
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient semi-hydrogenation in strained ultrathin PdCu shell and the atomic deciphering for the unlocking of activity-selectivity†
Fan
Xue
a,
Qiang
Li
 *a,
Weihua
Ji
b,
Mingxin
Lv
a,
Hankun
Xu
a,
Jianrong
Zeng
cd,
Tianyi
Li
e,
Yang
Ren
f,
Lihui
Zhou
g,
Xin
Chen
a,
Jinxia
Deng
a,
Kun
Lin
a and
Xianran
Xing
*a
*a,
Weihua
Ji
b,
Mingxin
Lv
a,
Hankun
Xu
a,
Jianrong
Zeng
cd,
Tianyi
Li
e,
Yang
Ren
f,
Lihui
Zhou
g,
Xin
Chen
a,
Jinxia
Deng
a,
Kun
Lin
a and
Xianran
Xing
*a
aBeijing Advanced Innovation Center for Materials Genome Engineering, Institute of Solid State Chemistry, University of Science and Technology Beijing, Beijing 100083, China. E-mail: qiangli@ustb.edu.cn; xing@ustb.edu.cn
bCollege of Materials Science and Engineering, Taiyuan University of Technology, Taiyuan 030024, China
cShanghai Synchrotron Radiation Facility, Shanghai Advanced Research Institute, Chinese Academy of Sciences, 201204 Shanghai, P. R. China
dShanghai Institute of Applied Physics, Chinese Academy of Sciences, 201800 Shanghai, P. R. China
eX-Ray Science Division, Argonne National Laboratory, Argonne, Illinois 60439, USA
fDepartment of Physics, City University of Hong Kong, Kowloon, Hong Kong 999077, China
gKey Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, China
First published on 27th June 2024
Abstract
Excellent ethylene selectivity in acetylene semi-hydrogenation is often obtained at the expense of activity. To break the activity-selectivity trade-off, precise control and in-depth understanding of the three-dimensional atomic structure of surfacial active sites are crucial. Here, we designed a novel Au@PdCu core–shell nanocatalyst featuring diluted and stretched Pd sites on the ultrathin shell (1.6 nm), which showed excellent reactivity and selectivity, with 100% acetylene conversion and 92.4% ethylene selectivity at 122 °C, and the corresponding activity was 3.3 times higher than that of the PdCu alloy. The atomic three-dimensional decoding for the activity-selectivity balance was revealed by combining pair distribution function (PDF) and reverse Monte Carlo simulation (RMC). The results demonstrate that a large number of active sites with a low coordination number of Pd–Pd pairs and an average 3.25% tensile strain are distributed on the surface of the nanocatalyst, which perform a pivotal function in the simultaneous improvement of hydrogenation activity and ethylene selectivity. Our work not only develops a novel strategy for unlocking the linear scaling relation in heterogeneous catalysis but also provides a paradigm for atomic 3D understanding of lattice strain in core–shell nanocatalysts.
Introduction
An ideal catalyst, which possesses high activity and high selectivity, is generally required to have strong activation of reactants but weak adsorption of intermediates.1,2 However, due to the coupling correlation between the adsorption strengths of reactants and intermediates on the surfaces of transition metals, the achievement of high selectivity is typically accompanied by a sacrifice of activity in heterogeneous catalytic reactions.3–5 Taking the selective hydrogenation of acetylene to ethylene as an example, which is an important industrial process for purifying alkene streams produced from petroleum cracking, Pd nanoparticles can achieve the complete conversion of acetylene at a lower temperature owing to their superior hydrogen activation capability, but tend to exhibit extremely low ethylene selectivity due to their strong adsorption of ethylene.6,7 Adding a second inert metal for this reaction, such as Cu, Ag, and Ga, is a common method to increase selectivity, which decreases Pd ensembles and dilutes Pd atoms in order to weaken the adsorption of ethylene; however, it is also accompanied by weaker hydrogen activation and requires a higher temperature to achieve high acetylene conversion.8–10 Therefore, it is desirable to develop a catalyst that is capable of maintaining high ethylene selectivity while also achieving full conversion of acetylene at a relatively low temperature.Core–shell nanocatalysts can provide a good platform for optimizing catalytic performance by integrating the structural design of shell parts and constraints of the core–shell interfaces.11–13 Specifically, the strain arising from lattice mismatch in the core and shell is transmitted from the metal-to-metal interface at several atomic layers to the surface of the catalyst to modulate the geometrical and electronic properties of active sites.14,15 It is intimately related to the adsorption and desorption behavior of reactants or intermediates in the catalytic process.16,17 For instance, tensile strain on the PdFe shell in Pd@PdFe catalysts contributes to the oxygen with optimal adsorption strength to enhance the activity for oxygen reduction reaction (ORR).18 In Au@Pd1Pt3 core–alloyed shell nanoparticles, tensile strain can still be transferred to the surface in a 2.3 nm Pd1Pt3 shell to increase formic acid oxidation reaction (FOR) activity.15 Hence, based on the fact that palladium-based bimetals with dispersed Pd sites can ensure high ethylene selectivity,19 exerting tensile strain on the surface of the nanoalloys may lead to an upward shift of the d-band center and improve the activation of hydrogen, promising to break the trade-off between activity and selectivity in the acetylene semi-hydrogenation. Nevertheless, the lattice strain exhibits spatial dependence.20 From the interface of the core and shell to the surface of the catalyst, the distribution of lattice strain is typically non-uniform, and the strain state that is particular to each active site on the surface is even more distinct.21 In order to accurately understand the relationship between surface strain and catalytic performance, as well as to effectively harness the interfacial strain effect, it is imperative to gain insight into the three-dimensional lattice strain of core–shell nanostructures at the atomic scale.
Considering the tunable elemental distribution of the PdCu alloy and the fact that Au is a superior driving force of tensile strain which has a large lattice mismatch and solid solubility with the Pd metal,22,23 herein, an Au@PdCu core–shell nanocatalyst is designed and synthesized. The intrinsic activity of the strained PdCu shell is considerably improved with the introduction of the Au core while maintaining high ethylene selectivity, which effectively breaks scaling relations in acetylene semi-hydrogenation. The three-dimensional lattice strain distribution of the PdCu shell and the local coordination environment of the surface Pd sites were revealed by combining the atom pair distribution function (PDF) and reverse Monte Carlo (RMC) methods,24–28 providing quantitative local structural evidence for the excellent catalytic performance. Superior hydrogenation activity in Au@PdCu core–shell nanocatalysts is attributed to the tensile strain on the surface active sites, whereas high ethylene selectivity results from the low Pd–Pd coordination number on the catalyst surface. DFT calculations further demonstrate that dispersed Pd sites under tensile strain optimize the adsorption of ethylene and hydrogen and contribute to the efficient semi-hydrogenation of acetylene. Our work opens a new avenue for designing efficient heterogeneous catalysts.
