DOI:
10.1039/D4SC03506B
(Edge Article)
Chem. Sci., 2024,
15, 12517-12522
Inherently chiral nor-heteracalixarenes: design and synthesis via enantioselective intramolecular Suzuki–Miyaura reaction†
Received
29th May 2024
, Accepted 29th June 2024
First published on 3rd July 2024
Abstract
There has been a recent upsurge in research aimed at synthesizing inherently chiral molecules devoid of point, axial, planar and helical chiralities. We present herein our design and enantioselective synthesis of a series of inherently chiral macrocycles. These compounds, termed nor-heteracalixaromatics, feature a biaryl bond that replaces one of the aryl-heteroatom-aryl linkages found in classic heteracalix[4]aromatics. Macrocyclization of linear achiral substrates via an intramolecular Suzuki–Miyaura cross-coupling reaction affords the 15-membered cyclophane without any chiral elements in high yields and enantioselectivities. Notably, the formation of the aryl–aryl bond does not induce axial chirality at the biaryl linkage. Instead, it restricts the free rotation of an aromatic ring located four bonds away, leading to the inherent chirality of the macrocycle. The intriguing chiroptical properties of these compounds made them promising platform for the development of CPL emitters.
Introduction
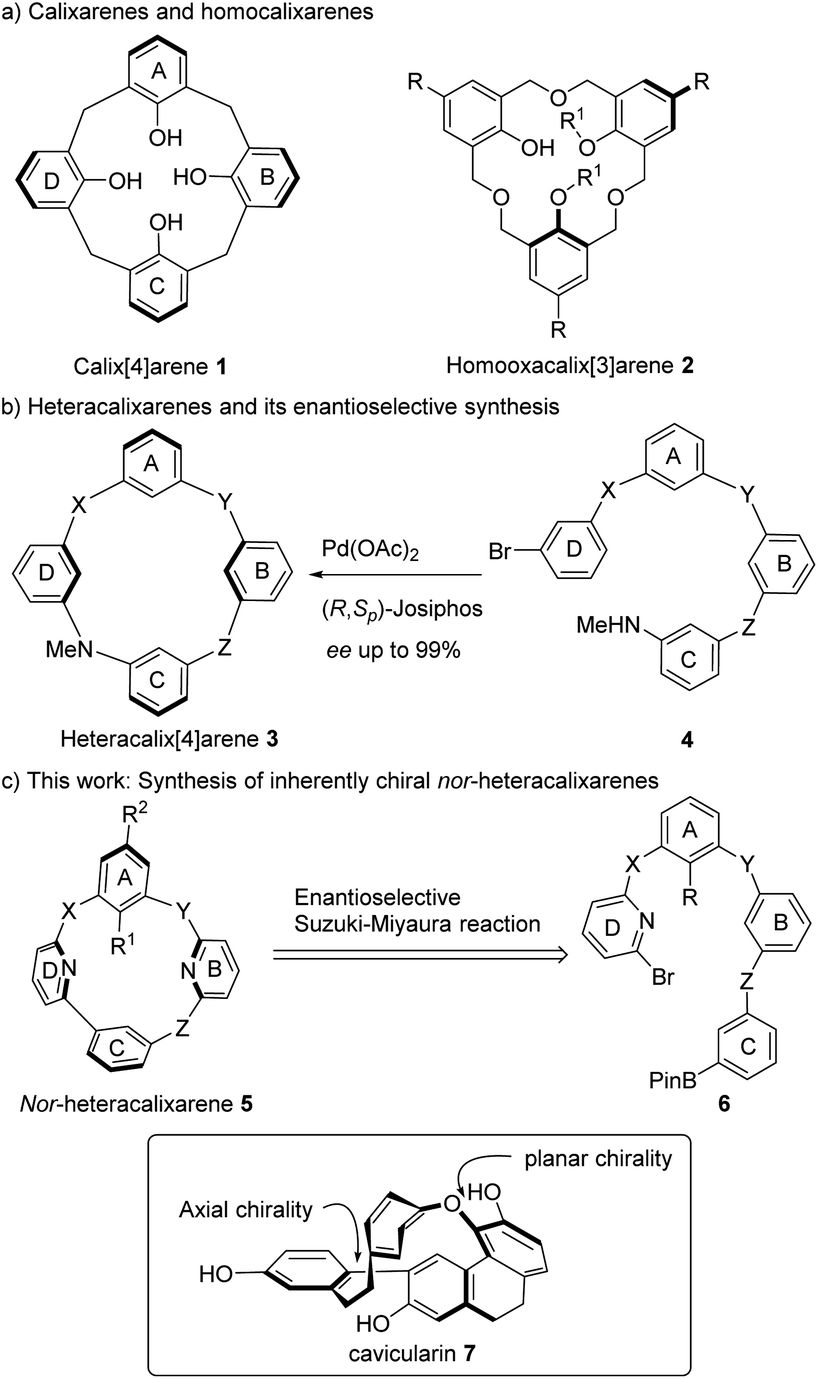
In 1994, Böhmer introduced the concept of inherent chirality to describe calixarenes devoid of point, axial, planar or helical chirality (Scheme 1a).1 Since then, this notion has been expanded to encompass various cyclic structures, such as saddle-shaped tetraphenylenes,2–9 tribenzocycloheptene derivatives,10,11 rotaxanes and catenanes,12 as well as other macrocycles characterized by a three-dimensional curvature architecture.13–16 Despite significant advancements in asymmetric synthesis, the catalytic enantioselective preparation of inherently chiral molecules has only recently gained attention.17,18 Two primary approaches, namely, desymmetrisation of macrocycles19–21 and enantioselective de novo construction of macrocycles,22–24 have emerged to access enantioenriched calixarenes and heteracalixarenes. While the latter strategy is regarded as more elegant and efficient, only two enantioselective macrocyclization reactions have been developed thus far. These methods include the Pd-catalyzed intramolecular Buchwald–Hartwig reaction22 (Scheme 1b) and chiral phosphoric acid-catalyzed intramolecular nucleophilic aromatic substitution (SNAr) reaction.24
