
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA corannulene-based metallobox for the encapsulation of fullerenes†
Susana
Ibáñez
 *a,
Carmen
Mejuto
a,
Katherin
Cerón
a,
Pablo J.
Sanz Miguel
b and
Eduardo
Peris
*a
*a,
Carmen
Mejuto
a,
Katherin
Cerón
a,
Pablo J.
Sanz Miguel
b and
Eduardo
Peris
*a
aInstitute of Advanced Materials (INAM), Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universitat Jaume I, Av. Vicente Sos Baynat s/n, Castellón, E-12071, Spain. E-mail: maella@uji.es; eperis@uji.es
bDepartamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), Universidad de Zaragoza-CSIC, 50009 Zaragoza, Spain
First published on 16th July 2024
Abstract
A corannulene-bis-N-imidazolium salt was used for the synthesis of two corannulene-bis-N-heterocyclic carbenes of dirhodium(I) complexes of formula (corannulene-di-NHC)[RhCl(COD)]2 and (corannulene-di-NHC)[RhCl(CO)2]2. Both complexes were characterized by spectroscopic techniques, and the electron-donating properties of the corannulene-di-NHC ligand were studied by means of infrared spectroscopy and cyclic voltammetry. The complex (corannulene-di-NHC)[RhCl(COD)]2 was used for the encapsulation of fullerenes C60 and C70, generating host–guest complexes with 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry, as evidenced by 1H NMR and ITC titrations. Then, a tetra-rhodium(I) metallo-rectangle supported by two corannulene-bis-imidazolylidene ligands and two cofacial 4,4′-bipyridine ligands was prepared and characterized. This metallobox is capable of quantitatively encapsulating fullerenes C60 and C70, forming complexes that are highly stable even at high temperatures. The molecular structure of the metallobox with encapsulated C60 reveals a perfect size and shape complementarity that benefits from the concave–convex π–π interaction between the polyaromatic surfaces of the host and the guest.
:1 stoichiometry, as evidenced by 1H NMR and ITC titrations. Then, a tetra-rhodium(I) metallo-rectangle supported by two corannulene-bis-imidazolylidene ligands and two cofacial 4,4′-bipyridine ligands was prepared and characterized. This metallobox is capable of quantitatively encapsulating fullerenes C60 and C70, forming complexes that are highly stable even at high temperatures. The molecular structure of the metallobox with encapsulated C60 reveals a perfect size and shape complementarity that benefits from the concave–convex π–π interaction between the polyaromatic surfaces of the host and the guest.
Introduction
Shape and size complementarity are very likely the most important factors to be considered in the design of artificial molecular receptors. This rule becomes even more significant when it comes to the design of molecular receptors for fullerenes. Fullerenes are electron-deficient spherical extended π-systems containing carbon atoms with sp2 hybridization, so together with shape compatibility, electron complementarity must be considered for the design of hosts for effective fullerene recognition.1 The search for synthetic receptors for the encapsulation of fullerenes has attracted great attention because fullerenes have shown applications in fields ranging from materials science2 to pharmacy.3 Polycyclic aromatic hydrocarbons (PAHs) are often used as the core building blocks for the design of molecular hosts for fullerenes, because they can produce strong π–π interactions with these spherical all-carbon guests. Examples of PAHs used for the construction of cages and boxes for the recognition of fullerenes include porphyrin,4 pyrene,5 anthracene6 and electron-deficient groups such as naphthalene diimide and perylene bisimide.7 Inducing nonplanarity in PAHs significantly modifies their properties. Corannulene,8 which can now be prepared on the kilogram scale,9 is most likely the best binding unit to be incorporated in synthetic molecular receptors for fullerenes. This is because the bowl-shaped structure of corannulene makes it especially suitable for establishing concave–convex π–π interactions with fullerenes.10 Most of the examples that have been described so far of corannulene-based receptors for fullerene encapsulation are based on molecular tweezers,11 which normally show strong binding with C60 and C70 because they benefit from their double concave π-extended character. On the other hand, examples of corannulene-containing molecular boxes and cages are very scarce12 and, to the best of our knowledge, only one was used for fullerene encapsulation.12d This is most likely due to the lack of corannulene-based polytopic ligands that could be used for the construction of metallosupramolecular assemblies.We recently became interested in the preparation of supramolecular organometallic complexes (SOCs)13 based on the use of bis- and tris-N-heterocyclic carbene (NHC) ligands connected by polycyclic aromatic hydrocarbons (PAHs).14 Most of the SOCs that we prepared were based on the use of Janus-di-NHC ligands that had been previously prepared in our laboratory.15 Among the NHC-based SOCs that we described, those with larger cavity sizes were used for the encapsulation for fullerenes C60 and C70,16 and the large binding affinities that we observed were partly explained as a consequence of an excellent shape and size complementarity, with the former one being greatly enhanced by the great adaptability of the shape of the PAH-panels of the host to the curved surfaces of the fullerenes. In the course of our research in the preparation of PAH-connected Janus-di-NHC ligands that could be used for the preparation of metallosupramolecular assemblies, we also described a corannulene-connected di-NHC ligand, which allowed us to prepare a corannulene-based NHC di-Au(I) complex.17 This complex showed good affinity with fullerene C60 in toluene solution. Built on these grounds, and by using a similar corannulene-connected di-NHC ligand, we now report a di-Rh(I) complex, and a double-concave tetra-Rh(I) metallobox. These two assemblies were used as molecular hosts for C60 and C70 and, as will be described below, the large binding constants and the great kinetic stability obtained with the metallobox are explained due to the perfect shape and dimensional match between the host and the spherical guests.