Results and discussion
Core–shell structure and lattice strain
The seed-mediated growth approach29 was employed for manufacturing Au@PdCu core–shell nanoparticles (NPs), as illustrated in Fig. 1a. In the synthesis, PdCl2 and Cu(acac)2 were co-reduced to form Pd–Cu alloys grown on the surface of pre-prepared Au seeds. Oleylamine was used as surfactant and reducing agent. Dispersed active sites can be ensured by a high feed ratio of Cu and Pd precursors, which weaken the adsorption of intermediates and enhance selectivity. To gain insight into the variation in the surface structure of the catalyst prior to and subsequent to the introduction of Au cores, PdCu and Au nanoparticles were synthesized employing a similar approach to that of Au@PdCu NPs. The XRD patterns of the prepared nanoparticles all exhibit typical face-centered cubic (FCC) structures (Fig. 1b). For Au@PdCu NPs, in addition to the diffraction peaks corresponding to Au, the new diffraction peak is also clearly seen to be located between the corresponding peak positions of pure FCC-structured Cu and Pd, indicating the successful construction of hierarchical structure of Au@PdCu alloy. The diffraction peaks of the (111) crystal plane about all nanoparticles were magnified in the inset of Fig. 1b. And the diffraction peak of the PdCu part in Au@PdCu NPs was observed to shift to a lower 2θ value compared to the PdCu nanoparticles, indicating that the PdCu part might be undergoing tensile strain. In contrast, the diffraction peak of the Au part in Au@PdCu NPs shifted to a greater 2θ value compared to the pure Au NPs due to the compressive strain from the PdCu part. These findings demonstrate that the interfacial structures of Au@PdCu nanoparticles can be compatible, which becomes the efficient transfer station of lattice interaction and electrons.30,31 The TEM images in Fig. S1† and 1c display the produced Au, PdCu, and Au@PdCu nanocrystals, which exhibit a quasi-spherical shape and have an average size of 10.22 ± 0.85, 12.96 ± 1.10, and 13.41 ± 1.07 nm, respectively (seen in Fig. S1† for the particle size histogram). To evaluate their catalytic performance, all the nanoparticles were uniformly dispersed on carbon black. After pretreatment, TEM and XRD showed consistent size and structure of NPs (Fig. S2†). | ||
| Fig. 1 Synthesis and characterization of Au@PdCu core–shell NPs. (a) Schematic diagram of the preparation of Au@PdCu NPs and the corresponding effect on the shift of the d-band center and the adsorption of reactants. (b) XRD patterns of Au, PdCu, and Au@PdCu NPs. The inset shows enlarged XRD patterns. (c–e) TEM image (c), HAADF-STEM image (d), and corresponding line-scans of the Au@PdCu NPs (e). (f) High-resolution HAADF-STEM image of Au@PdCu NPs. (g) Enlarged high-resolution HAADF-STEM image from the selected area in panel f. (h) Integrated pixel intensities of Au@PdCu taken along the olive and orange dashed rectangles in g. (i–l) HADDF-EDS elemental mapping of Au@PdCu NPs. | ||
Atomic-resolution aberration-corrected scanning transmission electron microscopy (STEM) was utilized to reveal the structural information of Au@PdCu nanocrystals. Fig. 1d shows the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of Au@PdCu nanocrystals, from which a darker shell around a bright core due to differences in atomic number can be resolved. Fig. 1e and i–l demonstrate the high-resolution EDS line scanning profile and elemental mappings of a representative Au@PdCu nanoparticle. Au and Pd signals are localized in the core and shell of the nanoparticles, respectively, confirming the formation of the core–shell structure. Whereas, the Cu signals are distributed throughout the nanoparticles, which may be the result of alloying some Cu atoms with the Au core, suggesting that copper plays an important role in the formation of highly compatible interfacial structures. A typical high-resolution HADDF-STEM image of an individual Au@PdCu nanoparticle is depicted in Fig. 1f. The enlarged image from the selected regions highlighted by the orange rectangle in Fig. 1f is shown in Fig. 1g. The integrated pixel intensity profiles are obtained from the outer PdCu (represented by the orange rectangle in Fig. 1g) and inner Au (indicated by the olive rectangle in Fig. 1g), as illustrated in Fig. 1h. For inner Au and outer PdCu, the averaged (111) spacings are measured to be 2.38 Å and 2.25 Å, respectively. Further, according to the formula for lattice mismatch,32 there is a 5.8% lattice mismatch between the gold core and the PdCu shell, and the PdCu shell endures a significant tensile strain. The (111) intergranular spacings of the pure Au and PdCu NPs are 2.40 Å and 2.18 Å, respectively (Fig. S3†). Taking the crystallographic spacing of PdCu nanoparticles without the Au core as a reference, the average tensile strain (3.21%) subjected to the PdCu shell can be calculated according to the strain equation.14 Strain-formed PdCu shells offer a highly conducive environment for enhancing catalytic performance; however, they present a formidable obstacle in terms of elucidating the local structure of the surface.
