
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the catalytic performances of metal-doped Ta2O5 catalysts for acidic oxygen evolution reaction with computations†
Congcong
Han
 ab and
Tao
Wang
*bcd
ab and
Tao
Wang
*bcd
aDepartment of Chemistry, Zhejiang University, Hangzhou 310058, Zhejiang Province, China
bCenter of Artificial Photosynthesis for Solar Fuels and Department of Chemistry, School of Science and Research Center for Industries of the Future, Westlake University, 600 Dunyu Road, Hangzhou 310030, Zhejiang Province, China. E-mail: twang@westlake.edu.cn
cInstitute of Natural Sciences, Westlake Institute for Advanced Study, 18 Shilongshan Road, Hangzhou 310024, Zhejiang Province, China
dDivision of Solar Energy Conversion and Catalysis at Westlake University, Zhejiang Baima Lake Laboratory Co., Ltd, Hangzhou 310000, Zhejiang, China
First published on 6th August 2024
Abstract
The design of stable and active alternative catalysts to iridium oxide for the anodic oxygen evolution reaction (OER) has been a long pursuit in acidic water splitting. Tantalum pentoxide (Ta2O5) has the merit of great acidic stability but poor OER performance, yet strategies to improve its intrinsic OER activity are highly desirable. Herein, by using density functional theory (DFT) calculations combined with aqueous stability assessment from surface Pourbaix diagrams, we systematically evaluated the OER activity and acidic stability of 14 different metal-doped Ta2O5 catalysts. Apart from the experimentally reported Ir-doped Ta2O5, we computationally identified Ru- and Nb-doped Ta2O5 catalysts as another two candidates with reasonably high stability and activity in acidic OER. Our study also underscores the essence of considering stable surface states of catalysts under working conditions before a reasonable activity trend can be computationally achieved.
1. Introduction
Water splitting for hydrogen (H2) production driven by sustainable energies plays a vital role in the roadmap of carbon neutrality due to the high energy density and zero emission during the utilization of H2.1–3 Currently, multiple strategies with different devices have been developed for effective water electrolysis, such as alkaline water electrolyzer (AWE),4 polymer electrolyte membrane water electrolyzer (PEM-WE),5 solid oxide water electrolyzer (SOWE)6 and anion exchange membrane water electrolyzer (AEM-WE).7 Among them, PEM-WE shows advantages of high current densities, high compatibility to intermittent electricity from sustainable energy and a compact cell with a small footprint.8 Unfortunately, the large-scale application of PEM-WEs is hindered by the heavy reliance on the scarce noble-metals platinum (Pt) and iridium (Ir) as the cathode for the hydrogen evolution reaction (HER) and anode oxygen evolution reaction (OER) catalyst, respectively.9–12 Indeed, this fundamental obstacle hampering the further development of PEM-WEs triggered extensive research efforts in developing earth-abundant catalysts as alternatives to Pt and IrOx.Currently, a group of non-noble metal oxides, particularly 3d transition metal oxides, have been experimentally identified as promising acidic OER catalysts,13–17 such as CoMnOx,18 γ-MnO2,19,20 Ti–MnO2,21 Co3O4,22,23 CoFePbOx,24 MnxSb1−xOz,25 and LaMn@Co-ZIF.26 In principle, the ideal OER catalyst should have high acidic stability, high activity, and low cost, while most candidates only meet two of the three merits. From the viewpoint of practical application, high acidic stability should be the highest priority. Among the reported non-noble metal oxides in the literature, tantalum pentoxide (Ta2O5) is extremely stable in acid but has poor OER activity.27,28 In principle, Ta2O5 would be an ideal candidate if adequate strategies could be used to improve its OER activity while preserving its high stability, despite having a wide bandgap in the bulk and poor charge transport.29 Noteworthily, economic viability should be considered before using Ta2O5 as a practical OER catalyst because Ta also has essential applications in resistive switching memories as a high-tech material.30 Indeed, doping has offered a promising avenue for improving the catalytic activity of materials by altering the d-orbital electronic structure, which governs bond formation or breakage involving oxygen-containing intermediates at active sites.31–33 Cationic doping and anionic doping have already been extensively explored to enhance the intrinsic OER activity of Ru- and Ir-based oxides.34–46 Doping with various metal elements, including Ti,47 Ni,48,49 Co,50 La,26 Fe,48,51 Zn,52 Cu,53 Ce,54 Mn,26,55 and Nb,56,57 has shown the ability to enhance the acidic OER activity and stability of pristine materials. Meanwhile, recent advancements in computations and machine learning have played a crucial role in understanding the fundamental principles governing acid-stable OER catalysts and also facilitated the rational design of materials with improved performance and durability.58–66
In this work, we focus on levering computational approaches combined with the doping strategy to understand the acidic OER mechanism and activity of metal-doped Ta2O5 (M = Ti, V, Cr, Mn, Fe, Co, Ni, Zr, Nb, Mo, Sn, Sb, Ru, and Ir). Our systematic density functional theory (DFT) calculations of the adsorbate evolution mechanism (AEM) on the 14 M-doped Ta2O5 catalysts, as well as aqueous stability evaluation, clearly revealed their stability and activity trend in acidic OER, where the Ir-, Ru-, and Nb-doped Ta2O5 systems were theoretically predicted to be promising candidates with good performances. Our stability analysis with surface Pourbaix diagrams indicates the essence of considering the stable surface state of catalysts under working conditions when conducting computational screening of electrocatalysts.
