
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomically precise metal nanoclusters combine with MXene towards solar CO2 conversion†
Yu-Shan
Cai
a,
Jia-Qi
Chen
a,
Peng
Su
a,
Xian
Yan
a,
Qing
Chen
a,
Yue
Wu
a and
Fang-Xing
Xiao
 *ab
*ab
aCollege of Materials Science and Engineering, Fuzhou University, New Campus, Minhou, Fujian Province 350108, China. E-mail: fxxiao@fzu.edu.cn
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, PR China
First published on 19th July 2024
Abstract
Atomically precise metal nanoclusters (NCs) have been deemed a new generation of photosensitizers for light harvesting on account of their quantum confinement effect, peculiar atom-stacking mode, and enriched catalytic active sites. Nonetheless, to date, precise charge modulation over metal NCs has still been challenging considering their ultra-short carrier lifetime and poor stability. In this work, we conceptually demonstrate the integration of metal NCs with MXene in transition metal chalcogenide (TMC) photosystems via a progressive, exquisite, and elegant interface design to trigger tunable, precise and high-efficiency charge motion over metal NCs, stimulating a directional carrier transport pathway. In this customized ternary heterostructured photosystem, metal NCs function as light-harvesting antennas, MXene serves as a terminal electron reservoir, and the TMC substrate provides suitable energy level alignment for retracting photocarriers of metal NCs, giving rise to a spatial cascade charge transport route and markedly boosting charge separation efficiency. The interface configuration and energy level alignment engineering synergistically contribute to the considerably enhanced visible-light-driven photocatalytic CO2-to-CO reduction performance of the metal NCs/TMCs/MXene heterostructure. The intermediate active species during the photocatalytic CO2 reduction are unambiguously determined, based on which the photocatalytic mechanism is elucidated. Our work will provide an inspiring idea to bridge the gap between atomically precise metal NCs and MXene in terms of controllable charge migration for solar-to-fuel conversion.
1 Introduction
Photocatalytic CO2 conversion to hydrocarbon fuels has been considered an effective strategy to solve the deteriorating energy crisis and mitigate the notorious greenhouse effect.1–4 However, semiconductor-based CO2 photocatalysis suffers from the inertness of CO2 molecules, slow charge transfer kinetics and rapid charge recombination, rendering it complicated and difficult to accurately regulate.5–8 Although there have been many investigations into CO2 photoreduction, high-efficiency solar CO2 conversion is far from satisfactory, which is ascribed mainly to the deficiency of applicable photocatalysts with robust and stable photoactivity,9–12 low CO2 reduction selectivity, and a complex charge transport route.7,13–15 Hence, it is highly desirable to explore novel artificial photosystems with desirable charge separation efficiency, selectivity, and photoactivity for solar CO2 conversion.16–18Metal nanoclusters (NCs, <2 nm), which consist of a precise number of metal atoms and thiolate ligands, have garnered enormous attention in recent years due to their unique atom-stacking pattern, abundant active sites, and favorable molecular-like discrete highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) energy levels, making them emerging photosensitizers for substantial light harvesting and conversion.19 Nevertheless, the ultra-short lifetime of photogenerated charge carriers, scarcity of matched semiconductor pairs with applicable energy levels, and difficulty in mediating charge migration concurrently hinder the widespread application of metal NCs in photocatalysis.20 Noteworthily, co-catalyst engineering demonstrates unprecedented merits in accurately tuning directional charge transport to the ideal active sites, which might provide an efficacious and convenient strategy to surmount the drawbacks of metal NCs and foster the construction of multifarious metal-NC-based composite photocatalytic CO2 reduction systems.21,22
In 2011, a novel category of two-dimensional (2D) transition metal carbide, nitride, or carbonitride (MXene) nanomaterials sparked enormous attention from academy communities.23 As a new generation of 2D materials, MXenes harbor the generic merits of rapid carrier mobility, abundant active sites, and excellent hydrophilicity due to their unique structural flexibility and diversity of elemental composition, featuring promising co-catalysts for photocatalysis.24,25 Recent studies have provided evidence that MXene is able to engender a Schottky barrier in photocatalysis to facilitate the spatial separation of photogenerated charge carriers, thus inhibiting charge recombination.26,27 Inspired by the merits of MXenes with excellent conductivity and well-defined 2D nano-architecture, we deduce that they could be judiciously used as co-catalysts to solve the inherent problems of the easy charge recombination and ultrashort charge lifetime of metal NCs by smartly constructing an MXene/metal NC heterojunction. This is rationalized by the fact that the 2D morphology of MXenes is beneficial for the uniform deposition of metal NCs to reduce the charge diffusion distance,28–30 and, moreover, the suitable energy level alignment formed between the Fermi level of MXene and the HOMO level of metal NCs benefits carrier migration. However, adornment with MXene alone does not provide sufficient impetus to withdraw the electrons photoexcited over metal NCs given their super-fast charge recombination rate. Involving second-component semiconductors with matched energy level alignment for fabricating metal NCs/semiconductor/MXene ternary heterostructured photosystems is expected to perfectly solve this problem by stimulating tunable charge transport among the building blocks. Transition metal chalcogenides (TMCs) represent the optimal semiconductor alternative owing to their large absorption coefficient, favorable energy level position, and enriched active sites, which would match well with both MXene and metal NCs by constructing a directional charge transport pathway, boosting interfacial charge separation.27,31,32
Herein, as a proof-of-concept investigation, 2D CdS is used as a bridging medium connecting MXene with gold nanoclusters (Aux NCs) by a facile and easily accessible electrostatic self-assembly method under ambient conditions, forming a well-defined CdS/Ti3C2Tx/Aux (CTA) heterostructure for photocatalytic CO2 reduction. In this exquisitely designed heterostructure, CdS and metal NCs concurrently serve as light-harvesting antennas and form a beneficial energy level alignment, while MXene attached on the outermost surface serves as a terminal electron-trapping reservoir, which synergistically contributes to the cascade electron flow from Aux NCs to CdS and eventual trapping by MXene, giving rise to a considerably prolonged charge lifetime and significantly boosting photocatalytic CO2 reduction performance under visible light irradiation. The photocatalytic mechanism was then elucidated with intermediate active species clearly determined during the photocatalytic CO2-to-CO conversion. Our work would provide new ideas for rationally constructing metal NC artificial photosystems and strategically mediating charge transport pathways over metal NCs for solar-to-fuel conversion.