Catalytic performance of acetylene semi-hydrogenation
The selective hydrogenation of acetylene, an industrial reaction that is very sensitive to surface structure, has been employed to study the relationship between surface local structure and catalytic performance. Acetylene conversion and ethylene selectivity among catalysts are illustrated in Fig. 2a as a function of reaction temperature. While both PdCu and Au@PdCu nanocatalysts show excellent ethylene selectivity (>90%), their levels of activity vary considerably. The Au@PdCu catalyst achieves full acetylene conversion at a lower temperature of 122 °C compared to the PdCu catalyst (153 °C). The corresponding activity and ethylene yield of the Au@PdCu nanocatalyst at 122 °C are 209.9 molC2H2 molPd−1 h−1 and 92.4%, which are approximately 3.3 and 2.2 times higher than those of the PdCu nanocatalyst, respectively (Fig. 2b). Additionally, a linear fit of ln (TOF) versus 1000/T was utilized to derive the apparent activation energy of the catalysts (Fig. 2c); the Au@PdCu nanocatalyst has a lower activation energy of 47.2 kJ mol−1 in comparison to the PdCu nanocatalyst. These results demonstrate that the introduction of an Au core to the PdCu shell can greatly enhance catalytic activity and maintain superior ethylene selectivity, giving it a competitive edge over other reported catalysts (Table S2† and Fig. 2d). Meanwhile, the Au@PdCu catalyst also has relatively good stability. The acetylene conversion and ethylene selectivity of the catalyst remained unchanged throughout the 40 hour stability test (Fig. 2e). XRD and TEM analyses verified that the size, shape, and crystal structure of the catalyst remained largely unchanged after the stability test, indicating the excellent structural stability of the Au@PdCu nanocatalyst (Fig. 2f and S4a†). A typical HRTEM image of the Au@PdCu nanocatalyst following the stability test is depicted in Fig. S4d.† Almost unchanged from prior to the test, the crystal plane spacing of the inner and outer layers is 2.34 Å and 2.22 Å, respectively, which correspond to the (111) crystal planes of Au and PdCu. That is, the stability test did not significantly alter the core–shell structure or lattice strain of the Au@PdCu nanocatalyst. In addition to structural stability, the capacity to resist carbon compound deposition is another indicator of catalyst stability.33 The thermogravimetric analysis test was conducted, and the result is illustrated in Fig. S4b.† The mass loss of about 1.5% suggests that Au@PdCu exhibits a relatively good resistance to the deposition of carbon compounds. Given that the Au@PdCu nanocatalyst's superior catalytic performance is probably attributed to the PdCu shell under tensile strain, it becomes imperative to investigate the catalyst's local structure and surface lattice strain. | ||
| Fig. 2 Catalytic evaluation of semi-hydrogenation of acetylene. (a) Plot of acetylene conversion and ethylene selectivity as a function of temperature for PdCu and Au@PdCu catalysts. (b) Activity and ethylene yield of different catalysts at 122 °C. (c) Arrhenius plots of PdCu and Au@PdCu catalysts. (d) Comparison of catalytic performance over different catalysts reported in the literature for semi-hydrogenation of acetylene. (e) Stability test for the Au@PdCu/C catalyst. (f) TEM image of Au@PdCu/C after the stability test. | ||
Electron structure and local structure
To gain a local structural understanding of the PdCu shell subjected to tensile strain, X-ray absorption spectroscopy (XAS) was used to reveal the electronic structure and local coordination environment of the Pd active sites.34,35 The normalized Pd K-edge X-ray absorption near-edge structure (XANES) shows that the adsorption edges of the Au@PdCu and PdCu nanocatalysts are close to the Pd foil (Fig. 3a), indicating that the palladium atoms are primarily in the metallic state. Additionally, compared with Pd foil and the PdCu catalyst, the absorption edge of Au@PdCu shifts towards lower energy, suggesting increased Pd electron density, which is favorable for improving hydrogen activation.36 However, Au@PdCu exhibits a lower absorption edge position in the Cu K-edge XANES spectra than PdCu (Fig. S5†), corresponding to a more electron-rich characterization of the Cu atom. Actually, within the Au@PdCu catalyst, electron transfer occurs not only between Cu and Pd but also between Au and Cu. The increase in Au@PdCu absorption intensity around 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 925 eV relative to Au NPs and Au foil in the Au L3-edge XANES spectra demonstrates the transfer of d electrons from Au to Cu (Fig. S6†).37 Copper assumes the role of an electron transfer mediator within the Au@PdCu catalyst.38 X-ray photoelectron spectroscopy (XPS) was further utilized to study the electronic structure of nanocatalysts.39 As shown in Fig. 3b, the binding energy of Pd 3d for the Au@PdCu catalyst is moved to a lower energy position than for the PdCu catalyst. The binding energy of Cu 2p in the Au@PdCu and PdCu catalysts does not exhibit notable alterations. However, the binding energy of Au 4f in the Au@PdCu catalyst exhibits a higher energy position when compared to Au NPs (Fig. S7†). These findings further illustrate that the elevated electron density of Pd can be attributed to Cu functioning as an electron transit station in the Au@PdCu catalyst.
925 eV relative to Au NPs and Au foil in the Au L3-edge XANES spectra demonstrates the transfer of d electrons from Au to Cu (Fig. S6†).37 Copper assumes the role of an electron transfer mediator within the Au@PdCu catalyst.38 X-ray photoelectron spectroscopy (XPS) was further utilized to study the electronic structure of nanocatalysts.39 As shown in Fig. 3b, the binding energy of Pd 3d for the Au@PdCu catalyst is moved to a lower energy position than for the PdCu catalyst. The binding energy of Cu 2p in the Au@PdCu and PdCu catalysts does not exhibit notable alterations. However, the binding energy of Au 4f in the Au@PdCu catalyst exhibits a higher energy position when compared to Au NPs (Fig. S7†). These findings further illustrate that the elevated electron density of Pd can be attributed to Cu functioning as an electron transit station in the Au@PdCu catalyst.