2. Methods and models
All calculations were carried out by spin-polarized density functional theory (DFT) as implemented in the Vienna Ab initio Simulation Package (VASP) interfaced with the atomistic simulation environment (ASE) within the projector augmented wave method (PAW).67,68 The generalized gradient approximation (GGA) in the scheme of revised-Perdew–Burke–Ernzerhof (RPBE) was employed to describe the exchange and correlation interaction.69,70 A plane-wave basis set with a kinetic energy cut-off of 500 eV is used for all calculations. The convergence criteria for electronic energy and forces during structural optimization are 1.0 × 10−5 eV and 0.02 eV Å−1, while those for frequency calculation are 1.0 × 10−7 eV and 0.02 eV Å−1, respectively.Ta2O5 has several crystalline structures, while the λ phase with the Pbam space group was chosen for further calculation in this work. The optimized bulk structure had lattice parameters of a = 6.29 Å, b = 7.47 Å, and c = 3.84 Å, which agreed well with experimental values (a = 6.22 Å, b = 7.36 Å, and c = 3.90 Å).71–74 The (100) surface was chosen as the model for OER mechanism simulation due to its high stability and wide experimental characterization, while both Ta (Ta16O40) and O (Ta16O42) terminations were considered. The p (1 × 2) slab contained four metal layers, while the top two layers were relaxed and the bottom two layers were fixed. A vacuum layer of 15 Å is applied in the z-direction of the slab to avoid interactions. A 3 × 3 × 1 gamma centered k-point meshes was used to sample the Brillouin zone. The vibrational frequencies of adsorbates on the surfaces were analyzed, while free energies were calculated within the harmonic approximation. The OER mechanism was simulated within the framework of the computational hydrogen electrode (CHE),75 which is detailed in the ESI.† The stability of the metal-doped Ta2O5 was evaluated by using formation energy (Ef) and cohesive energy (Ecoh), where a more negative value of Ef indicates higher synthetic feasibility and a more positive value of Ecoh indicates higher thermodynamic stability.48,76 More details for calculating Ef and Ecoh are summarized in the ESI.†
3. Results and discussion
3.1 Structure and stability of metal-doped Ta2O5 (100) surfaces
To evaluate the roles of transition metals in tuning the properties of the Ta2O5 catalyst for the high-performance OER, we applied the substitutional approach by replacing the surface Ta with different metals (M = Ti, V, Cr, Mn, Fe, Co, Ni, Zr, Nb, Mo, Sn, Sb, Ru, and Ir). As shown in Fig. 1a–d, the λ-Ta2O5 (100) surface has both Ta (Ta16O40) and O (Ta16O42) terminations. For the Ta-termination shown in Fig. 1c, there are two types of surface Ta atoms with five coordination environments. Therefore, we considered two substitutional models in Fig. 1e and f (M–Ta15O40). For the O-termination shown in Fig. 1b and d, there are also two types of Ta atoms with different coordination environments on the surface shown in Fig. 1d (M–Ta15O42). Based on the doping sites shown in Fig. 1e–h, we systematically evaluated the stabilities of various metals on these sites, where the most stable doping structures of each metal on both Ta- and O-terminations are summarized in Table S2.†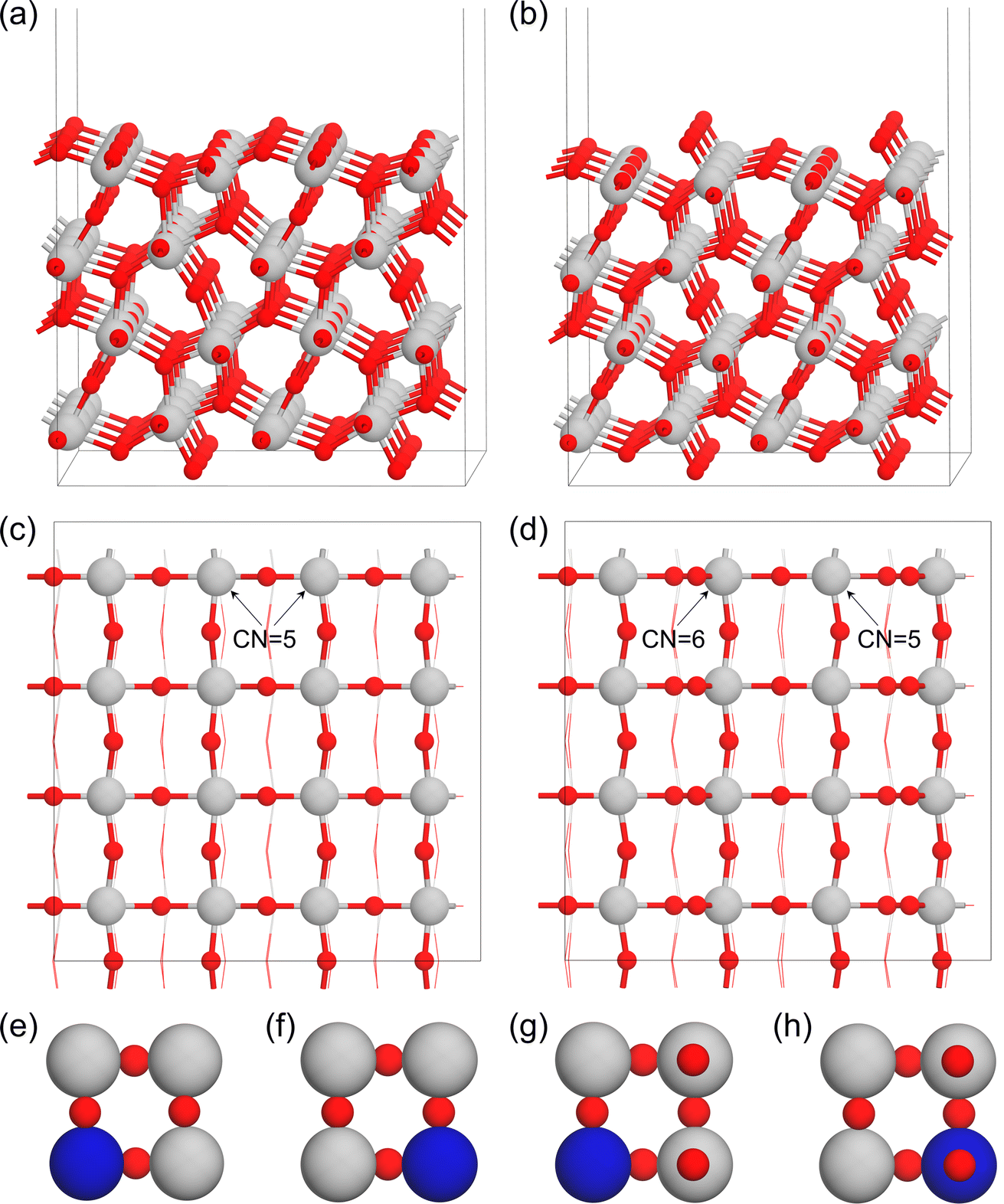 | ||
| Fig. 1 The side (a and b) and top (c and d) views of Ta- and O-terminations of Ta2O5, as well as the metal-doped Ta-termination (e and f) and O-termination (g and h) of Ta2O5. (The red, grey, and bright blue spheres represent O, Ta, and dopant atoms, respectively). | ||
In principle, the thermodynamic stability of the catalyst will determine whether it could be easily synthesized experimentally. Therefore, we calculated the formation energy (Ef) and cohesive energy (Ecoh) of all the doped systems. As shown in Fig. 2, the early transition metals (Zr, Ti, Nb, V, and Cr) doped systems generally have very negative values of Ef (Table S2†), indicating their high stabilities. Systems doped by the magnetic Fe, Co, and Ni metals, as well as the noble metal Ir and Ru metals, showed positive values of Ef, which indicates their relatively lower stabilities than the early transition metal doped Ta2O5. Considering that the Ir-doped Ta2O5 has been successfully synthesized and found to be stable and active for the OER experimentally,77 we anticipate that catalysts with lower Ef than Ir-doped Ta2O5 will show reasonable feasibility to be synthesized experimentally. In this respect, most of these 28 M-doped structures have reasonable synthetic feasibility, at least from a thermodynamic perspective. Meanwhile, the Ecoh values for all 28 M-doped structures shown in Fig. 2b and Table S2† are higher than 0.5 eV, suggesting their potential high stability upon synthesis.
