
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMore is different: progressive β-thiolation induced-porphyrin aggregation switches singlet oxygen photosensitization†
Mengliang
Zhu‡
ab,
Hang
Zhang‡
a,
Yuhang
Yao‡
a,
Mingpu
Wen
c,
Guangliu
Ran
d,
Yi
Yu
a,
Ruijing
Zhang
a,
Xing-Jie
Liang
 be,
Jing
Zhang
c,
Wenkai
Zhang
*d and
Jun-Long
Zhang
*a
be,
Jing
Zhang
c,
Wenkai
Zhang
*d and
Jun-Long
Zhang
*a
aBeijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China. E-mail: zhangjunlong@pku.edu.cn
bCAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology of China, Beijing 100190, P. R. China
cCollege of Materials Science and Opto-Electronic Technology, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
dCenter for Advanced Quantum Studies, Department of Physics and Applied Optics Beijing Area Major Laboratory, Beijing Normal University, Beijing 100875, P. R. China. E-mail: wkzhang@bnu.edu.cn
eUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 31st July 2024
Abstract
Incorporating sulfur atoms into photosensitizers (PSs) has been well-established to populate triplet states and increase singlet oxygen (1O2) production when exposed to light. In this work, we found that progressive thiolation of porphyrin β-periphery does promote intersystem crossing (ISC) between triplets and singlets, as seen in the excited state dynamics in dichloromethane or PS nanoparticles in water. However, in the latter case, more sulfur substitution deactivates 1O2 photosensitization, in contrast to the expected trend observed in dichloromethane. This observation was further supported by photocytotoxicity studies, where 1O2 photosensitization was switched off in living cells and multicellular spheroids despite being switched on in in vivo mice models. To understand the inconsistency, we performed molecular dynamics simulation and time-dependent density functional theory calculations to investigate possible aggregation and related excited states. We found that the extent of thiolation could regulate molecular packing inside nanoparticles, which gradually lowers the energy levels of triplet states even lower than that of 1O2 and, in turn, alters their energy dissipation pathways. Therefore, this study provides new insights into the design of metal-free PSs and sheds light on the excited state dynamics in aqueous media beyond the molecular level.
Introduction
Photodynamic therapy (PDT), which relies on oxygen activation upon light irradiation, has been an emerging treatment for tumors and dermatological conditions due to its inherent advantages such as high selectivity, non-invasiveness, and therapeutic efficacy.1–4 Typically, excitation of the photosensitizer (PS) leads to a singlet excited state which is subsequently converted to the triplet state through intersystem crossing (ISC), and it finally reacts with the surrounding molecular oxygen (3O2) to produce singlet oxygen (1O2).5–8 Thus, the excited state dynamics of PS is critical in determining oxidative events, cytotoxicity, and even death.9–11 Molecular engineering of PSs to modulate the excited state dynamics has been established; for example, the efficiency of the ISC process can be enhanced by introducing the heavy-atom effect,12,13 reducing the energy gap (ΔEST) between the lowest singlet (S1) and the lowest triplet (T1),14,15 or increasing the rigidity of PSs,16,17 according to Fermi's Golden rule.18 On the other hand, increasing attention has recently been devoted to molecular self-assembly or aggregate formation, demonstrating distinct optoelectronic, chemical, and biological properties from single molecules.19–22 These studies open an opportunity, namely “more is different”, to guide the molecular engineering of PSs for biomedicine purposes.23–26Introducing sulfur atoms into the backbone of PSs is promising to enhance ISC and populate the triplet states, probably circumventing the heavy metal toxicity.27 Yoon, Xiao, Lu, and others have demonstrated that thiocarbonyl PSs are readily obtained by the reaction of Lawesson's reagent with carbonyl PS analogs.28–31 Theoretical calculations suggested that sulfur substitution significantly promotes the spin–orbit coupling (SOC), narrows ΔEST between singlet and triplet states, and thus enhances the ISC rate.32,33 Despite the effectiveness of sulfur substitution, it has been observed that such PSs tend to aggregate in aqueous media,34 and whether and how this aggregation affects the excited state dynamics and 1O2 photosensitization has yet to be investigated.35–37
Porpholactones and their derivatives, in which one or two oxazolone moieties replace the pyrrole units in porphyrin, feature intriguing photophysical properties such as chlorophyll-like strong absorption at the near-infrared (NIR) region, and synthetic flexibility arising from the oxazolone moiety.38–40 Previously, we used Lawesson's reagent to prepare the porphothiolactone that shows a bathochromic shift of Q bands and remarkable fluorescence quenching, suggesting an enhanced ISC rate and narrowed HOMO–LUMO gap.41,42 In this work, we prepared a series of porphodilactones to investigate the effects of the extent of thiolation and regioisomerism on the molecular packing in crystals and photophysical properties. We then used DSPE-mPEG2000 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)2000])43 to prepare the water-soluble nanoparticles, allowing for the examination of the sulfur substitution-induced aggregation and photophysical properties in aqueous media. Notably, more sulfur substitution can switch off 1O2 photosensitization in aqueous media, in contrast to the expected trend observed in organic solvents. To better understand the excited state dynamics, we conducted transient absorption spectroscopic studies to investigate their excited state dynamics, along with the density functional theory (DFT) calculations and molecular dynamics (MD) simulations. We found that progressive β-thiolation indeed promotes the ISC processes and populating triplets in both CH2Cl2 and aqueous media. However, this is not sufficient to interpret the “switched-off” 1O2 photosensitization for more sulfur substitution. We then simulated sulfur substitution-induced aggregation, and importantly, the extent of thiolation significantly affects the energy levels of the aggregate models and 1O2 photosensitization, leading to different energy dissipation pathways (Scheme 1). Finally, we performed the PDT treatment in cells, 3D multicellular spheres, and in vivo mice models to demonstrate the photocytotoxicity of porphyrin PSs. Interestingly, lower photocytotoxicity was obtained in living cells and multicellular spheroids for more sulfur-substituted porphyrins, consistent with the switched-off 1O2 photosensitization in aqueous media. Therefore, these findings can provide broader guidance for evaluating innovative PSs from a single molecule and the possible molecular aggregates under physiological conditions, which may occur in preclinical and clinical studies for actual PDT treatments.