Results and discussion
The synthetic procedure used for the preparation of the corannulene-connected bis-imidazolium salt [A](PF6)2 is described in detail in the ESI file,† and follows a similar route as the one that we previously described for a related bis-azolium salt with tert-butyl wingtips.17 The corannulene bis-NHC rhodium complexes 1 and 2 were obtained following the synthetic procedure depicted in Scheme 1. The reaction between [A](PF6)2 and [RhCl(COD)]2 in refluxing acetone in the presence of K2CO3 allowed the preparation of the di-rhodium complex 1 in 68% yield. The tetra-carbonylated complex 2, was obtained by bubbling carbon monoxide through a methylene chloride solution of 1 at 0 °C (yield 85%). Both complexes were characterized by multinuclear NMR spectroscopy and mass spectrometry, and gave satisfactory elemental analysis. | ||
| Scheme 1 Synthesis of corannulene-based bis-NHC Rh(I) complexes 1 and 2. | ||
The 1H and 13C NMR spectra of 1 and 2 are consistent with the predicted Cs symmetry of the complexes. Single resonances due to the metallated carbene carbons are observed on the 13C NMR spectra, showing the characteristic 1JRh–C couplings (1, 190.5 ppm, 1JRh–C = 52 Hz; 2, 185.14 ppm, 1JRh–C = 54 Hz). The IR spectrum of 2 was carried out in CH2Cl2 solution, and showed two C–O stretching bands at 2083 and 2003 cm−1. The average value of the two stretching frequencies [ν(CO)av] is 2043.0 cm−1, from which a TEP value of 2055.0 cm−1 can be derived, by employing the well-known correlations.18 This value is very close to the one displayed by the planar pyrene-connected-di-NHC ligand (TEP = 2054 cm−1),19 indicating a very similar degree of electron-donating character. The cyclic voltammetry diagram of the bis-Rh(I) complex 1 shows an irreversible redox event at E = 1.07 V (vs. SCE), which is attributed to the one electron oxidation of Rh(I) to Rh(II) (see ESI† for full details). The analysis of the differential pulse voltammetry (DPV) generated for 1 indicates that only one rhodium redox event occurs, thus indicating that the bi-metallic complex contains two essentially decoupled rhodium centres.
We then studied the binding affinity between complex 1 with fullerenes C60 and C70. Toluene-d8 was chosen as solvent for the titrations due to the optimal solubility observed for both host and guests. The titrations were performed at a constant concentration of 1 (1 mM). As an illustrative example, Fig. 1 shows the aromatic region of the 1H NMR spectra resulting from the titration of 1 with C60. As can be observed from the series of spectra, the incremental addition of C60 to the solution of 1, induced a maximum downfield shift of 0.17 ppm of the corannulene proton signal assigned to Ha. The signals due to Hb and Hc also experienced downfield shifts, although to a much lesser extent. The chemical shift changes observed are consistent with a binding process that shows fast kinetics on the NMR timescale. The nonlinear analysis of the results provided by this titration were best fitted to a 2:1 binding model, in which two molecular of the dirhodium complex bind one molecule of C60. The determination of the association constant was performed assuming a statistical (non-cooperative) model,20 and the least square fitting returned a binding constant of K11 = 1146 ± 66 M−1. The same experiment was performed for the determination of the binding affinity of 1 with fullerene C70, for which the binding affinity obtained was K11 = 1293 ± 160 M−1, also assuming a statistical 2:1 binding model.