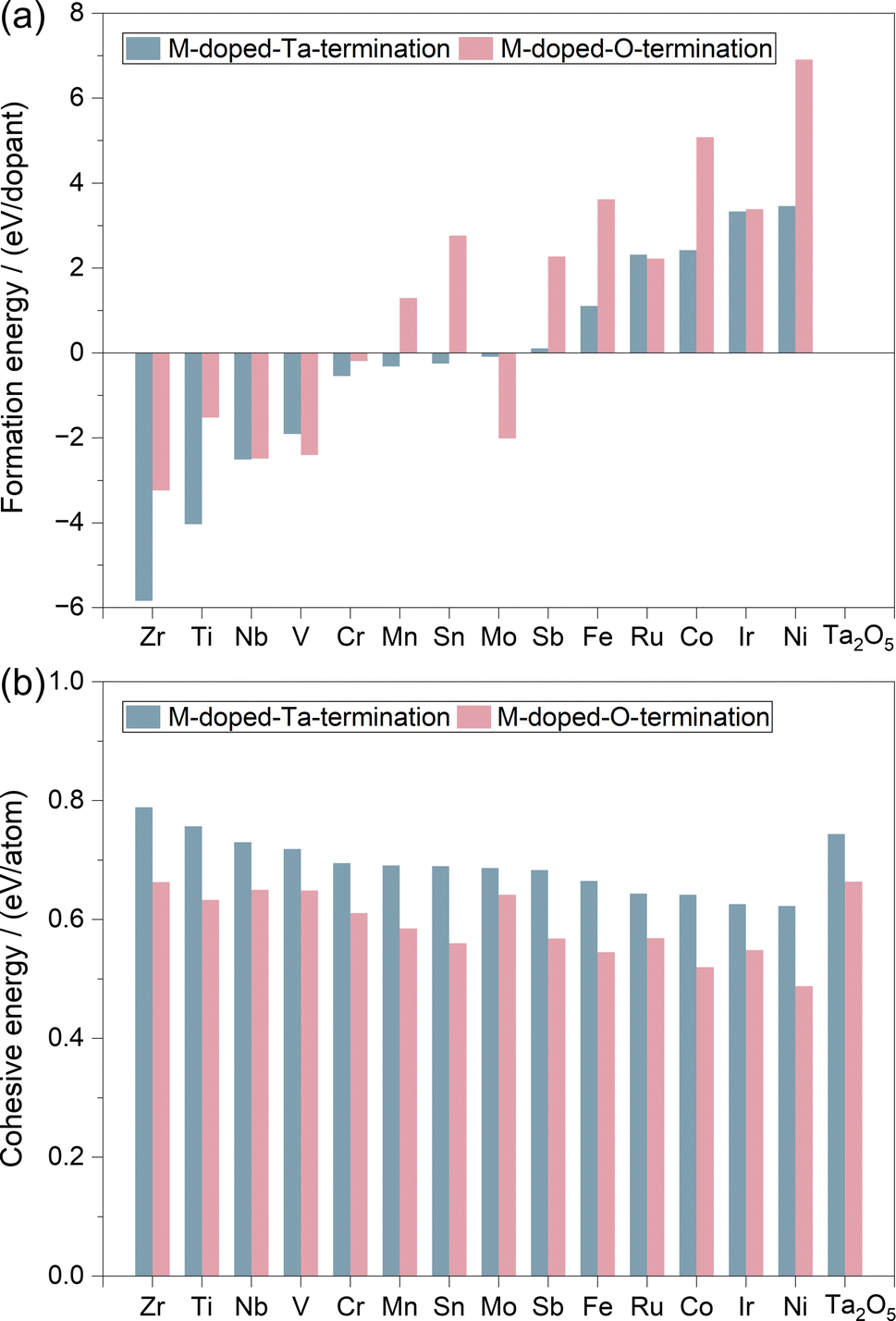 | ||
| Fig. 2 (a) Formation energy (Ef) and (b) cohesive energy (Ecoh) of M–Ta15O40 and M–Ta15O42. | ||
3.2 OER activity on the metal-doped Ta- and O-terminations of Ta2O5 (100)
To have a direct comparison of the OER activity among different catalysts, we systematically simulated the classical adsorbate evolution mechanism (AEM)78 on these M-doped Ta2O5 systems within the framework of computational hydrogen electrode (CHE) as developed by the Nørskov group79 (more details in the ESI†). As shown in Fig. 3a, the catalytic cycle starts with the deprotonation of H2O on the active site to form *OH species, followed by another deprotonation step to form the surface *O intermediate. Then, the O–O bond formation takes place via the reaction of *O with another H2O molecule to form the *OOH intermediate, accompanied by a deprotonation step. Finally, O2 is formed through the deprotonation of *OOH with the regeneration of the active site.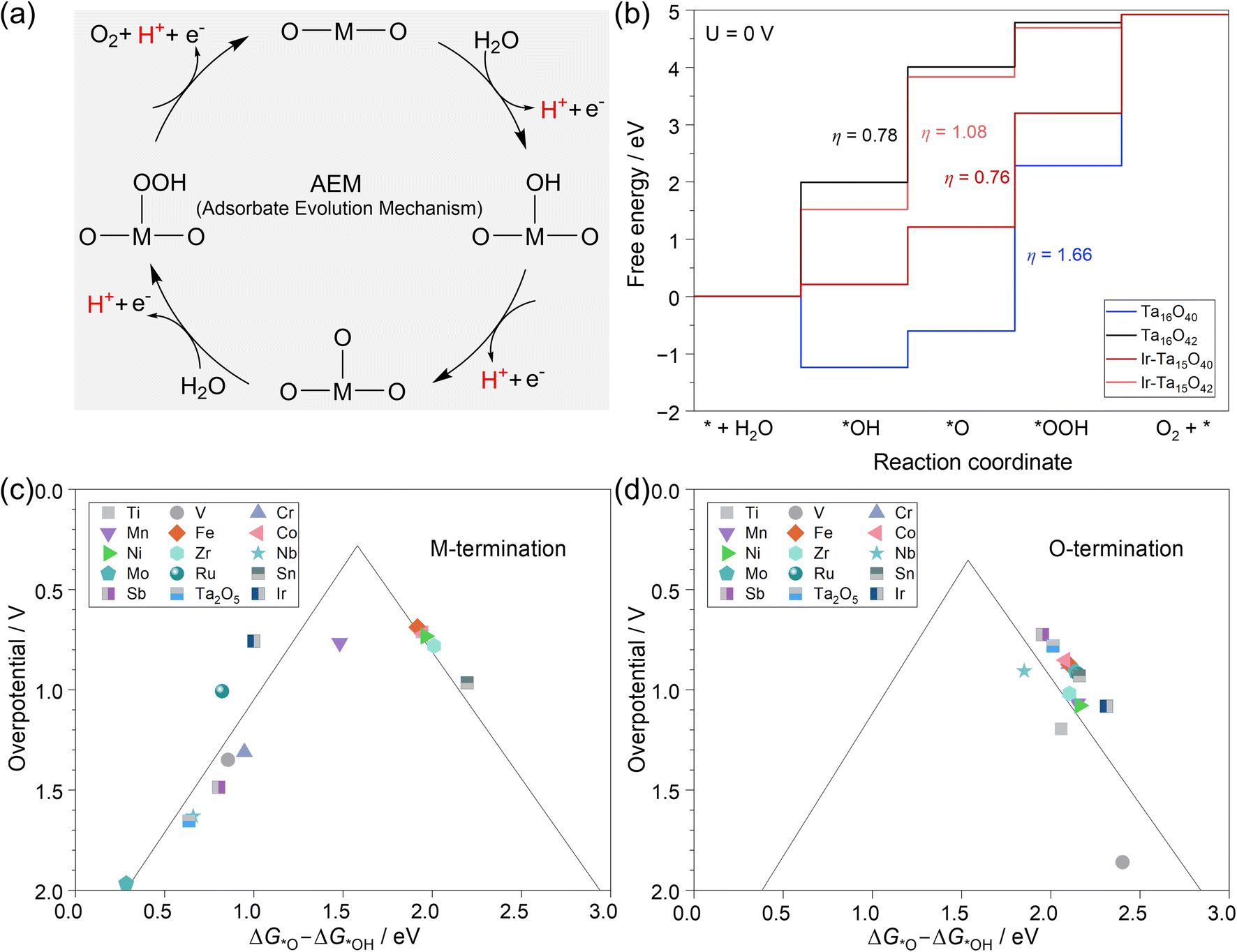 | ||
| Fig. 3 (a) Classical adsorbate evolution mechanism (AEM) for the oxygen evolution reaction; (b) free energy diagrams of the OER on the two pristine and Ir-doped terminations of the Ta2O5 (100) surface; theoretically predicted activity trends toward the OER on metal-doped (c) Ta termination (M–Ta15O40) and (d) O termination (M–Ta15O42) of the Ta2O5 (100) surface. | ||
Based on this mechanism, the reaction free energy change (ΔG) for each step was calculated at 0 V, pH = 0, and T = 298.15 K within the framework of the CHE model. Then, the potential-determining step (PDS) is typically the last step to go downhill in free energy as the potential increases. In other words, PDS is the step with the largest ΔG (ΔGPDS) among the four-step OER mechanism in Fig. 3a. The theoretical overpotential (η) can be expressed as (ΔGPDS)/e − 1.23 V. Using the pristine and Ir-doped