2 Experimental section
2.1 Preparation of Ti3C2Tx (MXene)33
Multilayered Ti3C2Tx was first synthesized by etching 1 g of Ti3AlC2 powder in a mixture of 1 g of lithium fluoride and 10 mL of hydrochloric acid (9 mol L−1) for 24 h at 35 °C. The product was washed with deionized water until the pH of the supernatant was above 5. The multilayered Ti3C2Tx powder was then added into 200 mL of deionized water and delaminated by bath sonication for 1 h under N2 flow. After centrifugation for 1 h at 3500 rpm, the dark green supernatant was collected. The concentration of the delaminated Ti3C2Tx was determined by filtering a known volume of the supernatant through a Celgard membrane and measuring the weight of the film after drying. The delaminated Ti3C2Tx colloid was then diluted to obtain a concentration of 0.5 mg mL−1.2.2 Preparation of CdS nanosheets
CdS nanosheets (NSs) were prepared via a hydrothermal method reported previously.33 In detail, 0.32 mmol CdCl2·2.5H2O, 2.0 mmol S powder and 12 mL of diethylenetriamine (DETA) were mixed and vigorously stirred to form a homogeneous suspension. Then, the mixture was transferred into a Teflon-lined stainless-steel autoclave with a capacity of 50 mL for 48 h at 80 °C. After cooling to room temperature, the yellowish precipitate was rinsed with deionized water and ethanol separately, and then dried in an oven at 60 °C for 24 h to obtain the CdS NS powder.2.3 Preparation of CdS NSs@MEA34
First, 0.1 g of CdS NSs was first dispersed in deionized H2O (100 mL) by sonication for 10 min, and then 9 mL of 2-mercaptoethylamine (MEA) (0.25 mol L−1) was added into the above mixture under stirring (1000 rpm) and stirred for 1 h under ambient conditions to promote the adsorption of MEA on the CdS NSs. Finally, the MEA-modified CdS NS substrates (CdS NSs@MEA) were rinsed with ethanol and fully dried in an oven at 333 K.2.4 Preparation of Aux@GSH NCs32
Gold(III) chloride trihydrate (40 mg) and L-glutathione (GSH, 46 mg) were thoroughly mixed in 50 mL of deionized water under ambient conditions. The mixture was continuously stirred until the appearance of a colorless solution and then heated at 343 K for 24 h. The aqueous solution of Aux clusters was stored at 277 K for further use.2.5 Preparation of CdS NSs@MEA/Ti3C2Tx
Typically, CdS NSs@MEA were dispersed in deionized water with a concentration of 1 mg mL−1 by ultrasonication. Ti3C2Tx colloid solution (0.06 mg mL−1) was then added into the as-prepared CdS NSs@MEA solution and stirred at room temperature for 5 h. Subsequently, the mixture was centrifuged and washed with deionized water (DI H2O). The precipitate obtained was dried in an oven at 60 °C to get the CdS-MXene heterostructures.2.6 Preparation of Aux@GSH/CdS NSs@MEA/Ti3C2Tx
First, 0.1 g of CdS NSs@MEA/Ti3C2Tx was dispersed in different volumes of Aux@GSH NCs aqueous solution (2, 4, 6, 9, 12, 15 mL) and stirred (1000 rpm) for 60 min. Then, the mixture was centrifuged and washed with deionized water and dried at 333 K in an oven to obtain the metal NCs/TMCs/MXene nanocomposites.2.7 Characterization
Zeta potential (ξ) measurements were operated by dynamic light scattering analysis (zeta sizer Nano ZS90) with five repeated measurements. The crystal structure was studied by X-ray diffraction (XRD, Miniflex600, Rigaku Corporation, Japan) using Cu Kα as the radiation source under 40 kV and 15 mA. Field-emission scanning electron microscopy (FESEM, Supra55, Carl Zeiss, Germany) was used to probe the morphology of the samples. Transmission electron microscopy (TEM), high-resolution (HR) TEM images and energy dispersive X-ray spectrum (EDS) were collected on a Tecnai G2 F20 transmission electron microscope with an accelerating voltage of 200 kV. UV-vis diffuse reflectance spectra (DRS, Cary50, Varian, America) were obtained using BaSO4 as the reflectance background ranging from 200 to 800 nm. X-ray photoelectron spectrometer (XPS) spectra were recorded on a photoelectron spectrometer (Escalab 250, Thermo Scientific, America), where the binding energy (BE) of the elements was calibrated based on the BE of carbon (284.8 eV). Fourier transform infrared (FTIR) spectra were collected by an infrared spectrophotometer (TJ270-30A, Tianjin, China). Raman spectra were studied with a Raman spectrometer (Dxr-2xi, Thermo Scientific, America). Brunauer–Emmett–Teller (BET) specific surface area and N2 adsorption experiments were carried out on a ASAP 2460. 13C NMR spectra were recorded on a Bruker Avance III 500 MHz spectrometer or a Bruker Avance NEO 600 MHz spectrometer. Photoluminescence spectra (PL) for solid samples were detected on an Edinburgh FS5 photoluminescence spectrophotometer and the excitation wavelength was set as 385 nm. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements were carried out on a Nicolet iS50FT-IR spectrometer (Thermo Fisher, USA).2.8 Photocatalytic CO2 reduction
