4 alkylation products in good to high yields with excellent regioselectivity (all >99:1). Due to the steric hindrance, tertiary Grignard reagent furnishes the product in modest yield (5d, 45%) but still only C-4 isomer is detected. It is notable that even for the smallest methyl Grignard reagent, the C-4/C-2 selectivity is superb (5f, 64%). Compared to the alkyl Grignard reagents, aryl and heteroaryl Grignard reagents are even harder nucleophiles, which tend to proceed via C-2 addition of pyridiniums. To our delight, aryl and heteroaryl Grignard reagents still allow for the C-4 arylation in good to high yields with excellent C-4 selectivity (5g–5j, 60–80%).
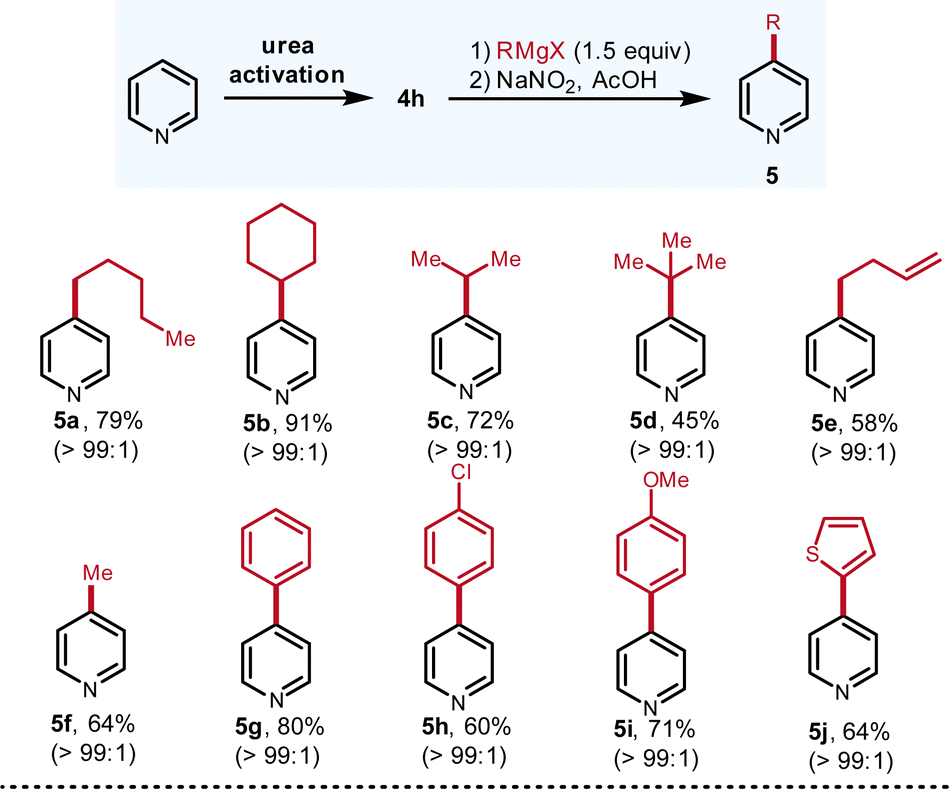 | ||
| Fig. 3 Scope of Grignard reagents. The yields refer to the isolated yields of pure compounds. The ratios were determined by 1H NMR and GC-MS. Due to the low boiling points, the yields of 5c and 5f were determined by 1H NMR. Please see ESI† for experimental details. | ||
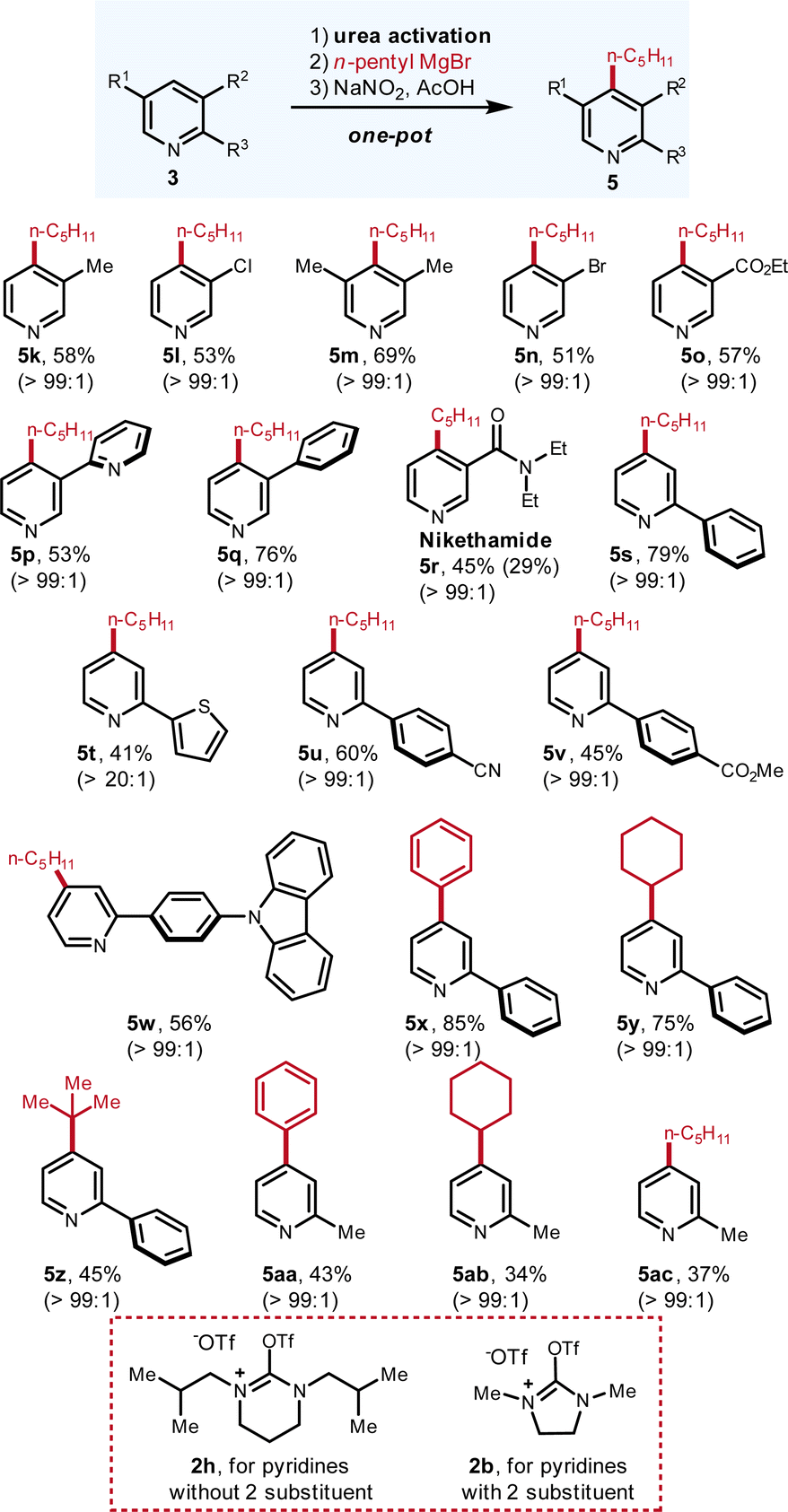 | ||
| Fig. 4 Scope of pyridines with Grignard as the nucleophile. The yields refer to the isolated yields of pure compounds in one pot from pyridine. The ratios were determined by 1H NMR and GC-MS. Please see ESI† for experimental details. | ||
Regarding the scope of pyridines, various C-3 and C-2 substituted pyridines are smoothly derivatized (Fig. 4). Numerous important functional groups such as –Cl (5l, 53%), –Br (5n, 51%), –ester (5o, 57%; 5v, 45%), –amide (5r, 45%) and –CN (5u, 60%) are well tolerated. For the di-substituted pyridine bearing two methyl groups at C-3 and C-5 positions, the C-4 is located in a very hindered environment but excellent C-4 selectivity (>99:1) and good yield were obtained (5m, 69%). For the 2,3-bipyridine substrate, the alkylation only occurs at the C-3-substituted pyridine with good yield and excellent selectivity (5p, 53%) whereas the functionalization at the other pyridine ring is not observed. Regarding the derivatization of 2-substituted pyridines, urea activation reagent should be switched from 2h to 2b. With 2b as the activation reagent, various 2-substituted pyridines can be effectively functionalized with Grignard reagents in good yields and excellent C-4 selectivity (5s–5ac, all >99:1).