Ta2O5 as examples, we plotted the potential free energy diagrams of the OER in Fig. 3b. It shows that each system has a different PDS and η, i.e., the PDS in the case of Ta terminations of Ta16O40 and Ir-doped Ta2O5 is *O → *OOH with the η values of 1.66 V and 0.76 V, while that in the case of the O-termination of Ta16O42 and Ir-doped Ta2O5 becomes *OH → *O with the η values of 0.78 V and 1.08 V, respectively.
As per the Sabatier principle, an ideal catalyst requires the binding strength of the key intermediate to be neither too strong nor too weak. Based on the framework established by the Nørskov group,27 there is a volcano-shaped relationship between ηOER and the value of ΔG*O – ΔG*OH among a group of materials. Our analysis in Fig. 3c and d also shows the volcanic OER activity trend among 28 M–Ta2O5 systems, but the two terminations have quite different trends. On the Ta termination in Fig. 3c and Table S3,† most of the cases are located at the left leg of the volcano due to the lower deprotonation energy of surface hydroxide, where the formation of *OOH species is the PDS. At the right leg of the volcano, the weak binding strength of *O makes the deprotonation of *OH species the PDS. Among these doped systems in the case of Ta termination, Fe-doping has the lowest η of 0.69 V, followed by Ti (0.71 V), Co (0.72 V), Ni (0.73 V), Ir (0.76 V), Mn (0.77 V), Sn (0.97 V), and Zr (0.78 V), while the rest of them are higher than 1.0 V.
In the case of the O-termination shown in Fig. 3d and Table S4,† all the metal-doped systems are located at the right leg of the volcano due to the weak binding strength with oxygen, while the deprotonation of *OH species is the PDS. Meanwhile, the activity trend of these 14 doping metals is quite different from that of the Ta termination. Among them, the Sb-doped O-termination has the lowest η of 0.72 V, followed by the pristine Ta15O42 (0.78 V).