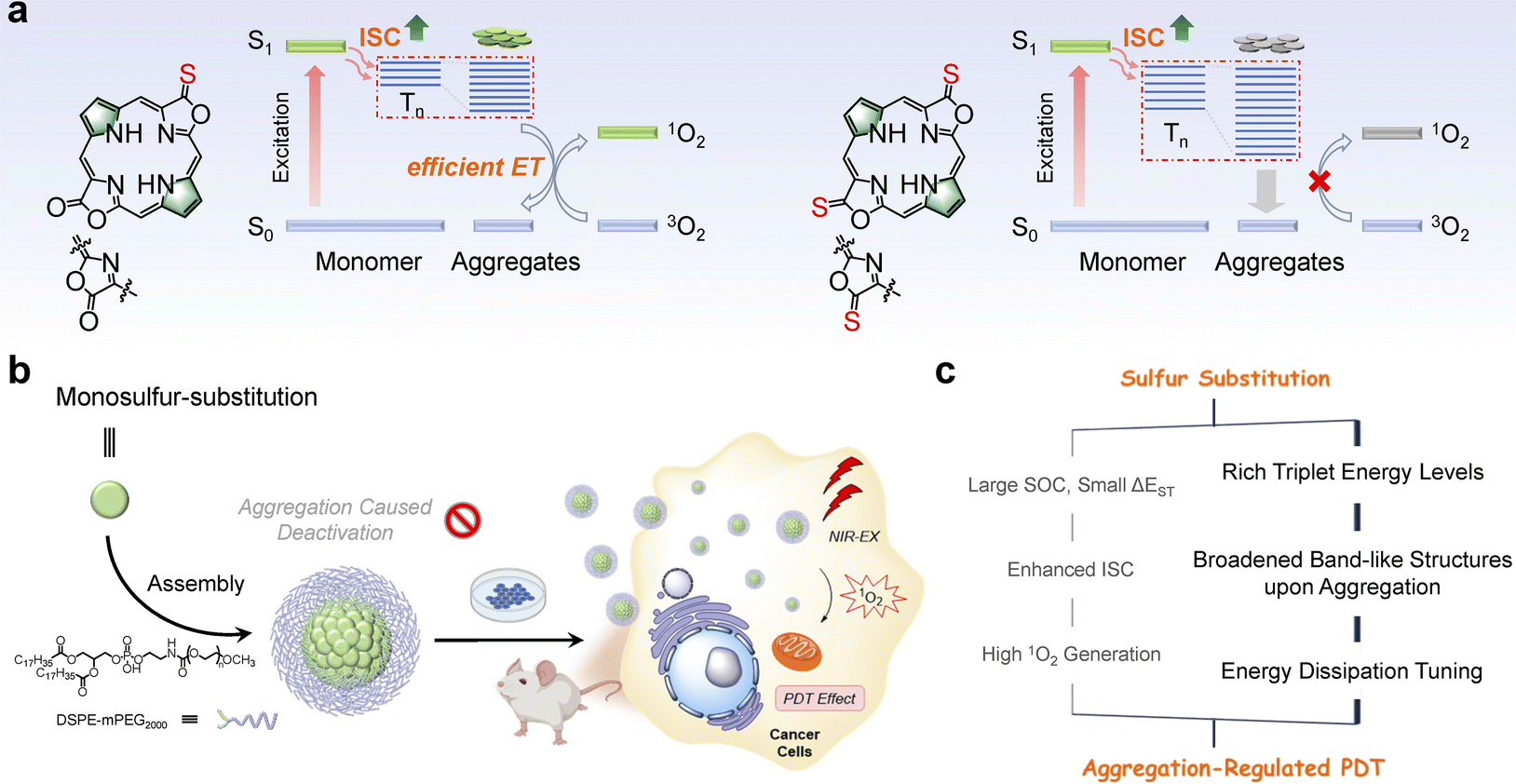 | ||
| Scheme 1 (a) Molecular structure of cis-O'S and cis-S'S and the excited state energy dissipation pathways in their aggregated states. (b) Schematic illustration of the cis-O'S self-assembly into nanospheres and subsequent PDT effect. (c) The proposed sulfur substitution strategy to obtain aggregation-regulated PDT effect by tuning excited state dynamics. | ||
Results and discussion
Synthesis, characterization, and photophysical properties
Lawesson's reagent is a classic thiolation agent that can replace oxygen atoms in the carbonyl group with sulfur atoms.44meso-C6F5-substituted porphodilactones (a mixture of cis- and trans-isomers) were mixed with Lawesson's reagent (20 equiv.) in toluene under reflux for 3 days to afford cis/trans-porphothiodilactone isomers with moderate yields of 20 and 15%, respectively (Fig. 1a). In a one-pot synthesis, the products with different extents of thiolation and different orientations of β-substituents (termed cis-O'S, cis-S'S, trans-O'S, and trans-S'S) at the opposite pyrrole positions could be isolated by silica gel chromatography. These new compounds were fully characterized by 1H and 19F NMR (nuclear magnetic resonance spectroscopy, Fig. S1–S4†), HR-MS (high-resolution mass spectrometry, Fig. S5†), and FT-IR (Fourier transform infrared spectroscopy, Fig. S6†). Single crystals of cis-O'S (CCDC: 2261655), cis-S'S (CCDC: 2261661), trans-O'S (CCDC: 2261656), and trans-S'S (CCDC: 2261662) were obtained by diffusing methanol into CHCl3 solution, respectively. Table S1† summarizes crystal structure data collection and refinement statistics. Fig. 1b shows the differing arrangements of lactone moieties in cis- and trans-porphothiodilactones. The bond length of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S in porphothiodilactones is approximately 1.616–1.651 Å, which is close to the CS bond length (1.665 Å) in porphothiolactone,42 but longer than that of CO bonds (1.214–1.261 Å) due to the smaller electronegativity of S.
S in porphothiodilactones is approximately 1.616–1.651 Å, which is close to the CS bond length (1.665 Å) in porphothiolactone,42 but longer than that of CO bonds (1.214–1.261 Å) due to the smaller electronegativity of S.
 | ||
| Fig. 1 (a) One-pot synthesis of porphothiodilactone isomers. (b) X-ray crystal structures of cis-/trans-O'S, and cis-/trans-S'S. Hydrogen atoms and solvents are omitted for clarity. (c) Absorption spectra of cis-/trans-O'O, cis-/trans-O'S, and cis-/trans-S'S in CH2Cl2. (d) Emission spectra of cis-/trans-O'O, cis-/trans-O'S, and cis-/trans-S'S in CH2Cl2 with excitation at 405 nm (A405 = 0.10). (e) Phosphorescence spectra of 1O2 obtained from air-saturated CH2Cl2 solutions of cis-/trans-O'O, cis-/trans-O'S, and cis-/trans-S'S. | ||
The introduction of sulfur atoms causes local distortion of the N4 macrocycle and affects the intermolecular packing modes. Cis- and trans-O'S crystallize in monoclinic cells with P21/c and P21/n space groups, respectively. Meanwhile, cis- and trans-S'S crystallize in tetragonal cells (I4/m space group) and orthorhombic cells (Pbca space group), respectively (Fig. S7†). Cis-O'S adopts mixed packing modes with two different orientations with a dihedral angle (φ) of 51.5°. The vertical distance between two π-stacked molecules is 5.250 Å with a slip angle (θ) of 29.8°. Cis-S'S is arranged in an edge-to-face manner (φ = 90.0°) with a distance of 3.065 Å between S atom and π plane and a vertical distance of 8.760 Å between two parallel molecules with a slip angle (θ) of 42.7°. The two packing modes in trans-O'S are also almost perpendicular (φ = 84.2°), and the distance between two π-stacked molecules is 5.336 Å (θ = 51.6°). There are four orientations of packing in the cell of trans-S'S (φ = 28.3°, 56.5°, 64.8°), and the distance between two π-stacked molecules is 4.192 Å (θ = 16.5°). Overall, the slip angles of π-stacked molecules are less than 54.7°, indicating J-aggregation.45 Besides, monosulfur substitution (cis- and trans-O'S) leads to a relatively loose packing (ρ = 1.535 or 1.645 g cm−3), and disulfur substitution (cis- and trans-S'S) leads to a relatively tight packing (ρ = 1.652 or 1.729 g cm−3) with more molecules and smaller spacing in cells. These results show that the replacement of O with S atoms influences the crystal structure and the aggregation of molecules.
The UV-vis absorption and emission spectra of cis/trans-porphothiodilactone derivatives were recorded in CH2Cl2 at room temperature (Fig. 1c and d). Photophysical data from this study are summarized in Table S2.† β-Thiolation of porphodilactones resulted in a significant red-shift of Soret bands compared to porphodilactones without S substitution. As the extent of thiolation increased, a stepwise red-shift in the lower energy Q band was observed. For cis-isomers, it started from 656 nm (cis-O'O), then shifted to 692 nm (cis-O'S), and finally reached 720 nm (cis-S'S). For trans-isomers, S substitution leads to a red-shift in Q bands, from 676 nm (trans-O'O) to 708 nm (trans-O'S) and 734 nm (trans-S'S), respectively. The cyclic voltammograms (CVs) showed a progressive decrease in HOMO–LUMO gap values as the extent of thiolation increased (1.89 eV for cis-O'O, 1.74 eV for cis-O'S, 1.58 eV for cis-S'S; 1.78 eV for trans-O'O, 1.59 eV for trans-O'S, 1.47 eV for trans-S'S, Table S3†). This suggested that S substitution is an effective strategy for remodeling energy levels of the frontier molecular orbitals and inducing NIR absorption (Fig. S8†).46,47
Regarding fluorescence, cis-/trans-O'S and cis-/trans-S'S, demonstrate weak fluorescence with minimal quantum yields (Φems < 0.1%), in contrast to the strong emission of porphodilactones (cis-/trans-O'O) in CH2Cl2 (Φems ∼15%).48 Then, we measured the 1O2 characteristic emission at ca. 1275 nm in air-saturated CH2Cl2 solution, as depicted in Fig. 1e. On measuring the 1O2 characteristic emission at ca. 1275 nm in air-saturated CH2Cl2 solution, the 1O2 quantum yields (ΦΔs) of 79–82% were measured (Table S2†) upon photoexcitation of cis-/trans-O'S and cis-/trans-S'S, much higher than those obtained by porphodilactones (cis-/trans-O'O, ΦΔ = 53–60%).41 Thus, the quenched fluorescence accompanied by the enhanced ability to 1O2 photosensitization demonstrates that β-thiolation enhances the ISC process.49 Additionally, regarding 1O2 generation, a further increase of sulfur substitution from 1 to 2 and the relative orientation of β-substituents have trivial effects.