 | ||
| Fig. 3 X-ray absorption spectra (XAS) and local structure. (a) Normalized Pd K-edge XANES spectra of PdCu, and Au@PdCu nanocatalysts in reference to Pd foil. (b) Pd 3d XPS spectra of PdCu/C and Au@PdCu/C catalysts. (c) k3-weighted Fourier transform for EXAFS spectra of Pd K-edge. (d) Comparison of Pd–Pd and Pd–Cu bond lengths, as well as Pd dispersity. (e) Pd K-edge WT-EXAFS contours of Pd foil, PdCu, and Au@PdCu catalysts. | ||
The Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra of PdCu, Au@PdCu, and Pd foil in R space are displayed in Fig. 3c. Although there is a little decrease in amplitude, the two Pd–Pd/Cu shells in the nanostructure of PdCu and Au@PdCu samples do not show any clear differences in the Fourier transformed EXAFS data of Pd K-edge. To further validate the differences in the bonding forms of Au@PdCu, PdCu, Au NPs, and Pd foil, EXAFS wavelet transform (WT) analysis was carried out.40 The Au L3-edge WT-EXAFS contours of Au NPs and Au@PdCu catalysts, as depicted in Fig. S8,† reveal that Au@PdCu exhibits a contribution from Au–Cu coordination in the low-k region, indicating alloying of some Cu atoms with the Au core. As illustrated in Fig. 3e, the WT analysis of the Pd foil, shows a single intensity maximum at approximately 10.1 Å−1, which corresponds to the Pd–Pd coordination contribution. In contrast, the maximum intensities of Au@PdCu and PdCu both exhibit a distinct negative shift to 8.7 Å−1 and 9.1 Å−1, respectively, which can be attributed to the coordination of Pd–Cu. Actually, the similar Pd K-edge WT-EXAFS contours of PdCu and Au@PdCu illustrate that the introduction of the Au core does not significantly change the local coordination environment of Pd and even maintains the dispersity of the Pd sites to some extent. Since the surface-active site palladium is surrounded by two near-neighbor coordination atoms (copper and palladium), we define the dispersity of the Pd site in terms of the ratio of the coordination number of Pd–Pd to the total coordination number of Pd–Pd (Cu). When the ratio is smaller, the dispersity of Pd is higher. The metal–metal bond lengths and metal coordination numbers (Tables S3–S5†) were extracted by fitting the EXAFS curves (Fig. S9–S17†). The Pd dispersity, Pd–Pd, and Pd–Cu bond lengths for PdCu and Au@PdCu nanocatalysts are depicted in Fig. 3d. The Pd–Cu bond length in the Au@PdCu catalyst showed no change compared to the PdCu catalysts; however, the Pd–Pd bond length notably increased, consistent with a significant improvement in hydrogenation activity. It has well known that dispersed active sites can enhance ethylene selectivity in the acetylene hydrogenation process.41 The Au@PdCu and PdCu catalysts both exhibit high Pd dispersities, which directly relate to their superior ethylene selectivity. The local structure determined by EXAFS appears to be closely related to the catalytic performance in the acetylene hydrogenation reaction. However, due to the fact that catalysis is essentially a surface-driven phenomenon, the limited spatial resolution of EXAFS makes it deficient in describing the distribution of surface active sites.42 The crucial surface structure information, which governs the adsorption and desorption behavior of the reactants, requires three-dimensional spatial peeling.
Surface structure and the 3D decoding of Pd–Pd pairs
The analysis of atomic pair distribution functions (PDF) based on total scattering data (both Bragg and diffuse scattering) has become a well-established method for determining the full-scale atomic structure of nanocatalysts over the past few decades.43,44 The peaks in PDF provide a graphical depiction of the real-space atomic pair distances as the Fourier transform of the total scattering. For revealing the 3D atomic arrangements of Au@PdCu and PdCu NPs, the PDF data were fitted utilizing the reverse Monte Carlo (RMC) technique, as illustrated in Fig. 4a and b. The 3D quantitative extractions of neighboring Pd–Pd coordination numbers and Pd–Pd bond lengths on the surface of PdCu and Au@PdCu NPs are illustrated in Fig. 4c, d, S19 and S20.