3.3 Surface structures and OER activity of M–Ta2O5 under working conditions
The above mechanism studies on the two terminations have clearly revealed that the OER activity is tightly related to the surface structure of M–Ta2O5. Then, the question becomes, what is the real surface structure of the catalyst under working conditions? To clarify this, we further applied the surface Pourbaix diagram to describe the surface states of each M–Ta2O5 system at different pH and working potentials.More specifically, the surface of M–Ta2O5 will inevitably be covered by different species during the OER, such as *O and *OH. We systematically calculated the binding of different species on the surface of pristine and 14 M-doped models to identify the most stable surface composition of each model at a wide range of pH and potentials. More computational details are shown in the ESI.† We use the electrochemical potential window (EPW) to describe the potential range in which a catalyst can maintain its pristine state at pH = 0.80 As shown in Fig. 4a, the pristine Ta2O5 (100) surface shows an EPW less than −1.1 V vs. SHE, while it will be covered by 1/4 monolayer (ML) *OH at −1.1 V to −0.1 V as well as 1/2 ML *OH at −0.1–2.0 V. For comparison, Ir-doped Ta2O5 (100) in Fig. 4b exhibits an EPW of less than 0.1 V, while it will be covered by 1/2 ML *OH at 0.1–1.6 V as well as 1/4 ML *OH + 1/4 ML *O at >1.6 V vs. SHE. Similar surface Pourbaix diagrams of the other M-doped systems are summarized in the ESI.† Clearly, it is crucial to consider the specific surface state of the catalyst under electrochemical operating conditions. In this respect, the OER mechanism investigation was conducted on the most stable surface state of each M-doped Ta2O5, where a potential of ∼1.60 V was chosen based on experimental setups.81 The calculated free energies of intermediates and the derived overpotentials are listed in Table S6.†
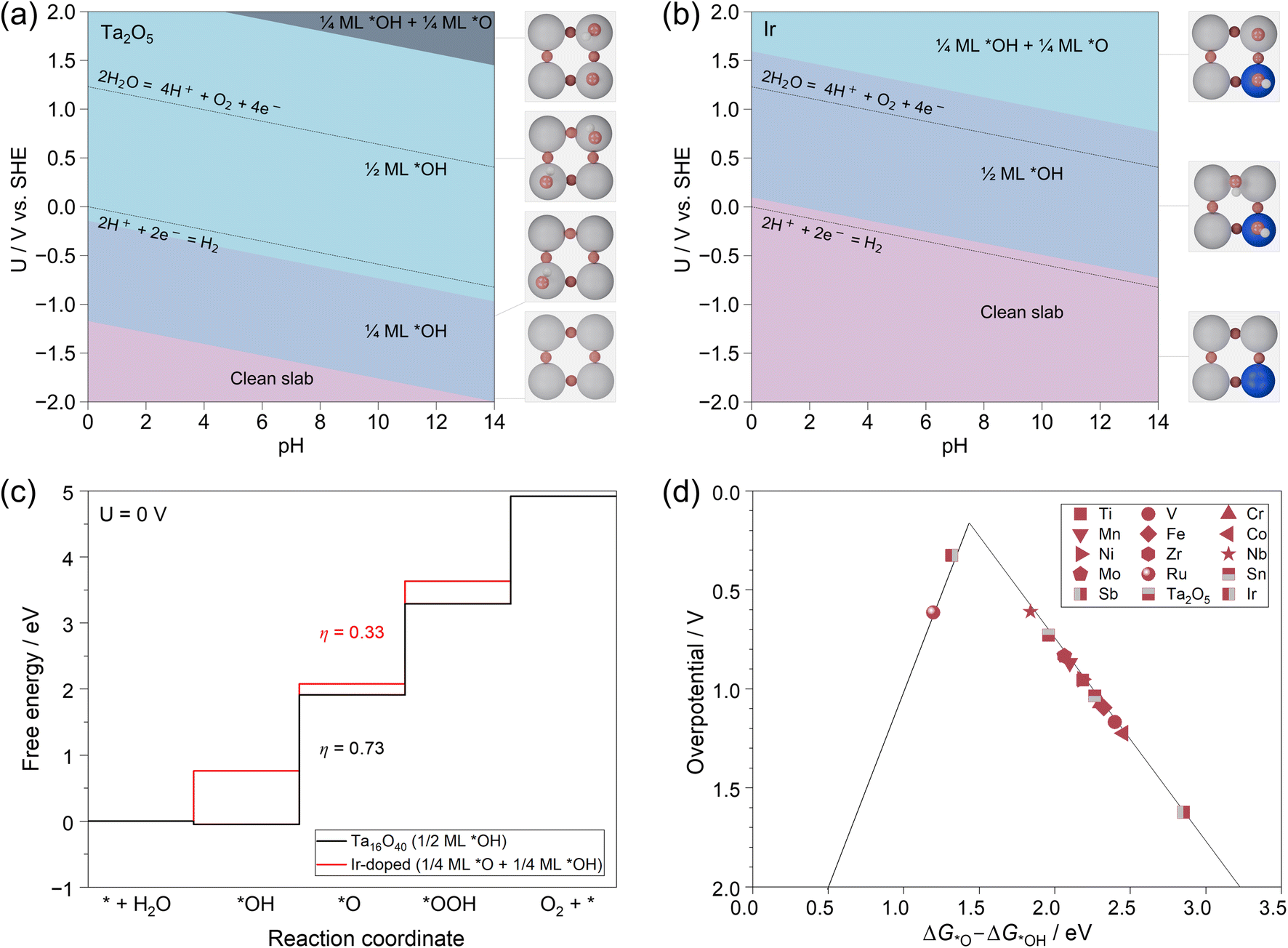 | ||
| Fig. 4 Surface Pourbaix diagrams of pristine (a) and Ir-doped (b) Ta2O5 (100) surface; free energy diagrams of the OER on the most stable surface states of pristine and Ir-doped Ta2O5 (100) surface at pH = 0 and U = 0 V (c); OER activity trend of M-doped Ta2O5 (100) at the most stable surface states determined by surface Pourbaix diagram analysis (d). | ||
As shown in Fig. 4c, the 1/2 ML *OH covered Ta-termination is the most stable state of the Ta2O5 (100) surface at pH = 0 and 1.6 V vs. SHE, where the theoretical overpotential is calculated to be 0.73 V with *OH → *O being the PDS. For the Ir-doped system, the surface is covered by 1/4 ML *O and 1/4 ML *OH at pH = 0 and 1.6 V vs. SHE, where the calculated η is only 0.33 V.