 |
| | Scheme 1 Inherently chiral calixarenes, homooxacalixarenes and nor-heteracalixarenes. | |
Due to their distinctive curved structure and pre-organized binding sites, calixarenes 1 have found extensive use as host molecules to mimic enzyme activities25,26 and to serve as chiral catalysts.27 To fully harness their potential, researchers continually design and synthesize new scaffolds with modulable cavity-shapes. In this context, homooxacalix[3]arenes 2, featuring the CH2OCH2 bridges instead of the CH2 bridges found in calixarenes, have emerged as a versatile platform for creating functional molecules (Scheme 1a). Our group has been actively engaged in the development of heteracalixarenes with tunable cavity size and conformations for molecular recognition.28,29 Continuing this endeavor, we became interested in the synthesis of meta-cyclophane 5 containing a biaryl unit (Scheme 1c). In line with the nomenclature used for homooxacalixarene, we named it nor-heteracalixarenes as one of the heteroatoms in heteracalixarenes 3 was deleted. It's worth noting that macrocycles containing both biaryl and biaryl ether linkages do exist, and cavicularin (7), bearing both axial and planar chirality, serves as a notable example. However, our designed molecules 5 lack these two chiral elements with the chirality arising solely from the hindered through-the-annulus rotation of one of the four (hetero)arenes, akin to inherent chirality observed in calixarenes. We planned to access enantioenriched nor-heteracalixarenes 5 from the linear achiral substrate 6via the formation of a biaryl bond. Herein, we report the first instance of an enantioselective intramolecular Suzuki–Miyaura reaction for constructing inherently chiral macrocycles. Notably, the chirality originates not from the newly formed carbon–carbon bond, but rather from the hindered rotation of an aromatic ring located four chemical bonds away. Interesting chiroptical properties of these macrocycles are also documented.