Photocatalytic reactions were conducted in an online photocatalytic CO2 reduction system using a Pyrex vessel. Photocatalytic CO2 reduction was performed by dispersing 10 mg of catalyst in 10 mL of aqueous solution containing 5 mL of acetonitrile (MeCN), 2 mL of DI H2O as a reaction solvent and 3 mL of triethanolamine (TEOA) as the sacrificial agent. The solution was constantly stirred with the help of a magnetic stirrer, preventing the settling of particles at the bottom of the reactor. Prior to irradiation, the reaction mixture was vacuumed to eliminate the dissolved gases, and then the photocatalytic reactor was filled with CO2 for 10 min. The photocatalytic system was thoroughly degassed and then irradiated by a 300 W Xe lamp (PLSSXE300D, Beijing Perfect Light Co. Ltd, China) equipped with a 420 nm cut-off optical filter (λ > 420 nm). A magnetic stirrer bar was continuously stirred at the bottom of the reactor to keep the catalyst in a suspended state during the whole experiment. The evolving CO and H2 were monitored periodically by an online gas chromatograph (Shimadzu GC-2014C using argon as carrier gas). Photoactivities were evaluated based on the amount of CO and H2 evolution in the first 2 h of the reaction. Recycling photocatalytic CO2 reduction reactions were carried out as follows. The photocatalytic system was thoroughly degassed again after the first run without separating the catalyst or supplementing anything. Subsequently, the thoroughly degassed system was irradiated again by a 300 W Xe lamp with a UV-CUT filter to cut off light with wavelength λ.2.9 Photoelectrochemical (PEC) measurements
PEC measurements were carried out with a conventional three-electrode system on electrochemical workstations (CHI 660E & Gamary Interface 1000 E). Pt foil, an Ag/AgCl electrode and the samples coated on FTO acted as the counter, reference and working electrodes, respectively. Information about the fabrication of the working electrodes is provided in ESI.† An aqueous solution of Na2SO4 (0.5 M, pH = 6.69) was utilized as the electrolyte, and the photoelectrodes were irradiated with visible light (λ > 420 nm) (FX300, Beijing Perfectlight Co. Ltd, China).3 Results and discussion
A flowchart for the fabrication of CdS/Ti3C2Tx/Aux heterostructure is exhibited in Scheme 1, which can be divided into three steps. Firstly, CdS NSs were prepared by a hydrothermal method and then modified with 2-mercaptoethylamine (MEA) to obtain a positively charged surface (Fig. S1†). Subsequently, CdS were integrated with negatively charged Ti3C2Tx nanosheets (Fig. S2a†) by electrostatic attraction to get the CdS/Ti3C2Tx (CT) nanocomposite. It is noteworthy that GSH ligands endow Aux NCs with a negatively charged surface. As a result, CdS/Ti3C2Tx/Aux ternary heterostructures are obtained by self-assembling negatively charged Aux@GSH NCs (Fig. S3 & 4a†) on the positively charged MEA-modified CT nanocomposite in an analogous integration mode via electrostatic interaction. Therefore, the complementary surface charge properties of CdS, Ti3AlC2 and Aux@GSH NC building blocks contribute to the construction of a CdS/Ti3C2Tx/Aux (CTA) heterostructure.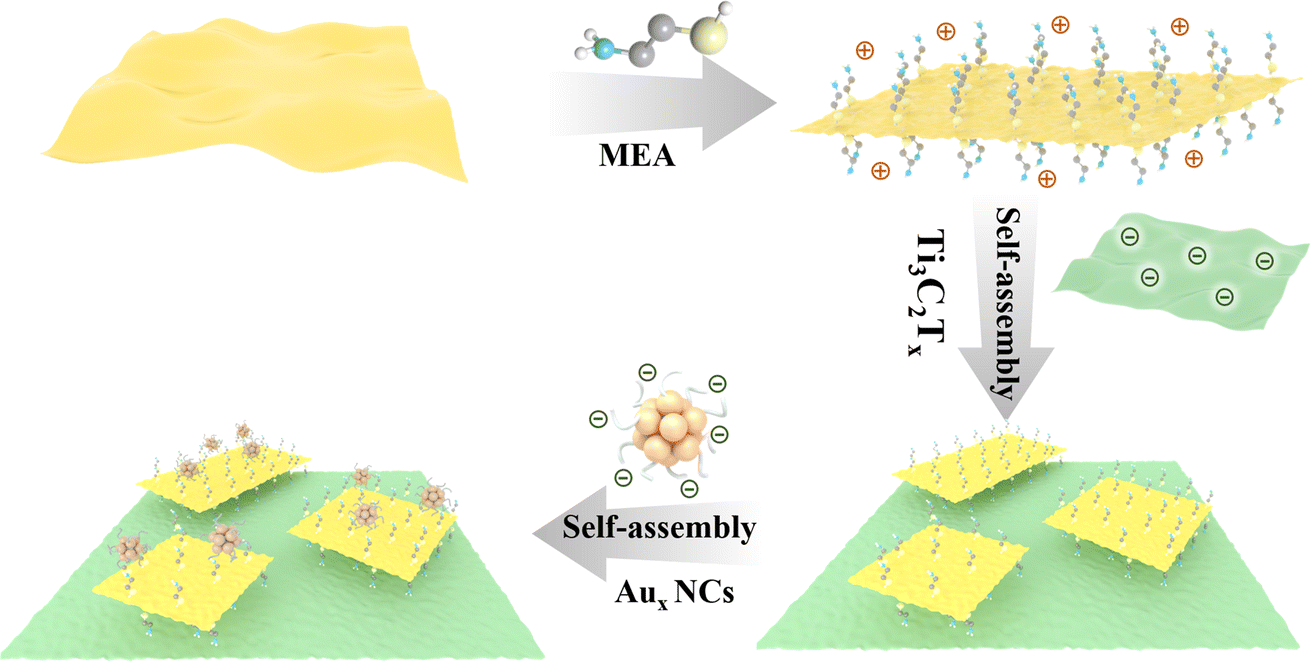 | ||
| Scheme 1 Schematic illustration of the self-assembly of CdS/Ti3C2/Aux nanocomposites. | ||
3.1 Characterization of CdS/Ti3C2Tx/Aux NCs heterostructure
X-ray powder diffraction (XRD) was used to explore the crystal structures of CdS, Ti3C2Tx, CT and CTA. As shown in Fig. S2c,† the as-prepared Ti3C2Tx nanosheets demonstrate a characteristic diffraction peak at 2θ = 6.7° corresponding to the (002) crystal plane, which is distinct from the XRD result of the Ti3AlC2 precursor.35 Note that the peak at 2θ = 38.9° attributable to the (104) crystal plane of Ti3AlC2 is almost invisible in the XRD pattern of Ti3C2Tx, indicating that the layer of Al atoms in Ti3AlC2 has been completely removed by etching and successful preparation of Ti3C2Tx. As can be seen from Fig. 1a, the peaks at 24.8°, 26.7°, 28.4°, 36.6°, 43.7°, 47.9° and 52° match well with the (100), (002), (101), (102), (110), (103) and (112) crystal planes of wurtzite phase CdS (PDF#65-3414). The XRD spectra of CT and CTA are basically consistent with CdS, but a new peak appears at 2θ = 20.7°, which can be attributed to the (004) crystal plane of Ti3C2Tx.36 No obvious peak of Aux@GSH NCs can be observed in the XRD result for CTA, probably due to the intrinsic amorphous property of Aux@GSH NCs. As displayed in Fig. 1b, two characteristic peaks at 300 and 600 cm−1 in the Raman spectrum of CdS are ascribed to the longitudinal-optical (LO) phonon of CdS and its first multiscattering (2LO),37 which is analogous to those of CT and CTA owing to the low deposition percentage of Ti3C2Tx and Aux@GSH NCs. | ||
| Fig. 1 (a) XRD results, (b) Raman, (c) FTIR, (d) DRS and (e) bandgap determination plots for CdS, CT, and CTA. High-resolution (f) S 2p, (g) Cd 3d, (h) Ti 2p, and (i) Au 4f spectra of (I) CdS, (II) CT, and (III) CTA. | ||
Fig. 1c shows the Fourier transform infrared (FTIR) spectra of CdS, CT and CTA, all of which show two peaks at 2917 and 2856 cm−1 that can be ascribed to the stretching vibration mode of –CH2 groups from the MEA molecules.20 The peak at 612 cm−1 in the FTIR spectra of CT and CTA originates from the Ti–O bond from the surface functional groups of Ti3AlC2,38 which matches the XRD result, indicating the successful integration of Ti3AlC2 with CdS in the composite. The single peak at 1630 cm−1 corresponds to the stretching vibration mode of the –COOH group from the GSH ligand of Aux NCs,20 which confirms the successful combination of Aux@GSH NCs with CT.