Photophysical properties in aqueous media
To explore the phototherapeutic potential of porphothiodilactones, cis-/trans-O'O, cis-/trans-O'S, and cis-/trans-S'S were prepared into nanoparticle (NP) formulations (cis-O'O@NPs, cis-O'S@NPs, cis-S'S@NPs, trans-O'O@NPs, trans-O'S@NPs, and trans-S'S@NPs) using DSPE-mPEG2000 as the biocompatible matrix (Fig. 2a). Transmission electron microscopy images show that there are spherical NPs present with diameters of approximately 10 nm (Fig. S9†), regardless of S substitutions or isomerization. Dynamic light scattering (DLS) analysis of porphothiodilactone NPs in water shows the average particle sizes with hydrodynamic diameters of ca. 13 nm (Fig. S10†). | ||
| Fig. 2 (a) Preparation of porphodilactone nanoparticles. (b) Absorption spectra of cis-O'O@NPs, cis-O'S@NPs, cis-S'S@NPs, trans-O'O@NPs, trans-O'S@NPs, and trans-S'S@NPs in aqueous solution. (c) Variation of SOSG emission intensity at 525 nm (λex = 488 nm) under light irradiation (700 nm, 5 mW cm−2, 0–30 min) in the presence of cis-O'O@NPs, cis-O'S@NPs, cis-S'S@NPs, and MB. (d) Variation of SOSG emission intensity at 525 nm (λex = 488 nm) under light irradiation (700 nm, 5 mW cm−2, 0–30 min) in the presence of trans-O'O@NPs, trans-O'S@NPs, trans-S'S@NPs, and MB. (e) Comparison of 1O2 quantum yields between porphodilactone molecules and nanoparticles. | ||
Then, the aggregation behaviors of PSs inside NPs were analyzed according to their absorption in water. As shown in Fig. 2b and S11,†cis-O'O@NPs, and cis-O'S@NPs exhibited broadened absorption without an apparent band shift, whereas cis-S'S@NPs showed clear red-shifted Q-bands (from 550 to 800 nm) compared to the molecular form. Such a shift is a characteristic absorption due to J-aggregation, leading to lower energy. Similarly, in the case of trans-isomers, trans-O'O@NPs, and trans-O'S@NPs demonstrated blue-shifted Q-bands or Q-band splitting in the 600–700 nm region, indicating the formation of H-aggregates. Interestingly, trans-S'S@NPs, just like cis-S'S@NPs, showed red-shifted Q-bands in nanoparticles. Thus, we found that, along with the increase in the extent of β-thiolation, porphothiodilactones may tend to form J-aggregates inside NPs.50
The trend of porphothiodilactone aggregation was also confirmed by the shifts of their characteristic absorption bands in tetrahydrofuran (THF)/water mixtures with varying ratios (v/v), as shown in Fig. S12.† As the water fraction increases, the absorption spectra of porphothiodilactones gradually align with those of nanoparticles. Additionally, the particles formed in water are significantly larger (hydrodynamic diameters of 160–350 nm) than those coated with DSPE-mPEG2000 (Fig. S13†), indicating that porphothiodilactones can aggregate without PEG protection. These results suggest that the encapsulation using the amphiphilic phospholipid polymer promotes and stabilizes the aggregation of the photosensitizers in small sizes.
Porphothiodilactone NPs as 1O2 photosensitizers were evaluated by employing the commercially available singlet oxygen sensor green (SOSG) as an indicator (Fig. 2c and d). Upon irradiation with the laser (700 nm, 5 mW cm−2), the strong emission of SOSG at 525 nm appeared in the presence of cis-O'O@NPs, trans-O'O@NPs, cis-O'S@NPs, or trans-O'S@NPs, indicating their remarkable capability towards 1O2 production (Fig. S14†). As expected, by using methylene blue (MB) as a reference, the 1O2 quantum yields of ca. 80% for cis-O'S@NPs and trans-O'S@NPs were obtained (Fig. 2e and Table S4†), much higher than those of cis-O'O@NPs (48%) and trans-O'O@NPs (62%).
However, the irradiation of cis-S'S@NPs or trans-S'S@NPs led to small spectral changes of SOSG with sharply declined 1O2 quantum yields of ca. 20%, suggesting much lower efficiency in sensitizing O2 compared to those in CH2Cl2 solution. It is worth noting that long-time (30 min) irradiation (700 nm, 5 mW cm−2) of all porphothiodilactone NPs does not lead to apparent changes in absorption (<5%, Fig. S15†), ruling out photostability as a cause of lower 1O2 photosensitization. Based on morphological studies and absorption in aqueous solution, we assumed that aggregation of PSs inside DSPE nanoparticles plays a critical role in tuning the excited states and, in turn, reacting with O2.
The photothermal effects of cis-O'S@NPs, cis-S'S@NPs, trans-O'S@NPs, and trans-S'S@NPs were investigated under laser irradiation (700 nm, 1 W cm−2). The temperature increased by ca. 15 °C, with the photothermal conversion efficiencies (η) being 18–26% (Fig. S16 and Table S4†), indicating the modest photothermal performance of porphothiodilactone NPs.
Excited state dynamics
To investigate the dynamics of triplet states for porphodilactone derivatives, we utilized nanosecond (ns) transient absorption (TA) spectroscopy in CH2Cl2. Our focus was on cis-isomers, as the regioisomerism has a negligible effect on the aggregation of these compounds. After photoexcitation at 355 nm, we observed excited state absorption (ESA) bands in the 450–650 nm spectral range which are attributed to T1 → Tn transitions in cis-O'O, along with ground state bleaching (GSB) centered at 405 and 652 nm (Fig. S17†). Notably, ns-TAs of cis-O'S and cis-S'S showed more ESA bands within the wavelength range of 450–700 nm. Furthermore, we determined the decay rates of the triplet states under degassed conditions to be 11 μs for cis-O'S and cis-S'S, representing a significant decrease compared to cis-O'O porphodilactone (39 μs, Fig. S18†).We then conducted femtosecond (fs) transient absorption studies to compare the excited state dynamics of cis-O'O, cis-O'S, cis-S'S in CH2Cl2 and cis-O'O@NPs, cis-O'S@NPs, cis-S'S@NPs in water. As shown in Fig. 3a, for cis-O'O in CH2Cl2 and cis-O'O@NPs in water, the initial TA spectra probe at 0.5 ps showed identical features as observed by excited state absorption (ESA, 500–650 nm) and ground state bleaching (GSB, 650–800 nm), indicating that the absorption feature stems from the localized excited (1LE) state. The fs-TA spectra decayed in nanoseconds, corresponding to the fluorescence of cis-O'O in CH2Cl2 and nonradiative relaxation process of cis-O'O@NPs in water,51 respectively, and the long-lived state was identified as the triplet state (3LE) by ns-TA spectroscopy.
 | ||
| Fig. 3 (a) Femtosecond transient absorption (fs-TA) difference spectra for cis-O'O recorded in CH2Cl2 (0.5–1 ps, 1–30 ps, and 50–3600 ps) and cis-O'O@NPs recorded in H2O (0.2–5 ps, 5–30 ps, and 50–3600 ps) monitored over different delay regimes. (b) Femtosecond transient absorption (fs-TA) difference spectra for cis-O'S recorded in CH2Cl2 (0.3–2 ps, 2–200 ps, and 200–3600 ps) and cis-O'S@NPs recorded in H2O (0.2–2 ps, 2–200 ps, and 200–3600 ps) monitored over different delay regimes. (c) Femtosecond transient absorption (fs-TA) difference spectra for cis-S'S recorded in CH2Cl2 (1–5 ps, 5–200 ps, and 200–3600 ps) and cis-S'S@NPs recorded in H2O (0.2–2 ps, 2–200 ps, and 200–3600 ps) monitored over different delay regimes. | ||
For cis-O'S in CH2Cl2 and cis-O'S@NPs in water, the initial TA spectra (∼0.3 ps) also feature the 1LE character, where a major ESA band and a major GSB band probe were centered at 650 and 700 nm, respectively. Within a few seconds (<2 ps), the singlet state efficiently forms, and the ISC process from the singlet to triplet states dominates during a period of 2–200 ps, accompanied by a pronounced increase centered on 650 nm in the ESA (Fig. 3b). A global analysis of the TA spectra unambiguously reveals that excited states under different conditions share similar properties, as indicated by the corresponding kinetic rate constants (k) of 3.7 × 1010 s−1 and 3.4 × 1010 s−1 obtained for cis-O'S in CH2Cl2 and cis-O'S@NPs in water (Table S5†), respectively. Over the following 200–3600 ps, the ESA and GSB bands exhibit slow decay to the ground state, consistent with the long lifetime obtained in ns-TA spectroscopy. Similarly, cis-S'S in CH2Cl2 and cis-S'S@NPs in water exhibited comparable kinetics in the excited states (k = 3.9 × 1010 s−1 and 4.1 × 1010 s−1, respectively), as shown in Fig. 3c. During 200–3600 ps, the features of ESA and GSB of photoexcited cis-S'S in CH2Cl2 exhibit a much faster decay (k = 1.8 × 108 s−1) towards the ground state than that seen in cis-S'S@NPs in water (unable to fit, Table S5†), probably due to slower relaxation of triplet states arising from aggregation of PSs in NPs.