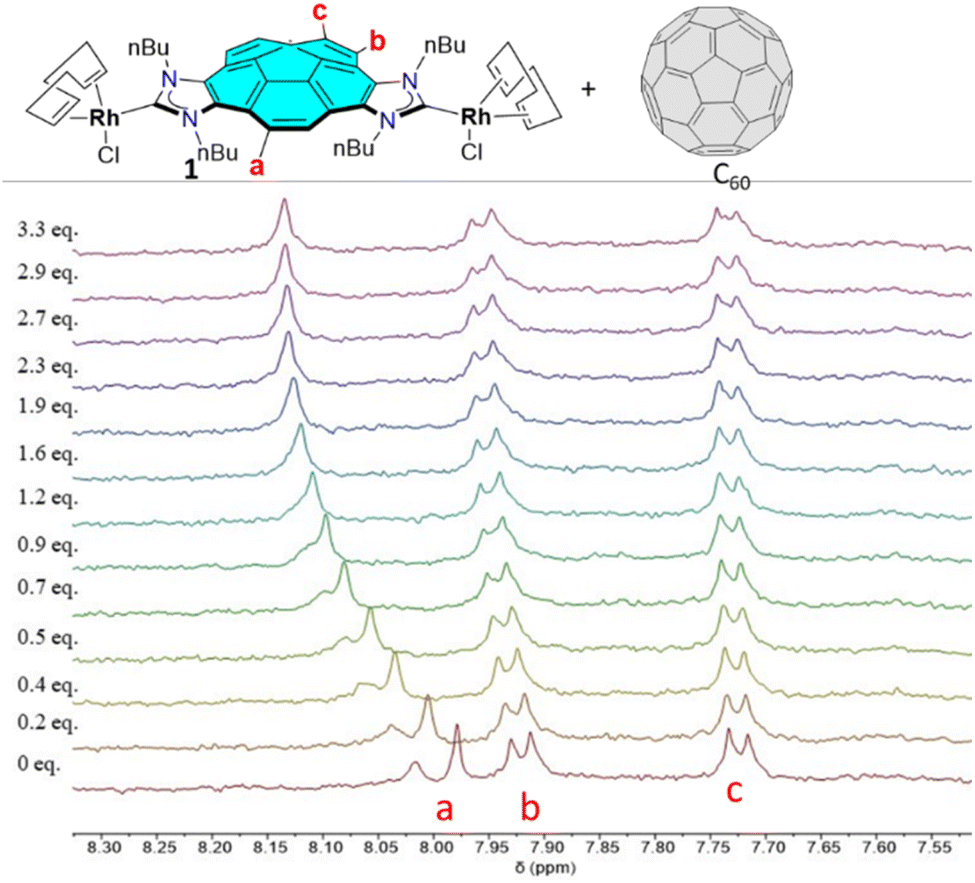 | ||
| Fig. 1 Aromatic region of the 1H NMR (500 MHz, toluene-d8, 298 K) spectra acquired during the titration of 1 with incremental amounts of fullerene C60. | ||
We also performed isothermal titration calorimetry (ITC) experiments in order to obtain additional information on the interaction of complex 1 with C60 and C70 (see Fig. S31 and S32 in ESI for full details†). Due to the reduced solubility of both fullerenes in toluene, the concentrations used were suboptimal considering the tentative binding affinities of about 1000 M−1, which we determined by 1H NMR titrations. The ITC titrations were performed by adding a toluene solution of the fullerene (2.7 mM) to a solution of 1 (0.6 mM) located in the cell. The experimentally obtained binding constant was fitted using the ‘independent’ binding model implemented in the instrument software. We obtained the best fit for a 2:1 binding model indicating the formation of a C60@12 type complex. The fit of the experimental data to the theoretical model allowed the determination of average value for the association constant, and the average binding enthalpy (ΔH) and entropy (ΔS) values. The average constant was (1.0 ± 0.5) × 103 M−1, thus in great agreement with the results obtained by 1H NMR titrations. The average ΔH value found was −1.7 ± 0.6 kcal mol−1, and ΔS was 8 cal mol−1 K−1. For the case of the ITC titrations of 3 with C70, we also obtained the best fit for a 2:1 binding model, and the least squares fit returned an average constant of 3565 M−1. The average ΔH value obtained was −0.5 ± 0.8 kcal mol−1, and ΔS was 14.5 cal mol−1 K−1. With these data in hand, we can conclude that the formation of the host–guest complex is entropically triggered, and that the positive entropy is due to desolvation of both, the host and the guest upon formation of C60@12.
We next decided to use 1 as a starting material for the formation of a double-concave tetra-Rh(I) metallobox. As shown in Scheme 2, the reaction of 1 with 4,4′-bipyridine in the presence of two equivalents of AgPF6 in CH2Cl2, leads to the corannulene-based metallobox 3 in 89% yield. Complex 3 was characterized by multinuclear NMR, UV-vis and fluorescence spectroscopy, and gave satisfactory elemental analysis. The Diffusion Ordered NMR spectrum (DOSY) of 3 shows that all proton resonances display the same diffusion coefficient in CD2Cl2 (7.04 × 10−10 m2 s−1) therefore indicating that this species forms a single assembly. The 1H NMR spectrum of the metallobox is consistent with the pseudo-C2v symmetry of the complex, which suggests that syn-conformation relative the two corannulene moieties. For example, the resonances due to the protons of the bipyridine ligands appear as four distinct doublets at 8.63, 8.52, 7.41 and 7.31 ppm, as a consequence of the presence of a mirror plane that bisects the molecule across the middle of the two opposite corannulene moieties, and another mirror plane that bisects the each of the two bipyridine linkers and lies perpendicular to the intraligand N,N axes. In the reaction shown in Scheme 2, another isomer of 3 could be potentially formed if the two corannulene moieties present on the metallobox were oriented in a relative anti-conformation, this is, with the two asymmetrically di-functionalized corannulene rings oriented in opposite directions, thus resulting in a C2-symmetry molecule. Given that only one assembly is formed, and considering the X-ray diffraction studies that we will explain below, we concluded that the reaction is selective in the formation of the syn-C2v-symmetry metallobox, which is the one depicted in Scheme 2.