Results and discussion
Optimization of reaction conditions
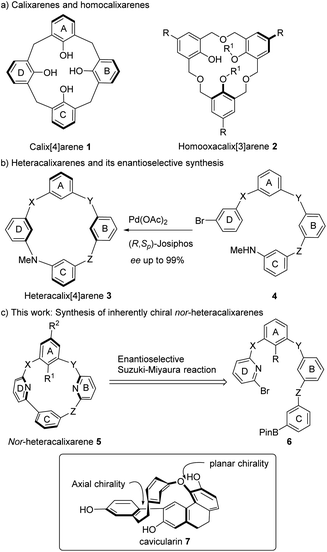
The Suzuki–Miyaura (SM) cross-coupling has emerged as one of the most powerful methods for constructing biaryl compounds30 and its intramolecular variant has proven effective in synthesizing biaryl-containing macrocycles.31–36 While asymmetric SM reaction has been developed to access enantioenriched atropisomers,37–41 its application to the synthesis of inherently chiral macrocycles has, to the best of our knowledge, not been explored. To validate our molecular design and the feasibility of the synthetic strategy, we first examined the cyclization of linear substrate 6a to racemic 5a. Pleasingly, heating a toluene solution of 6a (c 0.005 M) at 100 °C in the presence of Pd(PPh3)4 (30 mol%) and Cs2CO3 (3.0 equiv.) yielded the desired macrocycle 5a in 53% yield (ESI†). The relatively high Pd loading required for this reaction could be attributed to the presence of several tertiary amines and aza-arenes in the substrate, which may chelate the Pd catalyst. However, such chelation could, in return, pre-organize the substrate into a folded conformation, facilitating macrocyclization.42 Chiral HPLC analysis revealed the presence of two enantiomers which were isolated and were found to be configurationally stable at the reaction temperature (ESI†).
Encouraged by these results, the enantioselective cyclization of 5a was pursued. After initial screening of ligand structures and other reaction parameters (see ESI†), the monodentate H8-BINOL-derived phosphoramidite L1 stood out as the most promising candidate, affording 5a in 60% yield with 57% ee.43 To further enhance the reaction outcome, the structure of the phosphoramidite was varied (Scheme 2). Replacing one methyl group in L1 with benzyl (L2), naphthalene-1-ylmethyl (L3), 4-methylbenzyl (L4) and phenethyl group (L5) significantly increased the ee of 5a. However, ligand L6 incorporating an ortho-methylbenzyl group proved less efficient, yielding 5a in low yield and ee. Ligands (L7–L10) with substituents at 2,2′-positions of the H8-BINOL backbone are less efficient, indicating that steric hindrance around the coordinating phosphorous atom is detrimental to the enantioselectivity. Phosphoramidite L11, bearing a pyrrolidinyl group, was also an inefficient ligand. Additionally, the incorporation of a chiral amine (L12, L13), including the C2 symmetric one (L14), did not improve the enantioselectivity. Finally, the (R)-1,1′-spirobiindane-7,7′-diol derived phosphoramidites (R)-SIPHOS (L15)44,45 and L16 provided the desired product with much improved ee. With L15, macrocycle 5a was isolated in 70% yield with 90% ee.
 |
| | Scheme 2 Ligand screening for the synthesis of inherently chiral nor-heteracalixarene 5a. [a] Conditions: Pd(dba)2 (30 mol%), ligand (1.2 equiv.), Cs2CO3 (3.0 equiv.), toluene (c 0.005 M), 100 °C. Enantiomeric excess (ee) was determined by HPLC analysis on a chiral stationary phase. | |
Scope of the reaction
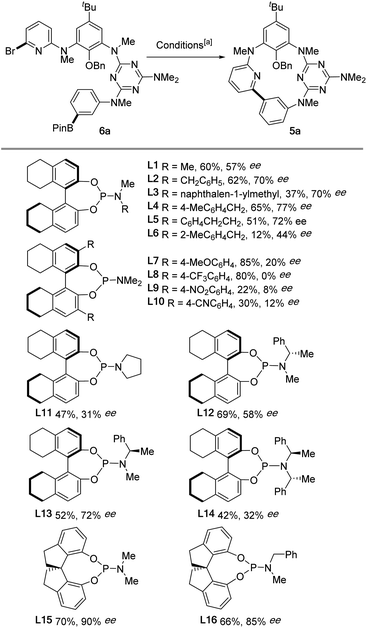
With the optimal conditions established, the scope of this enantioselective Suzuki–Miyaura reaction was next examined (Scheme 3). Varying the dialkylamino substituent at the C6 position of the 1,3,5-triazine (B ring) did not significantly alter the ee of the products (5a–5d, Scheme 3a), while substituents at the C-ring (5e, 5f, 5k, 5l) and D-ring (5m, 5n) were tolerated, with diminished enantioselectivity being observed in the latter two cases. Remarkably, the presence of an amino group (5n) remained compatible with the cyclization conditions. Macrocycles