
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible light induced cooperative carbonylation and (hetero)aryl migration: synthesis of multi-carbonyl compounds†
Hefei
Yang
ab,
Yuanrui
Wang
a,
Le-Cheng
Wang
ab and
Xiao-Feng
Wu
 *ab
*ab
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023 Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
bLeibniz-Institut für Katalyse e.V., 18059 Rostock, Germany. E-mail: Xiao-Feng.Wu@catalysis.de
First published on 14th August 2024
Abstract
Carbonylative transformation represents one of the most straightforward procedures for the synthesis of carbonyl-containing compounds. However, the carbonylative procedure toward 1,4-diketones is still limited which are key moieties with potent applications in various areas. Herein, we report a new strategy for the synthesis of multi-carbonyl compounds containing a 1,4-diketone skeleton through remote heteroaryl migration of traditionally restricted 1,3-migratory substrates utilizing carbon monoxide (CO) as the C1 synthon and diazonium compounds as the starting material.
Introduction
Carbonyl-containing compounds possess versatile reactive sites and are privileged building blocks for the construction of a wide range of useful chemicals. Among them, 1,4-diketones are widely used as synthetic precursors for the assembly of various bioactive compounds, including bioactive molecules such as pyrroles, furans, thiophenes, diols, diamines, cyclopentenones, etc.1 Many natural products also contain a 1,4-diketone skeleton. Examples include herquline A,2 amphidinolide F3 and maoecrystal V (Fig. 1(a)).4 Due to the recognized importance of the carbonyl group, it is therefore particularly important to develop new methods to produce these valuable multi-carbonyl compounds in synthetic chemistry. | ||
| Fig. 1 Study on multi-carbonyl compounds containing a 1,4-diketone skeleton. | ||
Compared with using complexed molecules for constructing challenging natural product skeletons, the use of simple molecules is more attractive in synthetic chemistry based on their readily availability. Carbon monoxide (CO) serves as a cost-effective and readily available C1 source, making it a significant synthon in carbonylation reactions.5 Radical mediated carbonylation is attracting growing interest because of its versatility for radical generation and relatively mild reaction conditions. Over the past few decades, there has been a systematic investigation into the radical-promoted difunctionalization reactions of unactivated alkenes, including remote functional group migration.6 Since the first report of radical-mediated aryl migration by Wieland in 1911,7 migration reactions have been extensively studied but are mainly limited to migration between two carbon atoms. In 1995, Kim et al. reported the migration of 1,2-aryl groups from carbon to N-centered radicals derived from azido groups.8 This type of reaction was subsequently enriched by Shi and Nevado et al.9 Migration reactions from the carbon center to the oxygen center and from the carbon center to carboxylated radicals have also developed over time.10 However, migration from carbon to acyl radicals has not been well achieved and needs to be explored and investigated (Fig. 1(b)).
Due to their high reactivity and wide range of applications, diazo compounds are highly regarded as carbene precursors under transition metal catalysis or direct light irradiation.11 In contrast, relevant research studies on the reactions of diazo compounds as radical precursors were less reported.12 We envision that the conversion of diazo compounds into carbon radicals through the proton-coupled electron transfer process13 might be a solution. Additionally, 1,3-migrating substrates are limited for remote functional group migration. Herein, we describe a mild and efficient protocol for the heteroaromatic group migration and carbonylation of unactivated alkenes with carbon monoxide and a variety of useful multi-carbonyl compounds containing a 1,4-dicarbonyl skeleton were prepared in good yields in general (Fig. 1(c)).
Results and discussion
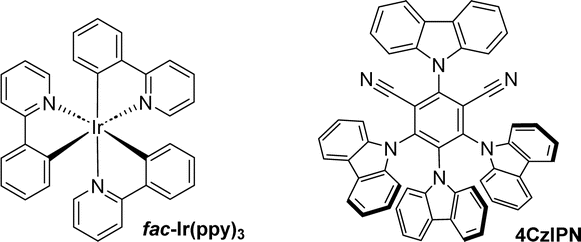
To realise this transformation, we studied the reaction using dimethyl 2-diazomalonate (1a) and benzothiazole-substituted tertiary alcohol (2a) as model substrates for the heteroaromatic group migration and carbonylation in the presence of 1.0 mol% fac-Ir(ppy)3. After exposure to CO at 40 bar with blue LEDs in N,N-dimethylformamide (DMF) at room temperature for 24 hours, the desired product 3a was obtained in 72% yield (Table 1, entry 1). Control reactions revealed that omission of the photocatalyst fac-Ir(ppy)3, or light, gave very low product 3a formation. Moreover, replacing fac-Ir(ppy)3 with 4CzIPN or changing the power of the lamp resulted in a decrease in the yield of the desired product 3a. Diverse solvents were examined for the reaction, and the desired product was identified, although the outcomes were found to be unsatisfactory (for details, please refer to the ESI† for more information).|
|
||
|---|---|---|
| Entry | Variation from the standard reaction conditions | Yield [%] |
| a Reaction conditions: 1a (0.6 mmol), 2a (0.2 mmol), photocatalyst (1 mol%), H2O (3 equiv.), DMF (2 mL) with blue LEDs (15 W) at room temperature for 24 h under CO (40 bar). Isolated yields. | ||
| 1 | None | 72 |
| 2 | No PC | 9 |
| 3 | No light | 2 |
| 4 | 4CzIPN instead of fac-Ir(ppy)3 | 53 |
| 5 | 7 W instead of 15 W | 60 |
| 6 | Other solvents instead of DMF | <60 |
|
||

After mastering the optimal conditions for the reaction, we began to examine the compatibility of this transformation with a series of different benzothiazole-substituted tertiary alcohols and dimethyl 2-diazomalonate (Fig. 2). The overall experimental results showed that the benzothiazole group preferentially migrated chemoselectively over the aryl group. When different electron-donating or electron-absorbing groups were used to replace the aryl group in the substrate, the carbonylated products were obtained in good yields with little variation in yields (3a–3g and 3n–3q). Surprisingly, halides, especially chlorides (3f), were compatible in this remote migration reaction, providing a basis for further modifications of the substrate. By changing the substitution site on the benzene ring, we also tested the tolerance of the reaction to spatial effects, which proved to have no significant effect on the reaction yields (3h–3j). When the aryl group is replaced by a double group, the target product can still be obtained in good yields (3k–3m). The tolerance of the reaction to aromatic heterocycles other than benzothiazole groups was then investigated. The migration of various substituted benzothiazoles proceeded smoothly, with electronic and spatial effects largely unaffected (3r–3t). In addition to substituted benzothiazoles, benzoxazoles and thiazoles, among others, also reacted smoothly and good yields were obtained (3u–3ab). Benzofuranyl and benzothiophene groups were slightly sensitive to reaction conditions and the established products were obtained in 44% yields (3ac–3ad). To our surprise, diaryl-substituted alcohols were also able to normalise the migration reaction. However, because of the migration efficiency, the target product was obtained in 36% yield (3ae). It's also worth mentioning that the reactions with 1-(benzo[d]thiazol-2-yl)-1-phenylpent-4-en-1-ol and 1-(benzo[d]thiazol-2-yl)-1-phenylprop-2-en-1-ol with diazo compound toward 1,5- and 1,3-aryl migration failed without the desired product being detectable. In the cases of 1-phenyl-1-(pyridin-2-yl)but-3-en-1-ol and 1-(1-methyl-1H-indol-2-yl)-1-phenylbut-3-en-1-ol, no targeted product could be detected.
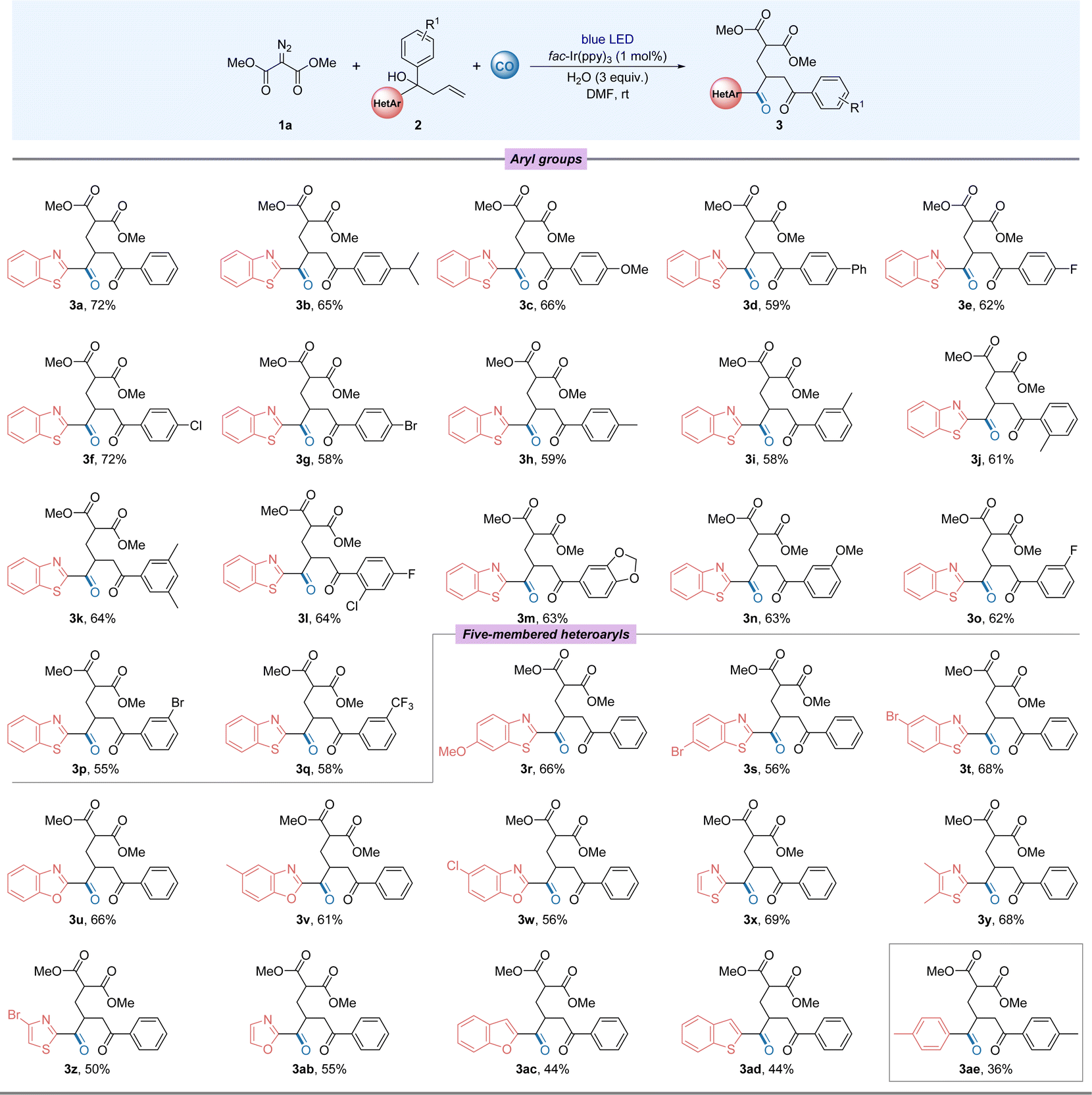 | ||
| Fig. 2 Substate scope: testing of the tertiary alcohols. Reaction conditions: 1a (0.6 mmol), 2 (0.2 mmol), fac-Ir(ppy)3 (1 mol%), H2O (3 equiv.), DMF (2 mL) with blue LEDs (15 W) at room temperature for 24 h under CO (40 bar), isolated yield. | ||
In order to further expand the product diversity of the reaction, diazo compounds were tested subsequently and decreased conversion was observed in some cases (Fig. 3). Considering the reduced activity of the diazo compounds compared to dimethyl 2-diazomalonate, the reaction was slightly optimised with an appropriate extension of the reaction time. Various alkyl alcohol substituted diazo esters were tested first and the results showed good compatibility (4a–4i). When the substituent group was a heterocycle, it showed sensitivity to the reaction and the target product could only be obtained in 36% yield (4g). Finally, we attempted to introduce various biomolecules into the reaction products and the corresponding products were obtained in moderate to good yields (4j–4n). Diazoacetylbenzene was also tested, but low yield was obtained.
 | ||
| Fig. 3 Substrate scope: testing of diazo compounds. Reaction conditions: 1 (0.6 mmol), 2a (0.2 mmol), fac-Ir(ppy)3 (1 mol%), H2O (3 equiv.), DMF (2 mL) with blue LEDs (15 W) at room temperature for 36 h under CO (40 bar), isolated yield.a The d.r. value was determined by 1H NMR. | ||
To better understand how the migration reaction occurs, we performed several control experiments as shown in Fig. 4. The model reaction was inhibited when a radical scavenger was added to the reaction under the standard conditions. In the presence of BHT (butylated hydroxytoluene), radical scavenging by-products were detected by GC-MS. The results of these radical quenching experiments suggest that the reaction may involve radical intermediates.
 | ||
| Fig. 4 Control experiments and synthetic applications. | ||
Additional control experiments were also carried out to obtain further mechanistic information. During the optimization of the reaction, it was found that the extra addition of H2O favored the conversion of diazonium compounds which promoted the reaction. To investigate the role of H2O, the experimental results were monitored by controlling the addition of H2O. When H2O was not present in the reaction system, the target product was obtained in 43% yield. After addition of 3 equivalents of D2O, the established product was obtained in a yield of 52% and 50% D was detected. The experimental results showed that the protons in the formation of carbon radicals from diazonium compounds partially originate from water in the reaction system. When carbon monoxide was not present in the system, the modelled reaction did not occur and only significant amounts of by-products were detected. This indicates that carbon monoxide plays an important role in the reaction by acting as a linker type of promoter.
To further demonstrate the value of the synthetic method, the products generated were further transformed. 1,4-Dicarbonyl compounds were used as synthetic building blocks for biologically active heterocycle preparation, and the corresponding pyrrole derivative 5 and thiophene derivative 6 were obtained in excellent yields using product 3a as a template substrate. Surprisingly, both products 5 and 6 showed excellent fluorescence under 365 nm light, which may further expand their applicability. The ester group of product 4a can be easily hydrolysed to afford the carboxylic acid derivative 7. With the research on carboxylic acid derivatives in recent years, a number of decarboxylation halogenations, decarboxylation arylations, decarboxylation borylations, decarboxylation unsaturations, etc. can be obtained by modifying the carboxylic acid group.14 These expressed reaction advantages may have potential applications in the future for drug developments. Concerning large-scale synthesis, this method can be easily expanded by performing the reaction with several vials parallelly in the same autoclave which is even more stable with the reaction outcome.
Based on the experimental results and literature,15 we proposed a possible reaction pathway for this transformation (Fig. 5). Initially, the diazonium compound was reduced using an excited state catalyst *fac-Ir(ppy)3 and undergoes a PCET process to release nitrogen to obtain alkyl radical I. Addition of alkyl radical I to the unactivated olefinic substrate 2a yields a new carbon radical intermediate II. At this point, the radical intermediate II can capture carbon monoxide under a carbon monoxide atmosphere to generate acyl radical III. Subsequent intramolecular addition of heteroaryl groups provides intermediate IV, which undergoes radical mobilisation to afford intermediate V. The final sequence yields the established product 4avia SET oxidation and deprotonation with simultaneous regeneration of the base state photocatalyst.
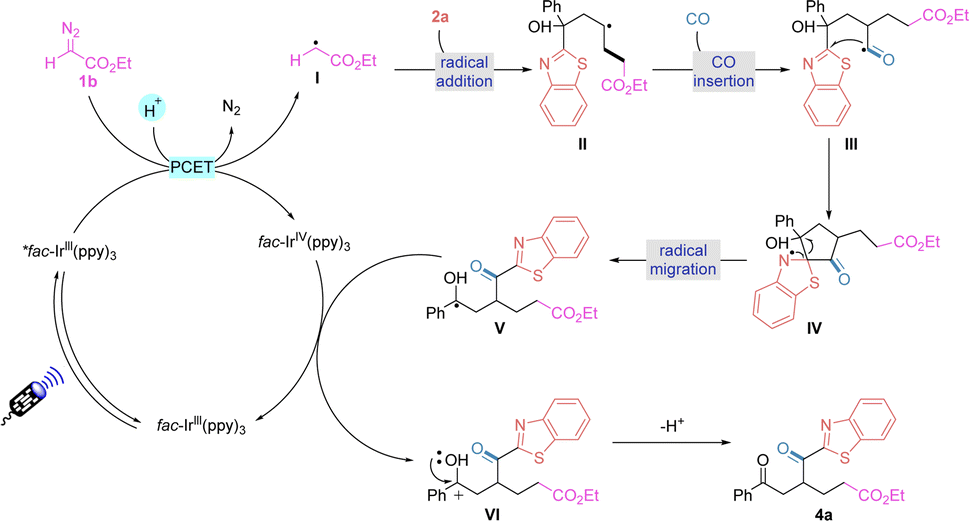 | ||
| Fig. 5 Proposed mechanism. | ||
Conclusions
In conclusion, we developed a new preparation strategy for multi-carbonyl compounds containing a 1,4-diketone skeleton. The reaction proceeds via radical-mediated difunctionalisation of unactivated alkenes using carbon monoxide (CO) as the C1 linker. By this procedure, remote heteroaryl migration of traditionally restricted 1,3-migrating substrates has been successfully achieved utilizing carbon monoxide (CO) as the C1 synthon, providing a new pathway for the subsequent use and study of carbon monoxide. In the future, efforts on 1,3-migration, 1,5-migration and others should be made.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
X.-F. W. conceived and directed the project. H. Y. performed all the experiments. X.-F. W. and H. Y. wrote and revised the manuscript. H. Y., Y. W., and L. C. W. prepared the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2023YFA1507500) and the International Partnership Program of the Chinese Academy of Sciences (Grant No. 028GJHZ2023045FN).Notes and references
- (a) Z.-L. Shen, K. K. Goh, H.-L. Cheong, C. H. Wong, Y.-C. Lai, Y.-S. Yang and T.-P. Loh, Synthesis of Water-Tolerant Indium Homoenolate in Aqueous Media and Its Application in the Synthesis of 1,4-Dicarbonyl Compounds via Palladium-Catalyzed Coupling with Acid Chloride, J. Am. Chem. Soc., 2010, 132, 15852–15855 CrossRef CAS PubMed; (b) X. Zhao, B. Li and W. Xia, Visible-Light-Promoted Photocatalyst-Free Hydroacylation and Diacylation of Alkenes Tuned by NiCl2·DME, Org. Lett., 2020, 22, 1056–1061 CrossRef CAS PubMed; (c) Y. Bai, S. Xiang, M. L. Leow and X.-W. Liu, Dual-Function Pd/NHC Catalysis: Tandem Allylation–Isomerization–Conjugate Addition that Allows Access to Pyrroles, Thiophenes and Furans, Chem. Commun., 2014, 50, 6168–6170 RSC; (d) A. Mattson, A. Bharadwaj and K. Scheidt, The Thiazolium-Catalyzed Sila-Stetter Reaction: Conjugate Addition of Acylsilanes to Unsaturated Esters and Ketones, J. Am. Chem. Soc., 2004, 126, 2314–2315 CrossRef CAS PubMed; (e) L. S. Bernardes, M. J. Kato, S. Albuquerque and I. Carvalho, Synthesis and Trypanocidal Activity of 1,4-Bis-(3,4,5-Trimethoxy-Phenyl)-1,4-Butanediol and 1,4-Bis-(3,4-Dimethoxyphenyl)-1,4-Butanediol, Bioorg. Med. Chem., 2006, 14, 7075–7082 CrossRef CAS PubMed; (f) R. F. Kaltenbach, D. A. Nugiel, P. Y. S. Lam, R. M. Klabe and S. P. Seitz, Stereoisomers of Cyclic Urea HIV-1 Protease Inhibitors: Synthesis and Binding Affinities, J. Med. Chem., 1998, 41, 5113–5117 CrossRef CAS PubMed; (g) G. Piancatelli, M. D'Auria and F. D'Onofrio, Synthesis of 1,4-Dicarbonyl Compounds and Cyclopentenones from Furans, Synthesis, 1994, 1994, 867–889 CrossRef.
- A. Furusaki and T. Furusaki, X-Ray Crystal Structure of Herquline, A New Biologically Active Piperazine from Penicillium Herquei Fg-372, J. Chem. Soc., Chem. Commun., 1980, 698a RSC.
- J. Kobayashi, M. Tsuda, M. Ishibashi, H. Shigemori, T. Yamasu, H. Yamasu and T. Sasaki, Amphidinolide F, A New Cytotoxic Macrolide from the Marine Dinoflagellate, J. Antibiot., 1991, 44, 1259–1261 CrossRef CAS PubMed.
- C. Zheng, I. Dubovyk, K. E. Lazarski and R. J. Thomson, Enantioselective Total Synthesis of (−)-Maoecrystal V, J. Am. Chem. Soc., 2014, 136, 17750–17756 CrossRef CAS PubMed.
- (a) B. Gabriele, Carbon Monoxide in Organic Synthesis-Carbonylation Chemistry, Wiley-VCH, Weinheim, 2021 CrossRef; (b) X.-F. Wu, B. Han, K. Ding and Z. Liu, The Chemical Transformations of C1 Compounds, Wiley-VCH, Weinheim, 2022 CrossRef; (c) M. Beller and X.-F. Wu, Transition Metal Catalysed Carbonylation Reactions: Carbonylative Activation of C-X Bonds, Springer, Berlin, 2013 CrossRef; (d) Z.-P. Bao, Y. Zhang, L.-C. Wang and X.-F. Wu, Difluoroalkylative carbonylation of alkenes to access carbonyl difluoro-containing heterocycles: convenient synthesis of gemigliptin, Sci. China: Chem., 2023, 66, 139–146 CrossRef CAS; (e) Y. Yuan and X.-F. Wu, Generation and Transformation of α-Oxy Carbene Intermediates Enabled by Copper-Catalyzed Carbonylation, Green Carbon, 2024, 2, 70–80 CrossRef.
- X. Wu, Z. Ma, T. Feng and C. Zhu, Radical-Mediated Rearrangements: Past, Present, and Future, Chem. Soc. Rev., 2021, 50, 11577–11613 RSC.
- H. Wieland, ÜBer Triphenylmethyl-Peroxyd. Ein Beitrag Zur Chemie der Freien Radikale, Chem. Ber., 1911, 44, 2550–2556 CrossRef CAS.
- S. Kim and J. Y. Do, 1,2-Radical Rearrangements of Aryl, Furanyl and Thiophenyl Groups from Carbon to Nitrogen, J. Chem. Soc., Chem. Commun., 1995, 1607–1608 RSC.
- (a) T. Zhou, F.-X. Luo, M.-Y. Yang and Z.-J. Shi, Silver-Catalyzed Long-Distance Aryl Migration from Carbon Center to Nitrogen Center, J. Am. Chem. Soc., 2015, 137, 14586–14589 CrossRef CAS PubMed; (b) W. Shu, A. Genoux, Z. Li and C. Nevado, γ-Functionalizations of Amines through Visible-Light-Mediated, Redox-Neutral C–C Bond Cleavage, Angew. Chem., Int. Ed., 2017, 56, 10521–10524 CrossRef CAS PubMed; (c) Y. Ning, A. Mekareeya, K. R. Babu, E. A. Anderson and X. Bi, Silver-Catalyzed C- to N-Center Remote Arene Migration, ACS Catal., 2019, 9, 4203–4210 CrossRef CAS.
- (a) Y. Zhu, Z. Zhang, R. Jin, J. Liu, G. Liu, B. Han and N. Jiao, DMSO-Enabled Selective Radical O–H Activation of 1,3(4)-Diols, Angew. Chem., Int. Ed., 2020, 59, 19851–19856 CrossRef CAS PubMed; (b) Z. Zhang, L. Zhang, X. Zhang, J. Yang, Y. Yin, Y. Jiang, C. Zeng, G. Lu, Y. Yang and F. Mo, Anodic Oxidation Triggered Divergent 1,2- and 1,4-Group Transfer Reactions of β-Hydroxycarboxylic Acids Enabled by Electrochemical Regulation, Chem. Sci., 2020, 11, 12021–12028 RSC.
- (a) Y. Xia, D. Qiu and J. Wang, Transition-Metal-Catalyzed Cross-Couplings through Carbene Migratory Insertion, Chem. Rev., 2017, 117, 13810–13889 CrossRef CAS PubMed; (b) H. M. L. Davies and J. R. Manning, Catalytic C–H Functionalization by Metal Carbenoid and Nitrenoid Insertion, Nature, 2008, 451, 417–424 CrossRef CAS PubMed; (c) C. Empel, C. Pei and R. M. Koenigs, Unlocking Novel Reaction Pathways of Diazoalkanes with Visible Light, Chem. Commun., 2022, 58, 2788–2798 RSC.
- (a) Y.-L. Su, G.-X. Liu, J.-W. Liu, L. Tram, H. Qiu and M. P. Doyle, Radical-Mediated Strategies for the Functionalization of Alkenes with Diazo Compounds, J. Am. Chem. Soc., 2020, 142, 13846–13855 CrossRef CAS PubMed; (b) X. Huang, R. D. Webster, K. Harms and E. Meggers, Asymmetric Catalysis with Organic Azides and Diazo Compounds Initiated by Photoinduced Electron Transfer, J. Am. Chem. Soc., 2016, 138, 12636–12642 CrossRef CAS PubMed; (c) F. Li, C. Pei and R. M. Koenigs, Photocatalytic gem-Difluoroolefination Reactions by a Formal C–C Coupling/Defluorination Reaction with Diazoacetates, Angew. Chem., Int. Ed., 2022, 61, e202111892 CrossRef CAS PubMed.
- (a) S. J. Mora, E. Odella, G. F. Moore, D. Gust, T. A. Moore and A. L. Moore, Proton-Coupled Electron Transfer in Artificial Photosynthetic Systems, Acc. Chem. Res., 2018, 51, 445–453 CrossRef CAS PubMed; (b) H. V. Huynh and T. J. Meye, Proton-Coupled Electron Transfer, Chem. Rev., 2007, 107, 5004–5064 CrossRef PubMed; (c) E. Hatcher, A. V. Soudackov and S. Schiffer, Proton-Coupled Electron Transfer in Soybean Lipoxygenase, J. Am. Chem. Soc., 2004, 126, 5763–5775 CrossRef CAS PubMed; (d) D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Proton-Coupled Electron Transfer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- (a) G. J. P. Perry, J. M. Quibell, A. Panigrahi and I. Larrosa, Transition-Metal-Free Decarboxylative Iodination: New Routes for Decarboxylative Oxidative Cross-Couplings, J. Am. Chem. Soc., 2017, 139, 11527–11536 CrossRef CAS PubMed; (b) X. Huang, W. Liu, J. M. Hooker and J. T. Groves, Targeted Fluorination with the Fluoride Ion by Manganese-Catalyzed Decarboxylation, Angew. Chem., Int. Ed., 2015, 54, 5241–5245 CrossRef CAS PubMed; (c) Z. Wang, C.-Y. Guo, C. Yang and J.-P. Chen, Ag-Catalyzed Chemoselective Decarboxylative Mono- and gem-Difluorination of Malonic Acid Derivatives, J. Am. Chem. Soc., 2019, 141, 5617–5622 CrossRef CAS PubMed; (d) H. Wang, C.-F. Liu, Z. Song, M. Yuan, Y. A. Ho, O. Gutierrez and M. J. Koh, Engaging α-Fluorocarboxylic Acids Directly in Decarboxylative C–C Bond Formation, ACS Catal., 2020, 10, 4451–4459 CrossRef CAS; (e) L. Candish, M. Teders and F. Glorius, Transition-Metal-Free, Visible-Light-Enabled Decarboxylative Borylation of Aryl N-hydroxyphthalimide Esters, J. Am. Chem. Soc., 2017, 139, 7440–7443 CrossRef CAS PubMed; (f) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Photoinduced Decarboxylative Borylation of Carboxylic Acids, Science, 2017, 357, 283–286 CrossRef CAS PubMed; (g) H. Cao, H. Jiang, H. Feng, J. M. C. Kwan, X. Liu and J. Wu, Photo-induced Decarboxylative Heck-Type Coupling of Unactivated Aliphatic Acids and Terminal Alkenes in the Absence of Sacrificial Hydrogen Acceptors, J. Am. Chem. Soc., 2018, 140, 16360–16367 CrossRef CAS PubMed; (h) Q.-Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L.-Q. Lu and W.-J. Xiao, Decarboxylative Alkynylation and Carbonylative Alkynylation of Carboxylic Acids Enabled by Visible-Light Photoredox Catalysis, Angew. Chem., Int. Ed., 2015, 54, 11196–11199 CrossRef CAS PubMed.
- (a) W. H. Urry and M. S. Kharasch, Factors Influencing the Course and Mechanism of Grignard Reactions. Xix. The Preparation of Substituted Bibenzyls from Grignard Reagents, Alkyl Halides, and Alkylbenzenes, In the Presence and Absence of Colbaltous Chloride, J. Org. Chem., 1948, 13, 101–109 CrossRef PubMed; (b) F. Bu, L. Lu, X. Hu, S. Wang, H. Zhang and A. Lei, Electrochemical Oxidative Decarboxylation and 1,2-Aryl Migration towards the Synthesis of 1,2-Diaryl Ethers, Chem. Sci., 2020, 11, 10000–10004 RSC; (c) Z. Cong, T. Miki, O. Urakawa and H. Nishino, Synthesis of Dibenz[b,f]oxepins via Manganese(III)-Based Oxidative 1,2-Radical Rearrangement, J. Org. Chem., 2009, 74, 3978–3981 CrossRef CAS PubMed; (d) X.-Q. Chu, Y. Zi, H. Meng, X.-P. Xu and S.-J. Ji, Radical Phosphinylation of α,α-Diaryl Allylic Alcohols with Concomitant 1,2-Aryl Migration, Chem. Commun., 2014, 50, 7642–7645 RSC; (e) X. Wu and C. Zhu, Radical-Mediated Remote Functional Group Migration, Acc. Chem. Res., 2020, 53, 1620–1636 CrossRef CAS PubMed; (f) Z. Wu, D. Wang, Y. Liu, L. Huan and C. Zhu, Chemo- and Regioselective Distal Heteroaryl ipso-Migration: A General Protocol for Heteroarylation of Unactivated Alkenes, J. Am. Chem. Soc., 2017, 139, 1388–1391 CrossRef CAS PubMed; (g) X. Tang and A. Studer, α-Perfluoroalkyl-β-alkynylation of Alkenes via Radical Alkynyl Migration, Chem. Sci., 2017, 8, 6888–6892 RSC; (h) L. Li, Z.-L. Li, Q.-S. Gu, N. Wang and X.-Y. Liu, A Remote C–C Bond Cleavage-Enabled Skeletal Reorganization: Access to Medium-/Large-Sized Cyclic Alkenes, Sci. Adv., 2017, 3, e1701487 CrossRef PubMed; (i) D. Alpers, K. P. Cole and C. R. J. Stephenson, Visible Light Mediated Aryl Migration by Homolytic C–N Cleavage of Aryl Amines, Angew. Chem., Int. Ed., 2018, 57, 12167–12170 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General comments, general procedure, analytical data and NMR spectra. See DOI: https://doi.org/10.1039/d4sc03221g |
| This journal is © The Royal Society of Chemistry 2024 |