The optical properties of CdS, CT and CTA were investigated by UV-vis diffuse reflectance spectroscopy (DRS). As shown in Fig. S4b,† Aux@GSH NCs demonstrate visible light absorption with a band edge approaching 450 nm,39 confirming the successful synthesis of Aux@GSH NCs. The absorption spectrum of Ti3C2Tx (Fig. S2d†) shows the appearance of characteristic peaks at 255 and 314 nm,40 and in particular the near-infrared (NIR) absorption at ca. 700–800 nm confirm the successful fabrication of MXene. From Fig. 1d, it is apparent that the light absorption band edge of the CdS NS substrate is around 550 nm owing to intrinsic band gap photoexcitation. When integrating MXene with CdS NSs, the absorption intensity of CT shows an enhancement in the range 550–800 nm compared with pure CdS NSs. This result agrees with the change in sample color from bright yellow to green, which confirms that MXene deposition can promote the light-capturing capacity of the nanocomposite. It is worth noting that the light absorption intensity of CTA is not remarkably altered compared with CT because of the low deposition percentage of Aux@GSH NCs. The band gap energy of these samples can be determined with the following formula:
| (αhv)n = A(hv − Eg) | (1) |
X-ray photoelectron spectroscopy (XPS) was utilized to explore the surface element composition and chemical states of CdS, CT and CTA. As depicted in Fig. S5,† the survey spectrum of CTA shows the signals of Cd 3d, Cd 4d, S 2p, S 2s, C 1s, Ti 2p, Au 4f and O 1s. Among them, the Cd & S signals come from CdS NSs, the C & Ti signals originate from Ti3C2Tx, and the Au signal stems from Aux@GSH NCs. In the high-resolution S 2p spectrum of CTA (Fig. 1f), two peaks of S 2p3/2 and S 2p1/2 are located at around 161.4 eV and 162.6 eV, respectively, neither of which are obviously changed compared with those of CT, but they demonstrate an evident red-shift in binding energy relative to those of CdS. As for CTA, the high-resolution Cd 3d spectrum shows Cd 3d3/2 and Cd 3d5/2 peaks at 411.8 eV and 405 eV, respectively,41 and the changes in binding energy of the high-resolution Cd 3d spectra for CdS, CT and CTA are consistent with that of the aforementioned S 2p spectrum (Fig. 1g). The change in position of these peaks in the high-resolution S 2p and Cd 3d spectra of CdS, CT and CTA can be attributed to the strong interaction between CdS and Ti3C2Tx, which facilitates the formation of an intimate interface and exerts a significant influence on the distribution of electron density in CT. The characteristic peaks at 460.2 eV/458.8 eV, 455.5 eV/462.3 eV, 461 eV and 463.1 eV in the high-resolution Ti 2p spectrum of CTA (Fig. 1h) match well with the Ti–C, Ti–Ox, Ti–x and Ti–O bonds, respectively.42 The presence of Ti–O and Ti–x species indicates that Ti3C2Tx features various functional groups on the surface, such as –O, –F and –OH, which can improve the adsorption capacity of CO2 or H2O.43 As shown in Fig. 1i, the high-resolution Au 4f spectrum of CTA shows two peaks which correspond to the Au+ and Au0 species from the generic core–shell nanostructure of Aux@GSH NCs,20 confirming the successful integration of Aux@GSH NCs into the nanocomposites.
An analysis of N2 adsorption/desorption isotherms and pore size distribution curves is shown in Fig. S7,† from which we can see that the three samples all exhibit type IV isotherms, and the mean pore size is between 2 and 50 nm, indicating they are mesoporous materials. As summarized in Table S2†, the specific surface area of CT (67.7794 m2 g−1) is much larger than that of CdS (36.08 m2 g−1) due to the introduction of 2D Ti3C2Tx. However, note that the specific surface area of CTA (54.53 m2 g−1) is slightly smaller than that of CT after introducing Aux@GSH NCs (ca. 1.36 nm, Fig. S4†) which blocks the surface pores of CT. Therefore, the remarkable enhancement in the photocatalytic performance of CTA illustrated in the latter part is independent of the specific surface area.
As shown in Fig. 2a, CdS exhibits a morphology of corrugated nanosheets stacking with each other. The elemental mapping and energy-dispersive X-ray spectroscopy (EDS) of CdS are consistent with the scanning electron microscopy (SEM) image (Fig. S8†). As displayed in Fig. 2b, the morphology of CT is similar to that of pure CdS, indicating that introducing 2D layered Ti3C2Tx with an average thickness of about 1.4 nm (Fig. S2e and f†) does not exert much impact on the morphology of the composite. The SEM image and EDS results of CT (Fig. S9†) suggest that Ti and C elements are evenly distributed on the surface of CdS, which implies the successful introduction of Ti3C2Tx. The morphology of CTA (Fig. 2c) is not apparently altered compared with CT and no Aux@GSH NCs can be directly observed on the surface, which is attributed mainly to the ultra-small size of the Aux@GSH NCs.
 | ||
| Fig. 2 Panoramic FESEM images of (a) CdS, (b) CT, and (c) CTA; TEM images of (d) CdS, (e) CT, and (f) CTA; HRTEM images of (g) CdS, (h) CT, and (i) CTA; (j–jr) TEM elemental mapping results of CTA with (j1) S, (j2) Ti, (j3) Au, (j4) F, (j5) Cd, (j6) C, (j7) O and (j8) N signals. | ||
The microstructure and morphology of CdS, CT and CTA were further probed by transmission electron microscopy (TEM) and high-resolution transmission electron microcopy (HRTEM). As shown in Fig. 2d, CdS exhibits an integrated 2D nanosheet structure, which is consistent with the SEM results, and the lattice finger with spacing of 0.336 nm (Fig. 2g) agrees well with the (002) crystal plane of hexagonal CdS. From the TEM image of CT (Fig. 2e), a layered structure of Ti3C2Tx can obviously be found in the area outlined by white dots. Meanwhile, we can see the clear boundaries marked by the dashed line, confirming the tight connection formed between MXene and the CdS substrate. Besides the (002) crystal plane of CdS, the (104) crystal plane of Ti3C2Tx can also be observed in the HRTEM image of CT (Fig. 2h). As shown in the TEM image of CTA (Fig. 2f), Aux@GSH NCs are uniformly dispersed on the CTA surface. It can be seen from HRTEM (Fig. 2i) that the lattice spacings of 0.318 nm and 0.248 nm correspond to the (101) and (006) crystal planes of hexagonal CdS and Ti3C2Tx, respectively, but no lattice fringe belonging to Aux@GSH NCs is observed because of its generic amorphous property. As shown in Fig. 2(j–j8), S, Cd, C, Ti, Au and N elements are uniformly distributed in the whole framework of the CTA nanocomposite, among which the N signal originates mainly from the GSH ligand, and the F signal derives from the surface functional group of Ti3C2Tx, confirming the successful dispersion of Ti3C2Tx and Aux@GSH NCs in the CTA nanocomposite.