 | ||
| Scheme 2 Synthesis of corannulene-based metallobox 3. | ||
The encapsulation of fullerenes C60 and C70 in 3 was performed in CD2Cl2 solution at room temperature. In both reactions, we observed that the gradual addition of the fullerene to the solution of the metallobox produced the gradual formation of the corresponding fullerene@3 host-guest complexes, as observed from the resulting 1H NMR spectra. As can be observed from the series of spectra shown in Fig. 2, addition of 0.5 equivalents of C60 to a solution of 3 results in the appearance of a new set of signals that are assigned to the formation of C60@3. The fact that both the signals due to 3 and C60@3 are simultaneously observed indicate that the equilibrium between 3 and C60@3 is slow on the NMR timescale. Subsequent addition of the 0.5 equivalents of C60 results in que quasi-quantitative formation of C60@3. As can be observed by comparing the 1H NMR spectra of between 3 and C60@3, it becomes evident that the signals due to the protons of the corannulene are slightly shifted upfield, and the resonances due to the four protons that are located on the same side of both corannulene moieties appear as a coupled AB system. On the other hand, the four doublets due to the pyridine protons in 3, are transformed into two doublets that are significantly shifted downfield in C60@3. A similar behavior can be observed for the sequence of 1H NMR spectra resulting from the encapsulation of C70 in 3 (see ESI† for details). The encapsulation of C60 and C70 within the cavity of 3 was also confirmed by 13C NMR spectroscopy. The 13C NMR of C60@3 (CD2Cl2) show the signal due to encapsulated C60 at 140.9 ppm. In the case of the 13C NMR spectrum of C70@3, four resonances at 148.7, 146.1, 145.3 and 143.3 ppm are observed due to the different carbon atoms of C70. For the determination of the association constants we took advantage that the exchange between the host and the host:guest complexes is slow on the NMR timescale, so that the equilibrium constants can be calculated by simply integrating the resonances associated to each of the species in solution at known initial concentrations of host and guests. The equilibrium constants obtained for the formation of C60@3 and C70@3 in CD2Cl2 were 8060 and 20000 M−1, respectively. Given the larger affinity of 3 for the encapsulation of C70, we also performed an experiment in which we dissolved the metallobox in CD2Cl2 with one equivalent of C60 and C70. Then the mixture was sonicated for 1 h. The NMR analysis of the solution indicated that only C70@3 was formed (see Fig. S35 in ESI†). While we envisioned that this encapsulating selectivity could eventually be used to purify C70 from mixtures of fullerenes, we did not find the right experimental conditions to release the molecule of C70 from the C70@3 complex without irreversibly dissembling the metallobox. The reversibility of the uptake/release process is the key point to accomplish the purification of fullerene molecules by using host–guest platforms, and this is the reason why the separation of mixtures of fullerenes remains a very challenging task.1a,21 To date, very few examples of supramolecular assemblies have shown to be effective in fullerene purification.4d,12d