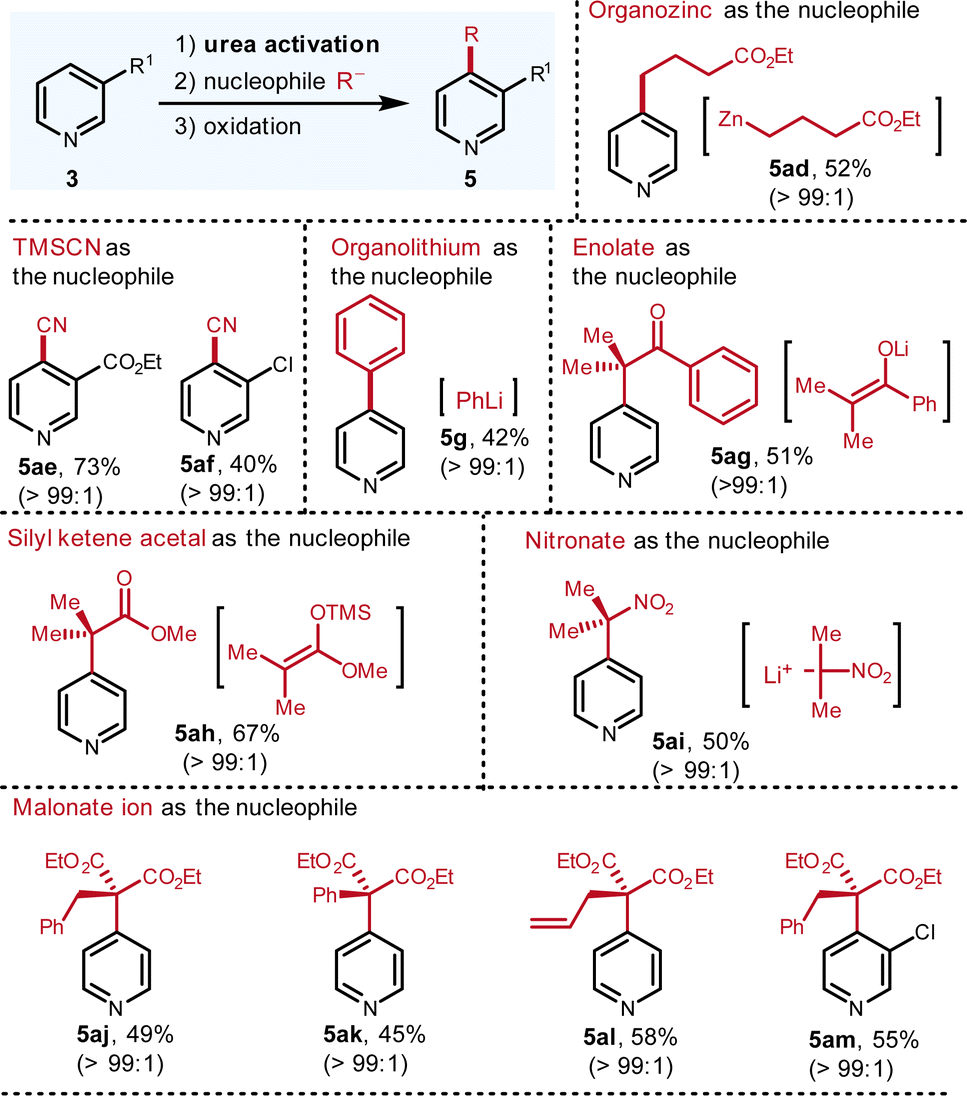
Although the Grignard reagents stand out as the most fundamentally important nucleophiles, to further expand the scope, other ionic nucleophiles were also assessed (Fig. 5). Organozinc reagent, TMSCN, organolithium reagent, silyl ketene acetal, nitronate and malonates are all workable nucleophiles, which elaborate C-4 substituted pyridines in excellent regioselectivity and good yields. Due to the weaker nucleophilicity of organozinc reagents, functionalized organozinc reagents with an ester group can be prepared. Via this platform, the desired addition proceeded smoothly with satisfactory result (5ad, 52%). The nucleophilic addition of cyanide to the pyridinium, known as the Reissert–Henze reaction, usually exhibits C-2 selectivity due to hard character of cyanide ion.12Via this protocol, only C-4 isomer is detected (5ae, 73%; 5af, 40%). In addition, when organolithium reagents are applied as the nucleophiles, due to their hardness, they tend to attack the activation reagents instead of the pyridinium rings considering that the activation reagent is the hardest position.3a To our delight, when phenyl lithium is employed in our protocol, the C-4 arylation product (5g, 42%) is isolated in good yield and excellent regioselectivity. To our knowledge, this is the first protocol to furnish C-4 arylation of pyridine with organolithium reagent.
 | ||
| Fig. 5 Other feasible nucleophiles for C-4 functionalization. The yields refer to the isolated yields of pure compounds. The ratios were determined by 1H NMR and GC-MS. Please see ESI† for experimental details. | ||
As a type of readily available nucleophile, the enolate smoothly participates this reaction to generate the product (5ag, 51%) with excellent selectivity. Besides the enolate from ketone, the silyl ketene acetal from the ester also reacts with pyridinium to deliver the alkylated pyridine (5ah, 67%). Both enolate and silyl ketene acetal nucleophiles furnish the alpha-pyridyl carbonyl compounds, which are useful synthons in organic synthesis. Furthermore, nitronate is capable to generate the desired C-4 alkylated pyridine (5ai, 50%). The nitro group can be reduced to radical intermediate or amine group, thereby making it versatile in multi-step synthesis. Finally, when the malonates serve as the nucleophiles, the corresponding products are also isolated in good yields and excellent regioselectivity (5aj–5am, 45–58%). The broad scope of ionic nucleophiles suggests that high electrophilicity, thanks to the two positive charges of the pyridiniums, are beneficial to incorporate a wide range of nucleophiles, thereby demonstrating the uniqueness of this guanidinium-type activation reagent.