3.2 Photocatalytic activities
The photoactivities of CdS, CT and CTA toward CO2 reduction under visible light irradiation were probed. As displayed in Fig. 3a, pure CdS demonstrates a CO yield of 19.458 μmol g−1 h−1. When Ti3C2Tx (0.06 mg mL−1) is integrated with CdS, the CO yield of CT0.06 increases to 66.792 μmol g−1 h−1, which is attributed to the merits of 2D Ti3C2Tx with excellent conductivity that not only promotes charge transfer and reduces carrier recombination, but also boosts the specific surface area of the CT nanocomposite to yield more active sites. When Aux@GSH NCs (1.2 mL) are self-assembled on the CT nanocomposite, the photocatalytic performance of CT0.06A1.2 is further enhanced with the CO generation rate reaching 117.327 μmol g−1 h−1, which is 6 and 2 times larger than those of pure CdS (19.458 μmol g−1 h−1) and CT0.06 (66.792 μmol g−1 h−1), respectively. This is mainly ascribed to the fact that the involvement of Aux@GSH NCs improves the light-trapping capacity of the CT0.06A1.2 nanocomposite and promotes interfacial charge separation and transfer. Notably, no CO can be detected when pure Aux@GSH NCs, pure Ti3C2Tx or Ti3C2Tx/Aux@GSH (T0.06A1.2) composite were utilized as the catalysts. The results are strong evidence for the indispensable role of CdS as a light-harvesting antenna and its applicable energy level alignment with Aux@GSH NCs, the advantageous photosensitization effect of Aux@GSH NCs, and the electron-trapping capability of Ti3C2Tx, which synergistically contribute to the significantly enhanced photocatalytic CO2 reduction performance of CTA under visible light irradiation.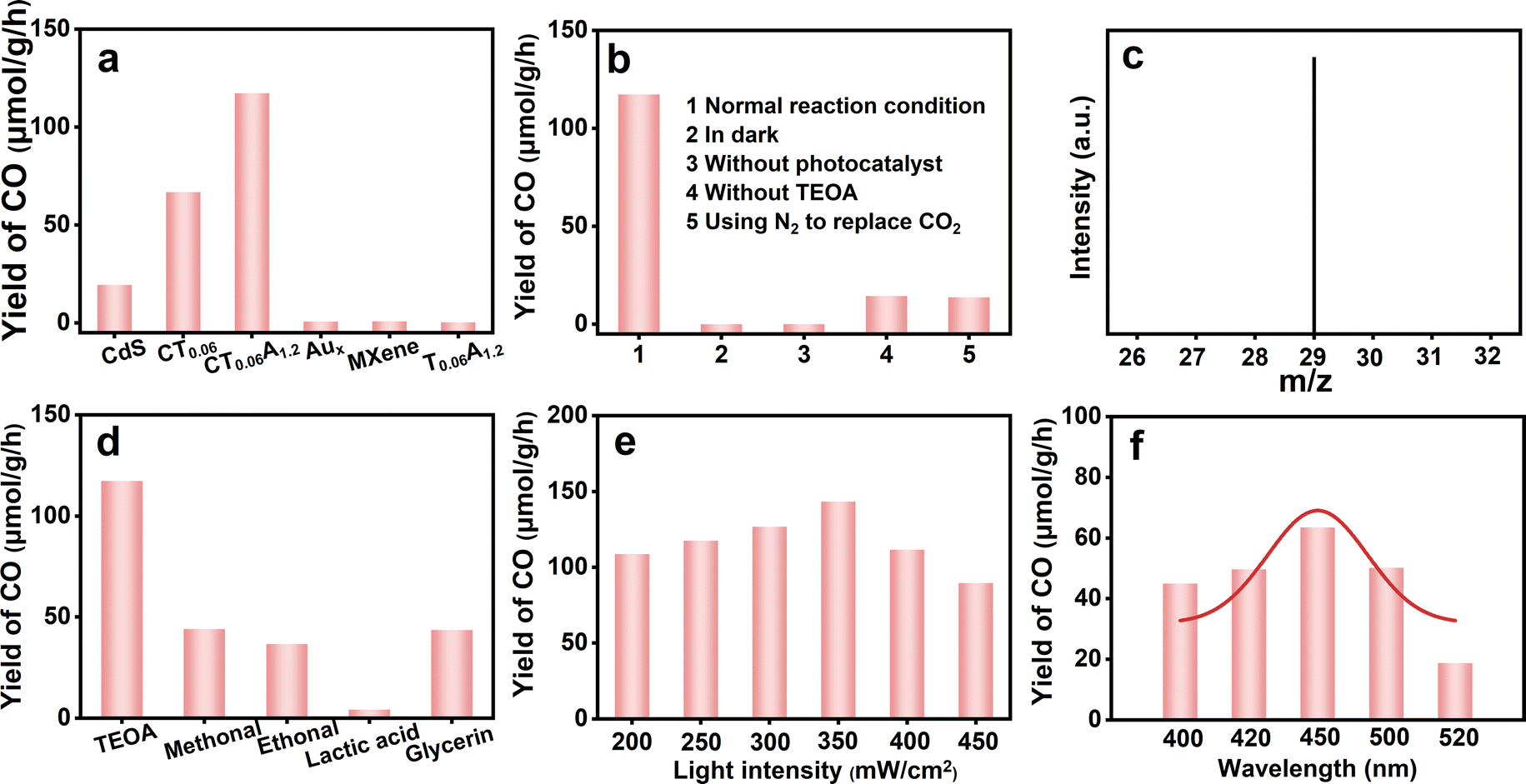 | ||
| Fig. 3 (a) CO2 photoreduction performances of CdS, CT0.06, CT0.06A1.2, CA1.2, pure Aux@GSH NCs, MXene and T0.06A1.2; (b) CO2 photoreduction performances of CT0.06A1.2 under different reaction conditions; (c) mass spectrum of 13CO (m/z = 29) produced over CT0.06A1.2 in the photocatalytic reduction of 13CO2; (d) photoactivities of CT0.06A1.2 using various sacrificial reagents; (e) photoactivities of CT0.06A1.2 under different light intensities; (f) wavelength-dependent CO2 photoreduction activities of CT0.06A1.2. | ||
As shown in Fig. 3b, no obvious CO2 reduction reaction occurs in the absence of catalyst or light, indicating that the reaction requires photoexcited catalyst and light to produce the charge carriers which trigger the CO2 photoreduction. That is, light and catalysts are the essential premise for CO2 photocatalysis. The CO yield of CT0.06A1.2 without using triethanolamine (TEOA) as a sacrificial agent (14.373 μmol g−1 h−1) is much lower than under normal conditions, highlighting the important role of a sacrificial reagent in remarkably promoting the CO2 conversion efficiency by quenching holes. It is worth noting that when N2 is used to replace CO2, there is a small amount of CO generation, which may stem from the trace CO2 that cannot be completely removed by vacuuming the system. Moreover, the carbon source of CO products was explored by isotope tracking control experiments and analyzed by gas chromatography-mass spectrometry (GC-MS). As shown in Fig. 3c, the peak at an m/z value of 29 corresponds to 13CO, which confirms that the CO product originates from CO2. Furthermore, we found that different sacrificial reagents also have a great impact on the CO2 reduction performance (Fig. 3d), among which the photosystem with added TEOA exhibits optimal photoactivity compared with the other counterparts. As exhibited in Fig. 3e, the CO yield of CT0.06A1.2 is heavily dependent on the light intensity. When the light intensity increases to 350 mW cm−2, the CO yield increases to its optimum (143.334 μmol g−1 h−1), confirming it is indeed a photocatalytic process. Besides, the CO2 photoreduction performance of CT0.06A1.2 under monochromatic light irradiation (Fig. 3f) also shows a volcanic variation trend when the wavelength changes from 400 to 520 nm, and reaches its highest value (63.513 μmol g−1 h−1) at 450 nm. This is completely consistent with the DRS results, indicating that the bandgap photoexcitation of the CdS substrate plays a dominant role in CO2 reduction.
Stability measurements (Fig. S10†) reveal that CT0.06A1.2 demonstrates improved photostability relative to pure CdS during the five successive photocatalytic reactions. Note that the photoactivity of CT0.06A1.2 is always larger than that of pristine CdS during photostability testing, once again confirming the synergy of MXene and Aux@GSH NCs in promoting charge separation. The structural integrity of CT0.06A1.2 after a cyclic photocatalytic CO2 reduction reaction was explored. As shown in Fig. S11 and S12,† the XRD results for CTA before and after the cyclic reaction are basically similar, and the morphology and elemental composition are not greatly changed according to the SEM and EDS results. The above results indicate that photocorrosion of CdS in the nanocomposite does not occur in our work.