Replacing S atom leads to a clear ISC process from the S1 to the T1 state, which is not present for cis-O'O in CH2Cl2 or water, making triplet generation more efficient. While increasing the S atom replacement from 1 to 2 does not significantly enhance the triplet state population, it does extend the lifetime in the nanoparticle form. We assume that the tendency for aggregation, as seen in the absorption and crystalline structure, is responsible for slowing the relaxation of triplet states. Interestingly, both cis-O'S@NPs and cis-S'S@NPs in water efficiently populate triplet states when exposed to light; however, only cis-O'S@NPs can effectively photosensitize 1O2 in a high quantum yield. Therefore, more than the current studies on excited state dynamics may be required to fully understand the deactivation of 1O2 production in cis-S'S@NPs in water.
Theoretical and computational studies
To gain insights into the triplet states of cis-O'S and cis-S'S isomers and their aggregates, we performed time-dependent density functional theory (TD-DFT) calculations to optimize the structures of porphodilactones and porphothiodilactones based on their crystal structures.52 We observed the impact of sulfur substitution on excited state features for cis- and trans-isomers, and SOC constants after thiolation exhibit a significant increase (Tables S6 and S7,†e.g. 1.10 cm−1 for cis-O'O, 11.9 cm−1 for cis-O'S, and 37.1 cm−1 for cis-S'S). The enhancement of SOC promotes the ISC process with increasing rate constants (kISC) from 5.03 × 106 s−1 for cis-O'S, to 4.63 × 108 s−1 and 8.75 × 108 s−1 for cis-O'S and cis-S'S, respectively. These calculated results are consistent with the experimental observation of fluorescence quenching and the enhanced 1O2 production after sulfur atom replacement of porphodilactones. Notably, sulfur atom replacement stabilizes the LUMO level and destabilizes the HOMO, thus reducing the energy gap and shifting the absorption to a long wavelength. This also leads to a strong lowering of the energies of the triplet states, resulting in the emergence of new ISC channels between the singlet and triplet excited states, as illustrated in Fig. S19–S20.† Importantly, this highlights the crucial role of sulfur substitution in promoting the population of the triplet states.To further investigate the effect of aggregation inside NPs on the population of triplet states, a molecular dynamics (MD) simulation was performed which provided insights into the distribution of conformational structures with short-range (<10 Å) ordering over a span of 200 ns.53,54 As illustrated in Fig. 4a, the molecules confined within a water box (50 × 50 × 50 Å) exhibit an aggregation tendency due to hydrophobicity and π–π interaction, resulting in distinct dimer and trimer structures with an inclination observed within the clusters. Remarkably, the cis-S'S molecules adopt nearly parallel conformations, featuring a dihedral angle of approximately 10.5° and an intermolecular distance of approximately 4.81 Å between the π-stacked dimers. The distribution of transition dipole moments within cis-S'S dimers follows a normal distribution, indicative of the simulation having reached the thermodynamic steady state. The obtained average angle of 19.2° (<54.7°) provides additional evidence for the propensity of cis-S'S to form J-aggregates at higher concentrations (Fig. 4b). In contrast, cis-O'O and cis-O'S display non-parallel aggregation patterns, characterized by dihedral angles of approximately 60° (Fig. S21†). Additionally, specific trans-O'O molecules display H-aggregation characteristics with a dihedral angle of less than 15° and a transition dipole moment angle larger than 54.7° (Fig. S22†).51 The simulation outcomes underscore the distinct aggregation tendency of porphodilactones within NPs, where monosulfur-substituted NPs are filled with mixed packing modes. In contrast, disulfur-substituted ones form a stable J-aggregation,55 consistent with UV-vis absorption in Fig. 2 and S11.†
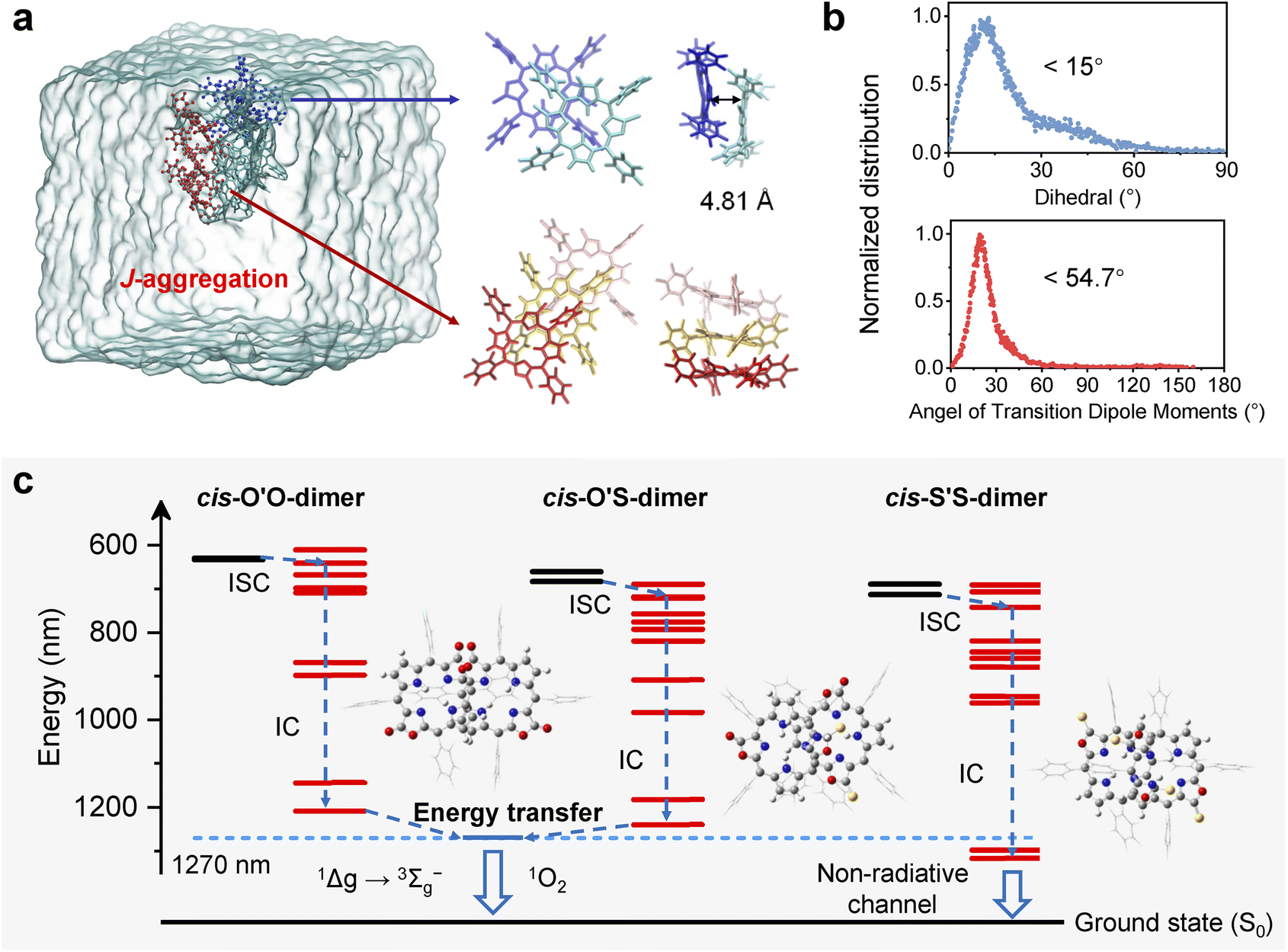 | ||
| Fig. 4 (a) Schematic illustration of the molecular aggregation of cis-S'S expected to persist in nanoparticles as deduced from equilibrated MD simulations in a water box (50 × 50 × 50 Å) and expected arrangements of the constituent aggregated dimers and trimers. (b) Normalized distribution of dihedral and transition dipole moment angle between cis-S'S dimers. (c) Depiction of the proposed excited state energy dissipation pathways and triplet energy levels of cis-O'O, cis-O'S, and cis-S'S dimers. | ||
To further elucidate the impact of aggregation on the PDT effect, we chose the dimer structures of cis-O'O, cis-O'S, and cis-S'S obtained by MD simulations. We then calculated the energy levels of singlet and triplet states (as shown in Fig. 4c). As shown in Fig. S23–S25,† these dimers exhibit more energy levels of triplet states below the S1 state than monomeric molecules. This is due to the π–π interactions and excitonic coupling among adjacent molecules in aggregation that cause the splitting of excited energy levels as per Kasha's rules.56 The formation of band-like structures can potentially introduce multiple pathways for deactivating triplets in porphothiodilactones.57
Specifically, we found that in the case of cis-S'S-dimer, the energy levels of the triplet state are inherently lower than that of 1O2 (1270 nm, 1Δg → 3Σg transition). In contrast, weak π–π interaction in non-parallel aggregation causes the energy levels for the triplet states of cis-O'S-dimer to remain higher than 1O2, promoting efficient intermolecular energy transfer and ultimately leading to 1O2 generation. This attenuation of 1O2 generation in cis-S'S NPs is particularly pronounced. On the other hand, the larger SOC parameter between T1 and S0 for cis-S'S-dimer (4.38 cm−1) was present, in comparison to cis-O'O-dimer (0.73 cm−1) and cis-O'S-dimer (3.02 cm−1). These results highlight different energy dissipation pathways observed between monosulfur- and disulfur-substituted porphodilactones inside NPs in water, which can be attributed to progressive β-thiolation that leads to porphyrinoid aggregation.