† The surface distribution of Pd–Pd coordination numbers on PdCu NPs is remarkably similar to that of Au@PdCu NPs (Fig. S19†). Furthermore, Au@PdCu and PdCu have average Pd–Pd coordination numbers of 3.58 and 3.29, respectively, on the outermost layer of nanoparticles (Fig. S20†). The surface features of low Pd–Pd coordination numbers on Au@PdCu and PdCu resemble those of single-atom alloys, which facilitate ethylene desorption and thus enhance selectivity. This is the structural origin at the local scale of the superior ethylene selectivity exhibited by both Au@PdCu and PdCu in acetylene semi-hydrogenation. However, Au@PdCu exhibits a significantly distinct surface Pd–Pd bond length distribution in comparison to PdCu (Fig. 4c). The average Pd–Pd bond length on the Au@PdCu surface is 2.767 Å, which is much larger than the 2.666 Å of PdCu (Fig. 4d). There is a correlation between the enhanced activity and the increased surface Pd–Pd bonds caused by the introduction of the Au core.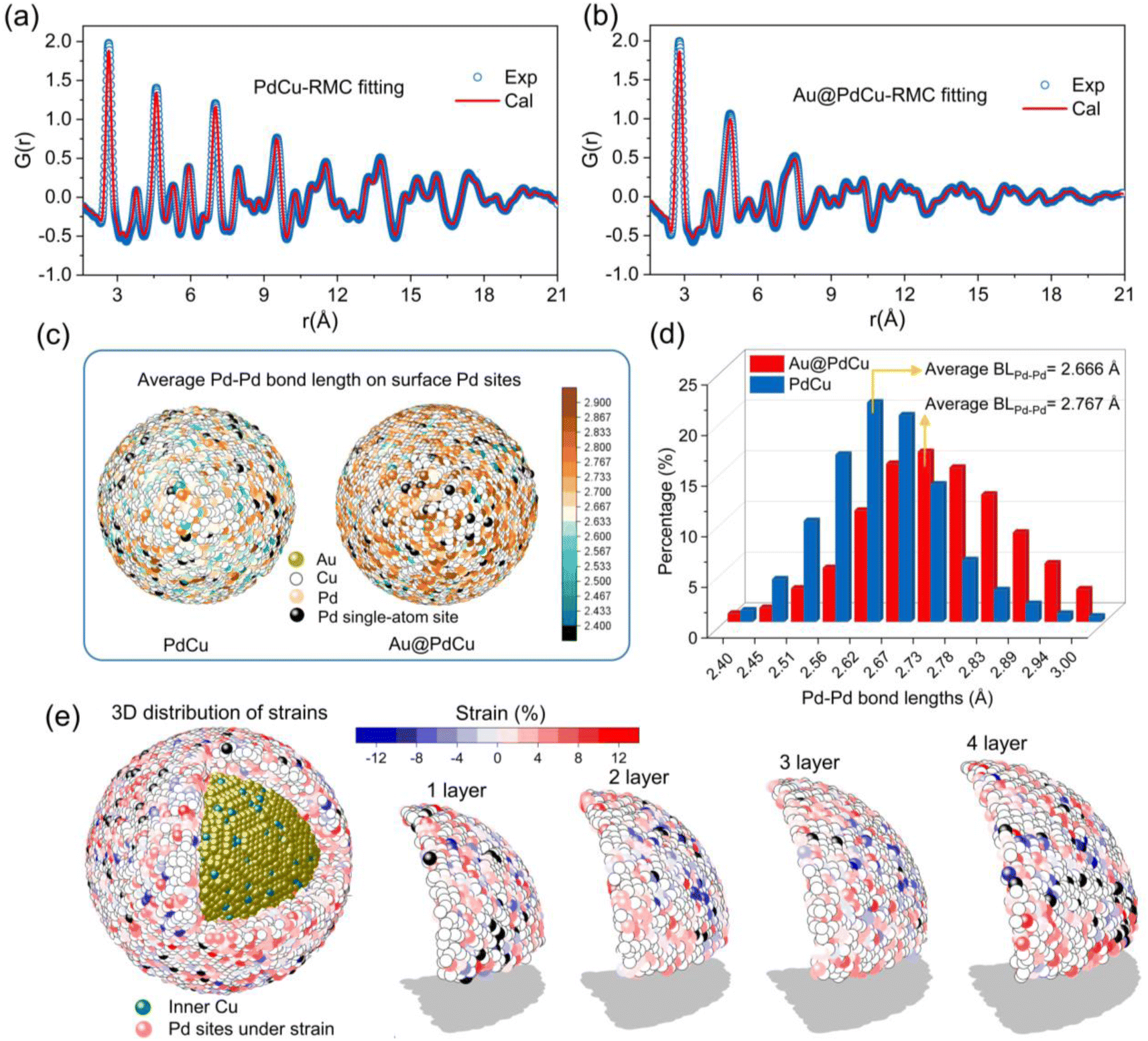 | ||
| Fig. 4 Reverse Monte Carlo (RMC) simulation for atomic pair distribution functions (PDF) and quantitative extractions for the neighboring coordination of Pd sites. (a and b) RMC fitting of PDF. (c) The 3D distribution of the average bond length of Pd–Pd on the Pd sites of PdCu and Au@PdCu NPs. (d) The comparison of surface Pd–Pd bond lengths of PdCu and Au@PdCu NPs. (e) The 3D distribution of surface strain on Pd sites within each layer of the PdCu shell for Au@PdCu NPs. | ||
It's well known that surface strain can effectively modulate the adsorption and desorption behavior of reactants or intermediates in heterogeneous catalytic processes, thus optimizing the catalytic performance.45 To date, however, the majority of strain research has been restricted to low-dimensional (2D) imaging or conventional diffraction, neither of which provides details of the complete 3D strain distribution in core–shell structures. Using the Pd–Pd bond length (2.68 Å) in PdCu NPs obtained by EXAFS as a benchmark, we employed DISCUS software46 to extract the Pd–Pd bond lengths of the Pd sites from the Au@PdCu structure model acquired by RMC fitting experimental PDF data. Further, according to the strain equation, strains were calculated for each Pd site. From inside to outside, we separated the PdCu shell in Au@PdCu nanoparticles into four layers, numbered 1, 2, 3, and 4 (Fig. 4e). A large number of Pd sites subject to tensile strain are distributed on the surface of each layer. Based on statistical analysis of the strain across all Pd sites in each layer in Fig. S21,† it was observed that the strain distribution on the PdCu shell was not uniform. Higher than the average strain in the middle two layers, the average strain of all Pd sites in the outermost and innermost layers of the PdCu shell is 3.25% and 3.21%, respectively.
DFT calculations
Based on the results of the coordination number and bond length of surface Pd sites in Fig. S20† and 4d, the surface models of PdCu alloy with different tensile strains were built for DFT calculations to understand the excellent catalytic performance of the Au@PdCu catalyst (Fig. S22†). With increasing tensile strain, as depicted in Fig. 5a, the center of the d-band of Pd gradually approaches the Fermi energy level. This implies that the tensile strain exerted by Au on the PdCu shells in Au@PdCu nanocatalysts leads to the rise of the d-band center of Pd and the reduction of the antibonding component, which facilitates the adsorption and activation of acetylene and hydrogen and is responsible for its excellent catalytic activity. To further elucidate the correlation between tensile strain and adsorption behavior, DFT calculations were performed on the adsorption energies of hydrogen and ethylene (Fig. S23 and S24†). Owing to the difference in the surface Pd–Pd coordination numbers (CN), the ethylene adsorption pattern on the Pd (111) model with CN of 9 tends to be strong di-σ binding, while the 0%-PdCu (111) model with CN of 3 exhibits weak π-bonding adsorption. The weak ethylene binding energies of 0.76 eV and 0.78 eV for the Pd sites in the 0%-PdCu (111) and 3.25%-PdCu (111) models, respectively, facilitate ethylene desorption and