Photoelectrochemical (PEC) measurements were performed to evaluate the roles of Ti3C2Tx and Aux@GSH in promoting interfacial charge separation. Photocurrents of CdS, CT0.06 and CT0.06A1.2 increase successively, which can be attributed to the following reasons: (1) Ti3C2Tx with superior conductivity facilitates electron transfer and suppresses the recombination of electron–hole pairs over CT0.06. (2) Aux@GSH NCs, as light-harvesting antennas, establish beneficial energy level alignment with CdS and Ti3C2Tx, further enhancing carrier migration and increasing carrier density, resulting in the highest photocurrent density of CT0.06A1.2 compared with CdS and CT0.06 (Fig. 4a). Electrochemical impedance spectroscopy (EIS) was utilized to probe interfacial charge transfer resistance. As shown in Fig. 4b, the semicircular arc radius of CT0.06A1.2 is successively smaller than those of CT0.06 and CdS, proving that CT0.06A1.2 harbours the lowest interfacial charge transfer resistance compared with the other counterparts. The specific interfacial charge resistance of the samples is summarized in Table S4†. Analogous results are found in the open-circuit photovoltage decay (OCVD) results (Fig. 4c). Specifically, CT0.06A1.2 shows the largest photovoltage and the longest decay rate in comparison with CT0.06 and CdS, which reflects its most efficient charge separation. As shown in Fig. S13,† the slope of the linear part of the Mott–Schottky (M–S) plots of CdS, CT0.06 and CT0.06A1.2 is utilized to calculate the carrier density according to the following formula:
 | (2) |
 | ||
| Fig. 4 (a) Transient photocurrent responses, (b) Nyquist plots of EIS results, (c) open-circuit-photovoltage decay curves, (d) M–S results and (e) carrier densities (ND) of CdS, CT0.06 and CT0.06A1.2 under visible light (λ > 420 nm) irradiation utilizing Na2SO4 aqueous solution (0.5 M, pH = 7) as the electrolyte; (f) solid-state PL spectra (λex = 385 nm) of CdS, CT0.06 and CT0.06A1.2. | ||
3.3 Photocatalytic mechanism
The active intermediate species formed during photocatalytic CO2 reduction over CdS and CT0.06A1.2 were explored through in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). Fig. 5a illustrates the in situ formation of unidentate carbonate (m-CO32−) (1540 cm−1, 1510 cm−1), bidentate carbonate (b-CO32−) (1650 cm−1, 1620 cm−1, 1338 cm−1), CO2− (1697 cm−1) and HCO3− (1473, 1223 cm−1) species under visible light irradiation, suggesting that CO2 molecules are adsorbed on the surface of the catalysts and are then converted into diverse intermediate active species.44 Additionally, as depicted in Fig. 5b, the adsorption bands of these species produced over CT0.06A1.2 exhibit enhanced intensity compared with pure CdS, indicating the superior CO2 activation capability of CT0.06A1.2. Noticeably, COOH* species at 1558 cm−1, featuring a key intermediate in the process of CO2-to-CO conversion, is observed in the DRFITS results of CdS and CT0.06A1.2.45 Meanwhile, CO* species with significantly enhanced intensity are observed at 1844 cm−1 and 1868 cm−1. Considering that CH4 is not produced in the photocatalytic products, we speculate that photocatalytic CO2 reduction over CdS and CT0.06A1.2 follows the pathway of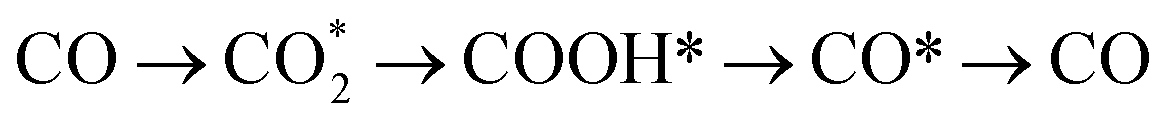 to convert CO2 into CO. Noteworthily, as illustrated in Fig. 5c, the saturated CO2 adsorption of CT0.06A1.2 exceeds that of CdS, which is also conducive to improving the photocatalytic activity of CT0.06A1.2.
to convert CO2 into CO. Noteworthily, as illustrated in Fig. 5c, the saturated CO2 adsorption of CT0.06A1.2 exceeds that of CdS, which is also conducive to improving the photocatalytic activity of CT0.06A1.2.
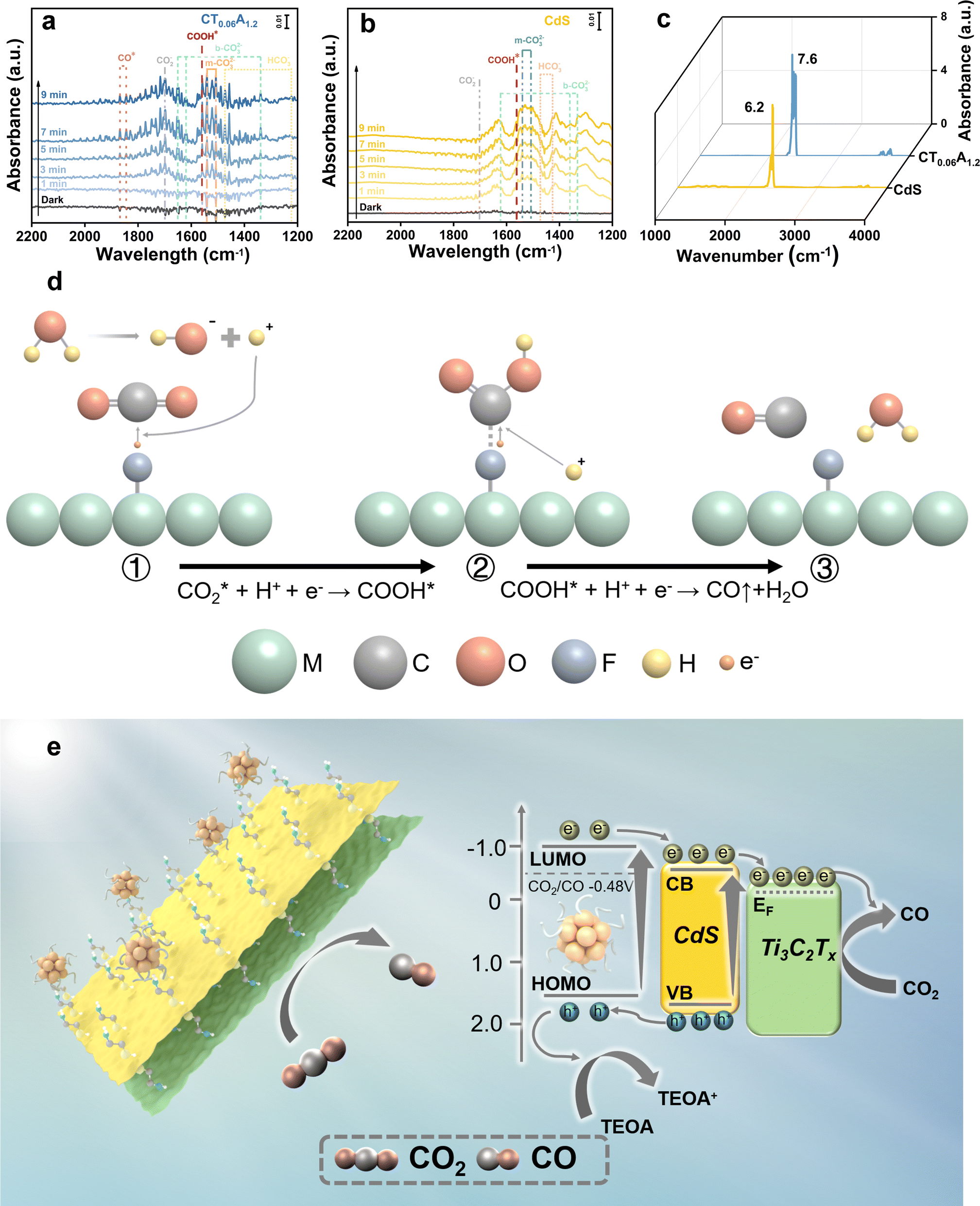 | ||
| Fig. 5 In situ DRIFTS analysis of photocatalytic CO2 reduction over (a) CdS, (b) CT0.06A1.2. The sorption equilibrium spectra of (c) CdS and CT0.06A1.2 after CO2 adsorption in the dark for 5 min; (d) CO2 photoreduction into CO via utilizing a single-metal-site based on carbine generation pathways. (e) Photocatalytic mechanism of CdS/Ti3C2Tx/AuxNCs heterostructure. | ||
To clarify the reaction mechanism, the energy level positions of CdS, MXene, and Aux@GSH NCs were explored. According to the M–S results of CdS (Fig. 4d), its flat band potential relative to a normal hydrogen electrode (ENHE = EAg/AgCl + 0.1976 V) is determined to be −0.44 V vs. NHE. In general, the conduction band (CB) position of n-type semiconductors is 0.1–0.2 V more negative than the flat band potential, by which the CB position of CdS is determined to be −0.54 V (vs. NHE, pH = 7). According to the bandgap determined with the DRS result, the valence band (VB) position of CdS is calculated to be 1.82 V (vs. NHE, pH = 7) with the formula EVB = ECB + Eg. On the other hand, based on the cyclic voltammetry (CV) results (Fig. S14a†) and UV-vis absorption spectra (Fig. S14b†), the LUMO and HOMO levels of Aux@GSH NCs are determined to be −1.0 and 1.75 V vs. NHE, respectively.46 Besides, as reported in previous work,47 the Fermi level of Ti3C2Tx is −0.04 V (vs. NHE). Therefore, according to the aforementioned energy level positions, a type II energy level configuration is established between CdS and Aux@GSH NCs while a Schottky barrier with a built-in electric field directed from CdS to Ti3C2Tx is formed.