In vitro and in vivo experiments
To explore the impact of thiolation on 1O2 photosensitization of cis-O'S@NPs and cis-S'S@NPs in living cells, we used 2′,7′-dichlorofluorescein diacetate (DCFH-DA) as a probe to measure intracellular ROS levels. In Fig. 5a, we observed bright green fluorescence in HeLa cells after exposure to cis-O'S@NPs upon photoirradiation (700 nm, 10 mW cm−2, 10 min) in contrast to weak intracellular fluorescence in cells treated with cis-S'S@NPs + hυ. This is consistent with the low efficiency of 1O2 generation of cis-S'S@NPs in water. Under dark conditions, there was trivial ROS generation when cells were treated with cis-O'S@NPs or cis-S'S@NPs. | ||
| Fig. 5 (a) Intracellular ROS level of HeLa cells treated with cis-O'S@NPs and cis-S'S@NPs under dark and light conditions (700 nm, 10 mW cm−2, 10 min), respectively. DCFH-DA was used as the fluorescent probe for ROS generation. Scale bar: 50 μm. (b) Photocytotoxicity of cis-O'S@NPs and cis-S'S@NPs (0–10 μM) on HeLa cells obtained by a CCK-8 assay. (c) Fluorescence images of Calcein AM/PI-stained HeLa cells treated with cis-O'S@NPs and cis-S'S@NPs under dark and light conditions, respectively. Scale bar: 200 μm. (d) Images of HeLa 3D MCSs treated with cis-O'S@NPs and cis-S'S@NPs (6 μM) under dark and light conditions, respectively. The images for day 1 were recorded before irradiation. Scale bar: 500 μm. | ||
Next, we assessed the photocytotoxicity of cis-O'S@NPs and cis-S'S@NPs on HeLa cells using a Cell Counting Kit-8 (CCK-8). After irradiation at 700 nm (10 mW cm−2) for 10 min, cis-O'S@NPs showed dose-dependent cytotoxicity with a low half-maximal inhibitory concentration (IC50) of 0.50 μM (Fig. 5b), whereas cis-S'S@NPs exhibited weak photocytotoxicity with an IC50 of 13.7 μM. There was no significant dark cytotoxicity observed for either cis-O'S@NPs or cis-S'S@NPs as cell viability remained above 80% even at a concentration of 10 μM (Fig. S26†). We also used calcein-AM and propidium iodide (PI) to identify live (green) and dead (red) cells (Fig. 5c). The results showed a high level of cell death with red fluorescence in cells treated with cis-O'S@NPs and subjected to photoirradiation, whereas a portion of cells remained alive in cis-S’S@NPs + hυ.
To evaluate their effectiveness on multicellular spheroids (MCSs), we conducted PDT experiments using cis-O'S@NPs or cis-S'S@NPs. MCSs are commonly used to simulate solid tumor microenvironments.58 After incubation with cis-O'S@NPs or cis-S'S@NPs, the MCSs were then divided into dark and light groups and exposed to photoirradiation (700 nm, 10 mW cm−2, 10 min).
As shown in Fig. 5e, the results showed that treatment with cis-O'S@NPs or cis-S'S@NPs significantly inhibited the growth of MCSs, while the control groups under dark incubation dramatically increased in volume after seven days (Fig. S27†). Additionally, the morphology of MCSs in the light groups changed significantly. On day 6, the MCSs treated with cis-O'S@NPs + hυ showed a remarkable collapse, indicating that cis-O'S@NPs had a higher PDT efficacy than cis-S'S@NPs under the same conditions. These findings suggest that the extent of β-thiolation effectively influences 1O2 photosensitization in living cells and MCSs.
We then extended our study to a mouse model (Fig. 6a). The mice were randomly divided into five groups: “control (PBS + hυ)”, “cis-O'S@NPs”, “cis-S'S@NPs”, “cis-O'S@NPs + hυ”, and “cis-S'S@NPs + hυ” with each group consisting of four mice. After 24 hours of tail vein injection, we irradiated the tumor sites using a 700 nm LED light for 10 min (200 mW cm−2). We monitored the tumor volumes and mice body weights every two days and analyzed photographs of dissected tumor tissues and tumor weights (Fig. 6b–d). A negligible difference in body weights in all groups was seen (Fig. 6e). We found that both “cis-O'S@NPs + hυ” and “cis-S'S@NPs + hυ” treatments resulted in a high inhibition of tumor growth (87.0% and 88.6%, respectively). This was in contrast to the control and dark-treated groups, which showed a distinct increase in tumor volume after 14 days of observation. TUNEL staining further confirmed the presence of apoptotic cells in the tumor tissues treated with both “cis-O'S@NPs + hυ” and “cis-S'S@NPs + hυ” (Fig. S28†). Furthermore, we conducted hematoxylin and eosin (H&E) staining on key organs but observed no destructive cell necrosis or inflammation lesions in any of the groups (Fig. S29†). Our study suggests that replacing the sulfur atom is an effective approach to enhance PDT treatment in vivo. However, we did not observe any significant difference in therapeutic effect between cis-O'S@NPs and cis-S'S@NPs. As the “switch-on/off” 1O2 photosensitization of cis-S'S is highly dependent on the molecular aggregation, we hypothesized that the possibility of nanoparticle dissociation and deaggregation of cis-O'S or cis-S'S in the tumor microenvironment or during circulation59,60 led to the recovery of PDT efficacy for monomeric cis-S'S.