Finally, we have summarized the theoretical overpotentials calculated from the most stable surface states of each M-doped Ta2O5 in Fig. 4d for comparison. It shows that most of the systems are located at the right leg of the volcano except the Ir-doped and Ru-doped Ta2O5. Among them, the Ir-doped Ta2O5 is closest to the peak of the volcano, which has the η of only 0.33 V and an optimum ΔG*O − ΔG*OH of ∼1.32 eV. Meanwhile, the Ru-doped and Nb-doped Ta2O5 also exhibit relatively low theoretical overpotentials of 0.62 V and 0.61 V. Noteworthily, the calculated η values of these catalysts are different from those of pure Ta- and O-terminations shown in Fig. 3, again indicating the essence of considering the stable surface state of catalysts under working conditions.
4. Conclusion
In summary, we conducted density functional theory calculations to systematically investigate the thermodynamic stability, aqueous stability, and acidic OER activity of various metal-doped (metal = Ti, V, Cr, Mn, Fe, Co, Ni, Zr, Nb, Mo, Sn, Sb, Ru, Ir) Ta2O5 (100) surfaces. Our calculated formation energies and cohesive energies of these 14 systems showed their reasonably high stability thermodynamically, thereby, a high experimental synthetic feasibility. Then, the most stable surface state of each M-doped Ta2O5 (100) surface at different pH and potential ranges was evaluated by using the surface Pourbaix diagrams. Further OER activity evaluations of these catalysts were based on the adsorbate evolution mechanism (AEM), where the Ir-doped Ta2O5 with the lowest theoretical overpotential of 0.33 V was computationally predicted to be most promising and consistent with experimental findings in the literature. Meanwhile, Ru- and Nb-doped catalysts were also predicted to be stable and active for acidic OER with theoretical overpotentials of 0.62 V and 0.61 V, respectively. Additionally, we note that electrochemical-driven pre-adsorption of species on the catalysts dramatically influence the OER activity. Therefore, it is crucial to analyze the surface state of a catalyst under electrochemical conditions with the surface Pourbaix diagram before conducting a detailed mechanism simulation. Our results not only adequately illustrated the general OER activity trend of metal-doped Ta2O5 catalysts but also provided theoretical guidance for further experimental studies of these systems.Data availability
Computational details and other results supporting the main conclusions are summarized in the ESI.† Other data supporting this article have been included as part of the ESI (dataset.txt).†Author contributions
T. W. conceived and supervised this project; C. C. H. performed all the DFT computations, data analysis, and figure drawing; all authors discussed the results and contributed to the writing of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFA0911900) and the National Natural Science Foundation of China (22273076); T. W. is thankful for the start-up packages from Westlake University and the Research Center for Industries of the Future (RCIF) at Westlake University for supporting this work. We thank Westlake University HPC Center for computation support. The authors thank Dr Wanghui Zhao for plotting Fig. 1.References
- S. Sharma and S. K. Ghoshal, Renewable Sustainable Energy Rev., 2015, 43, 1151–1158 CrossRef CAS.
- G. Nicoletti, N. Arcuri, G. Nicoletti and R. Bruno, Energy Convers. Manage., 2015, 89, 205–213 CrossRef.
- K. Ayers, N. Danilovic, R. Ouimet, M. Carmo, B. Pivovar and M. Bornstein, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 219–239 CrossRef CAS PubMed.
- M. David, C. Ocampo-Martínez and R. Sánchez-Peña, J. Energy Storage, 2019, 23, 392–403 CrossRef.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901 CrossRef CAS.
- Y. Xu, S. Cai, B. Chi and Z. Tu, Int. J. Hydrogen Energy, 2024, 50, 548–591 CrossRef.
- I. Vincent and D. Bessarabov, Renewable Sustainable Energy Rev., 2018, 81, 1690–1704 CrossRef CAS.
- S. A. Grigoriev, V. I. Porembsky and V. N. Fateev, Int. J. Hydrogen Energy, 2006, 31, 171–175 CrossRef CAS.
- O. Diaz-Morales, S. Raaijman, R. Kortlever, P. J. Kooyman, T. Wezendonk, J. Gascon, W. T. Fu and M. T. M. Koper, Nat. Commun., 2016, 7, 12363 CrossRef CAS PubMed.
- L. Zhang, K. Doyle-Davis and X. Sun, Energy Environ. Sci., 2019, 12, 492–517 RSC.
- F. Liao, K. Yin, Y. Ji, W. Zhu, Z. Fan, Y. Li, J. Zhong, M. Shao, Z. Kang and Q. Shao, Nat. Commun., 2023, 14, 1248 CrossRef CAS PubMed.
- J. Xu, H. Jin, T. Lu, J. Li, Y. Liu, K. Davey, Y. Zheng and S. Qiao, Sci. Adv., 2023, 9, eadh1718 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- M. Huynh, D. K. Bediako and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 6002–6010 CrossRef CAS PubMed.
- W. Xiong, H. Yin, T. Wu and H. Li, Chem.–Eur. J., 2023, 29, e202202872 CrossRef CAS PubMed.
- N. Wang, P. Ou, R. K. Miao, Y. Chang, Z. Wang, S. Hung, J. Abed, A. Ozden, H. Chen, H. Wu, J. E. Huang, D. Zhou, W. Ni, L. Fan, Y. Yan, T. Peng, D. Sinton, Y. Liu, H. Liang and E. H. Sargent, J. Am. Chem. Soc., 2023, 145, 7829–7836 CrossRef CAS PubMed.
- X. Zhang, C. Feng, B. Dong, C. Liu and Y. Chai, Adv. Mater., 2023, 35, 2207066 CrossRef CAS PubMed.
- A. Li, S. Kong, C. Guo, H. Ooka, K. Adachi, D. Hashizume, Q. Jiang, H. Han, J. Xiao and R. Nakamura, Nat. Catal., 2022, 5, 109–118 CrossRef CAS.
- A. Li, H. Ooka, N. Bonnet, T. Hayashi, Y. Sun, Q. Jiang, C. Li, H. Han and R. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 5054–5058 CrossRef CAS PubMed.
- S. Kong, A. Li, J. Long, K. Adachi, D. Hashizume, Q. Jiang, K. Fushimi, H. Ooka, J. Xiao and R. Nakamura, Nat. Catal., 2024, 7, 252–261 CrossRef CAS.