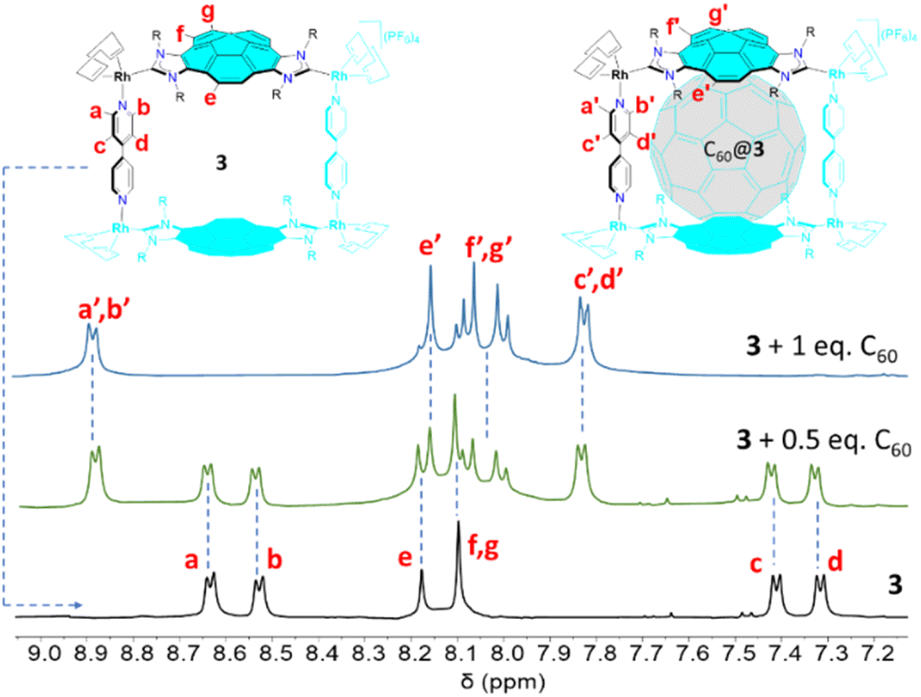 | ||
| Fig. 2 Partial section of the 1H NMR spectra (CD2Cl2, 500 MHz, 298 K) of the metallobox 3 (bottom), 3 + 0.5 equivalents of C60 (middle), and 3 + 1 equivalent of C60 (top). | ||
We also determined the binding constants of C60 with 3 in C2D2Cl4 at temperatures ranging from 30 to 90 °C, so that the thermodynamic parameters could be determined by using the corresponding Van't Hoff plots (see ESI† for details). We found that the equilibrium constant varied between 3150 and 13400 M−1, for the values determined at 30 and 90 °C, respectively. The thermodynamic values found were ΔH = 5.6 ± 0.5 kcal mol−1 and ΔS = 35 ± 2 cal mol−1 K−1. The positive value for the association enthalpy may be explained due to the solvation enthalpies of both C60 and 3 in C2D2Cl4. The large and positive entropy value is consistent with the desolvation of 3 and C60 upon formation of C60@3. As the room temperature binding constant between C70 and 3 is close to the upper limits for an accurate determination by 1H NMR spectroscopy,20b,22 we were unable to use this method for the determination of the thermodynamic parameters for the formation of C70@3.
Single crystals suitable for X-ray diffraction studies of C60@3 were obtained by slow diffusion of n-hexane into a concentrated solution of the complex in acetone (Fig. 3). The molecular structure consists of a tetra-rhodium(I) rectangle supported by two corannulene-bis-imidazolylidene ligands and two cofacial 4,4′-bipyridine ligands. The distances of the sides of the metallorectangle are 13.9 and 11.2 Å, measured as the Rh–Rh distances across the corannulene-bis-NHC and the bipyridine ligands, respectively. The relative configuration of the two corannulene-bis-NHC ligands is syn (their relative projections are superimposed), so that the symmetry of the molecule can be regarded as pseudo-C2v. The structure is bent, when observed from the top of the axle defined by the two centroids of the corannulene moieties (Fig. 3b), and the angle defined between the two Rh–Ccarbene coordination axes is of 138.2° (see simplified view of the molecule shown in Fig. 4a). As a consequence of the bent disposition of the corannulene-bis-NHC ligand, the centroid of the molecule of C60 is displaced from the plane defined by the four rhodium atoms by 1.6 Å (Fig. 4b). The average distance between the molecule of fullerene and the corannulene moiety is of 3.5 Å, calculated as the distance between the centroid of the central pentacyclic ring of the corannulene and the plane defined by six carbons of the closest hexacycle of the fullerene, therefore suggesting an optimum π–π-stacking interaction. The lateral view of the molecule (Fig. 3c), reveals that the two portals of the metallobox have different dimensions, as the disposition of the ‘non-cofacial’ corannulenes renders distances of 11.4 and 13.2 Å for the closest separation between the corannulene moieties on each side of the metallobox (Fig. 4b).