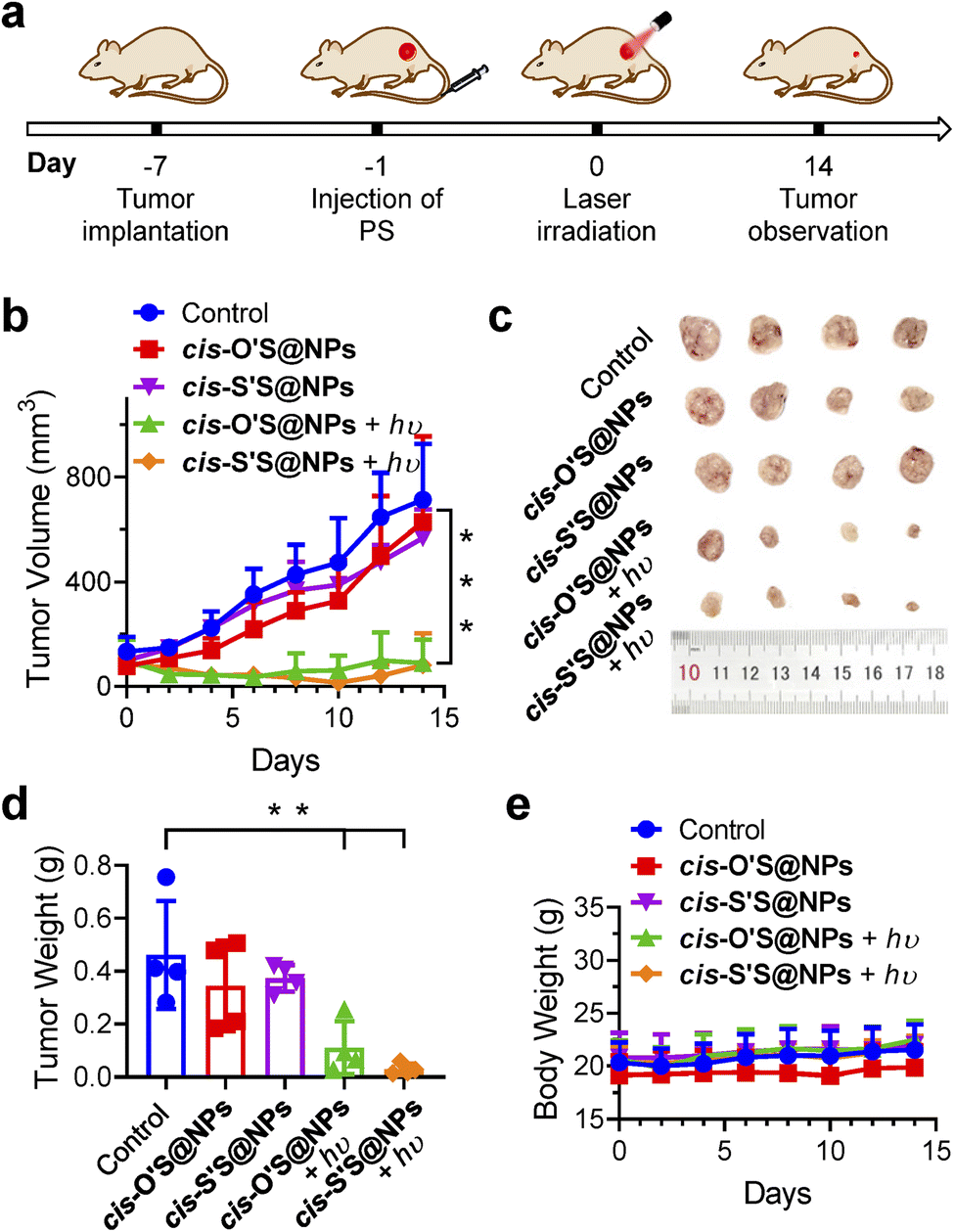 | ||
| Fig. 6 (a) Schematic illustration of the treatment regimen. (b) Tumor volume variation curves for the mice in control (PBS + hυ), cis-O'S@NPs, cis-S'S@NPs, cis-O'S@NPs + hυ, and cis-S'S@NPs + hυ groups during the treatment period (n = 4). ***p < 0.05. (c) Digital photographs of the tumors dissected from the mice in different groups. (d) Tumor weight of the mice in different groups at the end of treatment (mean ± SD, n = 4, **P < 0.05). (e) Body weight curves of the mice in different treatment groups (n = 4, mean ± SD). | ||
Considering the weak acidity of the tumor microenvironment (TME), we first examined the stability of porphothiodilactone nanoparticles (NPs) in a mildly acidic setting (pH ∼ 5.5). When the NPs were placed in a buffer solution with a pH of 5.5 for 24 h, their size increased significantly and exhibited a broad size distribution (Fig. S30†). Additionally, although the efficiency of photothermal conversion for the porphothiodilactone NPs is still low, we observed a thermal effect when we irradiated the tumor sites in mice. After 10 min of irradiation (700 nm, 200 mW cm−2), the temperature of the tumor areas injected with NPs increased by approximately 2 °C (Fig. S31†). This temperature elevation may induce the dissociation of the NPs, as supported by the photothermal heating curve in solutions and the increased particle size after irradiation (Fig. S32†). Nevertheless, the TME is quite complex and the response mechanisms of specific factors or targets still remains unclear, which needs more effort to understand the possible mechanism and develop switchable photosensitive systems related to aggregation and excited states in our future study.
Conclusions
In summary, we synthesized a range of porphothiodilactone derivatives to investigate the impact of β-thiolation on the photophysical properties and 1O2 photosensitization of porphyrinoids. By replacing oxygen with sulfur atoms at opposite pyrrole positions on the porphyrin periphery, we observed gradual redshifts in absorption and severely quenching emission. Our experiments, including transient absorption spectroscopy, showed that the different sulfur substitutions yielded similar excited state dynamics, promoting the ISC process and improving the population of triplet states. As a result, sulfur substitution effectively increased the 1O2 generation of porphyrinoids. Interestingly, we noted that mono- and disulfur substituted porphyrinoids had different 1O2 photosensitization in various media. This surprising discovery contradicts the traditional “more is better” approach and warrants further exploration.Through crystal structure analysis, characteristic absorption of J-aggregation, and MD simulation, we found that progressive thiolation of the β-periphery led to enhanced aggregation from loose to close packing. Furthermore, TD-DFT calculations of porphothiodilactone aggregation suggested that the energy levels of the triplet states of disulfur-substituted porphyrin aggregates are inherently lower than that of 1O2. In comparison, those of monosulfur-substituted congery have higher energy levels. This leads to different energy dissipation pathways at the excited states between mono- and disulfur-substituted porphodilactone aggregates, which may explain the switching of 1O2 photosensitization in aqueous media. Overall, our work challenges conventional wisdom and offers new insights into understanding the action mechanism of heteroatom-incorporated PSs beyond the molecule level.
Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
Conceptualization: M. Z., H. Z., and J.-L. Z. Data curation: M. Z., H. Z., and J.-L. Z. Investigation: M. Z. and H. Z. Project administration: M. Z., H. Z., and J.-L. Z. Synthesis section: M. Z., M. W., and Y. Yu. Calculation section: Y. Yao. Spectral section: M. Z. and G. R. Animal section: M. Z., H. Z., and R. Z. Writing – original draft: M. Z. and H. Z. Writing – review & editing: M. Z., H. Z., J. Z., X.-J. L., and J.-L. Z. Funding acquisition: J.-L. Z. Supervision: J. Z., M. Z., and J.-L. Z.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (21571007, 21621061, 21778002, and 21861162008) and the Chemistry and Chemical Engineering Guangdong Laboratory (1932002) is acknowledged. The measurements of fluorescence spectroscopy were performed at the Analytical Instrumentation Center of Peking University. This work was supported in part by the High-performance Computing Platform of Peking University. L.-J. Guo and W.-C. Xie are gratefully thanked for TEM measurements and biological experiments. All animal procedures were approved by the Institutional Animal Care and Use Committee of Sinoresearch (Beijing) Biotechnology Co., Ltd (protocol number: ZYZC202306011J).References
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Clinical development and potential of photothermal and photodynamic therapies for cancer, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef PubMed.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Recent progress in photosensitizers for overcoming the challenges of photodynamic therapy: from molecular design to application, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- T. C. Pham, V. N. Nguyen, Y. Choi, S. Lee and J. Yoon, Recent strategies to develop innovative photosensitizers for enhanced photodynamic therapy, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed.
- W. Fan, P. Huang and X. Chen, Overcoming the Achilles' heel of photodynamic therapy, Chem. Soc. Rev., 2016, 45, 6488–6519 RSC.
- E. L. Bastos, F. H. Quina and M. S. Baptista, Endogenous photosensitizers in human skin, Chem. Rev., 2023, 123, 9720–9785 CrossRef CAS PubMed.
- W. Hu, P. N. Prasad and W. Huang, Manipulating the dynamics of dark excited states in organic materials for phototheranostics, Acc. Chem. Res., 2021, 54, 697–706 CrossRef CAS PubMed.
- J. Chen, Y. Zhu, C. Wu and J. Shi, Nanoplatform-based cascade engineering for cancer therapy, Chem. Soc. Rev., 2020, 49, 9057–9094 RSC.