Based on the above analysis, the photocatalytic CO2 reduction mechanism of the CdS/Ti3C2Tx/Aux NCs heterostructure can thus be proposed. Under visible light irradiation, CdS NSs and Aux@GSH NCs are simultaneously photoexcited to generate electron–hole pairs, and consequently they are efficiently transferred due to the favorable energy level alignment and Schottky barrier generation. To be specific, holes photoexcited in the VB of CdS are transferred to the HOMO level of Aux@GSH NCs and then completely quenched by the hole scavenger (TEOA). Meanwhile, electrons photoexcited over Aux@GSH NCs migrate from the LUMO level to the CB position of CdS, and then transfer to the Fermi level of Ti3C2Tx, leading to a cascade electron transport pathway. Consequently, the spatial separation of electron–hole pairs over the CT0.06A1.2 heterostructure is considerably enhanced, giving rise to a markedly prolonged carrier lifetime. The ultimately separated photoelectrons on the surface of Ti3C2Tx participate in the photocatalytic CO2 reduction, which is specifically illustrated in Fig. 5d. Specifically, the carbon atom in the CO2 molecule, as a Lewis acid, is easily adsorbed on the –F functional group, which acts as a Lewis base on the surface of Ti3C2Tx.48 The acid–base interaction between fluorine and CO2 reduces the activation energy barrier of CO2 and accelerates the activation of CO2 molecules to form  .49 The activated
.49 The activated 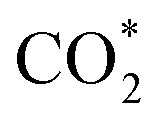 triggers a reaction with protons and photo-excited electrons, resulting in the formation of COOH* intermediates. Subsequent protonation further benefits conversion of COOH* to CO*, which is ultimately desorbed to generate CO.50
triggers a reaction with protons and photo-excited electrons, resulting in the formation of COOH* intermediates. Subsequent protonation further benefits conversion of COOH* to CO*, which is ultimately desorbed to generate CO.50
4 Conclusions
In summary, a cascade electron transport pathway is elaborately constructed by integrating CdS with Ti3C3Tx and Aux@GSH NCs in a controllable fashion to customize an elegant interface configuration via electrostatic self-assembly towards photocatalytic CO2 reduction. In this well-designed CdS/Ti3C2Tx/Aux ternary heterostructure, CdS and Aux@GSH NCs function as light-harvesting antennas and engender a favorable type II energy level alignment, while MXene serves as a terminal electron-trapping reservoir, which synergistically contributes to effective charge separation, thereby significantly enhancing the photocatalytic CO2-to-CO reduction performance under visible light irradiation. The photocatalytic mechanism of CO2 reduction along with generation of intermediate active species were determined by DRIFTS. Our work will provide an inspiring idea to combine MXene with atomically precise metal NCs to strategically mediate charge transport pathways for solar-to-fuel conversion.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Yu-Shan Cai performed the experiments, analyzed all data, and draft the manuscript. Jia-Qi Chen and Peng Su help to check the manuscript. Xian Yan, Qing Chen and Yue Wu carried out the DRIFTS experiments. Fang-Xing Xiao guided this work and correct the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support by the award Program for Minjiang scholar professorship is greatly acknowledged. This work was financially supported by the National Natural Science Foundation of China (No. 21703038, 22072025). The financial support from state key laboratory of structural chemistry, Fujian institute of research on the structure of matter, Chinese academy of science is acknowledged (No. 20240018).References
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed
.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649–1704680 CrossRef PubMed
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, Acs Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed
- M. Han, S. Zhu, S. Lu, Y. Song, T. Feng, S. Tao, J. Liu and B. Yang, Nano Today, 2018, 19, 201–218 CrossRef CAS
- W. Hou, W. H. Hung, P. Pavaskar, A. Goeppert, M. Aykol and S. B. Cronin, ACS Catal., 2011, 1, 929–936 CrossRef CAS
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS
- J. Yu, K. Wang, W. Xiao and B. Cheng, Phys. Chem. Chem. Phys., 2014, 16, 11492–11501 RSC
