
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the formation of a hetero-dimeric membrane transport complex with dual in vitro and in silico mutagenesis†
Nishadh
Rathod
 a,
M. Joanne
Lemieux
a,
Christophe
Chipot
bcd,
Benoît
Roux
b and
Howard S.
Young
*a
a,
M. Joanne
Lemieux
a,
Christophe
Chipot
bcd,
Benoît
Roux
b and
Howard S.
Young
*a
aDepartment of Biochemistry, University of Alberta, Edmonton, Alberta, Canada T6G 2H7. E-mail: hyoung@ualberta.ca; Fax: +1 (780) 492-3931; Tel: +1 (780) 492-3931
bDepartment of Biochemistry and Molecular Biology, University of Chicago, Chicago, USA 60637
cLaboratoire International Associé Centre National de la Recherche Scientifique et University of Illinois at Urbana-Champaign, Unité Mixte de Recherche no. 7019, Université de Lorraine, B.P. 70239, 54506 Vandœuvre-lès-Nancy Cedex, France
dTheoretical and Computational Biophysics Group, Beckman Institute, Department of Physics, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA
First published on 6th August 2024
Abstract
The reversible association of transmembrane helices is a fundamental mechanism in how living cells convey information and respond to physiological events. The cardiac calcium transport regulator phospholamban (PLN) is an example of a single-span transmembrane protein that populates a variety of reversible and competing oligomeric states. PLN primarily forms monomers and pentamers in the membrane, where the PLN pentamer is a storage form and the PLN monomer forms a hetero-dimeric inhibitory complex with SERCA. The binding affinity and free-energy of formation of the SERCA-PLN complex in a membrane have not been determined. As is the case for most transmembrane protein interactions, measuring these quantities experimentally is extremely challenging. In this study, we estimated binding affinities by employing in silico alchemical free-energy calculations for all PLN transmembrane alanine substitutions in a membrane bilayer. The binding affinities were calculated separately for the SERCA-PLN complex, a PLN monomer, and a PLN pentamer and compared to in vitro functional measurements of SERCA regulation by the PLN alanine substitutions. Initially, the changes in SERCA inhibition by PLN alanine substitutions were compared to the changes in free energy for the SERCA-PLN complex formed from the PLN monomer. However, the functional data for the PLN alanine substitutions were better explained by the formation of the SERCA-PLN complex directly from the PLN pentamer. This finding points to an inhibitory mechanism favoring conformational selection of SERCA and the interaction of a PLN pentamer with SERCA for ‘delivery’ of a PLN monomer to the inhibitory site. The implications of these findings suggest that the energetics of helix exchange between homo- and hetero-oligomeric signaling complexes is favored over an intermediate involving a free monomeric helix in the membrane bilayer.
Introduction
Transmembrane helix–helix interactions play a critical role in the folding, function, assembly, and oligomerization of many proteins. The reversible association of transmembrane helices is a fundamental mechanism in cellular signaling and how living cells convey information and respond to physiological events. While there are many single-pass transmembrane proteins that undergo reversible helix–helix interactions, some examples involved in signaling include the receptor tyrosine kinases, STIM proteins (stromal interaction molecule 1 & 2), and cadherins among many others. In these processes, the diversity of transmembrane helix interactions ranges from monomeric transmembrane helices in a lipid bilayer to helix–helix interactions that include both homo- and hetero-oligomeric complexes. The equilibrium between single transmembrane helices in a lipid bilayer and helix–helix interactions favors helix–helix association because transmembrane helices and their apolar side chains pack more efficiently with other transmembrane helices than they do with lipid acyl chains in the membrane bilayer.1 The competing effects of favorable helix–helix interactions and unfavorable helix–lipid interactions can contribute to helix self-association via a combination of favorable van der Waals interactions, hydrogen bonding, electrostatic interactions, and unfavorable lipophobic effects.2,3An example of a single-span transmembrane protein that populates a variety of reversible and competing association states (i.e., monomer, homo-oligomer, hetero-oligomer) is the cardiac calcium transport regulatory peptide phospholamban (PLN). PLN regulates the sarco-endoplasmic reticulum calcium ATPase (SERCA). SERCA is ubiquitously expressed in all eukaryotic cells, where it actively transports calcium ions from the cytosol to the lumen of the sarco-endoplasmic reticulum (SR/ER) against a concentration gradient.4 The structure of SERCA includes three cytoplasmic domains, the nucleotide-binding (N), phosphorylation (P), and actuator (A) domains, as well as ten transmembrane helices. SERCA progresses through an elaborate transport cycle that involves two main states, a calcium-bound E1 state and a calcium-free E2 state.5,6 PLN is a 52 amino acid transmembrane peptide that physically interacts with SERCA and regulates calcium transport activity (Fig. 1A). It is primarily expressed in ventricular muscle7,8 and mutations in the PLN gene are implicated in cardiomyopathies and heart failure.9–14 The structure of PLN includes an N-terminal cytoplasmic helix (residues 1–17), a short linker region (residues 18–25) and a transmembrane helix (residues 26–52). The transmembrane domain of PLN is primarily responsible for inhibiting the activity of SERCA, while the cytoplasmic domain of PLN is primarily responsible for regulating SERCA inhibition via phosphorylation (Ser16 & Thr17) and the β-adrenergic signaling pathway.
 | ||
| Fig. 1 Topology diagram for phospholamban (PLN) and structure of the SERCA-PLN complex. (A) The transmembrane domain is colored grey and the residues studied are circled in black. The leucine–isoleucine residues of PLN that are involved in pentamer formation are circled in grey. (B) Structure of SERCA-PLN complex (PDB: 4KYT) in cartoon format. PLN is shown as cyan. SERCA transmembrane domain is tan, transmembrane segments M4 and M5 are blue, the N domain is green, the P domain is magenta, and the A domain is yellow. | ||
The model for SERCA inhibition by PLN involves a PLN monomer binding to the transmembrane domain of SERCA involving transmembrane segments M2, M6, and M9 (hetero-dimeric SERCA-PLN complex; Fig. 1B). PLN inhibition of SERCA manifests as a change in the apparent calcium affinity (KCa) of SERCA such that the SERCA-PLN complex has lower affinity for calcium.8,15 By itself, PLN also forms a pentamer, which is suggested to be an inactive storage form of PLN in the membrane (homo-oligomeric PLN–PLN complex). The PLN pentamer is stabilized by a leucine–isoleucine zipper composed of residues Ile33, Leu37, Ile40, Leu44, Ile47 and Leu51.16 The model for SERCA regulation suggests that the PLN pentamer is in dynamic equilibrium with the PLN monomer, and the PLN monomer then associates with SERCA to cause inhibition.17–19 Thus, the helix–lipid and helix–helix interactions involved in this regulatory mechanism include an “active” PLN monomer in the membrane bilayer, a homo-pentameric PLN “storage” species, and an “inhibited” hetero-dimeric SERCA-PLN complex. However, the kinetics of monomer release from the PLN pentamer is slow (second timescale20) relative to the physiological window for formation of the inhibitory SERCA-PLN complex (millisecond timescale). Thus, the mechanism by which a PLN monomer transitions from the helix–helix interactions that stabilize the pentamer to the helix–helix interactions that stabilize the SERCA-PLN inhibitory complex still remains elusive.
The structure of the SERCA-PLN complex has been determined by X-ray crystallography.21–23 Despite this advance, the binding affinity and free-energy of formation of the SERCA-PLN complex in a membrane environment have not been determined. While measuring these quantities experimentally is extremely challenging, binding affinities can be estimated using in silico methods for free-energy calculations (ΔGbinding). Although, widespread application has been limited by the availability of computational resources. The recent offloading of free-energy perturbation (FEP) and thermodynamic integration (TI)24 calculations on GPUs in the NAMD simulation software25,26 has made these approaches more affordable and efficient, which is necessary for investigating membrane protein complexes. In this work, we carried out in silico alchemical free-energy perturbation calculations for all transmembrane alanine substitutions in PLN (from Ile18-Ala to Leu52-Ala). This was done in a lipid membrane for the SERCA-PLN complex, as well as the PLN monomer and PLN pentamer. We compared the simulation data to the in vitro functional analysis of the PLN alanine substitutions and their ability to inhibit SERCA. The PLN variants were classified for their ability to alter the apparent calcium affinity of SERCA (i.e. KCa parameters for SERCA). Loss-of-function PLN variants did not inhibit SERCA and gain-of-function PLN variants were super-inhibitors of SERCA. The changes in SERCA inhibition by PLN variants were compared to the changes in free energy (ΔΔG) for the formation of the SERCA-PLN complex from either the PLN monomer or the PLN pentamer. The functional data for the PLN variants were better explained by the formation of the SERCA-PLN complex directly from the PLN pentamer. This suggests an inhibitory mechanism that favors the interaction of a PLN pentamer with SERCA for the ‘delivery’ of a PLN monomer to the inhibitory site and selection of the appropriate conformation of SERCA.