- Z. Zhou, J. Song, L. Nie and X. Chen, Reactive oxygen species generating systems meeting challenges of photodynamic cancer therapy, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- Y. Gui, Y. Wang, D. Wang, Y. Qin, G. Song, D. Yan, B. Z. Tang and D. Wang, Thiophene π-bridge manipulation of NIR-II AIEgens for multimodal tumor phototheranostics, Angew. Chem., Int. Ed., 2024, 63, e202318609 CrossRef CAS PubMed.
- G. Feng, G. Q. Zhang and D. Ding, Design of superior phototheranostic agents guided by Jablonski diagrams, Chem. Soc. Rev., 2020, 49, 8179–8234 RSC.
- Q. Yao, J. Fan, S. Long, X. Zhao, H. Li, J. Du, K. Shao and X. Peng, The concept and examples of type-III photosensitizers for cancer photodynamic therapy, Chem, 2022, 8, 197–209 CAS.
- S. Monro, K. L. Colon, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Transition metal complexes and photodynamic therapy from a tumor-centered approach: challenges, opportunities, and highlights from the development of TLD1433, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- M. Zhu, H. Zhang, G. Ran, D. N. Mangel, Y. Yao, R. Zhang, J. Tan, W. Zhang, J. Song, J. L. Sessler and J. L. Zhang, Metal Modulation: An easy-to-implement tactic for tuning lanthanide phototheranostics, J. Am. Chem. Soc., 2021, 143, 7541–7552 CrossRef CAS PubMed.
- M. Liu, Y. Chen, Y. Guo, H. Yuan, T. Cui, S. Yao, S. Jin, H. Fan, C. Wang, R. Xie, W. He and Z. Guo, Golgi apparatus-targeted aggregation-induced emission luminogens for effective cancer photodynamic therapy, Nat. Commun., 2022, 13, 2179 CrossRef CAS PubMed.
- D. Liu, A. M. El-Zohry, M. Taddei, C. Matt, L. Bussotti, Z. Wang, J. Zhao, O. F. Mohammed, M. Di Donato and S. Weber, Long-lived charge-transfer state induced by spin-orbit charge transfer intersystem crossing (SOCT-ISC) in a compact spiro electron donor/acceptor dyad, Angew. Chem., Int. Ed., 2020, 59, 11591–11599 CrossRef CAS PubMed.
- H. Gu, W. Liu, W. Sun, J. Du, J. Fan and X. Peng, Single-molecule photosensitizers for NIR-II fluorescence and photoacoustic imaging guided precise anticancer phototherapy, Chem. Sci., 2022, 13, 9719–9726 RSC.
- H. Cao, B. Fang, J. Liu, Y. Shen, J. Shen, P. Xiang, Q. Zhou, S. C. De Souza, D. Li, Y. Tian, L. Luo, Z. Zhang and X. Tian, Photodynamic therapy directed by three-photon active rigid plane organic photosensitizer, Adv. Healthcare Mater., 2021, 10, e2001489 CrossRef PubMed.
- Z. Wang, M. Ivanov, Y. Gao, L. Bussotti, P. Foggi, H. Zhang, N. Russo, B. Dick, J. Zhao, M. Di Donato, G. Mazzone, L. Luo and M. Fedin, Spin–orbit charge-transfer intersystem crossing (ISC) in compact electron donor-acceptor dyads: ISC mechanism and application as novel and potent photodynamic therapy reagents, Chem.–Eur. J., 2020, 26, 1091–1102 CrossRef CAS PubMed.
- P. Xiao, W. Xie, J. Zhang, Q. Wu, Z. Shen, C. Guo, Y. Wu, F. Wang, B. Z. Tang and D. Wang, De novo design of reversibly pH-switchable NIR-II aggregation-induced emission luminogens for efficient phototheranostics of patient-derived tumor xenografts, J. Am. Chem. Soc., 2023, 145, 334–344 CrossRef CAS PubMed.
- H. Huang, D. Ma, Q. Liu, D. Huang, X. Zhao, Q. Yao, T. Xiong, S. Long, J. Du, J. Fan and X. Peng, Enhancing intersystem crossing by intermolecular dimer-stacking of cyanine as photosensitizer for cancer therapy, CCS Chem., 2022, 4, 3627–3636 CrossRef CAS.
- Z. Zhang, W. Xu, M. Kang, H. Wen, H. Guo, P. Zhang, L. Xi, K. Li, L. Wang, D. Wang and B. Z. Tang, An all-round athlete on the track of phototheranostics: subtly regulating the balance between radiative and nonradiative decays for multimodal imaging-guided synergistic therapy, Adv. Mater., 2020, 32 Search PubMed.
- C. Ji, Q. Gao, X. Dong, W. Yin, Z. Gu, Z. Gan, Y. Zhao and M. Yin, A size-reducible nanodrug with an aggregation-enhanced photodynamic effect for deep chemo-photodynamic therapy, Angew. Chem., Int. Ed., 2018, 57, 11384–11388 CrossRef CAS PubMed.
- Z. Deng, J. Zhang, J. Zhou, W. Shen, Y. Zuo, J. Wang, S. Yang, J. Liu, Y. Chen, C. C. Chen, G. Jia, P. Alam, J. W. Y. Lam and B. Z. Tang, Dynamic transition between monomer and excimer phosphorescence in organic near-infrared phosphorescent crystals, Adv. Mater., 2024, e2311384 CrossRef PubMed.
- Z. Zhuang, J. Li, P. Shen, Z. Zhao and B. Z. Tang, Exploring and leveraging aggregation effects on reactive oxygen species generation in photodynamic therapy, Aggregate, 2024, e540 CrossRef.
- K. Wei, Y. Wu, X. Zheng, L. Ouyang, G. Ma, C. Ji and M. Yin, A light-triggered J-aggregation-regulated therapy conversion: from photodynamic/photothermal therapy to long-lasting chemodynamic therapy for effective tumor ablation, Angew. Chem., Int. Ed., 2024, e202404395 CAS.
- C. Ji, L. Lai, P. Li, Z. Wu, W. Cheng and M. Yin, Organic dye assemblies with aggregation-induced photophysical changes and their bio-applications, Aggregate, 2021, 2, e39 CrossRef CAS.
- V. N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Heavy-atom-free photosensitizers: from molecular design to applications in the photodynamic therapy of cancer, Acc. Chem. Res., 2021, 54, 207–220 CrossRef CAS PubMed.
- Y. Xiao, X. Huang, J. Feng, Z. Ni, L. Gai, X. Xiao, X. Sui and H. Lu, A simple route toward triplet-forming thionated BODIPY-based photosensitizers, Dyes Pigm., 2022, 200, 110167 CrossRef CAS.
- V. N. Nguyen, S. J. Park, S. Qi, J. Ha, S. Heo, Y. Yim, G. Baek, C. S. Lim, D. J. Lee, H. M. Kim and J. Yoon, Design and synthesis of efficient heavy-atom-free photosensitizers for photodynamic therapy of cancer, Chem. Commun., 2020, 56, 11489–11492 RSC.
- J. Tang, L. Wang, A. Loredo, C. Cole and H. Xiao, Single-atom replacement as a general approach towards visible-light/near-infrared heavy-atom-free photosensitizers for photodynamic therapy, Chem. Sci., 2020, 11, 6701–6708 RSC.
- V. N. Nguyen, S. Qi, S. Kim, N. Kwon, G. Kim, Y. Yim, S. Park and J. Yoon, An Emerging Molecular design approach to heavy-atom-free photosensitizers for enhanced photodynamic therapy under hypoxia, J. Am. Chem. Soc., 2019, 141, 16243–16248 CrossRef CAS PubMed.
- V. N. Nguyen, Y. Yim, S. Kim, B. Ryu, K. Swamy, G. Kim, N. Kwon, C. Y. Kim, S. Park and J. Yoon, Molecular design of highly efficient heavy-atom-free triplet BODIPY derivatives for photodynamic therapy and bioimaging, Angew. Chem., Int. Ed., 2020, 59, 8957–8962 CrossRef CAS PubMed.
- S. Mai, M. Pollum, L. Martinez-Fernandez, N. Dunn, P. Marquetand, I. Corral, C. E. Crespo-Hernandez and L. Gonzalez, The origin of efficient triplet state population in sulfur-substituted nucleobases, Nat. Commun., 2016, 7, 13077 CrossRef CAS PubMed.
- V.-N. Nguyen, G. Baek, S. Qi, S. Heo, Y. Yim and J. Yoon, A lysosome-localized thionaphthalimide as a potential heavy-atom-free photosensitizer for selective photodynamic therapy, Dyes Pigm., 2020, 177, 108265 CrossRef CAS.