 | ||
| Fig. 3 (a)–(c) Show three perspectives of the molecular structure of C60@3, obtained from single crystal X-ray diffraction studies. Hydrogen atoms, omitted from the figure for clarity. Counter anions (PF6−) and solvent molecules (acetone), are also omitted. Carbon atoms are in blue, nitrogen atoms in red and rhodium atoms in yellow. | ||
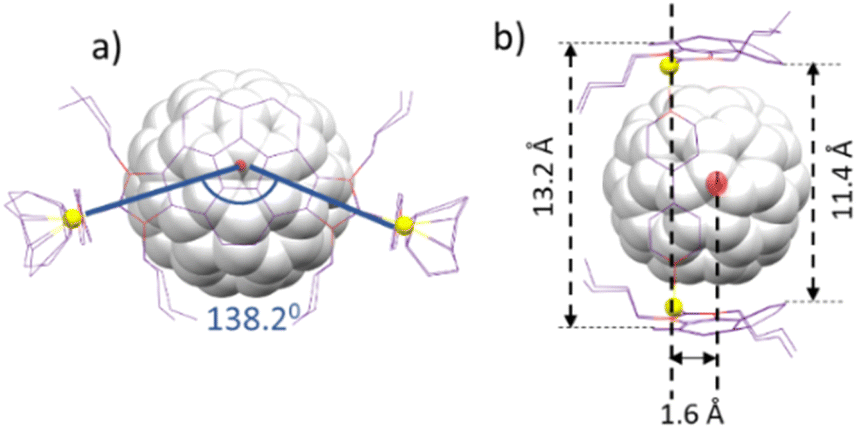 | ||
| Fig. 4 (a) and (b) Show two simplified representations of the molecular structure of C60@3, showing some of the metric parameters discussed in the text. | ||
Conclusion
In summary, by using a corannulene-bis-NHC ligand we prepared a di-rhodium(I) complex and a tetra-rhodium(I) metallobox. Both, the dimetallic complex 1 and the tetra-rhodium metallobox 3 were used for the encapsulation of fullerenes C60 and C70. The concave panels of the corannulene-based hosts enabled the receptors to bind fullerenes well, as the concave–convex interactions offer the best possible geometrical arrangement for an optimum host–guest contact. The corannulene-di-NHC complex 1 binds the fullerenes forming host–guest complexes of stoichiometry 1:2. On the other hand, metallobox 3 constitutes a very rare example of a corannulene-based metallosupramolecular assembly, with exceptional geometrical features that make it specially designed for fullerene encapsulation. To the best of our knowledge, complex 3 constitutes the first metallobox built with corannulene panels. The excellent shape and size complementarity between 3 and fullerenes C60 and C70 is reflected by the large binding constants obtained, and also by the high kinetic stability of the resulting host–guest complexes formed. The latter is explained by the fact that the free host and guests and the host–guest complexes show slow kinetics on the NMR timescale at temperatures as high as 80 °C, which is a good indication that the large free energy barrier (ΔG#) accompanying the complexation/decomplexation process is largely influenced by the complementarity of the entrance on the host and the size and shape of the guest. We think this study strongly suggests that our corannulene-based di-NHC ligand has the potential for extending the applications of metallosupramolecular assemblies with curved walls, and may help expanding the geometric diversity of artificial molecular cages. In addition, given the extraordinary applications of Janus-di-NHC ligands in catalysis and in materials chemistry, we envisage that the applicability of our new corannulene-di-NHC ligand can be extended well beyond the field of supramolecular chemistry.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
S. Ibáñez, C. Mejuto and K. Cerón prepared the complexes and perform the host–guest chemistry studies. P. Sanz did the crystallographic studies. S. Ibáñez and E. Peris designed and supervised the studies. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are very thankful to Professor Jay Siegel (University of Zurich/Tianjin University) for providing us the tetrabromo-corannulene compound, without which this work would not have been possible. We gratefully acknowledge financial support from the Ministerio de Ciencia y Universidades (PID2021-127862NB-I00 and PID2021-122406NB-I00), Generalitat Va-lenciana (CIPROM/2021/079) and Gobierno de Aragón (E42_23R). We are grateful to the Serveis Centrals d’Instrumentació Científica (SCIC-UJI) for providing with spec-troscopic facilities.Notes and references
- (a) C. Garcia-Simon, M. Costas and X. Ribas, Chem. Soc. Rev., 2016, 45, 40–62 RSC; (b) A. Sygula, Synlett, 2016, 27, 2070–2080 CrossRef CAS; (c) X. Chang, Y. Xu and M. von Delius, Chem. Soc. Rev., 2024, 53, 47–83 RSC.
- (a) M. Prato, J. Mater. Chem., 1997, 7, 1097–1109 RSC; (b) B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed; (c) G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS; (d) S. S. Babu, H. Möhwald and T. Nakanishi, Chem. Soc. Rev., 2010, 39, 4021–4035 RSC; (e) Y. Iwasa, Nature, 2010, 466, 191–192 CrossRef CAS PubMed; (f) V. Georgakilas, J. A. Perman, J. Tucek and R. Zboril, Chem. Rev., 2015, 115, 4744–4822 CrossRef CAS PubMed; (g) R. Ganesamoorthy, G. Sathiyan and P. Sakthivel, Sol. Energy Mater. Sol. Cells, 2017, 161, 102–148 CrossRef CAS; (h) J. H. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed; (i) A. Mateo-Alonso, G. Fioravanti, M. Marcaccio, F. Paolucci, D. C. Jagesar, A. M. Brouwer and M. Prato, Org. Lett., 2006, 8, 5173–5176 CrossRef CAS PubMed.