enhance ethylene selectivity, as compared to the Pd (111) model. This suggests that the superior ethylene selectivity exhibited by both the PdCu shell surface prior to and subsequent to the incorporation of the Au core is attributable to the comparable and low Pd–Pd coordination number on the PdCu shell surface. Moreover, the Pd sites in the 3.25%-PdCu (111) model adsorb hydrogen more strongly than those in the 0%-PdCu (111) model, with an adsorption energy of −0.44 eV, making it easier to hydrogenate and contributing to enhancing the catalytic activity. This also proves that tensile strain is the driving force behind the substantial increase in acetylene hydrogenation activity in the Au@PdCu nanocatalyst. Based on all the DFT calculations, the dispersed Pd sites under tensile strain contribute to breaking the trade-off between activity and selectivity, allowing for the complete conversion of acetylene at a lower temperature and maintaining excellent ethylene selectivity.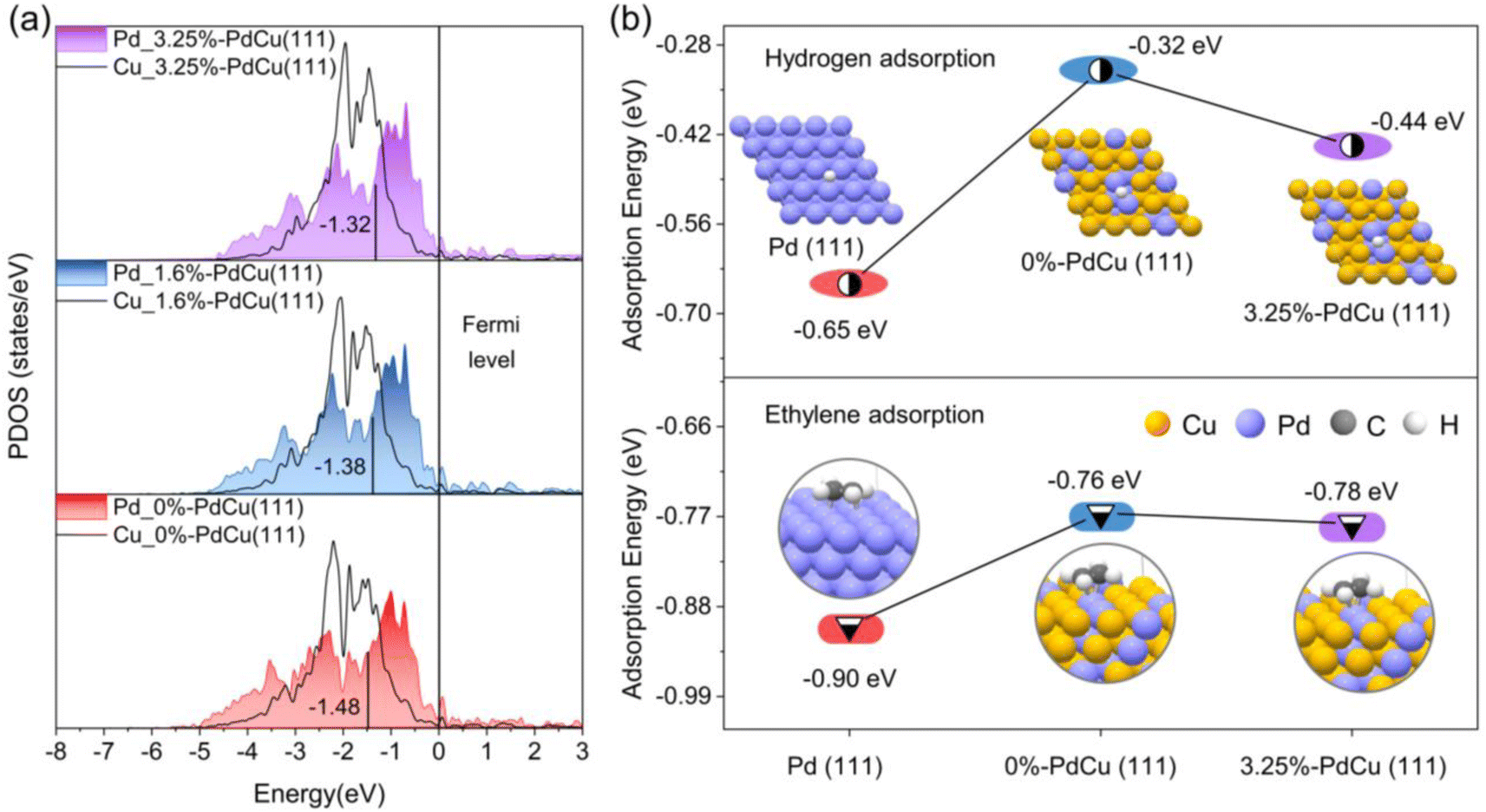 | ||
| Fig. 5 Density functional theory (DFT) calculations. (a) Projected density of states (PDOS) for the total d orbitals of Pd on PdCu surface models with different tensile strains. (b) Ethylene adsorption energy and hydrogen adsorption energy on Pd (111), 0% PdCu (111), and 3.25% PdCu (111) surfaces. | ||
Conclusions
In summary, we have successfully constructed core–shell NPs comprising an Au core and a PdCu alloy shell containing diluted Pd sites using a seed-mediated method. The Au@PdCu core–shell nanocatalyst shows excellent performance for acetylene semi-hydrogenation, achieving complete conversion of acetylene at a relatively mild temperature and maintaining a superior ethylene selectivity of 92.4%. The three-dimensional distribution of lattice strain and the local coordination environment of surface active sites in Au@PdCu core–shell nanoparticles are revealed by the combination of the atomic pair distribution function (PDF) and the reverse Monte Carlo (RMC) method. The quantitative extraction results demonstrate that a large number of active sites with a low Pd–Pd coordination number and in the tensile strain state existed on the surface. DFT calculations further validate that such active sites lead to optimal adsorption of ethylene and hydrogen, contributing to the simultaneous enhancement of hydrogenation activity and ethylene selectivity. This study highlights the importance of local structure insight into core–shell nanocatalysts and lays the structural foundation for the development of efficient heterogeneous catalysts.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
X. X. and Q. L. guided the experimental design and lead the study process. F. X. performed the majority of the experiments as well as data analysis. M. L. help with the catalytic testing. T. L. and Y. R. provided the necessary discussion and analysis for PDF data. W. J. and X. C. contributed to the DFT calculations. L. Z. help to obtain the HAADF-STEM images. J. D., H. X., J. Z. and K. L. participated in the carful discussion of local structure of nanocatalysts. The corresponding authors of this manuscript are X. X. and Q. L. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the National Key R&D Program of China (2020YFA0406202), the National Natural Science Foundation of China (22090042 and 22175018) and Guangxi BaGui Scholars Special Funding (2019M660446). This research used resources of the Advanced Photon Source (APS), a U. S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The total scattering experiments were performed at the beamline 11-ID-C of APS of Argonne National Laboratory. Au L3-edge and Pd K-edge X-ray absorption fine structure (XAFS) spectra were collected on the BL01B1 beamline at SPring-8. The authors thank the staff of beamline BL13SSW at Shanghai Synchrotron Radiation Facility (SSRF) for their support of Cu K-edge XAFS experiments.References
- P. Wang, F. Chang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Breaking scaling relations to achieve low-temperature ammonia synthesis through LiH-mediated nitrogen transfer and hydrogenation, Nat. Chem., 2017, 9, 64–70 CrossRef CAS PubMed
.