- M. D. Hernandez-Alonso, F. Fresno, S. Suarez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 RSC
- X. Li, M. Rui, J. Song, Z. Shen and H. Zeng, Adv. Funct. Mater., 2015, 25, 4929–4947 CrossRef CAS
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed
- Y. Dai, C. Li, Y. Shen, T. Lim, J. Xu, Y. Li, H. Niemantsverdriet, F. Besenbacher, N. Lock and R. Su, Nat. Commun., 2018, 9, 7 CrossRef PubMed
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed
- S. Zhou, Y. Liu, J. Li, Y. Wang, G. Jiang, Z. Zhao, D. Wang, A. Duan, J. Liu and Y. Wei, Appl. Catal., B, 2014, 158, 20–29 CrossRef
- Y. Wu, P. Wang, H. Che, W. Liu, C. Tang and Y. Ao, Angew. Chem., Int. Ed., 2024, 63, 202316410 CrossRef PubMed
- C. Xue, P. Wang, H. Che, W. Liu, B. Liu and Y. Ao, Appl. Catal., B, 2024, 340, 123259 CrossRef CAS
- Q. Zhang, T. Wu, H. Che, C. Tang, B. Liu and Y. Ao, Surf. Interfaces, 2024, 47, 104205 CrossRef CAS
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed
- S. Hou, M.-H. Huang and F.-X. Xiao, J. Mater. Chem. A, 2022, 10, 7006–7012 RSC
- Z.-Q. Wei, S. Hou, X. Lin, S. Xu, X.-C. Dai, Y.-H. Li, J.-Y. Li, F.-X. Xiao and Y.-J. Xu, J. Am. Chem. Soc., 2020, 142, 21899–21912 CrossRef CAS PubMed
- X.-Y. Fu, Z.-Q. Wei, S. Xu, X. Lin, S. Hou and F.-X. Xiao, J. Phys. Chem. Lett., 2020, 11, 9138–9143 CrossRef CAS PubMed
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed
- M. Khazaei, A. Ranjbar, M. Arai, T. Sasaki and S. Yunoki, J. Mater. Chem. C, 2017, 5, 2488–2503 RSC
- A. V. Mohammadi, J. Rosen and Y. Gogotsi, Science, 2021, 372, 1165–1180 Search PubMed
- J. Choi, Y.-J. Kim, S.-Y. Cho, K. Park, H. Kang, S. J. Kim and H.-T. Jung, Adv. Funct. Mater., 2020, 30, 2003998–2004007 CrossRef CAS
- R. Xiao, C. Zhao, Z. Zou, Z. Chen, L. Tian, H. Xu, H. Tang, Q. Liu, Z. Lin and X. Yang, Appl. Catal., B, 2020, 268, 118382–118393 CrossRef CAS
- M.-Y. Ye, Z.-H. Zhao, Z.-F. Hu, L.-Q. Liu, H.-M. Ji, Z.-R. Shen and T.-Y. Ma, Angew. Chem., Int. Ed., 2017, 56, 8407–8411 CrossRef CAS PubMed
- Y.-J. Yuan, Z. Shen, S. Wu, Y. Su, L. Pei, Z. Ji, M. Ding, W. Bai, Y. Chen, Z.-T. Yu and Z. Zou, Appl. Catal., B, 2019, 246, 120–128 CrossRef CAS
- Q. Liu, T. Chen, Y. Guo, Z. Zhang and X. Fang, Appl. Catal., B, 2016, 193, 248–258 CrossRef CAS
- J. Bai, R. Shen, Z. Jiang, P. Zhang, Y. Li and X. Li, Chin. J. Catal., 2022, 43, 359–369 CrossRef CAS
- X.-Y. Fu, Z.-Q. Wei, S. Xu, X. Lin, S. Hou and F.-X. Xiao, J. Phys. Chem. Lett., 2020, 11, 9138–9143 CrossRef CAS PubMed
- X. Xie, N. Zhang, Z.-R. Tang, M. Anpo and Y.-J. Xu, Appl. Catal., B, 2018, 237, 43–49 CrossRef CAS
- H. Liang, B.-J. Liu, B. Tang, S.-C. Zhu, S. Li, X.-Z. Ge, J.-L. Li, J.-R. Zhu and F.-X. Xiao, ACS Catal., 2022, 12, 4216–4226 CrossRef CAS
- J. Low, L. Zhang, T. Tong, B. Shen and J. Yu, J. Catal., 2018, 361, 255–266 CrossRef CAS
- B. Saini, K. Harikrishna, D. Laishram, R. Krishnapriya, R. Singhal and R. K. Sharma, ACS Appl. Nano Mater., 2022, 5, 9319–9333 CrossRef CAS
- K. Sun, J. Shen, Y. Yang, H. Tang and C. Lei, Appl. Surf. Sci., 2020, 505, 144042–144050 CrossRef CAS
- S.-C. Zhu, Z.-C. Wang, B. Tang, H. Liang, B.-J. Liu, S. Li, Z. Chen, N.-C. Cheng and F.-X. Xiao, J. Mater. Chem. A, 2022, 10, 11926–11937 RSC
- H. Choi, Y.-S. Chen, K. G. Stamplecoskie and P. V. Kamat, J. Phys. Chem. Lett., 2015, 6, 217–223 CrossRef CAS PubMed
- Y. Shao, C. Chen, Q. He, W. Wu, C. Li and Y. Gao, Nanomaterials, 2020, 10, 2544–2555 CrossRef CAS PubMed
- M. N. Meeran, N. Haridharan, M. Shkir, H. Algarni and V. R. M. Reddy, Chem. Phys. Lett., 2022, 809, 140150–140159 CrossRef
- X. Chen, Y. Guo, R. Bian, Y. Ji, X. Wang, X. Zhang, H. Cui and J. Tian, J. Colloid Interface Sci., 2022, 613, 644–651 CrossRef CAS PubMed
- M. Tahir, A. A. Khan, S. Tasleem, R. Mansoor, A. Sherryna and B. Tahir, J. Energy Chem., 2023, 76, 295–331 CrossRef CAS
- H. Yu, J. Li, Y. Zhang, S. Yang, K. Han, F. Dong, T. Ma and H. Huang, Angew. Chem., Int. Ed., 2019, 58, 3880–3884 CrossRef CAS PubMed
- J. Di, X. Zhao, C. Lian, M. Ji, J. Xia, J. Xiong, W. Zhou, X. Cao, Y. She, H. Liu, K. P. Loh, S. J. Pennycook, H. Li and Z. Liu, Nano Energy, 2019, 61, 54–59 CrossRef CAS
- L. Yu-Bing and X. Fang-Xing, J. Mater. Chem. A, 2022, 11, 589–599 Search PubMed
- J.-Y. Li, Y.-H. Li, F. Zhang, Z.-R. Tang and Y.-J. Xu, Appl. Catal., B, 2020, 118783–118792, DOI:10.1016/j.apcatb.2020.118783
- Y. Su, Y. Cheng, Z. Li, Y. Cui, C. Yang, Z. Zhong, Y. Song, G. Wang and L. Zhuang, J. Energy Chem., 2024, 88, 543–551 CrossRef CAS
- M. W. D. Hanson-Heine and N. A. Besley, Chem. Phys. Lett., 2015, 638, 191–195 CrossRef CAS
- J. Wu, X. Li, W. Shi, P. Ling, Y. Sun, X. Jiao, S. Gao, L. Liang, J. Xu, W. Yan, C. Wang and Y. Xie, Angew. Chem., Int. Ed., 2018, 57, 8719–8723 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03663h |
| This journal is © The Royal Society of Chemistry 2024 |