Results and discussion
Loss- and gain-of-function variants of PLN were initially reported based on alanine-scanning mutagenesis of PLN and SERCA functional measurements (e.g. ref. 17, 19 and 27). Loss-of-function variants were thought to disrupt physical interactions between PLN and SERCA that are required for inhibition. Based on the observation of PLN oligomeric states by SDS-PAGE, gain-of-function variants were thought to disrupt the PLN pentamer and lead to an increased concentration of monomers in the membrane that ultimately caused super-inhibition of SERCA.19,28 In this model for SERCA inhibition by PLN, the PLN pentamer releases a monomer, the monomer diffuses through the membrane until it encounters SERCA, and the monomer binds to SERCA and forms the inhibitory complex. However, as stated above, the slow kinetics for monomer release from the PLN pentamer is seemingly inconsistent with the faster timescale for SERCA conformational changes and PLN inhibition.20With this in mind, the primary objective of this study was to compare changes in free energy (ΔΔG), which reflect changes in binding affinity of PLN for SERCA, with the loss-of-function or gain-of-function behavior of PLN alanine substitutions. The expectation was that PLN loss-of-function variants will have lower binding affinity for SERCA (ΔΔG > 0), while PLN gain-of-function variants will have higher binding affinity for SERCA (ΔΔG < 0). The hypothesis we aimed to test was the formation of the SERCA-PLN complex from a PLN monomer. Towards this end, we performed alanine-scanning mutagenesis and functional analysis of PLN residues Ile18-Ala to Ile33-Ala using an in vitro membrane reconstitution system to determine the apparent calcium affinity (KCa) of SERCA in the presence of the PLN alanine variants. The remaining mutagenesis data included in this study for PLN residues Asn34-Ala to Leu52-Ala has been previously published.15,29 These functional data were then compared to in silico alanine-scanning mutagenesis of PLN using alchemical free-energy simulations and determination of ΔΔG for each alanine variant.
Apparent calcium affinity (KCa) of SERCA
SERCA was reconstituted into membrane vesicles (proteoliposomes) in the absence and presence of the PLN variants as previously described.15,29,30 Functional characterization of the proteoliposomes was assessed by the measurement of calcium-dependent ATPase activity of SERCA alone (negative control), SERCA in the presence of wild-type PLN (positive control), and SERCA in the presence of the PLN alanine variants. This activity assay has been validated against calcium transport measurements.31 The ATPase activity data were plotted as log calcium concentration versus specific activity, and the apparent calcium affinity (KCa) of SERCA was determined as the calcium concentration for half-maximal SERCA activity (Fig. S1 and Table S1†). This approach has been well-established for comparing KCa measurements between different variants of PLN (e.g. see ref. 14, 15, 29 and 30). To evaluate the status of the alanine substitutions relative to wild-type PLN, the KCa value determined for SERCA in the presence of wild-type PLN (KCa of 0.88 μM calcium) was subtracted from the KCa values determined for SERCA in the presence of the PLN variants (ΔKCa; Fig. 2). This was done for alanine substitutions Ile18-Ala to Ile33-Ala characterized in this study and Asn34-Ala to Leu52-Ala previously published.15,29 | ||
| Fig. 2 Changes in the apparent calcium affinity of SERCA in the presence of the alanine substitutions of PLN. To better delineate the changes in the apparent calcium affinity (KCa) of SERCA, we calculated ΔKCa for each alanine substitution. The ΔKCa values were calculated as the difference between the KCa value for the alanine substitution and the KCa value for SERCA in the presence of wild-type PLN. Negative ΔKCa values indicate loss-of-function variants that are weaker inhibitors of SERCA (e.g. N34A; SERCA has a higher affinity for calcium in the presence of this variant compared to wild-type PLN) and positive ΔKCa values indicate gain-of-function variants that are more potent inhibitors of SERCA (e.g. I47A; SERCA has a lower affinity for calcium in the presence of this variant compared to wild-type PLN). Error bars indicate SEM (n ≥ 4). Statistical significance for the KCa values is shown as asterisks (*) as in Fig. S1† (p < 0.01). | ||
Based on the ΔKCa values, the alanine variants were categorized as loss of function (ΔKCa < 0; loss of SERCA inhibition), neutral (ΔKCa ≈ 0), or gain of function (ΔKCa > 0; gain of SERCA inhibition). The dominant characteristic of the alanine variants was loss of function. The Asn34-Ala was the most severe loss-of-function variant, completely eliminating SERCA regulation by PLN.15,19 The KCa value determined for SERCA in the presence of wild-type PLN was 0.88 μM calcium compared to 0.45 μM calcium for SERCA in the presence of Asn34-Ala PLN (ΔKCa of −0.43). Another severe loss-of-function variant of PLN was Ile38-Ala (ΔKCa of −0.39). In total, 14 alanine variants of PLN resulted in KCa (Fig. S1 and Table S1†) and ΔKCa (Fig. 2) values that were indicative of loss of function (KCa values ranging from 0.45 for Asn34-Ala to 0.66 for Leu31-Ala & Cys46-Ala; ΔKCa values ranging from −0.43 to −0.22, respectively). Based on the crystal structure of the SERCA-PLN complex, most of these residues line the helical face of PLN that interacts with the inhibitory groove of SERCA.
In addition to the loss-of-function variants, a subset of alanine variants resulted in gain of function and more potent inhibition of SERCA (Fig. 2). The Leu37-Ala and Ile47-Ala were the most potent gain-of-function variants, markedly increasing SERCA inhibition relative to wild-type PLN. Compared to SERCA in the presence of wild-type PLN (KCa of 0.88 μM calcium), the Leu37-Ala and Ile47-Ala variants of PLN were super-inhibitors of SERCA (KCa of 1.57 and 1.87 μM calcium; ΔKCa of 0.69 and 0.99, respectively). Three other substitutions – Ile33-Ala, Leu43-Ala, and Leu44-Ala – were also more potent inhibitors of SERCA (ΔKCa of 0.28, 0.34, and 0.23, respectively). Finally, four additional substitutions resulted in mild gain of function – Met20-Ala, Lys27-Ala, Cys36-Ala, and Leu51-Ala (ΔKCa of 0.09, 0.12, 0.09, and 0.12, respectively). In the crystal structure of the SERCA-PLN complex, most of these residues line the helical face of PLN that is oriented away from the inhibitory groove of SERCA, with the exception of Lys27 and Met20.
Formation of the SERCA-PLN complex from a PLN monomer
Our next objective was to determine the changes in binding affinity for SERCA that accompany alanine substitution of each residue in PLN (Ile18-Ala to Leu52-Ala). To estimate binding affinities, we used the in silico method of alchemical free-energy simulations to determine the difference in free energy of binding for each of the alanine variants compared to wild-type PLN in the SERCA-PLN complex.21 The thermodynamic cycle for binding of a PLN monomer to SERCA and the alchemical transformations are shown in Fig. 3A. The thermodynamic cycle includes the formation of the SERCA-PLN complex from a PLN monomer for wild-type ( ) and the alanine variants (
) and the alanine variants ( ). The free-energy calculations in the thermodynamic cycle include alchemical transformation of the SERCA-bound complex (ΔGbound-alchemical) and the unbound PLN monomer (ΔGunbound-alchemical) in a membrane environment. We determined ΔGbound-alchemical and ΔGunbound-alchemical (Table S1†) using the alchemical free-energy perturbation method. The change in free energy of formation for the PLN variant (
). The free-energy calculations in the thermodynamic cycle include alchemical transformation of the SERCA-bound complex (ΔGbound-alchemical) and the unbound PLN monomer (ΔGunbound-alchemical) in a membrane environment. We determined ΔGbound-alchemical and ΔGunbound-alchemical (Table S1†) using the alchemical free-energy perturbation method. The change in free energy of formation for the PLN variant ( minus
minus  ) was equivalent to the difference in free energy between the bound and unbound alchemical transformations (ΔΔG = ΔGbound-alchemical minus ΔGunbound-alchemical).
) was equivalent to the difference in free energy between the bound and unbound alchemical transformations (ΔΔG = ΔGbound-alchemical minus ΔGunbound-alchemical).
 | ||
Fig. 3 Thermodynamic cycle describing the free energy of formation of the SERCA-PLN complex from the PLN monomer (A) or PLN pentamer (B). SERCA is shown in grey cartoon format, wild-type PLN in blue, and the alchemically transformed residues of PLN in red. Alchemical transformations were performed for alanine substitution of each residue in PLN in a lipid bilayer (ΔGunbound-alchemical) and in the SERCA-PLN complex in a lipid bilayer (ΔGbound-alchemical). The top horizontal process in each panel corresponds to the free energy for binding of SERCA to wild-type PLN  and the bottom horizontal process corresponds to the free energy for binding of SERCA to an alanine-substitution PLN and the bottom horizontal process corresponds to the free energy for binding of SERCA to an alanine-substitution PLN  . The left vertical process in each panel corresponds to the free energy for alchemically mutating a single residue in PLN to alanine in the unbound state (ΔGunbound-alchemical). The right vertical process in each panel corresponds to free energy for alchemically mutating a single residue in PLN to alanine in a SERCA-bound complex (ΔGbound-alchemical). The difference in the vertical processes equates to the difference in horizontal processes (ΔΔG). The ΔG and ΔΔG values are presented herein for all alanine substitutions of PLN from Ile18-Ala to Leu52-Ala (ESI Tables 1 and 2†). The ΔΔG values for the formation of the SERCA-PLN complex from the PLN monomer are shown in Fig. 4A. The ΔΔG values for the formation of the SERCA-PLN complex from the PLN pentamer are shown in Fig. 4B. . The left vertical process in each panel corresponds to the free energy for alchemically mutating a single residue in PLN to alanine in the unbound state (ΔGunbound-alchemical). The right vertical process in each panel corresponds to free energy for alchemically mutating a single residue in PLN to alanine in a SERCA-bound complex (ΔGbound-alchemical). The difference in the vertical processes equates to the difference in horizontal processes (ΔΔG). The ΔG and ΔΔG values are presented herein for all alanine substitutions of PLN from Ile18-Ala to Leu52-Ala (ESI Tables 1 and 2†). The ΔΔG values for the formation of the SERCA-PLN complex from the PLN monomer are shown in Fig. 4A. The ΔΔG values for the formation of the SERCA-PLN complex from the PLN pentamer are shown in Fig. 4B. | ||
The ΔΔG value for each PLN variant was indicative of higher affinity binding (ΔΔG < 0), neutral (ΔΔG ≈ 0), or lower affinity binding (ΔΔG > 0) to SERCA (Fig. 4A and Table S2†). A group of variants resulted in negative ΔΔG values indicative of higher affinity binding to SERCA (15 variants). The largest negative changes in ΔΔG were for Glu19-Ala, Gln23-Ala, Arg25-Ala, Asn30-Ala, Ile33-Ala, and Met50-Ala. Additional residues also exhibited negative changes in ΔΔG that were statistically significant but lower in magnitude by comparison (Fig. 4A). Another group of variants resulted in positive ΔΔG values indicative of lower affinity binding to SERCA (13 variants). The largest positive changes in ΔΔG were for Asn34-Ala, Ile38-Ala, and Ile48-Ala. Additional residues also exhibited positive changes in ΔΔG that were statistically significant but lower in magnitude by comparison (Fig. 4A). However, the calculation of the ΔΔG values depended on the chemical state of PLN, which in this thermodynamic cycle was a PLN monomer embedded in a lipid bilayer (Fig. 3A). Thus, determining the free-energy change between the bound and unbound states (ΔΔG = ΔGbound-alchemical minus ΔGunbound-alchemical) provided insights into the formation of the SERCA-PLN complex from a PLN monomer in a membrane environment.
 | ||
| Fig. 4 The ΔΔG values derived from the thermodynamic cycle for (A) formation of the SERCA-PLN complex from the PLN monomer (Fig. 3A) and (B) formation of the SERCA-PLN complex from the PLN pentamer (Fig. 3B). The ΔΔG values (kcal mol−1) were calculated as the difference between the SERCA-PLN complex (ΔGbound-alchemical) and the PLN monomer (A) or PLN pentamer (B) in a lipid bilayer (ΔGunbound-alchemical). Negative ΔΔG values (kcal mol−1) indicated alanine substitutions that have higher affinity for SERCA compared to wild-type PLN (e.g., R25A). Positive ΔΔG values (kcal mol−1) indicated alanine substitutions that have lower affinity for SERCA compared to wild-type PLN (e.g., I38A). The FEP values and associated errors were determined using ParseFEP and indicate Bennett Acceptance Ratio (BAR) free-energy estimates between the forward and backward transformations. | ||
To understand the effect of alanine variants on formation of the SERCA-PLN complex from the PLN monomer, we plotted ΔΔG (kcal mol−1) versus ΔKCa (μM calcium) for all variants (Fig. 5A). The objective was to correlate the changes in binding affinity (negative or positive changes in ΔΔG) with the loss-of-function or gain-of-function behavior of the alanine variants (negative or positive changes in ΔKCa). The expectation was that loss-of-function variants (ΔKCa < 0) should correlate with a lower binding affinity for SERCA (ΔΔG > 0) and gain-of-function variants (ΔKCa > 0) should correlate with a higher binding affinity for SERCA (ΔΔG < 0). Reduced binding of PLN to the inhibitory groove of SERCA should result in reduced inhibition and more efficient SERCA calcium transport. Similarly, increased binding of PLN to the inhibitory groove of SERCA should result in increased inhibition and less efficient SERCA calcium transport. This pattern fit many of the residues such as the most severe loss-of-function variants Asn34-Ala and Ile38-Ala (Fig. 5A; upper left quadrant), and gain-of-function variants such as Ile33-Ala, Leu43-Ala, and Leu44-Ala (Fig. 5A; lower right quadrant). However, there were some clear deviations from this pattern. Some loss-of-function variants were predicted to have higher affinity for SERCA, such as Glu19-Ala, Gln23-Ala, Arg25-Ala, and Met50-Ala (Fig. 5A; lower left quadrant). At first this seems implausible; however, it is interesting to consider that these latter variants lie at the membrane surfaces, where higher affinity binding might alter the way the transmembrane domain of PLN crosses the membrane bilayer and interacts with SERCA. Since SERCA inhibition depends on the orientation of PLN in the inhibitory groove,32 higher affinity binding could result in a non-productive complex and loss of function.
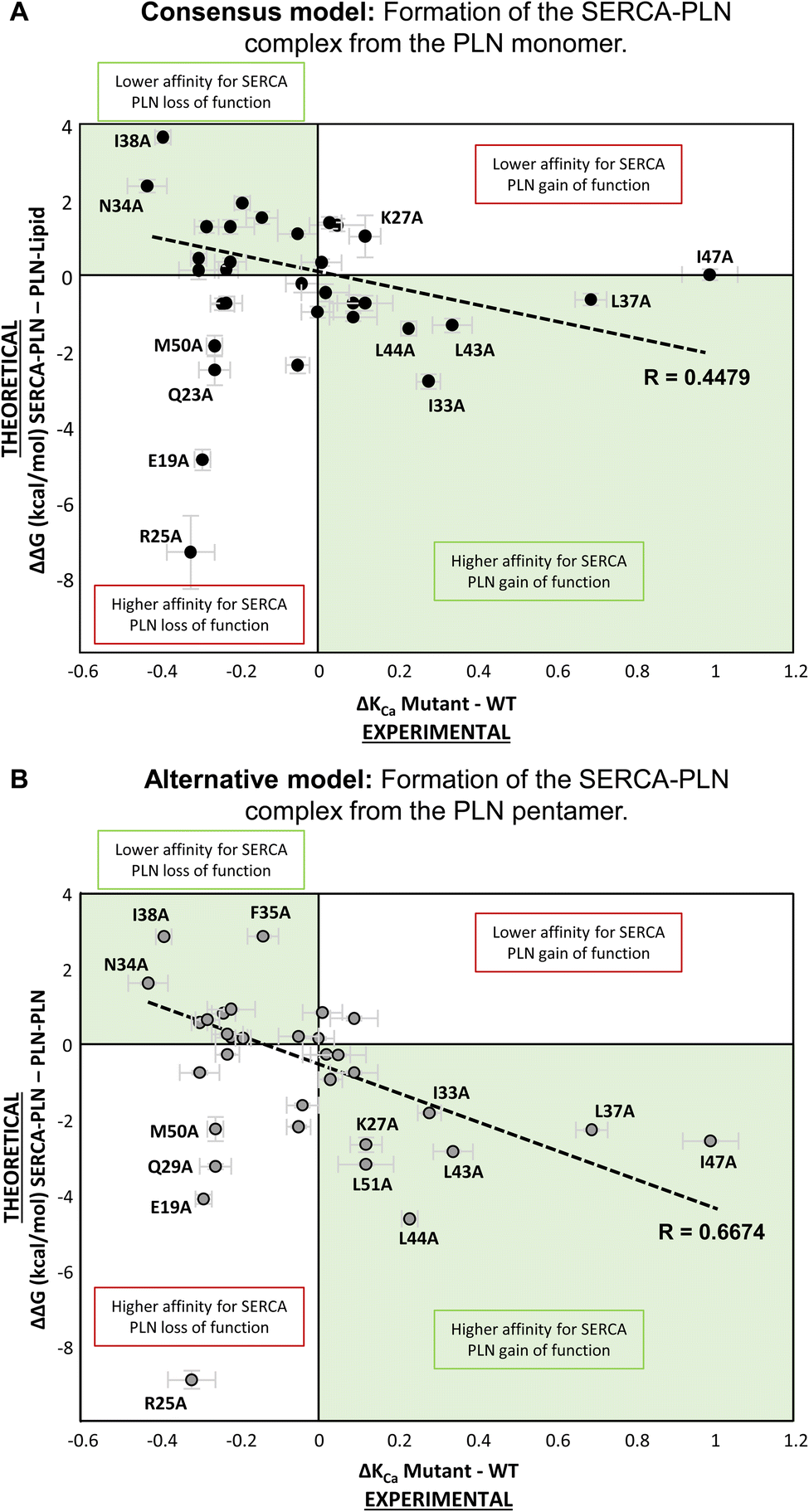 | ||
| Fig. 5 Scatter plots of ΔΔG (kcal mol−1) versus ΔKCa (μM) for each alanine substitution in PLN and the thermodynamic cycle from the PLN monomer (A) and thermodynamic cycle for the PLN pentamer (B). The expected regions are highlighted in green. Top left quadrant corresponds to alanine variants that have lower binding affinity for SERCA and result in loss of function compared to wild-type PLN (e.g., N34A). The bottom right quadrant corresponds to alanine variants that have higher binding affinity for SERCA and result in gain of function compared to wild-type PLN (e.g., L37A). The top right quadrant corresponds to alanine variants that have lower binding affinity for SERCA and result in gain of function compared to wild-type PLN. The bottom left quadrant corresponds to alanine variants that have higher binding affinity for SERCA and result in loss of function compared to wild-type PLN (e.g., R25A). Error bars indicate SEM for ΔKCa (n ≥ 4) and ΔΔG (BAR estimator implemented in ParseFEP). A linear regression fit is shown for each plot and the R values are indicated. Note that E19A, R25A, Q23A, and M50A were excluded from the regression fit because of their distinct behavior. | ||
From this comparison, there were several variants that remained difficult-to-explain outliers, particularly Lys27-Ala, Leu37-Ala, and Ile47-Ala. The Lys27-Ala variant is a well-known gain-of-function variant,15,27,33 yet the formation of the SERCA-PLN complex from the PLN monomer predicted this variant to have a lower affinity for SERCA (Fig. 5A; upper right quadrant). It seemed implausible that a PLN variant can result in both gain of function and lower affinity for SERCA. In addition, the two most potent super-inhibitors of SERCA, gain-of-function variants Leu37-Ala and Ile47-Ala (Fig. 2), did not appreciably alter the binding affinity for SERCA (Fig. 5A). These latter two residues, Leu37 and Ile47, have been reported to both stabilize the PLN pentamer and physically interact with SERCA.34 Thus, the hypothesis for formation of the SERCA-PLN complex from a PLN monomer did not explain the results for this subset of gain-of-function alanine variants of PLN.
Formation of the SERCA-PLN complex from a PLN pentamer
Since the formation of the SERCA-PLN complex from a PLN monomer did not explain some gain-of-function variants of PLN, we considered an alternative hypothesis for the formation of the SERCA-PLN complex from a PLN pentamer. As above, our objective was to determine how the alanine-scanning mutagenesis of PLN and SERCA functional measurements (ΔKCa) compared with changes in the binding affinity of PLN for SERCA (ΔΔG). The thermodynamic cycle for formation of the SERCA-complex directly from the PLN pentamer using in silico alchemical free-energy simulations is shown in Fig. 3B. The thermodynamic cycle includes the formation of the SERCA-PLN complex from a PLN pentamer for wild-type ( ) and alanine variants (
) and alanine variants (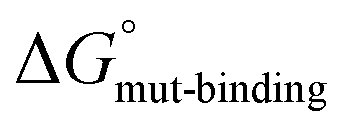 ). Analogous to Fig. 3A, the free-energy calculations in the thermodynamic cycle included the alchemical transformation of the SERCA-PLN complex (ΔGbound-alchemical) and the PLN pentamer in a membrane environment (ΔGunbound-alchemical). The unbound state in this thermodynamic cycle was the PLN pentamer in a membrane environment, where each residue may be involved in PLN–PLN and/or PLN-lipid interactions. We determined ΔGbound-alchemical and ΔGunbound-alchemical (Table S1†) using the alchemical free-energy perturbation method. As described above, the change in free energy of formation for the PLN variant (
). Analogous to Fig. 3A, the free-energy calculations in the thermodynamic cycle included the alchemical transformation of the SERCA-PLN complex (ΔGbound-alchemical) and the PLN pentamer in a membrane environment (ΔGunbound-alchemical). The unbound state in this thermodynamic cycle was the PLN pentamer in a membrane environment, where each residue may be involved in PLN–PLN and/or PLN-lipid interactions. We determined ΔGbound-alchemical and ΔGunbound-alchemical (Table S1†) using the alchemical free-energy perturbation method. As described above, the change in free energy of formation for the PLN variant ( minus
minus  ) was equivalent to the difference in free energy between the bound and unbound alchemical transformations (ΔΔG = ΔGbound-alchemical minus ΔGunbound-alchemical; Fig. 4B).
) was equivalent to the difference in free energy between the bound and unbound alchemical transformations (ΔΔG = ΔGbound-alchemical minus ΔGunbound-alchemical; Fig. 4B).
For a comparison of the effect of alanine variants on formation of the SERCA-PLN complex from the PLN pentamer, we plotted ΔΔG (kcal mol−1) versus ΔKCa (μM calcium) for all variants (Fig. 5B). As described above, the gain-of-function variants that remained unexplained, Lys27-Ala, Leu37-Ala, and Ile47-Ala, were better fit by the thermodynamic cycle involving the PLN pentamer. In fact, many of the gain-of-function variants were better explained (lower ΔΔG values) by formation of the SERCA-PLN complex from the PLN pentamer, including residues Lys27-Ala, Leu37-Ala, Leu43-Ala, Leu44-Ala, Ile47-Ala, and Leu51-Ala (Fig. 6). Most of these residues, with the exception of Lys27, were shown to be part of the Leu–Ile zipper involved in formation of the PLN pentamer.16,19 Furthermore, the ΔΔG values for super-inhibitory variants increased in magnitude in the formation of the SERCA-bound complex from the PLN pentamer indicating an improved correlation using this thermodynamic cycle as compared to the thermodynamic cycle involving the PLN monomer. As notable examples, the ΔΔG values decreased for Leu37-Ala (−0.7 → −2.3 kcal mol−1), Leu44-Ala (−1.4 → −4.6 kcal mol−1), and Ile47-Ala (0.0 → −2.6 kcal mol−1) indicating a better fit by the thermodynamic cycle involving the PLN pentamer. Unlike the model involving the PLN monomer (Fig. 3A), only a single variant, Met20-Ala, resulted in mild gain of function (ΔKCa > 0) and lower affinity for SERCA (ΔΔG > 0). Since Met20 is not part of the transmembrane domain of PLN, perhaps it is not surprising that this variant was not fit well by either thermodynamic cycle.
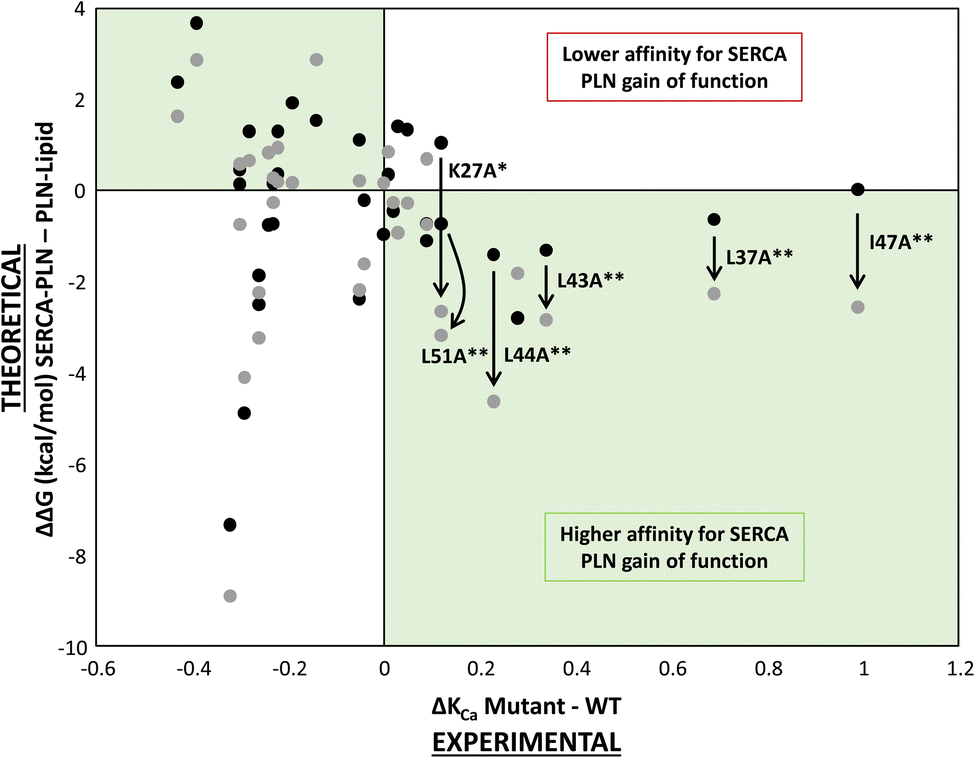 | ||
| Fig. 6 Overlay of the scatter plots of ΔΔG (kcal mol−1) versus ΔKCa for each alanine substitution in PLN and the thermodynamic cycle from the PLN monomer (black circles) and PLN pentamer (grey circles). This figure is an overlay of Fig. 5A and B. Notice the better fit between the ΔΔG and ΔKCa values for the thermodynamic cycle from the PLN pentamer including alanine substitutions K27A, L37A, L43A, L44A, I47A, and L51A. Statistical analysis was performed using an independent samples t-test (*p < 0.02; **p < 0.01). | ||
It is interesting to consider the structure of the SERCA-PLN complex (Fig. 7) and the location of key residues implicated in the formation of the SERCA-PLN complex from the PLN pentamer. Residues that exhibited lower affinity for SERCA and PLN loss of function included Asn34-Ala (N34A), Phe35-Ala (F35A), and Ile38-Ala (I38A). These residues face the inhibitory groove of SERCA (Fig. 7B). Residues that exhibited higher affinity for SERCA and PLN gain of function included Lys27-Ala (K27A), Ile33-Ala (I33A), Leu37-Ala (L37A), Leu43-Ala (L43A), Leu44-Ala (L44A), Ile47-Ala (I47A), and Leu51-Ala (L51A). Except for Lys27-Ala, these residues are peripheral to the inhibitory groove of SERCA (Fig. 7C). Finally, residues that exhibited higher affinity for SERCA and PLN loss of function included Glu19-Ala (E19A), Arg25-Ala (R25A), Gln29-Ala (Q29A), and Met50-Ala (M50A). While this was an unexpected behavior, these residues lie at the membrane surfaces suggesting that they play a role in orienting PLN in the inhibitory groove of SERCA (Fig. 7D).
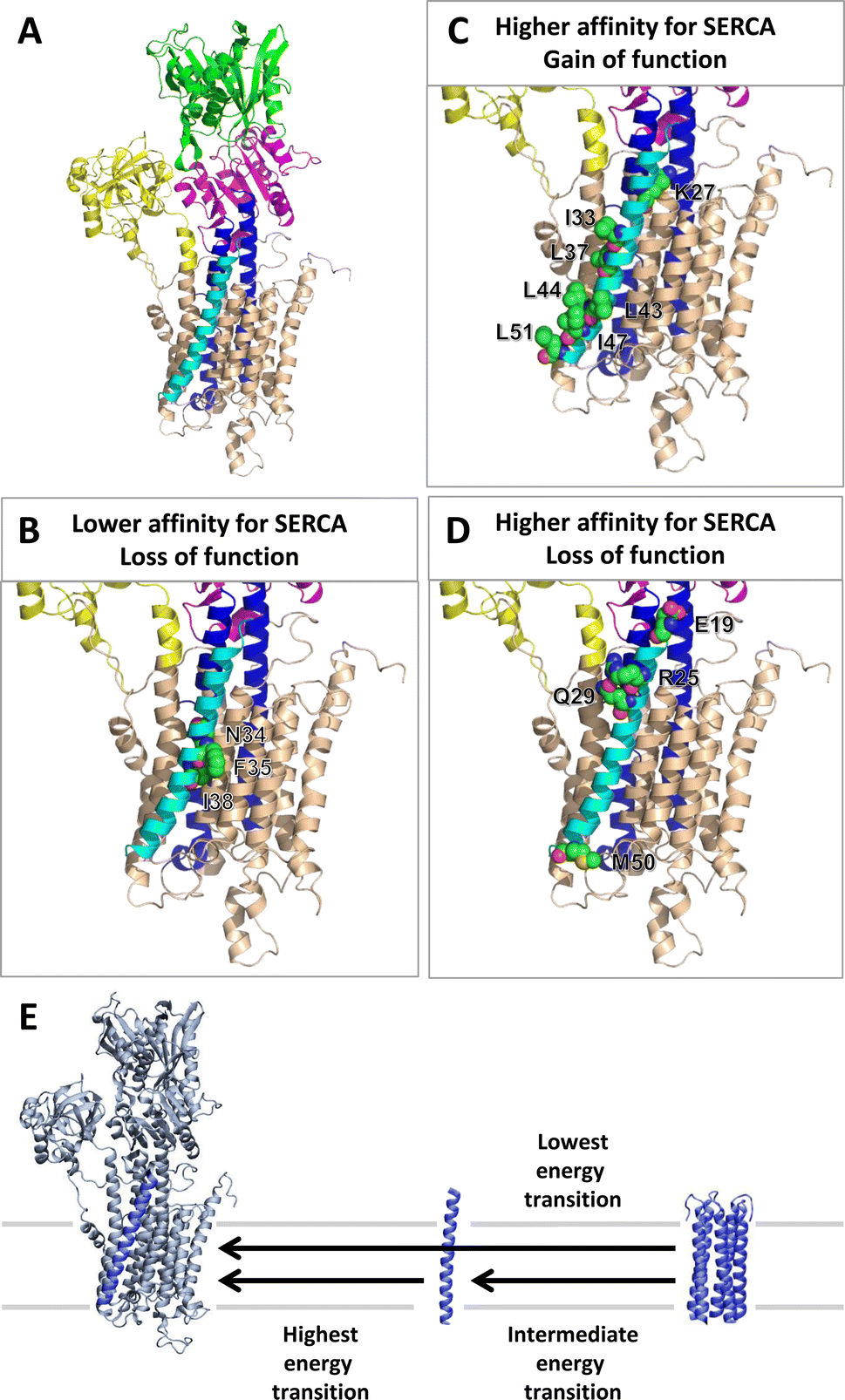 | ||
| Fig. 7 Structure of SERCA-PLN complex (PDB: 4KYT). (A) PLN is shown as cyan cartoon format. SERCA transmembrane domain is tan, transmembrane segments M4 and M5 are blue, the N domain is green, the P domain is magenta, and the A domain is yellow in cartoon format. The alanine substitutions labeled in Fig. 5B are shown for the three main quadrants: (B) lower affinity for SERCA, loss of function (N34A, F35A, I38A); (C) high affinity for SERCA, gain of function (K27A, I33A, L37A, L43A, L44A, I47A, L51A); and (D) higher affinity for SERCA, loss of function (E19A, R25A, Q29A, M50A). These residues are shown in sphere representation. (E) Scheme depicting relative energy states for the SERCA-PLN complex formed from the PLN monomer and PLN pentamer, as well as the release of a PLN monomer from the PLN pentamer. Arrows indicate the transitions between the different states. SERCA is shown in grey cartoon format and PLN is shown in blue. | ||
Conformational selection of SERCA by the PLN pentamer
Cellular signaling events are often mediated by the lateral association of membrane proteins that change state in response to chemical signals. Membrane protein self-association and helix–helix interactions make up a significant structural feature of the cellular signaling landscape. Despite the clear importance of membrane protein interactions, the measurement of binding affinities in a membrane environment has been challenging. To address this problem, we studied the interaction between PLN and SERCA using an in vitro functional assay and in silico computational approach. PLN was an attractive choice for the following reasons. First, there is a large amount of functional data for SERCA in the presence of PLN alanine variants.15,19,29 Second, PLN adopts a variety of helical association states in the membrane including a free monomer, a homo-pentameric state, and a hetero-oligomeric state when bound to SERCA. Third, there is an established model for SERCA regulation by PLN, where the PLN pentamer is in dynamic equilibrium with the PLN monomer, and the PLN monomer is the species that interacts with and inhibits SERCA.17–19 Finally, there are structures available for the dominant states including the SERCA-PLN complex,21 the PLN monomer,35 and the PLN pentamer.36The objective was to understand the free-energy changes that accompany helix–helix association in a membrane environment. To the best of our knowledge, this would be the first application of this method to a membrane protein complex. In the thermodynamic cycle for formation of the SERCA-PLN inhibitory complex (Fig. 3), the free-energy changes were calculated as the difference between the bound and unbound states. The SERCA-PLN complex was the bound state and the PLN monomer was the unbound state. This gave rise to a set of free-energy changes calculated for the array of PLN variants (Table S2† and Fig. 4). To reconcile these free-energy changes, we compared the ΔΔG values with the apparent calcium affinity (KCa) determined for SERCA in the presence of each PLN alanine substitution (Fig. 5). The expectation was changes in binding affinity (ΔΔG) of the PLN monomer should correlate with changes in function (ΔKCa), consistent with the existing model for SERCA regulation.
Although we expected our findings to support SERCA regulation by the PLN monomer, the free-energy changes did not offer a satisfactory fit to the functional data for the PLN variants. This led us to consider a second unbound state, the PLN pentamer, which gave rise to a different set of free-energy changes calculated for the array of PLN variants (Table S2† and Fig. 6). Surprisingly, using the PLN pentamer as the unbound state gave rise to free-energy changes (ΔΔG) that better fit the functional data (ΔKCa) (Fig. 6). This was particularly true for some of the gain-of-function variants of PLN, such as Lys27-Ala (K27A), Leu37-Ala (L37A), Leu43-Ala (L43A), Leu44-Ala (L44A), Ile47-Ala (I47A), Leu51-Ala (L51A) (Fig. 6). To further test this finding, we turned to another set of experimental data previously published by the MacLennan laboratory.19,27,37 These data included KCa measurements for SERCA in the presence of alanine variants of canine PLN. The only difference in the sequence of residues considered was Asn27 in canine PLN is Lys27 in human PLN. It should be noted that there are substantial differences in the KCa values determined by us (Fig. S1† and ref. 29) and those previously published (Fig. 1 in ref. 37). Nonetheless, these data were also better correlated by the thermodynamic cycle involving the PLN pentamer (Fig. S2†). Therefore, we concluded that the data support a model for SERCA regulation where the SERCA-PLN complex can form via an interaction between SERCA and the PLN pentamer (Fig. 7E). Note that this does not exclude formation of the SERCA-PLN complex from the PLN monomer, though complex formation from the PLN pentamer is energetically more favorable.
There is evidence in the literature for an active role for the PLN pentamer in SERCA regulation,21,38–41 and it has become increasingly clear that the pentameric form of PLN is required for normal cardiac contractility.15,19,42–45 If the PLN pentamer plays a direct role in the formation of the SERCA-PLN complex, what is the underlying mechanism that favors formation of the SERCA-PLN complex from the pentamer? The transition from the PLN pentamer to the monomer, and then from the PLN monomer to the SERCA-PLN complex involves a series of distinct molecular interactions. Depending on the location in the transmembrane domain of PLN, a particular residue may interact with another PLN, lipids, SERCA, or some combination of the three. The free-energy changes (ΔΔG) associated with these PLN states suggest that PLN–PLN interactions in the pentamer are a lower energy state and PLN-lipid interactions of the monomer are a higher energy state. By comparison, the free-energy changes associated with the SERCA-PLN complex suggest that it is an intermediate state between the PLN pentamer and monomer. Thus, PLN–PLN and SERCA-PLN (helix–helix) interactions are favored over PLN-lipid (helix-lipid) interactions (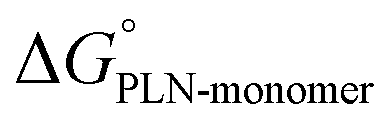 >
>  >
>  ). However, some alanine variants of PLN shift the energy landscape of the PLN pentamer, though PLN–PLN and SERCA-PLN interactions are still favored over PLN–lipid interactions. Generally, these residues are part of the leucine–isoleucine zipper that stabilizes the pentameric form of PLN, where alanine substitution of these residues destabilizes the pentamer and increases the probability of monomer release. These alanine variants of PLN tend to be super-inhibitors of SERCA.
). However, some alanine variants of PLN shift the energy landscape of the PLN pentamer, though PLN–PLN and SERCA-PLN interactions are still favored over PLN–lipid interactions. Generally, these residues are part of the leucine–isoleucine zipper that stabilizes the pentameric form of PLN, where alanine substitution of these residues destabilizes the pentamer and increases the probability of monomer release. These alanine variants of PLN tend to be super-inhibitors of SERCA.
Does super-inhibition of SERCA by PLN alanine variants offer insights into the formation of the SERCA-PLN complex? The original model for SERCA inhibition by PLN involved the calcium-free E2 conformation of SERCA and an inhibitory groove formed by transmembrane segments M2, M6, and M9 of SERCA.46,47 The inhibitory groove is “open” in the calcium-free E2 state and “closed” in the calcium-bound E1 state of SERCA, where PLN was thought to bind to the “open” inhibitory groove and impede groove closure in the E2 to E1 transition. However, the subsequent crystal structures of the SERCA-PLN and SERCA-SLN complexes revealed a different conformation of SERCA poised in a calcium-free E1-like state.21–23 In the calcium free E1-like conformation of SERCA,21 the inhibitory groove is partially closed and intermediate between the open E2 and closed E1 states. This raises the notion of differential recognition of the “open” and partially “closed” inhibitory grooves by PLN. The partially closed inhibitory groove in the calcium-free E1-like conformation of SERCA is compatible with binding of the PLN pentamer, while the open inhibitory groove in the E2 state of SERCA is too deep and narrow to engage the PLN pentamer. Therefore, we suggest that formation of the SERCA-PLN inhibitory complex via the PLN pentamer allows for conformational selection of SERCA in the calcium-free E1-like state for the formation of the correct (physiological) complex. In this scenario, super-inhibition of SERCA may result from loss of conformational selection and the interaction of a PLN monomer with the open inhibitory groove in the E2 conformation of SERCA.
Conclusion: implications for transmembrane helix assembly
Single-pass transmembrane proteins can form a variety of homo- and hetero-oligomeric assemblies that induce, amplify, or mitigate cellular signaling events. The ability of homo- and hetero-oligomeric assemblies to sense cellular events (e.g., changes in ion gradients or phosphorylation states) and change state are a fundamental feature of some cellular signaling systems. PLN is one example of a single-pass membrane protein that changes state from a homo-pentameric form to a hetero-oligomeric form with SERCA. The hetero-oligomeric interaction with SERCA results in SERCA inhibition, a shift in the apparent calcium affinity of SERCA, which is regulated by the phosphorylation of PLN and the cytosolic calcium concentration. The ability to transition from a homo-oligomeric state, the PLN pentamer, to a hetero-oligomeric state, the SERCA-PLN complex, is a fundamental feature of this regulatory axis. The results of this study support the notion that the homo-oligomeric form of PLN directs the formation of the “correct” hetero-oligomeric SERCA-PLN complex. This conformational selection may be a general phenomenon in cellular signaling pathways that involve the transition between homo- and hetero-oligomeric transmembrane assemblies, where transmembrane helix–helix interactions are often dynamic and dependent on the functional state of the protein.Experimental
Mutagenesis, expression, and purification of PLN
The protocol used for mutagenesis, expression and purification of PLN has been previously described.15,48 Residues Ile18 to Ile33 of PLN were individually mutated to alanine. The remaining alanine-scanning mutagenesis data included in this study for PLN residues Asn34-Ala to Leu52-Ala has been published.15,29 Further details can be found in the ESI.†Reconstitution of SERCA and PLN into proteoliposome
SERCA was purified from the SR of rabbit hind leg muscle and reconstituted into membrane vesicles (proteoliposomes) with purified recombinant human PLN as described.49 Further details can be found in the ESI.†Calcium-dependent ATPase activity assays
Proteoliposomes containing SERCA and PLN were then measured for their calcium-dependent ATPase activity using a coupled-enzyme assay as previously described.50 Further details can be found in the ESI.†Molecular modeling
Three different systems were prepared for in silico calculations. In the first system, the structure of the SERCA-PLN bound complex (PDB: 4KYT21) was used which contains three chains corresponding to one SERCA and two PLN. In this structure, the first PLN chain is in proximity to SERCA but only residues Pro21 to Val49 were resolved, while the second PLN chain was further away from SERCA and only residues Gln26 to Ile40 were resolved. All of the missing residues in the structure of SERCA as well as residues Ile18, Glu19, Met20, Met50, Leu51 and Leu52 in the first PLN chain were inserted using Modeller.51 For effective comparison to the functional data, differing residues were changed to the human PLN sequence. The second PLN chain was removed. This complex was embedded in a 256 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) pre-equilibrated lipid membrane box obtained from the CHARMM-GUI database.52 The acidic residues in the calcium-binding sites Glu771 and Glu908 were protonated while residues Glu309 and Asp800 were deprotonated based on PROPKA analysis.53,54 In the second and third systems, the structures of PLN monomer (PDB: 1FJK) and PLN pentamer (PDB: 2KYV), respectively, were embedded in the same lipid membrane box as above. In both PLN monomer and PLN pentamer, residues Met1 to Thr17 were removed and differing residues were changed to human wild-type sequence using Modeller to keep the structures consistent for comparison.51,55–57 Each system was solvated and ionized with 0.15 M KCl using solvate and autoionize VMD plugins.58 The protein structure file was generated using the VMD psfgen plugin58 using CHARMM36 force field topology.59,60 For the alchemical free energy perturbation (FEP) simulations for alanine scanning mutagenesis, structures for different alanine mutants of the model systems were generated using a hybrid topology file in combination with AlaScan,61 psfgen and Mutator VMD plugins.58Molecular dynamics (MD) and free-energy perturbation (FEP) simulations
NAMD software25,26,62 was used to carry out all of the in silico simulations with NVIDIA P100 Pascal GPUs and CHARMM36 force field parameter files.59,60 Each of the three model systems, i.e., SERCA-PLN complex, PLN monomer and PLN pentamer were first minimized, annealed, and equilibrated for overall 10 ns with unrestrained MD simulations prior to FEP calculations. The final dimension of all three systems upon equilibration were 110 Å × 110 Å × 150 Å. In the PLN pentamer system, alchemical FEP calculations in all five protomers were simultaneously performed and the stoichiometry was accounted for during the analysis step. Electrostatic and van der Waals cutoff distance was set to 11 Å with switching at 10 Å and pair list distance at 12 Å. Langevin piston pressure control was set to 1.01 bar and the temperature for Langevin calculations at 300 K. For periodic boundary conditions, the Particle Mesh Ewald method was applied for electrostatics and the wrapping of all coordinates was set to on. The alchemical FEP setup files were prepared using AlaScan61 with default parameters, the FEP calculation for each system was carried out with windows (λ) of width 0.05 or over 20 windows for forward transformation and the same procedure was repeated for the backward transformation. Each window consisted of 1 × 105 equilibration and 5 × 105 production steps of 1 fs each. The cumulative simulation time for each alanine alchemical FEP calculation was 24 ns. Alanine scanning FEP calculations for all residues in PLN from Ile18 to Leu52 (excluding Pro21) were performed separately and repeated for all three systems, SERCA-PLN complex, PLN monomer and PLN pentamer. The total cumulative simulation time was calculated to be 2.406 μs [(33 alanine mutations × 3 systems × 24 ns) + (10 ns equilibration × 3 systems)]. Alchemical transformation free energy (ΔGalchemical) was determined from FEP simulation for each alanine mutation using VMD ParseFEP plugin with default settings58,63 (Fig. S3 and S4†). Subsequently, values for ΔGbound-alchemical from SERCA-PLN complex system were subtracted for values from either the PLN monomer system or the PLN pentamer system (ΔGunbound-alchemical). This difference is equivalent to the difference in the free energy of formation of alanine-substituted SERCA-PLN complex compared to the wild-type SERCA-PLN complex (ΔΔG). ParseFEP performs the alchemical transformations bidirectionally, and the results of the forward and backward simulations are combined in the form of the Bennett acceptance-ratio (BAR) estimator of the free energy and statistical error (SEM).To examine the effect of equilibration time on the initial structures of the model systems, the three model systems (SERCA-PLN complex, PLN monomer and PLN pentamer) were equilibrated for 10, 25, 40, and 50 ns of unrestrained MD simulations. Each of these equilibrated model systems were used as replicates for FEP calculations as described in the preceding paragraph. This was done for a subset of transmembrane residues including L37A, L43A, L44A, I47A, and L51A (Table S3†). The overall conclusions were further supported by these calculations, showing a clear preference for the thermodynamic cycle from the PLN pentamer (Fig. 3B).
Data availability
Data for this article, including the ParseFEP log files, are available at Borealis-Dataverse at https://borealisdata.ca/dataset.xhtml?persistentId=doi:10.5683/SP3/7CKZXG. All other data supporting this article have been included as part of the main article or ESI.†Author contributions
Conceptualization – NR, MJL, CC, BR, HSY; methodology – NR, CC, BR, HSY; investigation – NR, HSY; visualization – NR, HSY; supervision – HSY; writing – NR, HSY; writing review & editing – NR, MJL, CC, BR, HSY.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
This work was funded by the Canadian Institutes of Health Research (PJT 180387 to HSY), the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL092321 to HSY), the Natural Sciences and Engineering Research Council of Canada (549297-2019 to MJL), the National Science Foundation (MCB-2309048 to BR), the Agence Nationale de la Recherche (ProteaseInAction and LOR-AI to CC) and the France and Chicago Collaborating in The Sciences (FACCTS) program. This research was enabled in part by support provided by Graham cluster at University of Waterloo (https://docs.alliancecan.ca/wiki/Graham) and Digital Research Alliance of Canada (https://alliancecan.ca).References
- M. Mravic, J. L. Thomaston, M. Tucker, P. E. Solomon, L. Liu and W. F. DeGrado, Packing of apolar side chains enables accurate design of highly stable membrane proteins, Science, 2019, 363, 1418–1423 CrossRef CAS PubMed.
- J. Lee and W. Im, Role of hydrogen bonding and helix-lipid interactions in transmembrane helix association, J. Am. Chem. Soc., 2008, 130, 6456–6462 CrossRef CAS PubMed.
- P. Lague, M. J. Zuckermann and B. Roux, Lipid-mediated interactions between intrinsic membrane proteins: dependence on protein size and lipid composition, Biophys. J., 2001, 81, 276–284 CrossRef CAS PubMed.
- D. E. Clapham, Calcium signaling, Cell, 1995, 80, 259–268 CrossRef CAS PubMed.
- C. Toyoshima, M. Nakasako, H. Nomura and H. Ogawa, Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution, Nature, 2000, 405, 647–655 CrossRef CAS PubMed.
- C. Toyoshima and H. Nomura, Structural changes in the calcium pump accompanying the dissociation of calcium, Nature, 2002, 418, 605–611 CrossRef CAS PubMed.
- D. H. MacLennan and E. G. Kranias, Phospholamban: a crucial regulator of cardiac contractility, Nat. Rev. Mol. Cell Biol., 2003, 4, 566–577 CrossRef CAS PubMed.
- N. Rathod, J. J. Bak, J. O. Primeau, M. E. Fisher, L. M. Espinoza-Fonseca, M. J. Lemieux and H. S. Young, Nothing Regular about the Regulins: Distinct Functional Properties of SERCA Transmembrane Peptide Regulatory Subunits, Int. J. Mol. Sci., 2021, 22, 8891 CrossRef CAS PubMed.
- K. Haghighi, F. Kolokathis, L. Pater, R. A. Lynch, M. Asahi, A. O. Gramolini, G. C. Fan, D. Tsiapras, H. S. Hahn, S. Adamopoulos, S. B. Liggett, G. W. Dorn II, D. H. MacLennan, D. T. Kremastinos and E. G. Kranias, Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human, J. Clin. Invest., 2003, 111, 869–876 CrossRef CAS PubMed.
- J. P. Schmitt, M. Kamisago, M. Asahi, G. H. Li, F. Ahmad, U. Mende, E. G. Kranias, D. H. MacLennan, J. G. Seidman and C. E. Seidman, Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban, Science, 2003, 299, 1410–1413 CrossRef CAS PubMed.
- M. M. DeWitt, H. M. MacLeod, B. Soliven and E. M. McNally, Phospholamban R14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy, J. Am. Coll. Cardiol., 2006, 48, 1396–1398 CrossRef CAS PubMed.
- K. Haghighi, F. Kolokathis, A. O. Gramolini, J. R. Waggoner, L. Pater, R. A. Lynch, G. C. Fan, D. Tsiapras, R. R. Parekh, G. W. Dorn II, D. H. MacLennan, D. T. Kremastinos and E. G. Kranias, A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1388–1393 CrossRef CAS PubMed.
- P. A. van der Zwaag, I. A. van Rijsingen, R. de Ruiter, E. A. Nannenberg, J. A. Groeneweg, J. G. Post, R. N. Hauer, I. C. van Gelder, M. P. van den Berg, P. van der Harst, A. A. Wilde and J. P. van Tintelen, Recurrent and founder mutations in the Netherlands-Phospholamban p.Arg14del mutation causes arrhythmogenic cardiomyopathy, Neth. Heart J., 2013, 21, 286–293 CrossRef CAS PubMed.
- H. S. Young, D. K. Ceholski and C. A. Trieber, Deception in simplicity: hereditary phospholamban mutations in dilated cardiomyopathy, Biochem. Cell Biol., 2015, 93, 1–7 CrossRef CAS PubMed.
- C. A. Trieber, J. L. Douglas, M. Afara and H. S. Young, The effects of mutation on the regulatory properties of phospholamban in co-reconstituted membranes, Biochemistry, 2005, 44, 3289–3297 CrossRef CAS PubMed.
- H. K. Simmerman, Y. M. Kobayashi, J. M. Autry and L. R. Jones, A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure, J. Biol. Chem., 1996, 271, 5941–5946 CrossRef CAS PubMed.
- J. M. Autry and L. R. Jones, Functional Co-expression of the canine cardiac Ca2+ pump and phospholamban in Spodoptera frugiperda (Sf21) cells reveals new insights on ATPase regulation, J. Biol. Chem., 1997, 272, 15872–15880 CrossRef CAS PubMed.
- R. L. Cornea, L. R. Jones, J. M. Autry and D. D. Thomas, Mutation and phosphorylation change the oligomeric structure of phospholamban in lipid bilayers, Biochemistry, 1997, 36, 2960–2967 CrossRef CAS PubMed.
- Y. Kimura, K. Kurzydlowski, M. Tada and D. H. MacLennan, Phospholamban inhibitory function is activated by depolymerization, J. Biol. Chem., 1997, 272, 15061–15064 CrossRef CAS PubMed.
- S. L. Robia, K. S. Campbell, E. M. Kelly, Z. Hou, D. L. Winters and D. D. Thomas, Forster transfer recovery reveals that phospholamban exchanges slowly from pentamers but rapidly from the SERCA regulatory complex, Circ. Res., 2007, 101, 1123–1129 CrossRef CAS PubMed.
- B. L. Akin, T. D. Hurley, Z. Chen and L. R. Jones, The structural basis for phospholamban inhibition of the calcium pump in sarcoplasmic reticulum, J. Biol. Chem., 2013, 288, 30181–30191 CrossRef CAS PubMed.
- C. Toyoshima, S. Iwasawa, H. Ogawa, A. Hirata, J. Tsueda and G. Inesi, Crystal structures of the calcium pump and sarcolipin in the Mg2+-bound E1 state, Nature, 2013, 495, 260–264 CrossRef CAS PubMed.
- A. M. Winther, M. Bublitz, J. L. Karlsen, J. V. Moller, J. B. Hansen, P. Nissen and M. J. Buch-Pedersen, The sarcolipin-bound calcium pump stabilizes calcium sites exposed to the cytoplasm, Nature, 2013, 495, 265–269 CrossRef CAS PubMed.
- R. W. Zwanzig, High-temperature equation of state by a perturbation method. I. Nonpolar gases, J. Chem. Phys., 1954, 22, 1420–1426 CrossRef CAS.
- H. Chen, J. D. C. Maia, B. K. Radak, D. J. Hardy, W. Cai, C. Chipot and E. Tajkhorshid, Boosting Free-Energy Perturbation Calculations with GPU-Accelerated NAMD, J. Chem. Inf. Model., 2020, 60, 5301–5307 CrossRef CAS PubMed.
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Henin, W. Jiang, R. McGreevy, M. C. R. Melo, B. K. Radak, R. D. Skeel, A. Singharoy, Y. Wang, B. Roux, A. Aksimentiev, Z. Luthey-Schulten, L. V. Kale, K. Schulten, C. Chipot and E. Tajkhorshid, Scalable molecular dynamics on CPU and GPU architectures with NAMD, J. Chem. Phys., 2020, 153, 044130 CrossRef CAS PubMed.
- Y. Kimura, M. Asahi, K. Kurzydlowski, M. Tada and D. H. MacLennan, Phospholamban domain Ib mutations influence functional interactions with the Ca2+-ATPase isoform of cardiac sarcoplasmic reticulum, J. Biol. Chem., 1998, 273, 14238–14241 CrossRef CAS PubMed.
- E. M. Kelly, Z. Hou, J. Bossuyt, D. M. Bers and S. L. Robia, Phospholamban oligomerization, quaternary structure, and sarco(endo)plasmic reticulum calcium ATPase binding measured by fluorescence resonance energy transfer in living cells, J. Biol. Chem., 2008, 283, 12202–12211 CrossRef CAS PubMed.
- C. A. Trieber, M. Afara and H. S. Young, Effects of phospholamban transmembrane mutants on the calcium affinity, maximal activity, and cooperativity of the sarcoplasmic reticulum calcium pump, Biochemistry, 2009, 48, 9287–9296 CrossRef CAS PubMed.
- D. K. Ceholski, C. A. Trieber and H. S. Young, Hydrophobic imbalance in the cytoplasmic domain of phospholamban is a determinant for lethal dilated cardiomyopathy, J. Biol. Chem., 2012, 287, 16521–16529 CrossRef CAS PubMed.
- S. Smeazzetto, G. P. Armanious, M. R. Moncelli, J. J. Bak, M. J. Lemieux, H. S. Young and F. Tadini-Buoninsegni, Conformational memory in the association of the transmembrane protein phospholamban with the sarcoplasmic reticulum calcium pump SERCA, J. Biol. Chem., 2017, 292, 21330–21339 CrossRef CAS PubMed.
- D. K. Weber, U. V. Reddy, S. Wang, E. K. Larsen, T. Gopinath, M. B. Gustavsson, R. L. Cornea, D. D. Thomas, A. De Simone and G. Veglia, Structural basis for allosteric control of the SERCA-Phospholamban membrane complex by Ca(2+) and phosphorylation, Elife, 2021, 10, e66226 CrossRef CAS PubMed.
- H. S. Young, L. R. Jones and D. L. Stokes, Locating phospholamban in co-crystals with Ca(2+)-ATPase by cryoelectron microscopy, Biophys. J., 2001, 81, 884–894 CrossRef CAS PubMed.
- R. L. Cornea, J. M. Autry, Z. Chen and L. R. Jones, Reexamination of the role of the leucine/isoleucine zipper residues of phospholamban in inhibition of the Ca2+ pump of cardiac sarcoplasmic reticulum, J. Biol. Chem., 2000, 275, 41487–41494 CrossRef CAS PubMed.
- S. Lamberth, H. Schmid, M. Muenchback, T. Vorherr, J. Krebs, E. Carafoli and C. Griesinger, NMR Solution Structure of Phospholamban, Helv. Chim. Acta, 2000, 83, 2141–2152 CrossRef CAS.
- R. Verardi, L. Shi, N. J. Traaseth, N. Walsh and G. Veglia, Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9101–9106 CrossRef CAS PubMed.
- D. H. MacLennan, Y. Kimura and T. Toyofuku, Sites of regulatory interaction between calcium ATPases and phospholamban, Ann. N. Y. Acad. Sci., 1998, 853, 31–42 CrossRef CAS PubMed.
- J. P. Glaves, J. O. Primeau, L. M. Espinoza-Fonseca, M. J. Lemieux and H. S. Young, The Phospholamban Pentamer Alters Function of the Sarcoplasmic Reticulum Calcium Pump SERCA, Biophys. J., 2019, 116, 633–647 CrossRef CAS PubMed.
- J. P. Glaves, J. O. Primeau, P. A. Gorski, L. M. Espinoza-Fonseca, M. J. Lemieux and H. S. Young, Interaction of a Sarcolipin Pentamer and Monomer with the Sarcoplasmic Reticulum Calcium Pump, SERCA, Biophys. J., 2020, 118, 518–531 CrossRef CAS PubMed.
- J. P. Glaves, C. A. Trieber, D. K. Ceholski, D. L. Stokes and H. S. Young, Phosphorylation and mutation of phospholamban alter physical interactions with the sarcoplasmic reticulum calcium pump, J. Mol. Biol., 2011, 405, 707–723 CrossRef CAS PubMed.
- D. L. Stokes, A. J. Pomfret, W. J. Rice, J. P. Glaves and H. S. Young, Interactions between Ca2+-ATPase and the pentameric form of phospholamban in two-dimensional co-crystals, Biophys. J., 2006, 90, 4213–4223 CrossRef CAS PubMed.
- G. Chu, L. Li, Y. Sato, J. M. Harrer, V. J. Kadambi, B. D. Hoit, D. M. Bers and E. G. Kranias, Pentameric assembly of phospholamban facilitates inhibition of cardiac function in vivo, J. Biol. Chem., 1998, 273, 33674–33680 CrossRef CAS PubMed.
- F. Funk, A. Kronenbitter, K. Hackert, M. Oebbeke, G. Klebe, M. Barth, D. Koch and J. P. Schmitt, Phospholamban pentamerization increases sensitivity and dynamic range of cardiac relaxation, Cardiovasc. Res., 2023, 119, 1568–1582 CrossRef CAS PubMed.
- D. K. Ceholski, C. A. Trieber, C. F. Holmes and H. S. Young, Lethal, hereditary mutants of phospholamban elude phosphorylation by protein kinase A, J. Biol. Chem., 2012, 287, 26596–26605 CrossRef CAS PubMed.
- T. Wittmann, M. J. Lohse and J. P. Schmitt, Phospholamban pentamers attenuate PKA-dependent phosphorylation of monomers, J. Mol. Cell. Cardiol., 2015, 80, 90–97 CrossRef CAS PubMed.
- C. Toyoshima, M. Asahi, Y. Sugita, R. Khanna, T. Tsuda and D. H. MacLennan, Modeling of the inhibitory interaction of phospholamban with the Ca2+ ATPase, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 467–472 CrossRef CAS PubMed.
- K. Seidel, O. C. Andronesi, J. Krebs, C. Griesinger, H. S. Young, S. Becker and M. Baldus, Structural characterization of Ca(2+)-ATPase-bound phospholamban in lipid bilayers by solid-state nuclear magnetic resonance (NMR) spectroscopy, Biochemistry, 2008, 47, 4369–4376 CrossRef CAS PubMed.
- J. L. Douglas, C. A. Trieber, M. Afara and H. S. Young, Rapid, high-yield expression and purification of Ca2+-ATPase regulatory proteins for high-resolution structural studies, Protein Expression Purif., 2005, 40, 118–125 CrossRef CAS PubMed.
- H. S. Young, J. L. Rigaud, J. J. Lacapere, L. G. Reddy and D. L. Stokes, How to make tubular crystals by reconstitution of detergent-solubilized Ca2(+)-ATPase, Biophys. J., 1997, 72, 2545–2558 CrossRef CAS PubMed.
- G. B. Warren, P. A. Toon, N. J. Birdsall, A. G. Lee and J. C. Metcalfe, Reconstitution of a calcium pump using defined membrane components, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 622–626 CrossRef CAS PubMed.
- B. Webb and A. Sali, Comparative Protein Structure Modeling Using MODELLER, Curr. Protoc. Bioinf., 2016, 54, 1–37 Search PubMed.
- S. Jo, T. Kim and W. Im, Automated builder and database of protein/membrane complexes for molecular dynamics simulations, PLoS One, 2007, 2, e880 CrossRef PubMed.
- C. R. Sondergaard, M. H. Olsson, M. Rostkowski and J. H. Jensen, Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values, J. Chem. Theory Comput., 2011, 7, 2284–2295 CrossRef CAS PubMed.
- M. H. Olsson, C. R. Sondergaard, M. Rostkowski and J. H. Jensen, PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- A. Sali and T. L. Blundell, Comparative protein modelling by satisfaction of spatial restraints, J. Mol. Biol., 1993, 234, 779–815 CrossRef CAS PubMed.
- A. Fiser, R. K. Do and A. Sali, Modeling of loops in protein structures, Protein Sci., 2000, 9, 1753–1773 CrossRef CAS PubMed.
- M. A. Marti-Renom, A. C. Stuart, A. Fiser, R. Sanchez, F. Melo and A. Sali, Comparative protein structure modeling of genes and genomes, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 291–325 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. MacKerell Jr. and R. W. Pastor, Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS PubMed.
- R. B. Best, X. Zhu, J. Shim, P. E. Lopes, J. Mittal, M. Feig and A. D. Mackerell Jr., Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS PubMed.
- V. Ramadoss, F. Dehez and C. Chipot, AlaScan: A Graphical User Interface for Alanine Scanning Free-Energy Calculations, J. Chem. Inf. Model., 2016, 56, 1122–1126 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, Scalable molecular dynamics with NAMD, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- P. Liu, F. Dehez, W. Cai and C. Chipot, A Toolkit for the Analysis of Free-Energy Perturbation Calculations, J. Chem. Theory Comput., 2012, 8, 2606–2616 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02915a |
| This journal is © The Royal Society of Chemistry 2024 |