- R. Frydendal, E. A. Paoli, I. Chorkendorff, J. Rossmeisl and I. E. L. Stephens, Adv. Energy Mater., 2015, 5, 1500991 CrossRef.
- J. Huang, H. Sheng, R. D. Ross, J. Han, X. Wang, B. Song and S. Jin, Nat. Commun., 2021, 12, 3036 CrossRef CAS PubMed.
- J. Yu, F. A. Garcés-Pineda, J. González-Cobos, M. Peña-Díaz, C. Rogero, S. Giménez, M. C. Spadaro, J. Arbiol, S. Barja and J. R. Galán-Mascarós, Nat. Commun., 2022, 13, 4341 CrossRef CAS PubMed.
- M. Chatti, J. L. Gardiner, M. Fournier, B. Johannessen, T. Williams, T. R. Gengenbach, N. Pai, C. Nguyen, D. R. MacFarlane, R. K. Hocking and A. N. Simonov, Nat. Catal., 2019, 2, 457–465 CrossRef CAS.
- L. Zhou, A. Shinde, J. H. Montoya, A. Singh, S. Gul, J. Yano, Y. Ye, E. J. Crumlin, M. H. Richter, J. K. Cooper, H. S. Stein, J. A. Haber, K. A. Persson and J. M. Gregoire, ACS Catal., 2018, 8, 10938–10948 CrossRef CAS.
- L. Chong, G. Gao, J. Wen, H. Li, H. Xu, Z. Green, J. D. Sugar, A. J. Kropf, W. Xu, X. Lin, H. Xu, L. Wang and D.-J. Liu, Science, 2023, 380, 609–616 CrossRef CAS PubMed.
- I. C. Man, H. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- Z. Wang, X. Guo, J. Montoya and J. K. Nørskov, npj Comput. Mater., 2020, 6, 160 CrossRef.
- H. Chen, G. Feng, W. Fan, J. Han, Y. Li and Q. Lai, Opt. Mater. Express, 2019, 9, 3132–3145 CrossRef CAS.
- M. Lee, C. B. Lee, D. Lee, S. R. Lee, M. Chang, J. H. Hur, Y. Kim, C. Kim, D. H. Seo, S. Seo, U. I. Chung, I. Yoo and K. Kim, Nat. Mater., 2011, 10, 625–630 CrossRef CAS PubMed.
- S. Sun, X. Zhou, B. Cong, W. Hong and G. Chen, ACS Catal., 2020, 10, 9086–9097 CrossRef CAS.
- S. R. Ede and Z. Luo, J. Mater. Chem. A, 2021, 9, 20131–20163 RSC.
- X. Zheng, Y. Chen, W. Lai, P. Li, C. Ye, N. Liu, S. X. Dou, H. Pan and W. Sun, Adv. Funct. Mater., 2022, 32, 2200663 CrossRef CAS.
- M. Retuerto, L. Pascual, F. Calle-Vallejo, P. Ferrer, D. Gianolio, A. G. Pereira, Á. García, J. Torrero, M. T. Fernández-Díaz, P. Bencok, M. A. Peña, J. L. G. Fierro and S. Rojas, Nat. Commun., 2019, 10, 2041 CrossRef PubMed.
- S. Hao, M. Liu, J. Pan, X. Liu, X. Tan, N. Xu, Y. He, L. Lei and X. Zhang, Nat. Commun., 2020, 11, 5368 CrossRef CAS PubMed.
- R. R. Rao, M. J. Kolb, L. Giordano, A. F. Pedersen, Y. Katayama, J. Hwang, A. Mehta, H. You, J. R. Lunger, H. Zhou, N. B. Halck, T. Vegge, I. Chorkendorff, I. E. L. Stephens and Y. Shao-Horn, Nat. Catal., 2020, 3, 516–525 CrossRef CAS.
- Y. Wen, P. Chen, L. Wang, S. Li, Z. Wang, J. Abed, X. Mao, Y. Min, C. T. Dinh, P. D. Luna, R. Huang, L. Zhang, L. Wang, L. Wang, R. J. Nielsen, H. Li, T. Zhuang, C. Ke, O. Voznyy, Y. Hu, Y. Li, W. A. Goddard III, B. Zhang, H. Peng and E. H. Sargent, J. Am. Chem. Soc., 2021, 143, 6482–6490 CrossRef CAS PubMed.
- L. Zhou, Y. Shao, F. Yin, J. Li, F. Kang and R. Lv, Nat. Commun., 2023, 14, 7644 CrossRef CAS PubMed.
- Y. Xue, J. Zhao, L. Huang, Y. Lu, A. Malek, G. Gao, Z. Zhuang, D. Wang, C. T. Yavuz and X. Lu, Nat. Commun., 2023, 14, 8093 CrossRef CAS PubMed.
- Z. Shi, J. Li, Y. Wang, S. Liu, J. Zhu, J. Yang, X. Wang, J. Ni, Z. Jiang, L. Zhang, Y. Wang, C. Liu, W. Xing and J. Ge, Nat. Commun., 2023, 14, 843 CrossRef CAS PubMed.
- X. Wang, H. Jang, S. Liu, Z. Li, X. Zhao, Y. Chen, M. G. Kim, Q. Qin and X. Liu, Adv. Energy Mater., 2023, 13, 2301673 CrossRef CAS.
- D. Galyamin, J. Torrero, I. Rodríguez, M. J. Kolb, P. Ferrer, L. Pascual, M. A. Salam, D. Gianolio, V. Celorrio, M. Mokhtar, D. G. Sanchez, A. S. Gago, K. A. Friedrich, M. A. Peña, J. A. Alonso, F. Calle-Vallejo, M. Retuerto and S. Rojas, Nat. Commun., 2023, 14, 2010 CrossRef CAS PubMed.
- S. Chen, S. Zhang, L. Guo, L. Pan, C. Shi, X. Zhang, Z. Huang, G. Yang and J.-J. Zou, Nat. Commun., 2023, 14, 4127 CrossRef CAS PubMed.
- X. Zheng, J. Yang, P. Li, Q. Wang, J. Wu, E. Zhang, S. Chen, Z. Zhuang, W. Lai, S. Dou, W. Sun, D. Wang and Y. Li, Sci. Adv., 2023, 9, eadi8025 CrossRef CAS PubMed.