- L. A. Ortiz-Rodriguez, S. J. Hoehn, A. Loredo, L. Wang, H. Xiao and C. E. Crespo-Hernandez, Electronic relaxation pathways in heavy-atom-free photosensitizers absorbing near-infrared radiation and exhibiting high yields of singlet oxygen generation, J. Am. Chem. Soc., 2021, 143, 2676–2681 CrossRef CAS PubMed.
- Z. Liu, Y. Gao, X. Jin, Q. Deng, Z. Yin, S. Tong, W. Qing and Y. Huang, Regioisomer-manipulating thio-perylenediimide nanoagents for photothermal/photodynamic theranostics, J. Mater. Chem. B, 2020, 8, 5535–5544 RSC.
- H. Abrahamse and M. R. Hamblin, New photosensitizers for photodynamic therapy, Biochem. J., 2016, 473, 347–364 CrossRef CAS PubMed.
- M. Zhu, H. Zhang, G. Ran, Y. Yao, Z. S. Yang, Y. Ning, Y. Yu, R. Zhang, X. X. Peng and J. Wu, Bioinspired design of seco-chlorin photosensitizers to overcome phototoxic effects in photodynamic therapy, Angew. Chem., Int. Ed., 2022, 61, e202204330 CrossRef CAS PubMed.
- Y. Ning, G. Q. Jin and J. L. Zhang, Porpholactone chemistry: an emerging approach to bioinspired photosensitizers with tunable near-infrared photophysical properties, Acc. Chem. Res., 2019, 52, 2620–2633 CrossRef CAS PubMed.
- Y. Ning, X. S. Ke, J. Y. Hu, Y. W. Liu, F. Ma, H. L. Sun and J. L. Zhang, Bioinspired orientation of beta-substituents on porphyrin antenna ligands switches ytterbium(III) NIR emission with thermosensitivity, Inorg. Chem., 2017, 56, 1897–1905 CrossRef CAS PubMed.
- X. S. Ke, Y. Chang, J. Z. Chen, J. Tian, J. Mack, X. Cheng, Z. Shen and J. L. Zhang, Porphodilactones as synthetic chlorophylls: relative orientation of beta-substituents on a pyrrolic ring tunes NIR absorption, J. Am. Chem. Soc., 2014, 136, 9598–9607 CrossRef CAS PubMed.
- Y. Yu, B. Czepukojc, C. Jacob, Y. Jiang, M. Zeller, C. Bruckner and J. L. Zhang, Porphothionolactones: synthesis, structure, physical, and chemical properties of a chemodosimeter for hypochlorite, Org. Biomol. Chem., 2013, 11, 4613–4621 RSC.
- H. Zhang, J. H. Wu, H. Z. Xue, R. Zhang, Z. S. Yang, S. Gao and J. L. Zhang, Biomimetically constructing a hypoxia-activated programmable phototheranostics at the molecular level, Chem. Sci., 2022, 13, 8979–8988 RSC.
- H. Khatoon and E. Abdulmalek, A focused review of synthetic applications of Lawesson's reagent in organic synthesis, Molecules, 2021, 26, 6937 CrossRef CAS PubMed.
- J. Sun, E. Zhao, J. Liang, H. Li, S. Zhao, G. Wang, X. Gu and B. Z. Tang, Diradical-featured organic small-molecule photothermal material with high-spin state in dimers for ultra-broadband solar energy harvesting, Adv. Mater., 2022, 34, 2108048 CrossRef CAS PubMed.
- Y. L. Lee, Y. T. Chou, B. K. Su, C. C. Wu, C. H. Wang, K. H. Chang, J. A. Ho and P. T. Chou, Comprehensive thione-derived perylene diimides and their bio-conjugation for simultaneous imaging, tracking, and targeted photodynamic therapy, J. Am. Chem. Soc., 2022, 144, 17249–17260 CrossRef CAS PubMed.
- B. A. Llewellyn, E. S. Davies, C. R. Pfeiffer, M. Cooper, W. Lewis and N. R. Champness, Thionated perylene diimides with intense absorbance in the near-IR, Chem. Commun., 2016, 52, 2099–2102 RSC.
- J.-Y. Hu, Z.-Y. Wu, K. Chai, Z.-S. Yang, Y.-S. Meng, Y. Ning, J. Zhang and J.-L. Zhang, β-Fluorinated porpholactones and metal complexes: synthesis, characterization and some spectroscopic studies, Inorg. Chem. Front., 2017, 4, 1539–1545 RSC.
- L. A. Ortiz-Rodriguez and C. E. Crespo-Hernandez, Thionated organic compounds as emerging heavy-atom-free photodynamic therapy agents, Chem. Sci., 2020, 11, 11113–11123 RSC.
- K. Cai, J. Xie and D. Zhao, NIR J-aggregates of hydroazaheptacene tetraimides, J. Am. Chem. Soc., 2013, 136, 28–31 CrossRef PubMed.
- Y. Wang, G. Xia, M. Tan, M. Wang, Y. Li and H. Wang, H-dimeric nanospheres of amphipathic squaraine dye with an 81.2% photothermal conversion efficiency for photothermal therapy, Adv. Funct. Mater., 2022, 32, 2113098 CrossRef CAS.
- S. Wang, W. Wu, P. Manghnani, S. Xu, Y. Wang, C. C. Goh, L. G. Ng and B. Liu, Polymerization-enhanced two-photon photosensitization for precise photodynamic therapy, ACS Nano, 2019, 13, 3095–3105 CrossRef CAS PubMed.
- S. Yang, J. Zhang, Z. Zhang, R. Zhang, X. Ou, W. Xu, M. Kang, X. Li, D. Yan, R. T. K. Kwok, J. Sun, J. W. Y. Lam, D. Wang and B. Z. Tang, More Is Better: Dual-acceptor engineering for constructing second near-infrared aggregation-induced emission luminogens to boost multimodal phototheranostics, J. Am. Chem. Soc., 2023, 145, 22776–22787 CrossRef CAS PubMed.
- Y. Yao, G. Ran, C. L. Hou, R. Zhang, D. N. Mangel, Z. S. Yang, M. Zhu, W. Zhang, J. Zhang, J. L. Sessler, S. Gao and J. L. Zhang, Nonaromatic organonickel(II) phototheranostics, J. Am. Chem. Soc., 2022, 144, 7346–7356 CrossRef CAS PubMed.
- M. Li, W. Sun, R. Tian, J. Cao, Y. Tian, B. Gurram, J. Fan and X. Peng, Smart J-aggregate of cyanine photosensitizer with the ability to target tumor and enhance photodynamic therapy efficacy, Biomaterials, 2021, 269, 120532 CrossRef CAS PubMed.
- J. Zhang, S. Mukamel and J. Jiang, Aggregation-induced intersystem crossing: rational design for phosphorescence manipulation, J. Phys. Chem. B, 2020, 124, 2238–2244 CrossRef CAS PubMed.
- Y.-F. Kang, W.-K. Chen, K.-X. Teng, L.-Y. Wang, X.-C. Xu, L.-Y. Niu, G. Cui and Q.-Z. Yang, Aggregation turns BODIPY fluorophores into photosensitizers: reversibly switching intersystem crossing on and off for smart photodynamic therapy, CCS Chem., 2022, 4, 3516–3528 CrossRef CAS.
- H. Yuan, Z. Han, Y. Chen, F. Qi, H. Fang, Z. Guo, S. Zhang and W. He, Ferroptosis photoinduced by new cyclometalated iridium (III) complexes and its synergism with apoptosis in tumor cell inhibition, Angew. Chem., Int. Ed., 2021, 133, 8255–8262 CrossRef.
- Y. Zhao, C. Yu, Y. Yu, X. Wei, X. Duan, X. Dai and X. Zhang, Bioinspired heteromultivalent ligand-decorated nanotherapeutic for enhanced photothermal and photodynamic therapy of antibiotic-resistant bacterial pneumonia, ACS Appl. Mater. Interfaces, 2019, 11, 39648–39661 CrossRef CAS PubMed.
- Y. Yuan, C. J. Zhang, M. Gao, R. Zhang, B. Z. Tang and B. Liu, Specific light-up bioprobe with aggregation-induced emission and activatable photoactivity for the targeted and image-guided photodynamic ablation of cancer cells, Angew. Chem., Int. Ed., 2014, 54, 1780–1786 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2261655, 2261661, 2261656 and 2261662. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03642e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |