
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient N2 electroreduction enabled by linear charge transfer over atomically dispersed W sites†
Jin
Wan
a,
Dong
Liu
a,
Chuanzhen
Feng
a,
Huijuan
Zhang
*ab and
Yu
Wang
 *ab
*ab
aThe School of Chemistry and Chemical Engineering, Chongqing University, 174 Shazheng Street, Shapingba District, Chongqing City, 400044, P. R. China
bCollege of Chemistry and Environmental Science, Inner Mongolia Normal University, Huhehaote, 010022, P. R. China. E-mail: zhanghj@cqu.edu.cn; wangy@cqu.edu.cn
First published on 9th July 2024
Abstract
Electrocatalytic nitrogen reduction reaction (NRR) presents a sustainable alternative to the Haber–Bosch process for ammonia (NH3) production. However, developing efficient catalysts for NRR and deeply elucidating their catalytic mechanism remain daunting challenges. Herein, we pioneered the successful embedding of atomically dispersed (single/dual) W atoms into V2−xCTyvia a self-capture method, and subsequently uncovered a quantifiable relationship between charge transfer and NRR performance. The prepared n-W/V2−xCTy shows an exceptional NH3 yield of 121.8 μg h−1 mg−1 and a high faradaic efficiency (FE) of 34.2% at −0.1 V (versus reversible hydrogen electrode (RHE)), creating a new record at this potential. Density functional theory (DFT) computations reveal that neighboring W atoms synergistically collaborate to significantly lower the energy barrier, achieving a remarkable limiting potential (UL) of 0.32 V. Notably, the calculated UL values for the constructed model show a well-defined linear relationship with integrated-crystal orbital Hamilton population (ICOHP) (y = 0.0934x + 1.0007, R2 = 0.9889), providing a feasible activity descriptor. Furthermore, electronic property calculations suggest that the NRR activity is rooted in d–2π* coupling, which can be explained by the “donation and back-donation” hypothesis. This work not only designs efficient atomic catalysts for NRR, but also sheds new insights into the role of neighboring single atoms in improving reaction kinetics.
Introduction
Ammonia (NH3) plays a vital role in our daily life, serving as a fundamental raw material for the manufacture of fertilizers, pharmaceuticals, and synthetic fibers.1–3 Currently, the production of NH3 heavily relies on the traditional Haber–Bosch process, which operates under harsh production conditions (300–500 °C and 200–300 atm), leading to high energy consumption and environmental pollution.4,5 Thus, it is critical to explore sustainable and environmentally friendly approaches for NH3 production. Electrocatalytic nitrogen (N2) fixation demonstrates significant potential as an alternative to conventional processes, offering a straightforward method for converting N2 to NH3 in a mild environment.6–8 Nevertheless, the electrocatalytic nitrogen reduction reaction (NRR, N2 + 6H+ + 6e− = 2NH3) suffers from sluggish kinetics due to the presence of inert N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds (bond energy = 941 kJ mol−1), as well as the presence of competing hydrogen evolution reaction (HER) in the electrolyte, limiting the NH3 yield and faradaic efficiency (FE).9–11 To address the above challenges, the designed catalysts must exhibit high activity, selectivity and stability.
N bonds (bond energy = 941 kJ mol−1), as well as the presence of competing hydrogen evolution reaction (HER) in the electrolyte, limiting the NH3 yield and faradaic efficiency (FE).9–11 To address the above challenges, the designed catalysts must exhibit high activity, selectivity and stability.
The emergence of atomically dispersed catalysts opens up opportunities for efficient NH3 production due to their maximum atom utilization and outstanding catalytic properties.12–14 Until now, numerous single atom catalysts (SACs), such as those incorporating Fe, Ru, Cu, Mo, and Mn atoms have been proven to be promising electrocatalysts for NH3 production.15–22 However, the simultaneous improvement of NH3 yield and FE in the presence of multiple reaction intermediates remains a challenge for SACs. Furthermore, SACs with a single active site are rarely able to break the linear scaling relationship between the reaction intermediates.10,23 By introducing another active site, dual atom catalysts (DACs) are equipped with more flexible active sites and interatomic synergies, thereby tuning the adsorption of intermediates and solving the drawbacks of SACs.24–26 Recently, researchers mainly focus on various substrates, including carbon materials, carbon nitride compounds, and some organic porous materials, due to their abundance of available loading sites.27–29 Unfortunately, the strong interactions between metal atoms may cause lattice distortions in these flexible substrates, resulting in poor stability. On the other hand, the presence of multiple reaction intermediates leads to unsatisfactory activity and complicates understanding of the reaction mechanism. Most of the studies emphasized that the catalytic activity originates from the electronic modulation between the dual atoms, but quantitative analysis of the relationship between charge transfer and activity remains ambiguous.30,31 Thus, it is particularly crucial to develop a robust conducting substrate for loading dual atoms to achieve efficient NRR activity and gain deep insights into NRR catalytic mechanism.
MXene, a cutting-edge two-dimensional material, holds immense potential for electrocatalytic applications. V2CTx MXene, a structural analog of Ti3C2Tx, can be synthesized by selectively etching V2AlC MAX with hydrofluoric acid (HF). Boasting exceptional electrical conductivity and robust support for metal atom loading, V2CTx MXene, despite its current limited research, holds promising potential for significant advancements across a wide range of applications.32–35 Due to the highly corrosive nature of HF, some V atoms adjacent to aluminum (Al) atoms are also etched away during the etching process, forming V single vacancies or vacancy clusters, providing opportunities for embedding foreign atoms.36,37 These defects are inherently unstable and active, allowing metal atoms to be readily trapped under natural conditions after the addition of metal precursors.38,39 Moreover, surface functional groups (T stands for –F, –O, and –OH) facilitate the electrosorption of metallic precursors, promoting the subsequent anchoring of metal atoms.12,38 The embedded metal atoms in V vacancies generate strong C–metal bonds, sustaining the generation of various intermediates.
In view of these issues, we present a self-capture method for successfully synthesizing W atom-dispersed catalysts (defined as n-W/V2−xCTy, n = 1, 2) under mild conditions. The W atoms were successfully anchored into the V vacancies, forming stable W dual atom catalysts with a loading of ∼1.39 wt%. The W atoms in n-W/V2CTx are surrounded by three C atoms, forming strong W–C bonds, as confirmed by aberration-corrected electron microscopy and synchrotron radiation characterization. Upon application to NRR, the prepared n-W/V2−xCTy exhibited superior performance with NH3 yield up to 121.8 μg h−1 mg−1 and a FE of 34.2% at −0.1 V (versus reversible hydrogen electrode (RHE)). This performance surpasses the catalytic capabilities of V2−xCTy, WSAC/V2−xCTy, and W NPs/V2−xCTy, clearly indicating that the incorporation of neighboring W atoms in n-W/V2−xCTy significantly boosted its NRR catalytic performance. Drawing from the experimental characterization results, three theoretical structural models were developed, including pure V2−xCTy, WSAC/V2−xCTy, and WDAC/V2−xCTy. Our density functional theory (DFT) calculations suggest that the incorporation of the neighboring active site significantly reduces the limiting potential (UL) compared to pure V2−xCTy and WSAC/V2−xCTy (0.32, 0.81, and 0.71 V for WDAC/V2CTx, pure V2−xCTy, and WSAC/V2CTx, respectively). Furthermore, we constructed theoretical atomic models of W triple atom catalysts (WTAC/V2−xCTy) and W quadruple atom cluster catalysts (WQAC/V2−xCTy) to investigate the intrinsic relationship between charge transfer and NRR performance. Remarkably, these calculated UL values exhibit a linear relationship with integrated-crystal orbital Hamilton population (ICOHP) (y = 0.0934x + 1.0007, R2 = 0.9889). The synergistic interaction of W dual atoms can provide more d electrons to the 2π* orbitals of N2, intensifying the disruption of the NN bond and thus displaying outstanding NRR performance.
Results and discussion
As illustrated in Fig. 1a, multi-layer V2−xCTy sheets were obtained by etching V2AlC with HF solution. This process involves breaking the Al–V bonds and concurrent removal of some neighboring V atoms, ultimately leading to the formation of V single vacancies or vacancy clusters on the surface. The existence of V vacancy clusters allows dispersed W atoms to be embedded in adjacent vacancies, forming monatomic group structures. Subsequently, few-layer V2−xCTy nanosheets were fabricated by stripping multi-layer V2−xCTy sheets in tetrabutylammonium hydroxide (TBAOH) solution and then heat-treated to control the V surface primarily covered by O functional groups. Compared to –F and –OH groups, –O groups exhibit preferential adsorption of metal precursors, favoring the capture of single atoms.40,41 Finally, WCl6 powder was added to an aqueous solution containing V2−xCTy with V vacancies. The unstable and reactive V vacancies efficiently capture free W ions, resulting in the formation of W atom-dispersed catalysts.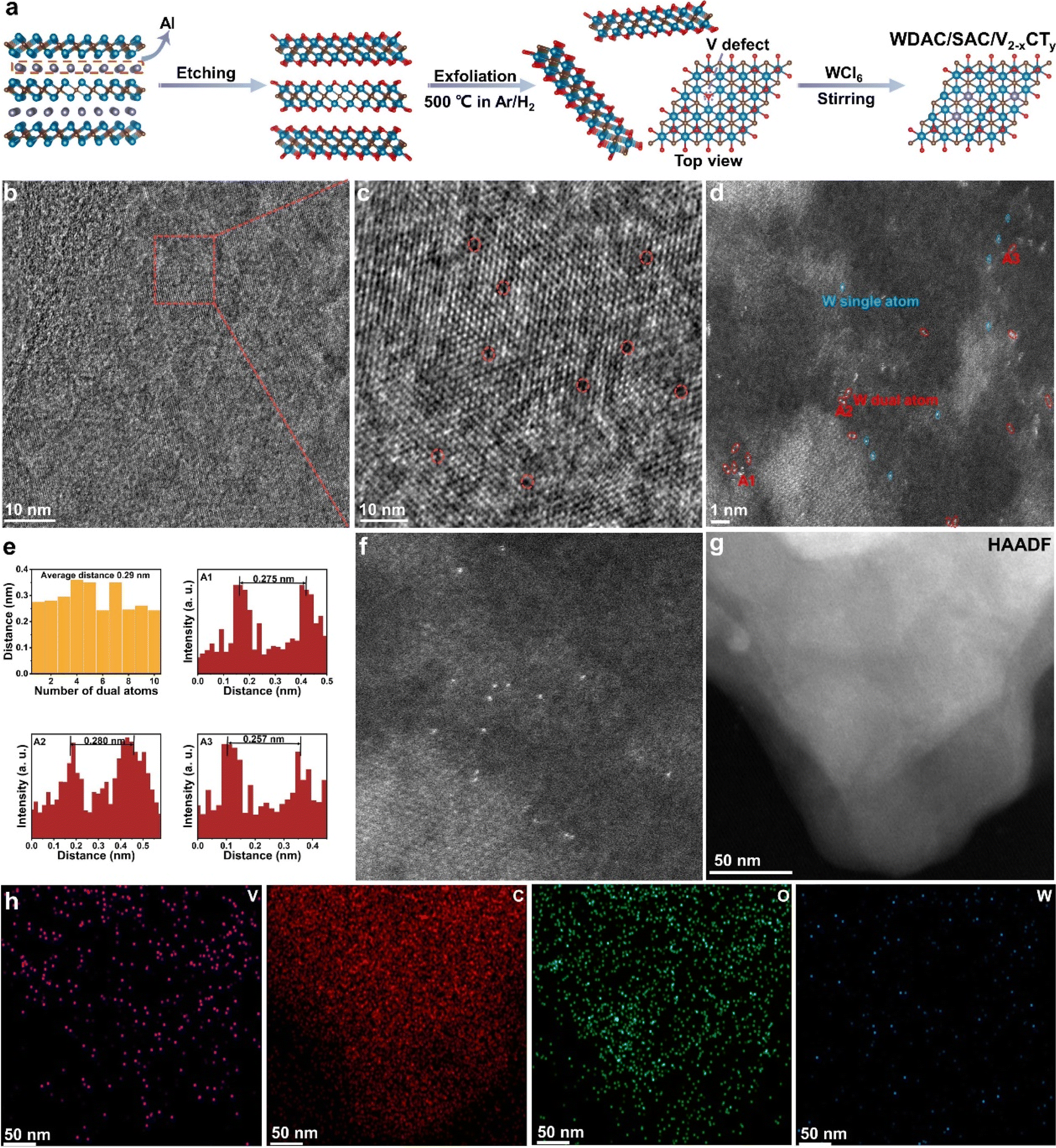 | ||
| Fig. 1 (a) Schematic diagram of the synthesis of n-W/V2−xCTy. (b and c) HRTEM image of the V2−xCTy layer and V vacancies on the V2−xCTy surface. (d) Atomic-resolution HAADF-STEM image of atomically dispersed W atoms on the V2−xCTy surface. (e) Intensity profiles obtained for WDAC/V2−xCTy. (f) Atomic-resolution HAADF-STEM image of single W atoms on the V2−xCTy surface. (g and h) HAADF-STEM image and corresponding EDS mapping of n-W/V2−xCTy. | ||
Scanning electron microscopy (SEM) images in Fig. S1 and S2† depict the changes in V2−xCTy nanosheets before and after exfoliation. After etching V2AlC MAX phases, the characteristic accordion-like structure of multi-layer V2−xCTy is visible. Fig. S2† displays the distinct two-dimensional morphology of few-layer V2−xCTy, implying successful exfoliation of multi-layer V2−xCTy nanosheets. Atomic force microscopy (AFM) images (Fig. S3†) suggest that the thickness of exfoliated V2−xCTy nanosheets ranges from 4 to 6 nm. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and its energy-dispersive X-ray spectroscopy (EDS) elemental mapping in Fig. S4† demonstrate uniform distributions of elements (C, O, V), affirming the successful preparation of few-layer V2−xCTy nanosheets. Additionally, V vacancies are vividly displayed by high-resolution TEM (HRTEM) in Fig. 1b and c. Selecting an area on the surface of V2−xCTy and zooming in, we can clearly observe lattice fringes and some dark holes within the lattice, which are identified as V defects.
Upon the addition of the WCl6 precursor, the X-ray diffraction (XRD) pattern of n-W/V2−xCTy shows no significant difference from that of V2−xCTy, and no signals associated with crystalline W species are observed for n-W/V2−xCTy (Fig. S5†). To examine the dispersion of W atoms and the surface morphology of n-W/V2−xCTy, aberration-corrected HAADF-STEM was applied. The results reveal uniform distribution of W atom pairs and isolated W atoms on the V2−xCTy surface, accompanied by distinct lattice fringes (Fig. 1d and S6†). The intensity profiles confirm that the average spacing of neighboring W atoms is separated approximately 0.29 nm (Fig. 1e), confirming the homogeneous dispersion of the metal pairs and matching the theoretical structural modeling of the distance between W dual atoms (2.92 Å). Furthermore, the HAADF-STEM and EDS results demonstrate that the W atoms are evenly dispersed on the V2−xCTy surface (Fig. 1g and h), without any detectable W nanoparticles, aligning with the XRD analysis. For comparative purposes, we modulated the concentration of HF, wherein a reduced HF concentration mitigates the disruption of the Al–V bonds. Consequently, this strategy lessens the formation of V vacancy clusters, ultimately leading to the majority of W atoms being securely embedded within single V vacancies. Fig. 1f and Fig. S7† present aberration-corrected HAADF-STEM and EDS images of WSAC/V2−xCTy, revealing a uniform distribution of isolated W atoms on the V2−xCTy surface. Notably, the ICP-OES analysis (Table S1†) reveals that the W atomic content in n-W/V2−xCTy stands at 1.39 wt%, whereas for WSAC/V2−xCTy, it is measured at 0.89 wt%. In addition, we prepared W nanoparticles loaded on the surface of V2−xCTy (named W NPs/V2−xCTy) using a comparable approach by adding excess WCl6 precursor. The ICP-OES results reveal that the W loading for W NPs/V2−xCTy is 5.12 wt%. HAADF-STEM clearly observes uniform arrangement of W nanoparticles on the V2−xCTy surface, while EDS results indicate a higher content of W than n-W/V2−xCTy (Fig. S8 and S9†).
To delve deeper into the atomic structure and coordination state of W atoms dispersed in n-W/V2−xCTy, we performed X-ray absorption near-edge structure (XANES), X-ray photoelectron spectroscopy (XPS), and extended X-ray absorption fine structure (EXAFS). Fig. 2a presents the XANES spectra of the W L3-edge relative to standard W foil, WO3 and WC. The absorption threshold of n-W/V2−xCTy is positioned between WC and WO3, suggesting that the single W atom possessed a positive valence state (+4 to +6). The electronic structure and local coordination environment of W atoms were further analyzed using X-ray photoelectron spectroscopy (XPS) (Fig. S10†). The W 4f XPS spectrum of n-W/V2−xCTy shows two peaks at 38.0 eV and 35.9 eV, corresponding to W 4f5/2 and W 4f7/2, respectively, corroborating the XANES results that the valence state of W atoms is near +6.42 As shown in Fig. 2b, the EXAFS Fourier-transform (FT) curve of W in n-W/V2−xCTy exhibits a prominent peak at 1.3 Å, assigned to the W–C bond.43 An intense W–W characteristic peak at 2.6 Å in the W foil is absent in n-W/V2−xCTy, indicating the sole existence of atomically dispersed W in n-W/V2−xCTy. In addition, a wavelet transform (WT) was applied to W EXAFS, revealing the relationship between R and k space, as shown in Fig. 2d–f.44,45 The WT EXAFS spectrum of n-W/V2−xCTy displays an intensity maximum at ∼7 Å−1, correlating with the W–C bond. Unlike the W foil, no intensity contour is observed around 13 Å−1, further validating the atomic dispersion of W atoms. To gain a profound understanding of the atomic structure and coordination state of W atoms, we performed EXAFS curve fitting. Our simulated EXAFS data, based on the WDAC/V2−xCTy structure model, demonstrated a commendable alignment with the experimental findings. The fitted spectra expose the R space and k space of the n-W/V2−xCTy system, which are presented in Fig. 2c and S11,† respectively. The inset of Fig. 2c offers a visual representation of the atomic structure model, highlighting the dual W atoms embedded within the V2−xCTy lattice. Moreover, when we applied the same fitting procedure to the theoretical model of WSAC/V2−xCTy, the outcomes exhibited a similar trend to that of WDAC/V2−xCTy, attributed to their identical coordination environments (Fig. S12 and S13†). The EXAFS fittings disclose that the coordination number of isolated W atoms for n-W/V2−xCTy is 3.6 ± 0.7, and the bond length is 1.97 ± 0.02 Å (Table S2†). This suggests that the W sites are primarily triply coordinated with C atoms.
 | ||
| Fig. 2 (a) W K-edge XANES spectra of n-W/V2−xCTy, WO3, WC, and W foil. (b) FT-EXAFS curves of n-W/V2−xCTy, WO3, WC, and W foil. (c) FT-EXAFS fitting results of n-W/V2−xCTy (inset: theoretical model of WDAC/V2−xCTy). (d–g) WT-EXAFS of n-W/V2−xCTy, WO3, WC, and W foil, respectively. | ||
The electrochemical NRR performance of n-W/V2−xCTy was investigated under ambient conditions within an H-type electrolytic cell. The NH3 yield and its by-product (hydrazine (N2H4)) were measured using a UV-vis spectrophotometer, and the corresponding calibration curves are shown in Fig. S14 and S15.† To begin, linear sweep voltammetry (LSV) was first conducted in N2- and Ar-saturated 0.05 M H2SO4 electrolyte (Fig. 3a). The LSV curves reveal higher current density in N2-saturated electrolyte compared to those in Ar-saturated between 0 and −0.7 V (versus RHE), suggesting that n-W/V2−xCTy is active for the NRR. Subsequently, the NH3 yields and corresponding FEs of n-W/V2−xCTy were evaluated at various applied potentials by chronoamperometry tests (Fig. S16a†) and UV-Vis absorption spectroscopy (Fig. 3b and S17†). As shown in Fig. 3c, the NH3 yield and FE reach the maximum values of 121.8 μg h−1 mg−1 and 34.2% at −0.1 V (versus RHE). It is noteworthy that as the applied potentials shift towards more negative values, there is a discernible decline in both the yield of NH3 and its FE. This phenomenon is primarily attributed to the intensified competition between the HER and the desired reaction on the catalyst surface. Moreover, n-W/V2−xCTy demonstrates exceptional selectivity for NH3 production, as verified by the absence of detectable N2H4 (Fig. S16b†). To validate the accuracy of our detection method, we further quantified the NH3 production using 1H nuclear magnetic resonance (1H NMR) analysis (Fig. S18†). The results showed that the NH3 yield and FE of n-W/V2−xCTy stood at 124.2 μg h−1 mg−1 and 34.9% at −0.1 V versus RHE, respectively. These results were in excellent agreement with those obtained through the indophenol blue method (Fig. 3d and S19†). We further tested NO3− concentration in the system (Fig. S20 and S21†). Based on the calibration curve, the calculated NO3− yield is 0.68 μg mL−1, which is essentially negligible in the context of our experimental results. Since N-containing species (TBAOH) were utilized during the material synthesis, we conducted a test to evaluate the nitrogen reduction reaction (NRR) performance of n-W/V2−xCTy at −0.1 V (versus RHE) under an Ar atmosphere. As depicted in Fig. S22,† the calculated NH3 yields and FEs of n-W/V2−xCTy were 4.2 μg h−1 mg−1 and 4.5%, respectively, implying negligible ammonia production.
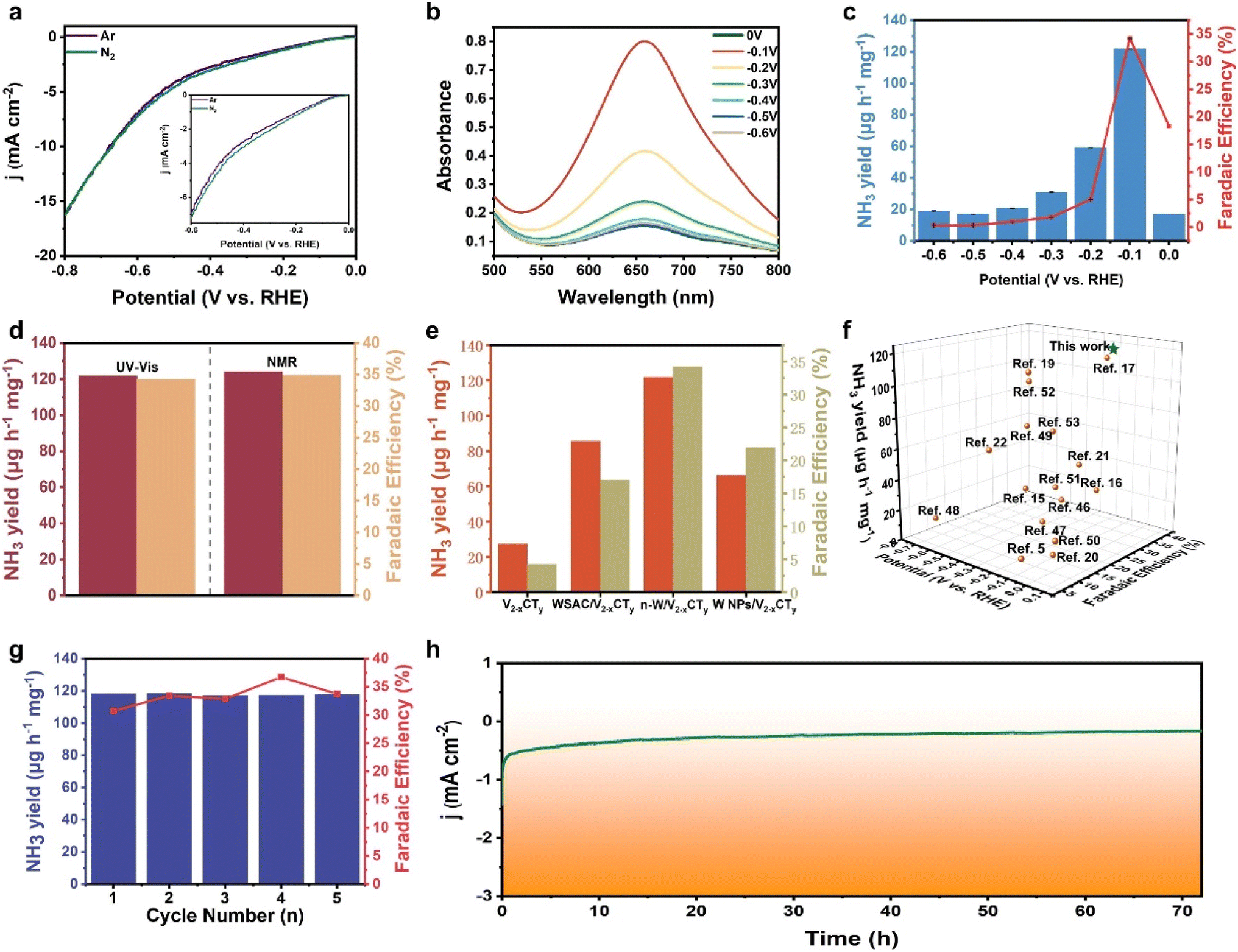 | ||
| Fig. 3 (a) LSV curves in Ar- and N2-saturated 0.05 M H2SO4 solution. (b) UV-Vis absorption spectra of the electrolytes at different potentials. (c) NH3 yields and FEs at different applied potentials. (d) NH3 yields and FEs measured on n-W/V2−xCTy by NMR and UV-vis methods at −0.1 V (versus RHE). (e) NH3 yields and FEs of V2−xCTy, WSAC/V2−xCTy, n-W/V2−xCTy, and W NPs/V2−xCTy at −0.1 V (versus RHE). (f) Comparison of NH3 yields and FEs of recently developed NRR electrocatalysts at respective potentials. (g) NH3 yield and FE in cycle tests of n-W/V2−xCTy at −0.1 V (versus RHE). (h) Chronoamperometric curve of n-W/V2−xCTy during 72 h electrolysis. | ||
To highlight the superior performance of the n-W/V2−xCTy electrocatalyst, we examined the NRR activity of all comparative samples (including pure V2−xCTy nanomeshes, WSAC/V2−xCTy, and W-NPs/V2−xCTy) across varying applied potentials. By analyzing the UV-Vis absorption spectra (Fig. S23–S28†), the optimal NH3 yield and FE for each sample were determined and are presented in Fig. 3e. It can be concluded without doubt that the electrocatalyst of n-W/V2−xCTy provides the highest NRR activity. This phenomenon clearly indicates that the incorporation of neighboring W atoms in n-W/V2−xCTy significantly boosted its NRR catalytic performance. Moreover, we compared the catalytic performance of n-W/V2−xCTy with that of state-of-the-art NRR SACs (Fig. 3f and Table 1). n-W/V2−xCTy possesses outstanding NRR performance at minimal potentials, presenting significant application potential. Besides high activity, an ideal NRR catalyst should be stable during the catalytic process, enabling sustained NH3 production. We first investigated the NRR stability of n-W/V2−xCTy by conducting consecutive electrolysis recycling and a 72 h long-term electrocatalytic test at a potential of −0.1 V (versus RHE). As shown in Fig. 3g and S29,† the NH3 yield and FE of n-W/V2−xCTy varied slightly over 5-cycle tests, demonstrating the excellent stability of n-W/V2−xCTy toward NRR. The long-term electrocatalytic test result indicates a negligible decay in current density over 72 h, also suggesting good stability (Fig. 3h). In addition, the XRD and XPS analysis results of the n-W/V2−xCTy electrocatalyst remained essentially unchanged before and after our stability test, further highlighting their structural stability (Fig. S30 and S31†). These results collectively reveal the superior stability of n-W/V2−xCTy for NRR.