with different alkoxy groups (5g–5j), including the smallest methoxy group (5i), at the A-ring were prepared in high yields and ees. Similarly, N-allyl substituted macrocycle 5o was prepared without issues. The inherent chirality of compounds 5 results from the hindered rotation of the A ring through the annulus, suggesting that the size of the A ring substituent could be a key factor in conferring configurational stability. Gratifyingly, we found that the presence of a bulky tert-butyl group at the A ring is not mandatory for the inherent chirality of the nor-heteracalixarene system. Indeed, compounds 5p and particularly 5q, with a single methyl substituent at the low rim of the macrocycle, were obtained in excellent yields and ees. Cyclization of a substrate containing an (R)-prolinol unit 6r afforded the cyclophane 5r in moderate yield with an excellent diastereoselectivity (dr 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Noteworthily, starting from the same linear precursor 6r using ent-L15 as the ligand under otherwise standard conditions, macrocyclic compound 5r was synthesized in 50% yield with a reversed d.r. of 1:20 (ESI†). These results indicated that the substrate chirality has a little impact on the enantioselectivity of the cyclization. However, replacing the pyridine with a benzene ring (ring D) significantly reduced the enantioselectivity of the reaction (5s, 5t). In the case of 5t, the racemic cyclophane 5t was obtained in 70% yield, indicating that the coordination of pyridine nitrogen (D ring) to Pd likely played a crucial role in controlling the enantioselectivity of the process.
:1). Noteworthily, starting from the same linear precursor 6r using ent-L15 as the ligand under otherwise standard conditions, macrocyclic compound 5r was synthesized in 50% yield with a reversed d.r. of 1:20 (ESI†). These results indicated that the substrate chirality has a little impact on the enantioselectivity of the cyclization. However, replacing the pyridine with a benzene ring (ring D) significantly reduced the enantioselectivity of the reaction (5s, 5t). In the case of 5t, the racemic cyclophane 5t was obtained in 70% yield, indicating that the coordination of pyridine nitrogen (D ring) to Pd likely played a crucial role in controlling the enantioselectivity of the process.
 |
| | Scheme 3 Enantioselective synthesis of inherently chiral nor-heteracalixarenes and X-ray molecular structures of (cS)-5p, (cR)-5w and classic azaheteracalix[4]arenes. [a] Conditions: Pd(dba)2 (30 mol%), L15 (1.2 equiv.), Cs2CO3 (3.0 equiv.), toluene (c 0.005 M), 100 °C. Enantiomeric excess (ee) was determined by HPLC analysis on a chiral stationary phase. [b] Using ent-L15 as the ligand under otherwise identical conditions. | |
To further extend the reaction scope, we examined the synthesis of oxabridged nor-heteracalixarenes. As shown in Scheme 3b, cyclophanes containing one biaryl ether linkage (5u–5aa) were obtained in high yields and ees from the corresponding linear achiral substrates. Changing the ring D from pyridine to triazine diminished the ee of the product (5ab), suggesting a negative effect of an additional coordination site. Additionally, 5ac bearing two biaryl ether linkages was obtained in 59% yield with 64% ee. The absolute configuration of 5p and 5w was assigned to be (cS) and (cR),46 respectively on the basis of their X-ray structures (Scheme 3c).47 The absolute configurations of other inherently chiral macrocycles were assigned accordingly by comparing their electronic circular dichroism (ECD) with that of the (cS)-5p and (cR)-5w (ESI†).