- (a) P. Mroz, A. Pawlak, M. Satti, H. Lee, T. Wharton, H. Gali, T. Sarna and M. R. Hamblin, Free Radical Biol. Med., 2007, 43, 711–719 CrossRef CAS; (b) Z. Hu, C. H. Zhang, Y. D. Huang, S. F. Sun, W. C. Guan and Y. H. Yao, Chem. Biol. Interact., 2012, 195, 86–94 CrossRef CAS PubMed; (c) C. Pesado-Gómez, J. S. Serrano-García, A. Amaya-Flórez, G. Pesado-Gómez, A. Soto-Contreras, D. Morales-Morales and R. Colorado-Peralta, Coord. Chem. Rev., 2024, 501, 215550 CrossRef; (d) F. Cataldo and T. D. Ros, Medicinal Chemistry and Pharmacological Potential of Fullerenes and Carbon Nanotubes, Springer, 2008 CrossRef; (e) S. Bosi, T. Da Ros, G. Spalluto and M. Prato, Eur. J. Med. Chem., 2003, 38, 913–923 CrossRef CAS PubMed; (f) Z. Markovic and V. Trajkovic, Biomaterials, 2008, 29, 3561–3573 CrossRef CAS PubMed; (g) R. Partha and J. L. Conyers, Int. J. Nanomed., 2009, 4, 261–275 CAS; (h) E. Castro, A. H. Garcia, G. Zavala and L. Echegoyen, J. Mater. Chem. B, 2017, 5, 6523–6535 RSC.
- (a) K. Tashiro and T. Aida, Chem. Soc. Rev., 2007, 36, 189–197 RSC; (b) C. S. Wood, C. Browne, D. M. Wood and J. R. Nitschke, ACS Cent. Sci., 2015, 1, 504–509 CrossRef CAS PubMed; (c) F. J. Rizzuto and J. R. Nitschke, Nat. Chem., 2017, 9, 903–908 CrossRef CAS PubMed; (d) C. Garcia-Simon, M. Garcia-Borras, L. Gomez, T. Parella, S. Osuna, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Nat. Commun., 2014, 5, 5557 CrossRef CAS; (e) W. J. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS; (f) W. Brenner, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 75–78 CrossRef CAS PubMed; (g) C. Fuertes-Espinosa, A. Gómez-Torres, R. Morales-Martínez, A. Rodríguez-Fortea, C. García-Simón, F. Gándara, I. Imaz, J. Juanhuix, D. Maspoch, J. M. Poblet, L. Echegoyen and X. Ribas, Angew. Chem., Int. Ed., 2018, 57, 11294–11299 CrossRef CAS PubMed; (h) F. J. Rizzuto, D. M. Wood, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 11008–11011 CrossRef CAS PubMed; (i) D. M. Wood, W. J. Meng, T. K. Ronson, A. R. Stefankiewicz, J. K. M. Sanders and J. R. Nitschke, Angew. Chem., Int. Ed., 2015, 54, 3988–3992 CrossRef CAS PubMed.
- (a) S. Bera, S. Das, M. Melle-Franco and A. Mateo-Alonso, Angew. Chem., Int. Ed., 2023, 62, e202216540 CrossRef CAS PubMed; (b) T. K. Ronson, A. B. League, L. Gagliardi, C. J. Cramer and J. R. Nitschke, J. Am. Chem. Soc., 2014, 136, 15615–15624 CrossRef CAS PubMed.
- (a) T. K. Ronson, B. S. Pilgrim and J. R. Nitschke, J. Am. Chem. Soc., 2016, 138, 10417–10420 CrossRef CAS PubMed; (b) N. Kishi, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2014, 53, 3604–3607 CrossRef CAS PubMed.
- (a) Q. H. Ling, J. L. Zhu, Y. Qin and L. Xu, Mater. Chem. Front., 2020, 4, 3176–3189 RSC; (b) X. M. Chang, S. M. Lin, G. Wang, C. D. Shang, Z. L. Wang, K. Q. Liu, Y. Fang and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 15950–15960 CrossRef CAS PubMed; (c) K. Mahata, P. D. Frischmann and F. Würthner, J. Am. Chem. Soc., 2013, 135, 15656–15661 CrossRef CAS.
- (a) Y. T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed; (b) E. Nestoros and M. C. Stuparu, Chem. Commun., 2018, 54, 6503–6519 RSC; (c) M. C. Stuparu, Acc. Chem. Res., 2021, 54, 2858–2870 CrossRef CAS PubMed.
- A. M. Butterfield, B. Gilomen and J. S. Siegel, Org. Process Res. Dev., 2012, 16, 664–676 CrossRef CAS.
- (a) L. N. Dawe, T. A. AlHujran, H. A. Tran, J. I. Mercer, E. A. Jackson, L. T. Scott and P. E. Georghiou, Chem. Commun., 2012, 48, 5563–5565 RSC; (b) E. S. Larsen, G. Ahumada, P. R. Sultane and C. W. Bielawski, Chem. Commun., 2022, 58, 6498–6501 RSC.
- (a) V. H. Le, M. Yanney, M. McGuire, A. Sygula and E. A. Lewis, J. Phys. Chem. B, 2014, 118, 11956–11964 CrossRef CAS PubMed; (b) C. Muck-Lichtenfeld, S. Grimme, L. Kobryn and A. Sygula, Phys. Chem. Chem. Phys., 2010, 12, 7091–7097 RSC; (c) A. Sygula, F. R. Fronczek, R. Sygula, P. W. Rabideau and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 3842–3843 CrossRef CAS PubMed; (d) C. M. Alvarez, L. A. Garcia-Escudero, R. Garcia-Rodriguez, J. M. Martin-Alvarez, D. Miguel and V. M. Rayon, Dalton Trans., 2014, 43, 15693–15696 RSC; (e) M. Yanney, F. R. Fronczek and A. Sygula, Angew. Chem., Int. Ed., 2015, 54, 11153–11156 CrossRef CAS; (f) M. Yanney and A. Sygula, Tetrahedron Lett., 2013, 54, 2604–2607 CrossRef CAS; (g) A. Sygula, M. Yanney, W. P. Henry, F. R. Fronczek, A. V. Zabula and M. A. Petrukhina, Cryst. Growth Des., 2014, 14, 2633–2639 CrossRef CAS; (h) C. M. Alvarez, G. Aullon, H. Barbero, L. A. Garcia-Escudero, C. Martinez-Perez, J. M. Martin-Alvarez and D. Miguel, Org. Lett., 2015, 17, 2578–2581 CrossRef CAS; (i) A. Sacristán-Martín, D. Miguel, A. Diez-Varga, H. Barbero and C. M. Alvarez, J. Org. Chem., 2022, 87, 16691–16706 CrossRef PubMed.
- (a) C. Y. Shao, Y. N. Zhao, S. K. Han, F. Huang, W. J. Guo, H. Jiang and Y. Wang, Org. Chem. Front., 2023, 10, 1412–1422 RSC; (b) F. Huang, L. S. Ma, Y. K. Che, H. Jiang, X. B. Chen and Y. Wang, J. Org. Chem., 2018, 83, 733–739 CrossRef CAS PubMed; (c) T. K. Ronson, Y. Wang, K. Baldridge, J. S. Siegel and J. R. Nitschke, J. Am. Chem. Soc., 2020, 142, 10267–10272 CrossRef CAS PubMed; (d) W. Sun, Y. Wang, L. Ma, L. Zheng, W. Fang, X. Chen and H. Jiang, J. Org. Chem., 2018, 83, 14667–14675 CrossRef CAS PubMed.
- A. Pöthig and A. Casini, Theranostics, 2019, 9, 3150–3169 CrossRef PubMed.
- S. Ibáñez, M. Poyatos and E. Peris, Acc. Chem. Res., 2020, 53, 1401–1413 CrossRef PubMed.
- M. Poyatos and E. Peris, Dalton Trans., 2021, 50, 12748–12763 RSC.
- (a) V. Martinez-Agramunt and E. Peris, Inorg. Chem., 2019, 58, 11836–11842 CrossRef CAS PubMed; (b) V. Martinez-Agramunt, T. Eder, H. Darmandeh, G. Guisado-Barrios and E. Peris, Angew. Chem., Int. Ed., 2019, 58, 5682–5686 CrossRef CAS PubMed; (c) V. Martinez-Agramunt, D. G. Gusev and E. Peris, Chem.–Eur. J., 2018, 24, 14802–14807 CrossRef CAS.
- C. Mejuto, L. Escobar, G. Guisado-Barrios, P. Ballester, D. Gusev and E. Peris, Chem.–Eur. J., 2017, 23, 10644–10651 CrossRef CAS PubMed.
- (a) D. L. Nelson and I. P. Nolan, Chem. Soc. Rev., 2013, 6723–6753 RSC; (b) A. R. Chianese, X. W. Li, M. C. Janzen, J. W. Faller and R. H. Crabtree, Organometallics, 2003, 22, 1663–1667 CrossRef CAS; (c) R. A. Kelly III, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202–210 CrossRef.
- S. Gonell, M. Poyatos and E. Peris, Chem.–Eur. J., 2014, 20, 9716–9724 CrossRef CAS PubMed.
- (a) L. K. S. von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637 RSC; (b) A. J. Lowe, F. M. Pfeffer and P. Thordarson, Supramol. Chem., 2012, 24, 585–594 CrossRef CAS.
- C. Fuertes-Espinosa, C. Garcia-Simon, M. Pujals, M. Garcia-Borras, L. Gomez, T. Parella, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Chem, 2020, 6, 169–186 Search PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis and characterization of new compounds, calculation of binding affinities, X-ray diffraction details. CCDC 2352467. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03661a |
| This journal is © The Royal Society of Chemistry 2024 |