- Y. Wang, X. Lei, B. Zhang, B. Bai, P. Das, T. Azam, J. Xiao and Z.-S. Wu, Angew. Chem., Int. Ed., 2024, 63, e202316903 CrossRef CAS PubMed.
- X. Ping, Y. Liu, L. Zheng, Y. Song, L. Guo, S. Chen and Z. Wei, Nat. Commun., 2024, 15, 2501 CrossRef CAS PubMed.
- H. Li, Y. Xu, N. Lv, Q. Zhang, X. Zhang, Z. Wei, Y. Wang, H. Tang and H. Pan, ACS Sustainable Chem. Eng., 2023, 11, 1121–1132 CrossRef CAS.
- Y. Peng, H. Hajiyani and R. Pentcheva, ACS Catal., 2021, 11, 5601–5613 CrossRef CAS.
- Z. Wu, F. Chen, B. Li, S. Yu, Y. Z. Finfrock, D. M. Meira, Q. Yan, P. Zhu, M. Chen, T. Song, Z. Yin, H. Liang, S. Zhang, G. Wang and H. Wang, Nat. Mater., 2023, 22, 100–108 CrossRef CAS PubMed.
- Y. Tian, S. Wang, E. Velasco, Y. Yang, L. Cao, L. Zhang, X. Li, Y. Lin, Q. Zhang and L. Chen, iScience, 2020, 23, 100756 CrossRef CAS PubMed.
- P. Ye, K. Fang, H. Wang, Y. Wang, H. Huang, C. Mo, J. Ning and Y. Hu, Nat. Commun., 2024, 15, 1012 CrossRef CAS PubMed.
- D. Zhang, M. Li, X. Yong, H. Song, G. I. N. Waterhouse, Y. Yi, B. Xue, D. Zhang, B. Liu and S. Lu, Nat. Commun., 2023, 14, 2517 CrossRef CAS PubMed.
- Y. Feng, S. Si, G. Deng, Z. Xu, Z. Pu, H. Hu and C. Wang, J. Alloys Compd., 2022, 892, 162113 CrossRef CAS.
- T. Audichon, S. Morisset, T. W. Napporn, K. B. Kokoh, C. Comminges and C. Morais, ChemElectroChem, 2015, 2, 1128–1137 CrossRef CAS.
- S. Chen, H. Huang, P. Jiang, K. Yang, J. Diao, S. Gong, S. Liu, M. Huang, H. Wang and Q. Chen, ACS Catal., 2020, 10, 1152–1160 CrossRef CAS.
- N. Todoroki, R. Kudo, K. Hayashi, M. Yokoi, N. Naraki and T. Wadayama, ACS Catal., 2023, 13, 11433–11440 CrossRef CAS.
- W. Hu, S. Chen and Q. Xia, Int. J. Hydrogen Energy, 2014, 39, 6967–6976 CrossRef CAS.
- G. T. K. K. Gunasooriya and J. K. Nørskov, ACS Energy Lett., 2020, 5, 3778–3787 CrossRef CAS.
- J. Peng, L. Giordano, T. C. Davenport and Y. Shaohorn, Chem. Mater., 2022, 34, 7774–7787 CrossRef CAS.
- S. Jo, M.-C. Kim, K. B. Lee, H. Choi, L. Zhang and J. I. Sohn, Adv. Energy Mater., 2023, 13, 2301420 CrossRef CAS.
- A. Badreldin, O. Bouhali and A. Abdel-Wahab, Adv. Funct. Mater., 2023, 34, 2312425 CrossRef.
- X. Mao, L. Wang and Y. Li, J. Phys. Chem. Lett., 2023, 14, 170–177 CrossRef CAS PubMed.
- J. Moon, W. Beker, M. Siek, J. Kim, H. S. Lee, T. Hyeon and B. A. Grzybowski, Nat. Mater., 2024, 23, 108–115 CrossRef CAS.
- H. Park, Y. Kim, S. Choi and H. J. Kim, J. Energy Chem., 2024, 91, 645–655 CrossRef CAS.
- Z. Fang, S. Li, Y. Zhang, Y. Wang, K. Meng, C. Huang and S. Sun, J. Phys. Chem. Lett., 2024, 15, 281–289 CrossRef CAS PubMed.
- J. A. Esterhuizen, A. Mathur, B. R. Goldsmith and S. Linic, J. Am. Chem. Soc., 2024, 146, 5511–5522 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- L. A. Aleshina and S. V. Loginova, Crystallogr. Rep., 2002, 47, 415–419 CrossRef CAS.
- S. H. Lee, J. S. Kim, J. S. Kim, S. J. Kim and G. S. Park, Phys. Rev. Lett., 2013, 110, 1–5 Search PubMed.
- Y. Yang and Y. Kawazoe, Phys. Rev. Mater., 2018, 2, 034602 CrossRef CAS.
- C. Han and T. Wang, Energy Fuels, 2023, 37, 13624–13644 CrossRef CAS.
- T. Murase, H. Irie and K. Hashimoto, J. Phys. Chem. B, 2004, 108, 15803–15807 CrossRef CAS.
- S. Cao, H. Chen, Y. Hu, J. Li, C. Yang, Z. Chen, S. Wei, S. Liu, Z. Wang, D. Sun and X. Lu, Chem. Eng. J., 2023, 461, 141936 CrossRef CAS.
- Y. Zheng, J. Vernieres, Z. Wang, K. Zhang, D. Hochfilzer, K. Krempl, T. Liao, F. Presel, T. Altantzis, J. Fatermans, S. B. Scott, N. M. Secher, C. Moon, P. Liu, S. Bals, S. Van Aert, A. Cao, M. Anand, J. K. Nørskov, J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2022, 7, 55–64 CrossRef CAS.
- C. Rong, K. Dastafkan, Y. Wang and C. Zhao, Adv. Mater., 2023, 35, 2211884 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- W. Yang, Z. Jia, B. Zhou, L. Wei, Z. Gao and H. Li, Commun. Chem., 2023, 6, 6 CrossRef CAS PubMed.
- X. Han, X. Ling, D. Yu, D. Xie, L. Li, S. Peng, C. Zhong, N. Zhao, Y. Deng and W. Hu, Adv. Mater., 2019, 31, 1905622 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03554b |
| This journal is © The Royal Society of Chemistry 2024 |