| Catalyst | Electrolyte | Potential (V vs. RHE) | NH3 yield (μg h−1 mg−1) | FE (%) | Ref |
|---|---|---|---|---|---|
| n-W/V2−xCTy | 0.05 M H2SO4 | −0.1 | 121.8 | 34.2 | This work |
| Fe–S–C 700 | 0.1 M KOH | −0.1 | 8.8 | 6.1 | 5 |
| Ru SAs/Ti3C2O | 0.1 M HCl | −0.2 | 27.56 | 23.3 | 46 |
| Fe1Sx@TiO2 | 0.1 M HCl | −0.2 | 18.3 | 17.3 | 47 |
| SA-Ru@rGO/NC | 0.1 M HCl | −0.3 | 110.1 | 17.9 | 19 |
| W–NO/NC | 0.5 M LiClO4 | −0.7 | 12.62 | 8.35 | 48 |
| Mn–O3N1/PC | 0.1 M HCl | −0.35 | 66.41 | 8.91 | 22 |
| PdCu/NC | 0.05 M H2SO4 | −0.45 | 69.2 | 24.8 | 49 |
| MoSA/CMF-S | 0.1 M HCl | −0.2 | 46.6 | 28.9 | 21 |
| PdFe1 | 0.5 M LiClO4 | −0.2 | 111.9 | 37.8 | 17 |
| FeSA-NSC-900 | 0.1 M HCl | −0.4 | 30.4 | 21.9 | 15 |
| Ru SAs/g-C3N4 | 0.5 M NaOH | 0.05 | 23 | 8.3 | 50 |
| Zn/Fe–N–C | 0.1 M PBS | −0.3 | 30.5 | 26.5 | 51 |
| Fe-(O–C2)4 | 0.1 M KOH | −0.1 | 32.1 | 29.3 | 16 |
| Fe–B/N–C | 0.1 M HCl | −0.4 | 100.1 | 23 | 52 |
| Cu SAs/TiO2 | 0.5 M K2SO4 | −0.05 | 6.26 | 12.88 | 20 |
| Rh SA/GDY | 0.005 M H2SO4, 0.1 M K2SO4, 0.01 M ascorbic acid | −0.2 | 74.15 | 20.36 | 53 |
In order to validate the electrocatalytic activity of n-W/V2−xCTy and decode the origin of its NRR performance, DFT calculations were performed to investigate the reaction mechanisms of NRR as well as the electronic properties of n-W/V2−xCTy and V2−xCTy. Based on the experimental characterization results, we constructed theoretical structural models of pure V2−xCTy, WSAC/V2−xCTy, and WDAC/V2−xCTy. Under ambient conditions, N2 molecules may adsorb on the catalyst either in side-on or end-on configurations. We first calculated the adsorption energy for different adsorption modes and the results are shown in Fig. 4a. The end-on configuration possesses a lower adsorption energy, which implies that the active site prefers to adsorb N2 in an end-on configuration. According to the above results, we probed into the NRR route with an end-on configuration. Distal and alternative pathways were envisaged for the electrocatalytic conversion of N2 to NH3 (Fig. 4c and S32†). During the reaction sequences, the pre-reacted NN* can be transformed into NNH* by capturing a proton/electron (H+/e−) pair, a step that ordinarily serves as a potential-limiting step (PDS) due to the involvement of inert NN. The Gibbs free energy changes (ΔG) associated with this step on pure V2−xCTy and WSAC/V2−xCTy are 0.81 and 0.71 eV, respectively, and the initial hydrogenation procedure can occur spontaneously on WDAC/V2−xCTy due to the presence of the doubly active site. Subsequently, the adsorbed NNH* intermediate can be hydrogenated into either NNH2* or NHNH*. For the distal pathway, NNH2* is further hydrogenated into the NNH3* intermediate, and this step poses the greatest challenge for WDAC/V2−xCTy, featuring an energy barrier of 0.61 eV (Fig. S32†). In the ensuing stages, one NH3 molecule is emitted, followed by the hydrogenation of the N* intermediate via the NH* → NH2* → NH3* sequence until the release of the second NH3 molecule. By comparing the Gibbs free energy diagrams, it becomes apparent that WDAC/V2−xCTy favors the alternative pathway, while the first hydrogenation step remains the most challenge for both pure V2−xCTy and WSAC/V2−xCTy (Fig. 4c). On the surface of WDAC/V2−xCTy, the NHNH* species proceeds through the NHNH* → NHNH2* → NH2NH2* → 2NH3* sequence, with the generation of NH2NH2* being identified as the PDS (0.32 eV). Briefly, we computed the limiting potential (UL) value for these three catalysts, yielding UL values of 0.81, 0.71, and 0.32 V for pure V2−xCTy, WSAC/V2−xCTy, and WDAC/V2−xCTy, respectively. As the theoretical simulation of reaction pathways relies solely on a vacuum model, we employed both implicit and explicit solvation models to investigate the PDS of the NRR process on WDAC/V2−xCTy, aiming to validate the credibility of our theoretical calculations. As detailed in Table S3,† all PDSs identified using the implicit solvation model exhibit exothermic characteristics. However, in the vacuum model, the hydrogenation step of NHNH2* → NH2NH2* emerges as the determining PDS, possessing a limiting potential of 0.32 V. To further validate the energy requirements of this critical step, we employed the explicit solvation model. As illustrated in Fig. S33,† the introduction of water molecules results in an energy requirement of 0.25 eV for the NHNH2* → NH2NH2* step, which closely aligns with the limiting potential obtained from the vacuum model. This slight difference in overpotentials suggests that the theoretical calculations based on the vacuum model provide reliable insights into the reaction mechanism. The adsorption energies (Eads) of N2 and H on these three catalysts are presented in Fig. 4b. Considering the selectivity criterion, a catalyst with more negative Eads suggests a greater preference for the corresponding pre-processed reaction, thereby offering improved selectivity. Surprising, all three systems show more negative Eads for N2.
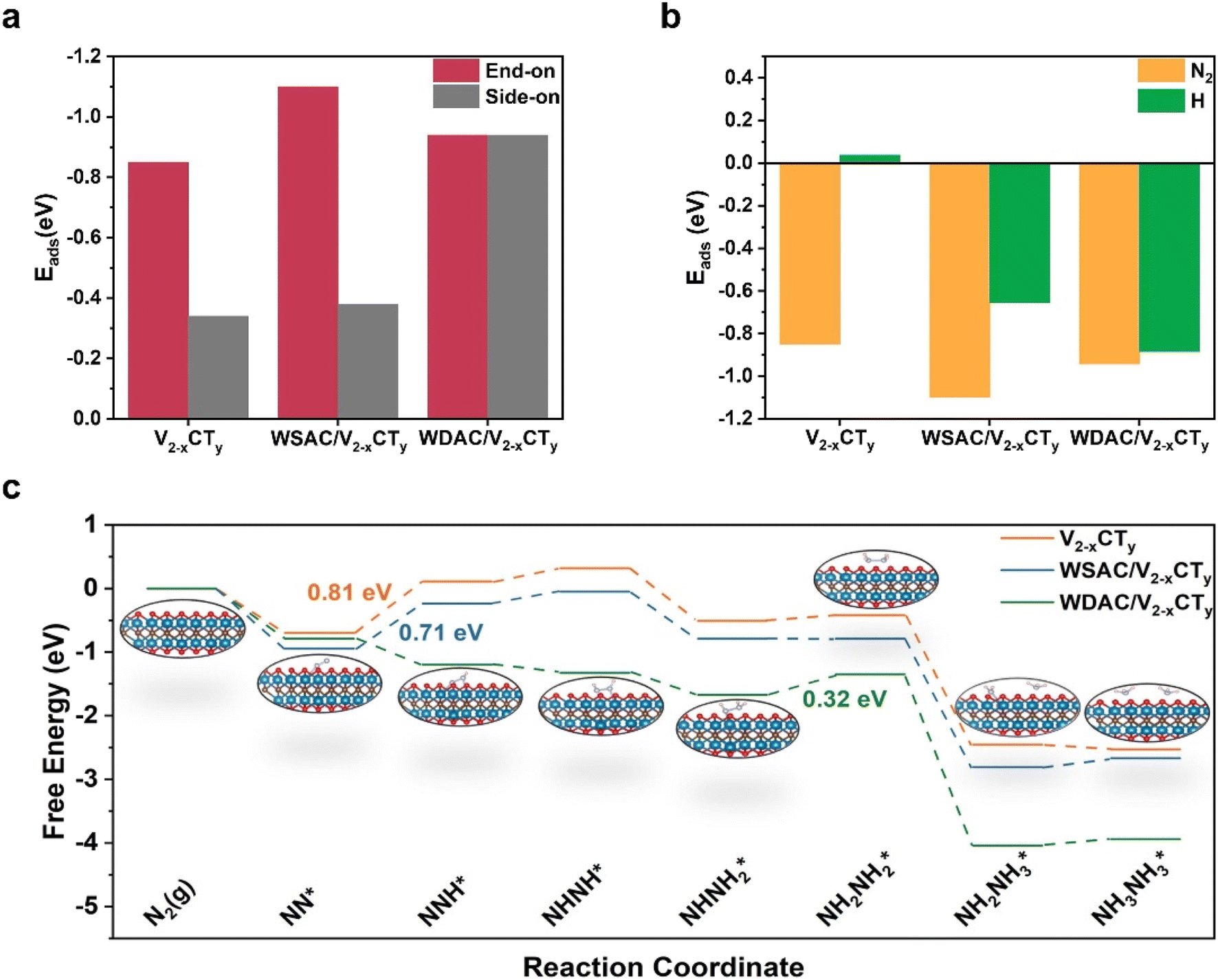 | ||
| Fig. 4 (a) The adsorption energies of N2 on V2−xCTy, WSAC/V2−xCTy, and WDAC/V2−xCTy in side-on and end-on configurations, respectively. (b) The adsorption energies of V2−xCTy, WSAC/V2−xCTy, and WDAC/V2−xCTy for N2 and H, respectively. (c) Gibbs free energy diagrams of NRR via alternating pathways on WDAC/V2−xCTy, WSAC/V2−xCTy, and V2−xCTy, respectively. | ||
We delved deeper into understanding the origin of the active site's electrocatalytic behavior by examining the charge density difference, partial density of states (PDOS), and integrated-crystal orbital Hamilton population (ICOHP). As illustrated in Fig. 5b and S37,† N2 molecules adsorbed on the active sites in an end-on configuration with substantial electron transfer. The adsorbed N2 can interact with the active sites through the so-called “donation and back-donation” process, where the active sites can donate electrons into the antibonding orbitals of N2, while N2 donates electrons back to the d orbitals of metal atoms. Bader charge calculations were employed to quantify the relationship between the extent of charge transfer and the elongation NN bond. Particularly, the W atoms of WDAC/V2−xCTy contribute more electrons to the antibonding orbitals of N2 compared to WSAC/V2−xCTy and V2−xCTy, causing a noticeable elongation of the N2 bond (1.20 Å vs. 1.12 Å for free N2). Importantly, WSAC/V2−xCTy can offer more electrons to N2 than pure V2−xCTy, and the introduction of another W atom augments the electron donor for N2 (0.169 e−, 0.187 e−, and 0.732 e− for V2−xCTy, WSAC/V2−xCTy, and WDAC/V2−xCTy, respectively). More electrons are supplied to the antibonding orbitals of N2, promoting the activation of N2. The PDOS of free N2 and these three systems is displayed in Fig. S38 and S39.† Referring to the molecular orbitals of free N2, as N2 adsorbed onto the active site, the d orbitals of W atoms push electrons to the antibonding orbitals of N2, causing a negative shift of the partially occupied 2π* orbital close to the Fermi level. The ICOHP results further illustrate the activation of N2 on the active site (Fig. 5a). The computed ICOHP for WDAC/V2−xCTy is more negative than those for WSAC/V2−xCTy and pure V2−xCTy, suggesting that the interaction between N and W is more robust in WDAC/V2−xCTy, leading to superior activation of N2 on the WDAC/V2−xCTy surface.
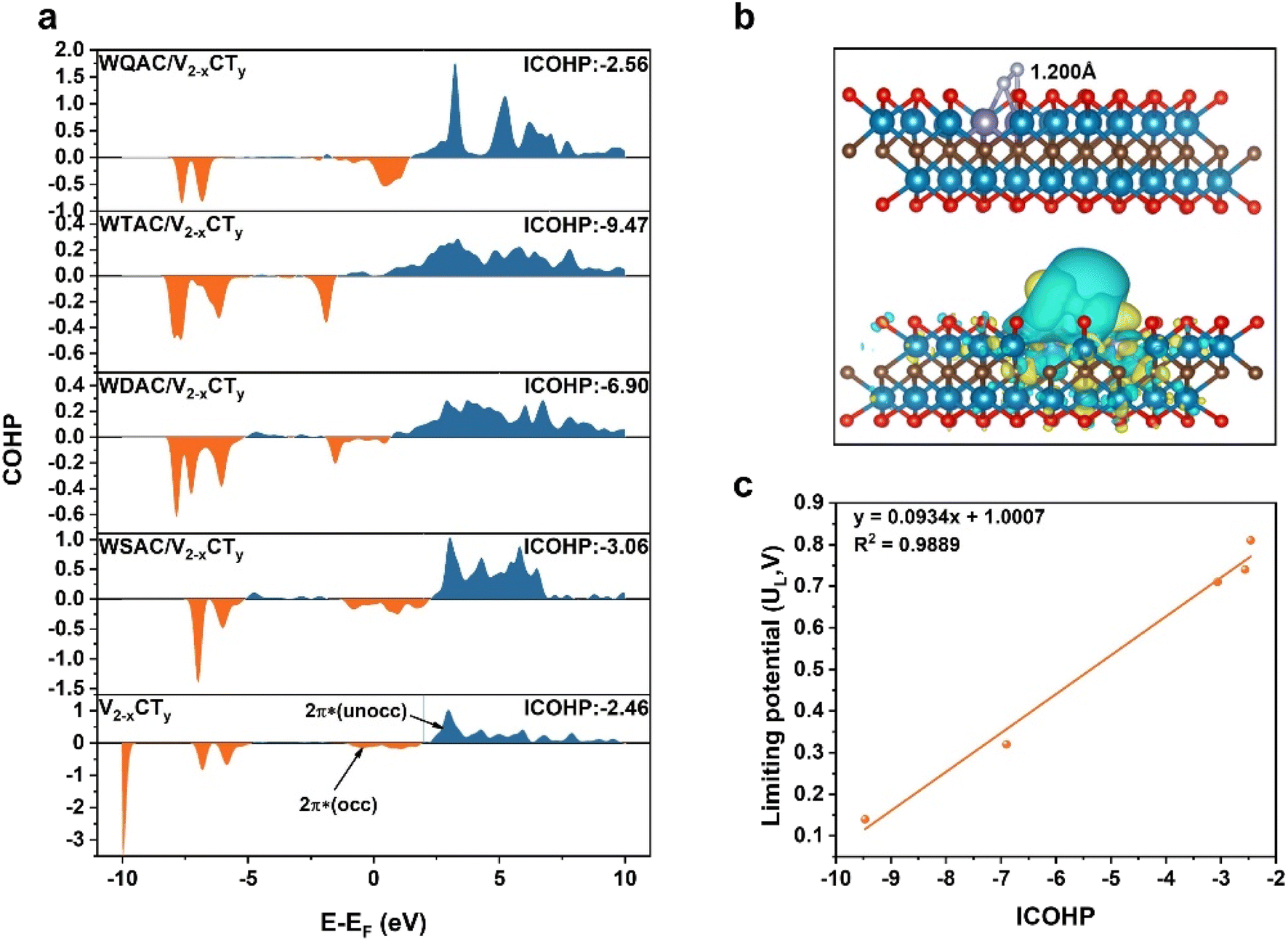 | ||
| Fig. 5 (a) The COHPs of N2 on V2−xCTy, WSAC/V2−xCTy, WDAC/V2−xCTy, WTAC/V2−xCTy, and WQAC/V2−xCTy surfaces (orange and blue denote bonding and antibonding states, respectively). (b) Optimized structures and charge density differences of N2 on the WDAC/V2−xCTy surface (cyan and yellow represent charge depletion and accumulation, respectively. The isosurface value is 0.001 e Å−3). (c) The line relationship between ICOHP and limiting potential. | ||
To quantify the relationship between the charge transfer and NRR performance. We further constructed theoretical atomic models of W triple atom catalysts (WTAC/V2−xCTy) and W quadruple atom cluster catalysts (WQAC/V2−xCTy) for comparison. The adsorption and activation of N2 on the surfaces of WTAC/V2−xCTy and WQAC/V2−xCTy were first examined. The results show that N2 also tends to adsorb on these two catalyst surfaces in an end-on configuration, and the calculated UL values for WTAC/V2−xCTy and WQAC/V2−xCTy are 0.14 and 0.74 V, respectively (Fig. S34S36†). Electronic property calculations show that WTAC/V2−xCTy and WQAC/V2−xCTy also follow the rule that strong d–2π* coupling leads to higher NRR activity (Fig. S37 and S39†). By correlating the ICOHP values with the calculated UL values, the results reveal a strong linear relationship between the two (y = 0.0934x + 1.0007, R2 = 0.9889), providing a practical descriptor for predicting the NRR performance (Fig. 5c).
Conclusions
In summary, n-W/V2−xCTy was rationally synthesized by a self-capture approach. HAADF-STEM and EXAFS results revealed that W single/dual atoms were successfully embedded in V single vacancies or vacancy clusters, forming stable W–C bonds with three neighboring C atoms. A notable NH3 yield of 121.8 μg h−1 mg−1 and a substantial FE of 34.2% at −0.1 V (versus RHE) were achieved, establishing a new record at this potential level. This exceptional performance exceeds the catalytic capabilities of V2−xCTy, WSAC/V2−xCTy, and W NPs/V2−xCTy, unequivocally demonstrating that the integration of neighboring W atoms in n-W/V2−xCTy substantially enhances its NRR catalytic performance. Moreover, the exceptional stability of n-W/V2−xCTy with a negligible decay of current density over 72 h primarily stems from the robust W–C bond. DFT calculations demonstrated that the additional W atom contributes to the improvement of the reactivity of WDAC/V2−xCTy by donating more d electrons to the 2π* orbitals of N2. This donation results in the better activation of the inert NN bonds and lowering of the reaction energy barrier (the calculated UL values for pure V2−xCTy, WSAC/V2−xCTy, and WDAC/V2−xCTy are 0.81, 0.71, and 0.32 V, respectively). Furthermore, a quantitative investigation of the correlation between the calculated UL and the amount of charge transfer was conducted. The results indicated that there exists a well-defined linear relationship between the calculated UL and ICOHP values (y = 0.0934x + 1.0007, R2 = 0.9889). Our finding opens up new avenues for the design of higher activity atomic catalysts and provides novel insights into understanding the catalytic mechanism of NRR.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Author contributions
Y. W. conceived and supervised the research; J. W. and C. Z. F. synthesized the catalysts and performed the catalysis experiments; J. W. performed DFT calculations; D. L. and H. J. Z. provided experimental measurements and STEM images; Y. W., C. Z. F., H. J. Z. and J. W. analyzed the data and wrote the paper; Y. W. discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities (0301005202017, 2018CDQYFXCS0017, and 106112017CDJXSYY0001), Thousand Young Talents Program of the Chinese Central Government (Grant No. 0220002102003), National Natural Science Foundation of China (NSFC, Grant No. 22371022, 22271029, U19A20100, 21971027, 21373280, and 21403019), Beijing National Laboratory for Molecular Sciences (BNLMS) and Hundred Talents Program at Chongqing University (Grant No. 0903005203205), The State Key Laboratory of Mechanical Transmissions Project (SKLMT-ZZKT-2017M11), Natural Science Foundation of Chongqing (Grant No. cstc2019jcyj-msxmX0426), and Science and Technology Research Project of Education Agency in Chongqing (Grant No. KJZDK201800102).References
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm and W. F. Schneider, Science, 2018, 360, eaar6611 CrossRef.
- L. Wang, M. Xia, H. Wang, K. Huang, C. Qian, C. T. Maravelias and G. A. Ozin, Joule, 2018, 2, 1055–1074 CrossRef CAS.
- B. C. Cui, S. Licht, B. Wang, F.-F. Li, J. Lau and S. Liu, Science, 2014, 345, 637–640 CrossRef.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- M. I. Ahmed, L. J. Arachchige, Z. Su, D. B. Hibbert, C. Sun and C. Zhao, ACS Catal., 2022, 12, 1443–1451 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, 146 CrossRef.
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS.
- H. S. Kim, J. Choi, J. Kong, H. Kim, S. J. Yoo and H. S. Park, ACS Catal., 2020, 11, 435–445 CrossRef.
- J. Wan, Y. Wang, W. Tian, H. Zhang and Y. Wang, Appl. Surf. Sci., 2021, 569, 151020 CrossRef CAS.
- X. Guo, J. Gu, S. Lin, S. Zhang, Z. Chen and S. Huang, J. Am. Chem. Soc., 2020, 142, 5709–5721 CrossRef CAS PubMed.
- M. Li, Y. Cui, X. Zhang, Y. Luo, Y. Dai and Y. Huang, J. Phys. Chem. Lett., 2020, 11, 8128–8137 CrossRef CAS.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- J. Li, M. F. Stephanopoulos and Y. Xia, Chem. Rev., 2020, 120, 11699–11702 CrossRef CAS PubMed.
- Y. Li, Y. Ji, Y. Zhao, J. Chen, S. Zheng, X. Sang, B. Yang, Z. Li, L. Lei, Z. Wen, X. Feng and Y. Hou, Adv. Mater., 2022, 34, e2202240 CrossRef PubMed.
- S. Zhang, M. Jin, T. Shi, M. Han, Q. Sun, Y. Lin, Z. Ding, L. R. Zheng, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Angew Chem. Int. Ed. Engl., 2020, 59, 13423–13429 CrossRef CAS.
- X. Li, P. Shen, Y. Luo, Y. Li, Y. Guo, H. Zhang and K. Chu, Angew Chem. Int. Ed. Engl., 2022, 61, e202205923 CrossRef CAS PubMed.
- H. C. Tao, C. Choi, L. X. Ding, Z. Jiang, Z. S. Hang, M. W. Jia, Q. Fan, Y. N. Gao, H. H. Wang, A. W. Robertson, S. Hong, Y. S. Jung, S. Z. Liu and Z. Y. Sun, Chem, 2019, 5, 204–214 CAS.
- Y. Chen, R. Xu, Y. Li, L. Cai, Y. Yang, Y. Zheng, C. Zuo, H. Huang, Z. Wen and Q. Wang, Chem. Commun., 2023, 59, 5403–5406 RSC.
- Z. Q. Zhao, K. Li, J. Liu, J. J. Mao and Y. Q. Lin, Small, 2023, 19, e2206626 CrossRef.
- L. Li, W. Yu, W. Gong, H. Wang, C.-L. Chiang, Y. Lin, J. Zhao, L. Zhang, J.-M. Lee and G. Zou, Appl. Catal., B, 2023, 321, 122038 CrossRef CAS.
- L. Han, M. Hou, P. Ou, H. Cheng, Z. Ren, Z. Liang, J. A. Boscoboinik, A. Hunt, I. Waluyo, S. Zhang, L. Zhuo, J. Song, X. Liu, J. Luo and H. L. Xin, ACS Catal., 2020, 11, 509–516 CrossRef.
- Z. W. Chen, J.-M. Yan and Q. Jiang, Small Methods, 2019, 3, 1800291 CrossRef.
- W. H. Li, J. Yang and D. Wang, Angew Chem. Int. Ed. Engl., 2022, 61, e202213318 CrossRef CAS PubMed.
- A. Pedersen, J. Barrio, A. Li, R. Jervis, D. J. L. Brett, M. M. Titirici and I. E. L. Stephens, Adv. Energy Mater., 2021, 12, 2102715 CrossRef.
- L. J. Arachchige, Y. Xu, Z. Dai, X. Zhang, F. Wang and C. Sun, J. Phys. Chem. C, 2020, 124, 15295–15301 CrossRef.
- M. Zhang, Z. Hu, L. Gu, Q. Zhang, L. Zhang, Q. Song, W. Zhou and S. Hu, Nano Res., 2020, 13, 3206–3211 CrossRef CAS.
- P. Peljo, L. Murtomaki, T. Kallio, H. J. Xu, M. Meyer, C. P. Gros, J. M. Barbe, H. H. Girault, K. Laasonen and K. Kontturi, J. Am. Chem. Soc., 2012, 134, 5974–5984 CrossRef CAS PubMed.
- Y. Zhang, J. Hu, C. Zhang, Y. Liu, M. Xu, Y. Xue, L. Liu and M. K. H. Leung, J. Mater. Chem. A, 2020, 8, 9091–9098 RSC.
- Y. N. Gong, C. Y. Cao, W. J. Shi, J. H. Zhang, J. H. Deng, T. B. Lu and D. C. Zhong, Angew Chem. Int. Ed. Engl., 2022, 61, e202215187 CrossRef CAS PubMed.
- W. Zhao, C. Luo, Y. Lin, G.-B. Wang, H. M. Chen, P. Kuang and J. Yu, ACS Catal., 2022, 12, 5540–5548 CrossRef CAS.
- J. Wan, Y. Wang, H. Zhang and Y. Wang, Chem. Eng. J., 2023, 470, 144151 CrossRef CAS.
- E. Ghasali, Y. Orooji, A. Azarniya, M. Alizadeh, M. Kazem-zad and T. Ebadzadeh, Appl. Surf. Sci., 2021, 542, 148538 CrossRef CAS.
- F. Liu, J. Zhou, S. Wang, B. Wang, C. Shen, L. Wang, Q. Hu, Q. Huang and A. Zhou, J. Electrochem. Soc., 2017, 164, A709–A713 CrossRef CAS.
- A. VahidMohammadi, A. Hadjikhani, S. Shahbazmohamadi and M. Beidaghi, ACS Nano, 2017, 11, 11135–11144 CrossRef CAS.
- S. Zhou, Y. Zhao, R. Shi, Y. Wang, A. Ashok, F. Heraly, T. Zhang and J. Yuan, Adv. Mater., 2022, 34, e2204388 CrossRef PubMed.
- D. Zhao, Z. Chen, W. Yang, S. Liu, X. Zhang, Y. Yu, W.-C. Cheong, L. Zheng, F. Ren, G. Ying, X. Cao, D. Wang, Q. Peng, G. Wang and C. Chen, J. Am. Chem. Soc., 2019, 141, 4086–4093 CrossRef CAS PubMed.
- Z. Li, Y. Cui, Z. Wu, C. Milligan, L. Zhou, G. Mitchell, B. Xu, E. Shi, J. T. Miller, F. H. Ribeiro and Y. Wu, Nat. Catal., 2018, 1, 349–355 CrossRef CAS.
- Z. Zhang, H. Li, G. Zou, C. Fernandez, B. Liu, Q. Zhang, J. Hu and Q. Peng, ACS Sustainable Chem. Eng., 2016, 4, 6763–6771 CrossRef CAS.
- Z. Fu, N. Wang, D. Legut, C. Si, Q. Zhang, S. Du, T. C. Germann, J. S. Francisco and R. Zhang, Chem. Rev., 2019, 119, 11980–12031 CrossRef CAS PubMed.
- I. Persson, J. Halim, T. W. Hansen, J. B. Wagner, V. Darakchieva, J. Palisaitis, J. Rosen and P. O. Å. Persson, Adv. Funct. Mater., 2020, 30, 1909005 CrossRef CAS.
- J. Yan, L. Kong, Y. Ji, J. White, Y. Li, J. Zhang, P. An, S. Liu, S. T. Lee and T. Ma, Nat. Commun., 2019, 10, 2149 CrossRef PubMed.
- W. Chen, J. Pei, C. T. He, J. Wan, H. Ren, Y. Wang, J. Dong, K. Wu, W. C. Cheong, J. Mao, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, e1800396 CrossRef.
- H. Funke, A. C. Scheinost and M. Chukalina, Phys. Rev. B, 2005, 71, 094110 CrossRef.
- H. Funke, M. Chukalina and A. C. Scheinost, J. Synchrotron Radiat., 2007, 14, 426–432 CrossRef CAS PubMed.
- G. Chen, M. Ding, K. Zhang, Z. Shen, Y. Wang, J. Ma, A. Wang, Y. Li and H. Xu, ChemSusChem, 2022, 15, e202102352 CrossRef CAS PubMed.
- J. Chen, Y. Kang, W. Zhang, Z. Zhang, Y. Chen, Y. Yang, L. Duan, Y. Li and W. Li, Angew Chem. Int. Ed. Engl., 2022, 61, e202203022 CrossRef CAS PubMed.
- Y. Gu, B. Xi, W. Tian, H. Zhang, Q. Fu and S. Xiong, Adv. Mater., 2021, 33, e2100429 CrossRef.
- L. Han, Z. Ren, P. Ou, H. Cheng, N. Rui, L. Lin, X. Liu, L. Zhuo, J. Song, J. Sun, J. Luo and H. L. Xin, Angew Chem. Int. Ed. Engl., 2021, 60, 345–350 CrossRef CAS PubMed.
- B. Yu, H. Li, J. White, S. Donne, J. Yi, S. Xi, Y. Fu, G. Henkelman, H. Yu, Z. Chen and T. Ma, Adv. Funct. Mater., 2019, 30, 1905665 CrossRef.
- L. Zhang, G. Fan, W. Xu, M. Yu, L. Wang, Z. Yan and F. Cheng, Chem. Commun., 2020, 56, 11957–11960 RSC.
- Y. Zhao, S. Zhang, C. Han, Q. Lu, Q. Fu, H. Jiang, L. Yang, Y. Xing, Q. Zheng, J. Shen, L. Yan and X. Zhao, Chem. Eng. J., 2023, 468, 143517 CrossRef CAS.
- H. Zou, W. Rong, S. Wei, Y. Ji and L. Duan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 29462–29468 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03612c |
| This journal is © The Royal Society of Chemistry 2024 |