Scope of radical precursors
Besides the ionic nucleophiles, radical intermediates can also be applied to functionalize C-4 of pyridiniums via this platform (Fig. 6). The classical Minisci reaction conditions were first investigated. Although the Minisci reaction has been extensively studied in the past five decades,3b,c efficient methods to solve the regioselectivity and overreaction issues are still scarce.13a To obtain single regioisomer, other reactive sites are usually blocked with substituents. Regarding the pyridines with multiple available sites, regioisomer mixtures are inevitably produced. To the delight, with our urea activation reagent, only the C-4 alkylated pyridines are observed for the unbiased pyridine. A bonus for this radical functionalization is that derivatized pyridines are directly obtained without an extra rearomatization step. A wide range of alkyl carboxylic acids and pyridines are suitable, which all deliver the products in good yields with excellent C-4 selectivity. Secondary (5b, 70%; 5an, 35%; 5aq, 56%; 5ar, 61%; 5at, 33%), tertiary (5ao, 46%; 5as, 64%) and primary (5ap, 48%) carboxylic acids all afford the alkylated pyridines. Due to the compatibility issue of Grignard reagents, some products generated from radical addition cannot be synthesized by the ionic addition (5an, 5as), which further illustrates the advantages of radical addition. A wide variety of halogen substituted pyridines are tolerated (5au–5aw), which provide functional handles for further modification. The most important feature of this protocol is that when aryl boronic acids are employed as the radical precursors, the desired C-4 arylations proceed in good yields and excellent regioselectivity (5ax–5bb). This is in striking contrast to the previous protocols engaging radical species, none of which can achieve the C-4 radical arylations.13 Furthermore, alkanes with the alcohol functional groups proceed well to derivatize the pyridines in satisfactory yields (5bc, 5bd).14 It is reasonable to propose that in combination with this urea-activation platform, other Minisci-type reactions with different radical progenitors that suffer from the regioselectivity issues can also be enabled C-4 selective.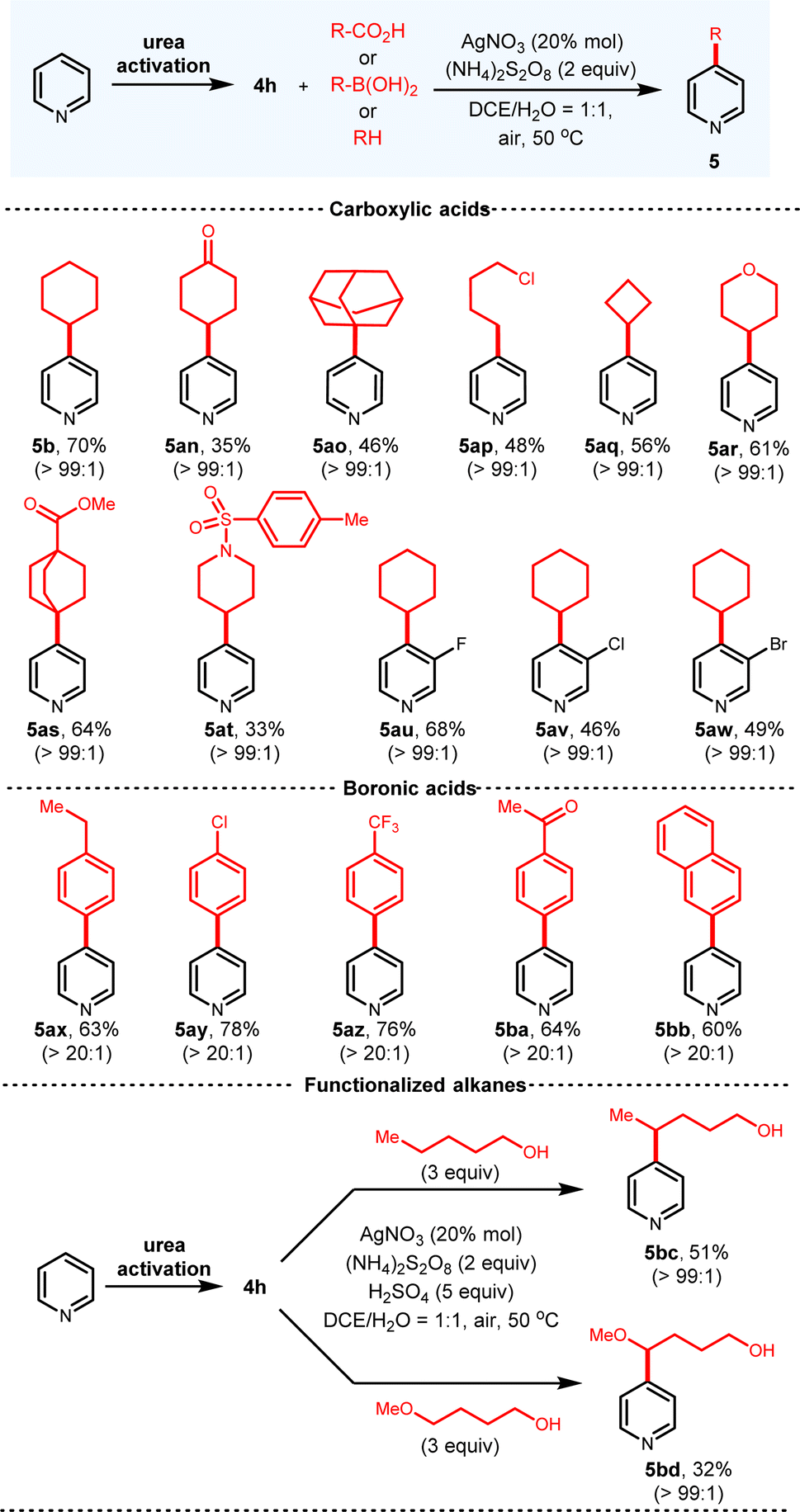 | ||
| Fig. 6 Scope of radical precursors for C-4 functionalization. The yields refer to the isolated yields of pure compounds. The ratios were determined by 1H NMR and GC-MS. | ||
Late-stage functionalization of drug-like molecules
Late-stage functionalization (LSF) has been recognized as an efficient tool to rapidly construct diverse molecular libraries of drug-like molecules.15 However, due to the structural complexity of drug compounds, only very robust reactions under mild conditions can be leveraged for the LSF. Therefore, the suitability to LSF is useful to evaluate the practicality of new organic reactions. To this end, various drug-related structures were subjected to this C-4 derivatization protocol (Fig. 7). Nicoboxil-derived pyridine contains the motif of an ester and easily oxidizable ether, which afforded the corresponding alkylated pyridine in good yield (5be, 47%). As a multi-halogenated drug, the pyridine derivative of triclosan was converted to the alkylated analogue smoothly (5bf, 49%). In addition, Vitamin E is tolerated under the standard conditions although it is well-known that Vitamin E tends to be oxidized due to the electron-richness of fully-substituted aromatic ring (5bg, 55%). The compatibility with Vitamin E demonstrates the mildness of the oxidative rearomatization step of this protocol. Besides, the pyridine-containing precursor of Empagliflozin was transformed into the desired product in high yield despite the presence of an electron-rich phenol-type ether and the reactive diaryl methylene (5bh, 89%). As a densely functionalized drug, the pyridine analogue of Mecarbinate with an ester and an indole ring was succesffully alkylated in good yield (5bi, 69%). The derivative of drug Oxaprozin, which possesses an ester, an oxazole and the enolizable methylene, is also feasible for the C-4 alkylation (5bj, 47%). Highly functionalized drug Febuxostat, which accomodates –CN, ester, thiazole andoxidizable benzylic methylene, was straightforwardly alkylated (5bk, 50%). The drug Loratadine with C2 and C3 substituents was directly functionalized as well in good yield (5bl, 72%). Besides, the pyridine analogue of the drug Isoxepac with a ketone, an oxidizable benzylic ethylene, an ester and an acidic benzylic methylene was compatible (5bm, 52%). The tolerance of the ketone, ester and acidic proton further illustrates that the activated pyridinium is very reactive towards the Grignard addition. Moreover, sulfonamide-bearing drug Probenecid-derived pyridine and pyrazole-containing drug Rimonabant precursor can also be functionalized smoothly (5bn, 64%; 5bo, 60%).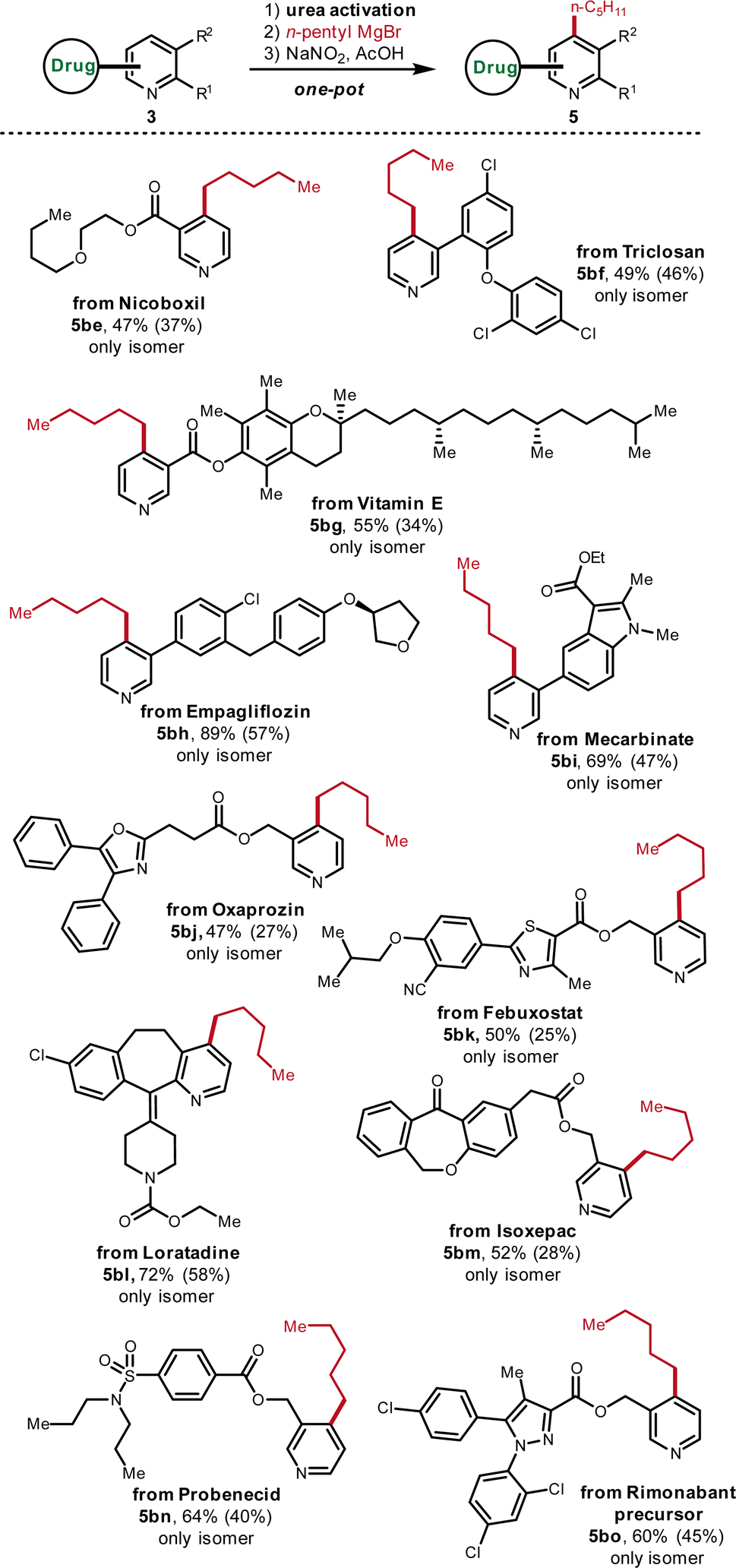 | ||
| Fig. 7 Late-stage functionalization of drug-like molecules. All the yields refer to isolated one-pot yields from pyridines based on the recovered starting materials. The yields in the bracket are the isolated yields. | ||
Target-oriented synthetic applications
To further demonstrate the synthetic applications of this protocol in real contexts, we performed the target-oriented synthesis of three biologically active compounds (Fig. 8). Starting from simple pyridine, carboxylic acid 6 underwent decarboxylation to generate the radical intermediate, which delivered 5at with only the desired C-4 isomer isolated in 33% yield. By subjecting 5at to another radical addition, 8 was prepared in 68% yield. Based on the previously established route, the target fungicide can be synthesized from 8 (Fig. 8a). Moreover, VU6001966 has been identified as the glutamate receptor modulator with IC50 = 78 nM. Previous synthetic route comprises 8 steps from expensive 2,5-difluoro-4-iodopyridine as the staring material.16 In comparison, with aryl boronic acid as the aryl radical donor, we completed the synthesis of VU6001966 in three steps from cost-effective 3-fluoropyridine. Under Baran's boron-Minisci conditions, pyridinium 9 reacted with 10 to generate C-4 arylated pyridine 11 in 61% yield, which reacted with alcohol 12 through the SNAr mechanism and allowed for the synthesis of 13 in 66% yield. With 13 in hand, another radical formylation afforded the target VU6001966 in 34% yield (Fig. 8b). In addition, small molecule LJI308 behaves as the inhibitor for RSK, which is capable to promote tumor relapse.17 Regarding the synthesis of LJI308, the ionic arylation of the pyridinium was deployed as the key step. Functionalized aryl bromide 15 was first converted to the Grignard reagent 16, which then reacted with the pyridinium salt. Following the nucleophilic addition and oxidation, 17 was smoothly synthesized in one-pot manner. A further Suzuki-coupling between 17 and 18 delivered the desired molecule in 68% isolated yield (Fig. 8c). Remarkably, all these routes are highly modulated, which can efficiently access different analogues of the biologically active molecules, thereby benefiting the structure–activity relationship investigation. These target-oriented synthesis suggest the synthetic potential of this pyridine C-4 functionalization platform in real synthetic contexts.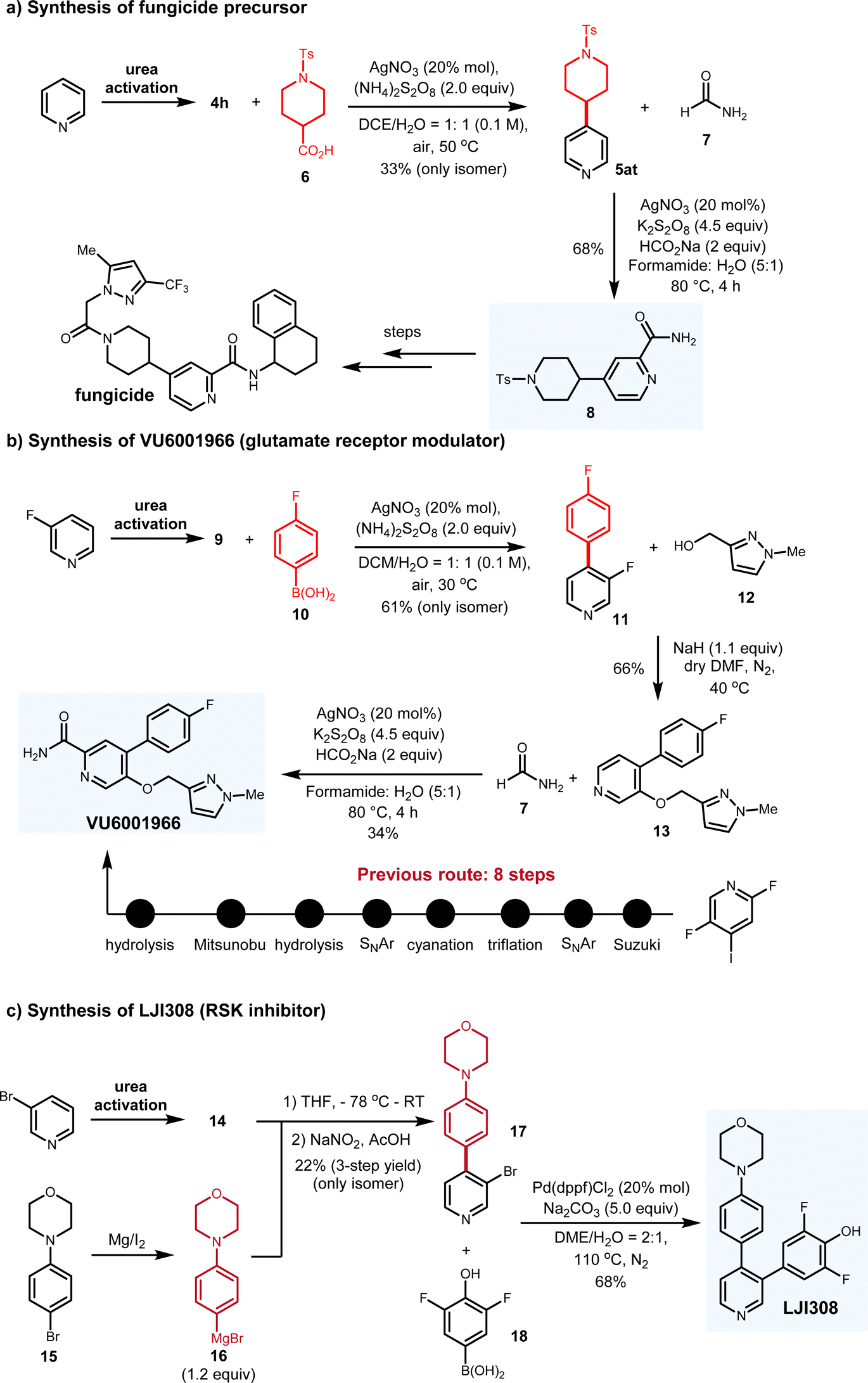 | ||
| Fig. 8 Target-oriented synthesis of biologically active molecules via this C-4 functionalization as key steps. | ||
Conclusions
In summary, we have developed a practical and general C-4 functionalization strategy of unbiased pyridines by identifying a readily synthesized substituted urea as the pyridine activation reagent. Via this reagent, pyridines can be converted to highly electrophilic pyridiniums with both C-2 and C-6 positions shielded. Benefited from these features, this strategy can accommodate both ionic and radical nucleophiles and is suitable for both alkylation and arylation. Moreover, a wide range of ionic nucleophiles including Grignard, organozinc, organolithium, cyanide, enolate, silyl ketene acetal, nitronate and malonate with variable nucleophilicity are compatible. As for radical species, abundant carboxylic acids, boronic acids and alkanes can serve as the radical precursors. Most notably, the C-4 selective radical arylation of pyridiniums with high regioselectivity (>20:1) is uncovered for the first time. Considering the significance of pyridine derivatives, the operational simplicity, the generality and robustness of this protocol, as well as the ready availability of starting materials and reagents, it is anticipated that this protocol would find broad use in organic synthesis. Applications of this method towards C-4 heterofunctionalization and derivatization of other heteroaromatics beyond pyridines are forthcoming.
Data availability
Experimental details and characterization data can be found in the ESI.†Author contributions
Q. Shi performed the optimization, investigated the scope of substrate and conducted the synthetic application experiments; X. Huang and R. Yang took part in the reaction development and synthesized several substrates; W. Liu prepared the manuscript, guided and supervised the project. All the authors discussed the experimental results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22201311), Guangdong Basic and Applied Basic Research Foundation (2023A0505050100) and Sun Yat-sen University. The authors thank Dr Long Jiang from the Instrumental Analysis & Research Center of Sun Yat-sen University for single-crystal X-ray diffraction measurements and structural analysis.Notes and references
- For selected reviews, see
(a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed
; (b) G. Yang and W. Zhang, Chem. Soc. Rev., 2018, 47, 1783–1810 RSC
- For selected reviews, see
(a) K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302–9332 CrossRef CAS PubMed
- For selected reviews, see
(a) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed
- For selected examples of C-2 functionalization of pyridines, see
(a) A. B. Charette, M. Grenon, A. Lemire, M. Pourashraf and J. Martel, J. Am. Chem. Soc., 2001, 123, 11829–11830 CrossRef CAS PubMed
- For selected examples of C-3 functionalization of pyridines, see
(a) H. Cao, Q. Cheng and A. Studer, Angew. Chem., Int. Ed., 2023, 62, e202302941 CrossRef CAS PubMed
- For selected examples of C-4 functionalization of pyridines with heteroatom nucleophile as the linchpin, see
(a) M. C. Hilton, R. D. Dolewski and A. McNally, J. Am. Chem. Soc., 2016, 138, 13806–13809 CrossRef CAS PubMed
- For ionic C-4 functionalization, see
(a) Q. Chen, X. M. du Jourdin and P. Knochel, J. Am. Chem. Soc., 2013, 135, 4958–4961 CrossRef CAS PubMed
- For radical C-4 functionalization, see
(a) M. Vellakkaran, T. Kim and S. Hong, Angew. Chem., Int. Ed., 2022, 61, e202113658 CrossRef CAS PubMed
- Q. Shi, Y. Huang and W. H. Liu, Precis. Chem., 2023, 1, 316–325 CrossRef CAS
-
(a) N. Bormann, J. S. Ward, A. K. Bergmann, P. Wenz, K. Rissanen, Y. Gong, W.-B. Hatz, A. Burbaum and F. F. Mulks, Chem.–Eur. J., 2023, 29, e202302089 CrossRef CAS PubMed
-
(a) D. Wang, L. Désaubry, G. Li, M. Huang and S. Zheng, Adv. Synth. Catal., 2021, 363, 2–39 CrossRef CAS
-
(a) W. K. Fife, J. Org. Chem., 1983, 48, 1375–1377 CrossRef CAS
- For regioselectivity issues of radical additions, please see:
(a) F. O'Hara, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2013, 135, 12122–12134 CrossRef PubMed
- M. Wang, C. Yin and P. Hu, Org. Lett., 2021, 23, 722–726 CrossRef CAS PubMed
- For reviews on the late-stage functionalization, see
(a) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem, 2021, 5, 522–545 CrossRef CAS PubMed
- K. A. Bollinger, A. S. Felts, C. J. Brassard, J. L. Engers, A. L. Rodriguez, R. L. Weiner, H. P. Cho, S. Chang, M. Bubser, C. K. Jones, A. L. Blobaum, C. M. Niswender, P. J. Conn, K. A. Emmitte and C. W. Lindsley, ACS Med. Chem. Lett., 2017, 8, 919–924 CrossRef CAS PubMed
- A. H. Davies, K. Reipas, K. Hu, R. Berns, N. Firmino, A. L. Stratford and S. E. Dunn, Oncotarget, 2015, 6(24), 20570–20577 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03739a |
| This journal is © The Royal Society of Chemistry 2024 |