Structural elucidation
Structurally, these compounds exhibit a shallower curvature compared to the related heteracalix[4]arenes.22 The C2 substituted benzene ring (ring A) stands orthogonal to the annulus defined by the other three (het)arenes (B, C and D). In the azabridged macrocycle 5p, all three nitrogen atoms align quasi co-planar with the adjacent heteroarenes (ring B and D), but not with the benzene rings (rings A and C). Interestingly, in compounds 5u–5aa featuring a biaryl ether linkage, the oxygen atom is no longer fully conjugated with the triazine ring (ring B), but forms a cross-conjugated system with both the triazine (ring B) and the benzene rings (ring C). This change renders the three (het)arenes (ring B, C and D) more planar than those in 5p, as illustrated by the X-ray structure of (cR)-5w in Scheme 3c. The dihedral angle between the ring A and ring C is 68° for 5p and 89° for 5w, in contrast to the much-reduced angle of 41° observed in our previous ABCD-type heteracalixarenes (CCDC 1996984†).22 Moreover, the dihedral angle of the biaryl bond is 44° for 5p and 27° for 5w, indicating a significant ring strain in these cyclophanes. We have also noticed that, unlike the azabridged macrocycles 5a–5r, the aza/oxabridged macrocycles 5u–5aa have generally opposite optical rotations (Scheme 3c and ESI†). This reversal in optical rotation, resulting from only one heteroatom difference in the bridging positions of the inherently chiral macrocyclic skeleton, is intriguing.
Conformational stability
As can be seen from the X-ray structures, the inherent chirality of these nor-heteracalixarenes arises from the hindered rotation of ring A, with any racemization of the macrocycle attributed to the flipping of the C2-alkoxy substituent through the annulus. Interestingly, the C5 substituent in ring A does not appear to play a critical role in determining the configurational stability of the inherent chirality. In fact, compound 5q, lacking the C5-alkyl substituent, is configurationally stable. The ee value of 5q remained unchanged after heating the macrocycle solution in o-dichlorobenzene at 160 °C for 24 h (Fig. 1 and ESI†). However, the macrocycle 5ad lacking the substituent at C2 cannot be resolved into enantiomers at ambient temperature by HPLC presumably due to the fast ring inversion process. To further support the notion that the chirality of 5 results from the hindered through-the-annulus rotation of the A ring, a free phenol derivative 5ae (90% ee) was prepared by boron tribromide-mediated selective O-debenzylation of 5a for comparison (ESI†). While 5a is configurationally stable even at 160 °C, the determined enantiomerization barrier is about 24 kcal mol−1 at 298 K for 5ae (Fig. 1, for the kinetic study, see ESI†). These results clearly indicate that the inherent chirality results from the hindered rotation of the A-ring and that the size of the C2 substituent determines the stability of the chirality.
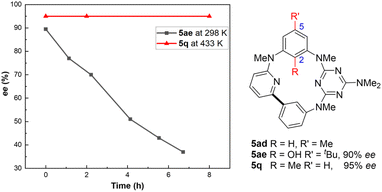 |
| | Fig. 1 Importance of the C2 substituent in the configurational stability of the nor-heteracalixarene. ee of 5ae was measured in iPrOH solution while 5q was in o-dichlorobenzene solution. | |
Chiroptical properties
The chiroptical properties of some of these macrocycles were probed using UV, fluorescence, ECD and circularly polarized luminescence (CPL) spectroscopy (Fig. 2 and ESI†). Compounds 5a, 5c, 5d, and 5p exhibited almost identical emission maxima (λem) at 410 nm in CH3CN solution, with photoluminescence quantum yields (PLQYs) of 21.5%, 15.6%, 11.0%, and 18.3%, respectively. The relatively high fluorescence quantum yield could be attributed to the rigidity of the nor-heteracalixarene macrocycles. The gabs and glum values of these compounds fall within the range of 10−3, similar to their heteracalix[4]arene counterparts (ESI†).21 Moreover, the CPL brightness (BCPL) values were determined to be 0.40 (5a), 0.38 (5c), 0.35 (5d), and 0.65 (5p) (ESI†). These encouraging results, coupled with their tunable conformational structures, make them promising candidates for CPL-related applications.
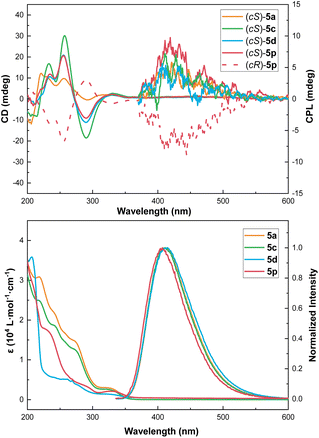 |
| | Fig. 2 ECD (top left, c 5 × 10−6), CPL (top right, c 1 × 10−4), UV-vis (bottom left, c 2 × 10−5), and fluorescence (bottom right, c 5 × 10−6) spectra of 5a, 5c, 5d, and 5p at 25 °C in CH3CN. | |
Conclusions
In summary, we have designed and synthesized a new class of enantioenriched nor-heteracalixaromatics devoid of point, axial, planar and helical chiralities. We have developed a method for macrocyclization using the intramolecular Suzuki–Miyaura reaction, which leads to inherently chiral macrocycles in high yields and enantioselectivities. Remarkably, the chirality originates not from the newly formed carbon–carbon bond, but rather from the restricted rotation of an aromatic ring positioned four bonds away. These nor-heteracalixaromatics, which exhibit interesting chiroptical properties, are promising platforms for the development of valuable CPL emitters.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and the ESI,† as well as from the authors upon request.
Author contributions
Y. F. J., S. T., J. Z. and M. X. W. conceived and designed the experiments. Y. F. J. carried out the experiments. S. T., J. Z. and M. X. W. wrote the manuscript. All authors contributed to the reviewing and editing of the manuscript and ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the National Natural Science Foundation of China (21920102001, 22171160, and 22371161) and EPFL (Switzerland) for financial support.
Notes and references
- V. Böhmer, D. Kraft and M. Tabatabai, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 17–39 CrossRef.
- J.-W. Han, X.-S. Peng and H. N. C. Wong, Natl. Sci. Rev., 2017, 4, 892–916 CrossRef CAS.
- S. Link and C. Sparr, Chem. Soc. Rev., 2018, 47, 3804–3815 RSC.
- T. Shibata, T. Chiba, H. Hirashima, Y. Ueno and K. Endo, Angew. Chem., Int. Ed., 2009, 48, 8066–8069 CrossRef CAS PubMed.
- C. Cheng, X. Zuo, D. Tu, B. Wan and Y. Zhang, Chem. Commun., 2021, 57, 2939–2942 RSC.
- Y. Luo, S. Cheng, Y. Peng, X. Long, J. Li, Y. He, C. Gan, S. Luo and Q. Zhu, CCS Chem., 2022, 4, 3427–3435 Search PubMed.
- Y. Luo, X. Wang, W. Hu, Y. Peng, C. Wang, T. Yu, S. Cheng, J. Li, C. Gan, S. Luo and Q. Zhu, CCS Chem., 2023, 5, 982–993 CrossRef CAS.
- D. Zhang, J. Zhou, T. Qin and X. Yang, Chem Catal., 2024, 4, 100827 CrossRef CAS.
- J. Zhou, M. Tang and X. Yang, Chin. J. Chem., 2024, 42, 1953–1959 CrossRef.
- Y.-K. Tahara, R. Matsubara, A. Mitake, T. Sato, K. S. Kanyiva and T. Shibata, Angew. Chem., Int. Ed., 2016, 55, 4552–4556 CrossRef CAS PubMed.
- J.-H. Li, X.-K. Li, J. Feng, W. Yao, H. Zhang, C.-J. Lu and R.-R. Liu, Angew. Chem., Int. Ed., 2024, 63, e202319289 CrossRef CAS PubMed.
- E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chem. Soc. Rev., 2018, 47, 5266–5311 RSC.
- K. Katagiri, T. Ikeda, A. Muranaka, M. Uchiyama, M. Tominaga and I. Azumaya, Tetrahedron: Asymmetry, 2009, 20, 2646–2650 CrossRef CAS.
- H. Zhou, Y.-F. Ao, X.-D. Wang and Q.-Q. Wang, J. Am. Chem. Soc., 2022, 144, 16767–16772 CrossRef CAS PubMed.
- A. Dalla Cort, L. Mandolini, L. Pasquini and L. Schiaffino, New J. Chem., 2004, 28, 1198–1199 RSC.
- A. Szumna, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC.
- G. E. Arnott, Chem.–Eur. J., 2018, 24, 1744–1754 CrossRef CAS PubMed.
- M. Tang and X. Yang, Eur. J. Org Chem., 2023, 26, e202300738 CrossRef CAS.
- Y.-Z. Zhang, M.-M. Xu, X.-G. Si, J.-L. Hou and Q. Cai, J. Am. Chem. Soc., 2022, 144, 22858–22864 CrossRef CAS PubMed.
- X. Zhang, S. Tong, J. Zhu and M.-X. Wang, Chem. Sci., 2023, 14, 827–832 RSC.
- X.-Y. Wang, X.-Y. Zhang, S. Tong, Q.-H. Guo, M.-L. Tan, C.-J. Li and M.-X. Wang, CCS Chem., 2024, 6, 1198–1210 CrossRef CAS.
- S. Tong, J.-T. Li, D.-D. Liang, Y.-E. Zhang, Q.-Y. Feng, X. Zhang, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 14432–14436 CrossRef CAS PubMed.
- K. Ishibashi, H. Tsue, H. Takahashi and R. Tamura, Tetrahedron: Asymmetry, 2009, 20, 375–380 CrossRef CAS.
- X.-C. Li, Y. Cheng, X.-D. Wang, S. Tong and M.-X. Wang, Chem. Sci., 2024, 15, 3610–3615 RSC.
- S. B. Nimse and T. Kim, Chem. Soc. Rev., 2013, 42, 366–386 RSC.
- R. Kumar, A. Sharma, H. Singh, P. Suating, H. S. Kim, K. Sunwoo, I. Shim, B. C. Gibb and J. S. Kim, Chem. Rev., 2019, 119, 9657–9721 CrossRef CAS PubMed.
- M. Durmaz, E. Halay and S. Bozkurt, Beilstein J. Org. Chem., 2018, 14, 1389–1412 CrossRef CAS PubMed.
- M.-X. Wang, Acc. Chem. Res., 2012, 45, 182–195 CrossRef CAS PubMed.
- D.-X. Wang and M.-X. Wang, Acc. Chem. Res., 2020, 53, 1364–1380 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. M. Elder and D. H. Rich, Org. Lett., 1999, 1, 1443–1446 CrossRef CAS PubMed.
- A.-C. Carbonnelle and J. Zhu, Org. Lett., 2000, 2, 3477–3480 CrossRef CAS PubMed.
- R. Lépine and J. Zhu, Org. Lett., 2005, 7, 2981–2984 CrossRef PubMed.
- Y. Jia, M. Bois-Choussy and J. Zhu, Angew. Chem., Int. Ed., 2008, 47, 4167–4172 CrossRef CAS PubMed.
- J. Dufour, L. Neuville and J. Zhu, Chem.–Eur. J., 2010, 16, 10523–10534 CrossRef CAS PubMed.
- Z. Wang, M. Bois-Choussy, Y. Jia and J. Zhu, Angew. Chem., Int. Ed., 2010, 49, 2018–2022 CrossRef CAS PubMed.
- A. N. Cammidge and K. V. L. Crépy, Chem. Commun., 2000, 1723–1724 RSC.
- J. Yin and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 12051–12052 CrossRef CAS.
- O. Baudoin, Eur. J. Org Chem., 2005, 4223–4229 CrossRef CAS.
- C. Li, D. Chen and W. Tang, Synlett, 2016, 27, 2183–2200 CrossRef CAS.
- G. Hedouin, F. Hazra, S. Gallou and S. Handa, ACS Catal., 2022, 12, 4918–4937 CrossRef CAS.
- J. Blankenstein and J. Zhu, Eur. J. Org Chem., 2005, 1949–1964 CrossRef CAS.
- J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486–2528 CrossRef CAS PubMed.
- Y. Fu, J.-H. Xie, A.-G. Hu, H. Zhou, L.-X. Wang and Q.-L. Zhou, Chem. Commun., 2002, 480–481 RSC.
- A.-G. Hu, Y. Fu, J.-H. Xie, H. Zhou, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2002, 41, 2348–2349 CrossRef CAS.
- The italic “c” in cS and cR stands for curvature. For the nomenclature of calixarenes, see ref. 14. 5w was assigned as cR configured due to the higher priority of oxygen vs. nitrogen atom.
- Deposition numbers CCDC 2333560 (5p) and CCDC 2338518 (5w)† contain the supplementary crystallographic data for this paper.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Jieping
Zhu
*a,
Jieping
Zhu