- M. M. Montemore and J. W. Medlin, Scaling relations between adsorption energies for computational screening and design of catalysts, Catal. Sci. Technol., 2014, 4, 3748–3761 RSC
- J. Pérez-Ramírez and N. López, Strategies to break linear scaling relationships, Nat. Catal., 2019, 2, 971–976 CrossRef
- G. Sun, Z. J. Zhao, R. Mu, S. Zha, L. Li, S. Chen, K. Zang, J. Luo, Z. Li, S. C. Purdy, A. J. Kropf, J. T. Miller, L. Zeng and J. Gong, Breaking the scaling relationship via thermally stable Pt/Cu single atom alloys for catalytic dehydrogenation, Nat. Commun., 2018, 9, 4454 CrossRef PubMed
- H. Jin, R. Zhao, P. Cui, X. Liu, J. Yan, X. Yu, D. Ma, W. Song and C. Cao, Sabatier Phenomenon in Hydrogenation Reactions Induced by Single-Atom Density, J. Am. Chem. Soc., 2023, 145, 12023–12032 CrossRef CAS PubMed
- Q. Gao, Z. Yan, W. Zhang, H. S. Pillai, B. Yao, W. Zang, Y. Liu, X. Han, B. Min, H. Zhou, L. Ma, B. Anaclet, S. Zhang, H. Xin, Q. He and H. Zhu, Atomic Layers of B2 CuPd on Cu Nanocubes as Catalysts for Selective Hydrogenation, J. Am. Chem. Soc., 2023, 145, 19961–19968 CrossRef CAS PubMed
- S. Wei, X. Liu, C. Wang, X. Liu, Q. Zhang and Z. Li, Atomically Dispersed Pd-N1C3 Sites on a Nitrogen-Doped Carbon Nanosphere for Semi-hydrogenation of Acetylene, ACS Nano, 2023, 17, 14831–14839 CrossRef CAS PubMed
- G. X. Pei, X. Y. Liu, A. Wang, A. F. Lee, M. A. Isaacs, L. Li, X. Pan, X. Yang, X. Wang, Z. Tai, K. Wilson and T. Zhang, Ag Alloyed Pd Single-Atom Catalysts for Efficient Selective Hydrogenation of Acetylene to Ethylene in Excess Ethylene, ACS Catal., 2015, 5, 3717–3725 CrossRef CAS
- X. Xu, Q. Wang, L. Xie, Y. Liu, D. Li, J. Feng and X. Duan, Thermal effect optimization endows a selective and stable PdCu single atom alloy catalyst for acetylene hydrogenation, AIChE J., 2023, 69, e18042 CrossRef CAS
- M. Armbrüster, K. Kovnir, M. Behrens, D. Teschner, Y. Grin and R. Schlögl, Pd−Ga Intermetallic Compounds as Highly Selective Semihydrogenation Catalysts, J. Am. Chem. Soc., 2010, 132, 14745–14747 CrossRef PubMed
- K. Kim, J. Byun, H. Kim, K.-S. Lee, H. S. Lee, J. Kim, T. Hyeon, J. J. Kim and J. W. Han, Systematic Approach to Designing a Highly Efficient Core–Shell Electrocatalyst for N2O Reduction, ACS Catal., 2021, 11, 15089–15097 CrossRef CAS
- Q. Sun, X.-Q. Zhang, Y. Wang and A.-H. Lu, Recent progress on core–shell nanocatalysts, Chin. J. Catal., 2015, 36, 683–691 CrossRef CAS
- H. Jo, D. H. Wi, T. Lee, Y. Kwon, C. Jeong, J. Lee, H. Baik, A. J. Pattison, W. Theis, C. Ophus, P. Ercius, Y. L. Lee, S. Ryu, S. W. Han and Y. Yang, Direct strain correlations at the single-atom level in three-dimensional core–shell interface structures, Nat. Commun., 2022, 13, 5957 CrossRef PubMed
- Z. Hou, C. Cui, Y. Li, Y. Gao, D. Zhu, Y. Gu, G. Pan, Y. Zhu and T. Zhang, Lattice-Strain Engineering for Heterogenous Electrocatalytic Oxygen Evolution Reaction, Adv. Mater., 2023, 35, e2209876 CrossRef PubMed
- B. P. Williams, M. Yaguchi, W. S. Lo, C. R. Kao, L. K. Lamontagne, B. T. Sneed, C. N. Brodsky, L. Y. Chou, C. H. Kuo and C. K. Tsung, Investigating lattice strain impact on the alloyed surface of small Au@PdPt core–shell nanoparticles, Nanoscale, 2020, 12, 8687–8692 RSC
- A. Khorshidi, J. Violet, J. Hashemi and A. A. Peterson, How strain can break the scaling relations of catalysis, Nat. Catal., 2018, 1, 263–268 CrossRef
- P. Moseley and W. A. Curtin, Computational Design of Strain in Core–Shell Nanoparticles for Optimizing Catalytic Activity, Nano Lett., 2015, 15, 4089–4095 CrossRef CAS PubMed
- X. Li, X. Li, C. Liu, H. Huang, P. Gao, F. Ahmad, L. Luo, Y. Ye, Z. Geng, G. Wang, R. Si, C. Ma, J. Yang and J. Zeng, Atomic-Level Construction of Tensile-Strained PdFe Alloy Surface toward Highly Efficient Oxygen Reduction Electrocatalysis, Nano Lett., 2020, 20, 1403–1409 CrossRef CAS PubMed
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Selective Hydrogenation over Supported Metal Catalysts: From Nanoparticles to Single Atoms, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed
- G. Wu, X. Han, J. Cai, P. Yin, P. Cui, X. Zheng, H. Li, C. Chen, G. Wang and X. Hong, In-plane strain engineering in ultrathin noble metal nanosheets boosts the intrinsic electrocatalytic hydrogen evolution activity, Nat. Commun., 2022, 13, 4200 CrossRef CAS PubMed
- S. Yang, F. Liu, C. Wu and S. Yang, Tuning Surface Properties of Low Dimensional Materials via Strain Engineering, Small, 2016, 12, 4028–4047 CrossRef CAS PubMed
- Y. Ding, F. Fan, Z. Tian and Z. L. Wang, Atomic Structure of Au−Pd Bimetallic Alloyed Nanoparticles, J. Am. Chem. Soc., 2010, 132, 12480–12486 CrossRef CAS PubMed
- F.-R. Fan, D.-Y. Liu, Y.-F. Wu, S. Duan, Z.-X. Xie, Z.-Y. Jiang and Z.-Q. Tian, Epitaxial Growth of Heterogeneous Metal Nanocrystals: From Gold Nano-octahedra to Palladium and Silver Nanocubes, J. Am. Chem. Soc., 2008, 130, 6949–6951 CrossRef CAS PubMed
- Q. Li, H. Zhu, X. Chen, H. Liu, Y. Ren, Y. Chen, K. Ohara, L. Zhou, J. Chen, J. Deng, J. Miao, K. Lin, X. Kuang and X. Xing, Local Structure Insight into Hydrogen Evolution Reaction with Bimetal Nanocatalysts, J. Am. Chem. Soc., 2022, 144, 20298–20305 CrossRef CAS PubMed
- D. A. Keen, Total scattering and the pair distribution function in crystallography, Crystallogr. Rev., 2020, 26, 143–201 CrossRef
- Q. Li, Y. Ren, Q. Zhang, L. Gu, Q. Huang, H. Wu, J. Sun, Y. Cao, K. Lin and X. Xing, Chemical order-disorder nanodomains in Fe3Pt bulk alloy, Natl. Sci. Rev., 2022, 9, nwac053 CrossRef CAS PubMed
- F. Xue, Q. Li, M. Lv, Y. Song, T. Yang, X. Wang, T. Li, Y. Ren, K. Ohara, Y. He, D. Li, Q. Li, X. Chen, K. Lin and X. Xing, Atomic Three-Dimensional Investigations of Pd Nanocatalysts for Acetylene Semi-hydrogenation, J. Am. Chem. Soc., 2023, 145, 26728–26735 CrossRef CAS PubMed
- F. Xue, Q. Li, M. Lv, S. Weng, T. Li, Y. Ren, Y. Liu, D. Li, Y. He, Q. Li, X. Chen, Q. Zhang, L. Gu, J. Deng, J. Chen, L. He, X. Kuang, J. Miao, Y. Cao, K. Lin and X. Xing, Decoding Active Sites for Highly Efficient Semihydrogenation of Acetylene in Palladium–Copper Nanoalloys, Nano Lett., 2024, 24, 6269–6277 CrossRef CAS PubMed
- Z. L. Zhao, L. Y. Zhang, S. J. Bao and C. M. Li, One-pot synthesis of small and uniform Au@PtCu core–alloy shell nanoparticles as an efficient electrocatalyst for direct methanol fuel cells, Appl. Catal., B, 2015, 174–175, 361–366 CrossRef CAS
- T. M. Clarke and J. R. Durrant, Charge Photogeneration in Organic Solar Cells, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS PubMed
- Y. Zhang, Y. Lin, T. Duan and L. Song, Interfacial engineering of heterogeneous catalysts for electrocatalysis, Mater. Today, 2021, 48, 115–134 CrossRef CAS
- J. Zhao, B. Chen and F. Wang, Shedding light on the role of misfit strain in controlling core–shell nanocrystals, Adv. Mater., 2020, 32, 2004142 CrossRef CAS PubMed
- R. Ma, Y. He, J. Feng, Z.-Y. Hu, G. Van Tendeloo and D. Li, A facile synthesis of Ag@PdAg core–shell architecture for efficient purification of ethene feedstock, J. Catal., 2019, 369, 440–449 CrossRef CAS
- J. Timoshenko and B. Roldan Cuenya, In Situ/Operando Electrocatalyst Characterization by X-ray Absorption Spectroscopy, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed
- T. Ricciardulli, S. Gorthy, J. S. Adams, C. Thompson, A. M. Karim, M. Neurock and D. W. Flaherty, Effect of Pd Coordination and Isolation on the Catalytic Reduction of O2 to H2O2 over PdAu Bimetallic Nanoparticles, J. Am. Chem. Soc., 2021, 143, 5445–5464 CrossRef CAS PubMed
- F. Huang, M. Peng, Y. Chen, X. Cai, X. Qin, N. Wang, D. Xiao, L. Jin, G. Wang, X. D. Wen, H. Liu and D. Ma, Low-Temperature Acetylene Semi-Hydrogenation over the Pd1–Cu1 Dual-Atom Catalyst, J. Am. Chem. Soc., 2022, 144, 18485–18493 CrossRef CAS PubMed
- T. E. R. Fiuza and D. Zanchet, Supported AuCu Alloy Nanoparticles for the Preferential Oxidation of CO (CO-PROX), ACS Appl. Nano Mater., 2020, 3, 923–934 CrossRef CAS
- M. Kuhn and T. K. Sham, Charge redistribution and electronic behavior in a series of Au–Cu alloys, Phys. Rev. B, 1994, 49, 1647–1661 CrossRef CAS PubMed
- S. Alayoglu and G. A. Somorjai, Nanocatalysis II: In Situ Surface Probes of Nano-Catalysts and Correlative Structure–Reactivity Studies, Catal. Lett., 2015, 145, 249–271 CrossRef CAS
- Z. Yu, S. Xu, Y. Feng, C. Yang, Q. Yao, Q. Shao, Y.-f. Li and X. Huang, Phase-Controlled Synthesis of Pd–Se Nanocrystals for Phase-Dependent Oxygen Reduction Catalysis, Nano Lett., 2021, 21, 3805–3812 CrossRef CAS PubMed
- F. Huang, Y. Deng, Y. Chen, X. Cai, M. Peng, Z. Jia, P. Ren, D. Xiao, X. Wen, N. Wang, H. Liu and D. Ma, Atomically Dispersed Pd on Nanodiamond/Graphene Hybrid for Selective Hydrogenation of Acetylene, J. Am. Chem. Soc., 2018, 140, 13142–13146 CrossRef CAS PubMed
- H. Zhu, Y. Huang, J. Ren, B. Zhang, Y. Ke, A. K. Jen, Q. Zhang, X. L. Wang and Q. Liu, Bridging Structural Inhomogeneity to Functionality: Pair Distribution Function Methods for Functional Materials Development, Crystallogr. Rev., 2021, 8, 2003534 CAS
- N. K. Zimmerli, C. R. Müller and P. M. Abdala, Deciphering the structure of heterogeneous catalysts across scales using pair distribution function analysis, Trends Chem., 2022, 4, 807–821 CrossRef CAS
- B. Ingham, X-ray scattering characterisation of nanoparticles, Crystallogr. Rev., 2015, 21, 229–303 CrossRef
- T. He, W. Wang, F. Shi, X. Yang, X. Li, J. Wu, Y. Yin and M. Jin, Mastering the surface strain of platinum catalysts for efficient electrocatalysis, Nature, 2021, 598, 76–81 CrossRef CAS PubMed
- T. Proffen and R. B. Neder, DISCUS: a program for diffuse scattering and defect-structure simulation, J. Appl. Crystallogr., 1997, 30, 171–175 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03291h |
| This journal is © The Royal Society of Chemistry 2024 |