
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvancing the enzymatic toolkit for 2′-fluoro arabino nucleic acid (FANA) manipulation: phosphorylation, ligation, replication, and templating RNA transcription†
Yingyu
Liu‡
 ab,
Jun
Wang‡
ab,
Yashu
Wu
bc and
Yajun
Wang
*bc
ab,
Jun
Wang‡
ab,
Yashu
Wu
bc and
Yajun
Wang
*bc
aCollege of Pharmaceutical Sciences, Soochow University, Suzhou 215123, China
bHangzhou Institute of Medicine (HIM), Chinese Academy of Sciences, Hangzhou, Zhejiang 310000, China. E-mail: wangyj@ibmc.ac.cn
cThe Cancer Hospital of the University of Chinese Academy of Science (Zhejiang Cancer Hospital), Hangzhou, Zhejiang 310022, China
First published on 24th June 2024
Abstract
2′-Fluoro arabino nucleic acid (FANA), classified as a xeno nucleic acid (XNA), stands as a prominent subject of investigation in synthetic genetic polymers. Demonstrating efficacy as antisense oligonucleotides (ASOs) and exhibiting the ability to fold into functional structures akin to enzymes and aptamers, FANA holds substantial promise across diverse biological and therapeutic domains. Owing to structural similarities to DNA, the utilization of naturally occurring DNA polymerases for DNA-mediated FANA replication is well-documented. In this study, we explore alternative nucleic acid processing enzymes typically employed for DNA oligonucleotide (ON) phosphorylation, ligation, and amplification, and assess their compatibility with FANA substrates. Notably, T4 polynucleotide kinase (T4 PNK) efficiently phosphorylated the 5′-hydroxyl group of FANA using ATP as a phosphate donor. Subsequent ligation of the phosphorylated FANA with an upstream FANA ON was achieved with T4 DNA ligase, facilitated by a DNA splint ON that brings the two FANA ONs into proximity. This methodology enabled the reconstruction of RNA-cleaving FANA 12-7 by ligating two FANA fragments amenable to solid-phase synthesis. Furthermore, Tgo DNA polymerase, devoid of 3′ to 5′ exonuclease activity [Tgo (exo-)], demonstrated proficiency in performing polymerase chain reaction (PCR) with a mixture of dNTPs and FANA NTPs (fNTPs), yielding DNA–FANA chimeras with efficiency and fidelity comparable to traditional DNA PCR. Notably, T7 RNA polymerase (T7 RNAP) exhibited recognition of double-stranded fA–DNA chimeras containing T7 promoter sequences, enabling in vitro transcription of RNA molecules up to 649 nt in length, even in the presence of highly structured F30 motifs at the 3′ end. Our findings significantly expand the enzymatic toolkit for FANA manipulation, encompassing phosphorylation, ligation, chimeric amplification, and templating T7 RNAP-catalyzed RNA transcription. These advancements are poised to expedite fundamental research, functional evolution, and translational applications of FANA-based XNA agents. They also have the potential to inspire explorations of a broader range of non-natural nucleic acids that can be routinely studied in laboratories, ultimately expanding the repertoire of nucleic acid-based biomedicine and biotechnology.
Introduction
Artificial genetic polymers, also known as xeno nucleic acids (XNAs), feature distinct backbone sugar structures differing from the ribose and deoxyribose present in RNA and DNA.1,2 Despite these noncanonical backbone structures, most XNAs retain the Watson–Crick base pairing capability with DNA, RNA, and among themselves, serving as a cornerstone for the development of XNA-based modulating agents3–5 and the evolution of functional XNA molecules such as aptamers6 and XNAzymes.7–9 Due to their non-natural sugar moieties, XNAs exhibit unique chemophysical properties, including expanded conformational diversity, heightened resistance to nucleases, tolerance to extreme chemical conditions, and an extended affinity range when hybridized with DNA or RNA. These distinctive properties have propelled research interest in XNAs beyond the original exploration of prebiotically plausible genetic materials.10,11 The development of XNA-based alternatives seeks to address challenges encountered by DNA and RNA, particularly in achieving full realization of their applicational potentials in biomedicine and biotechnology. Key areas of focus include enhancing biostability and conformational stability, thereby opening avenues for novel applications in various domains.122′-Fluoro arabino nucleic acid (FANA, as depicted in Scheme 1), initially developed as an antisense oligonucleotide with enhanced RNA substrate engagement capable of eliciting RNase H-mediated RNA degradation, stands out as one of the most extensively studied XNAs.13,14 With advancements in polymerase engineering facilitating XNA replication, FANA has been evolved into aptamers with ligand-binding activities and FANAzymes with catalytic RNA cleavage activities, achieved through folding into intricate secondary and tertiary structures.6,7,15 These FANA-based agents exhibited significantly enhanced biostability under simulated physiological conditions and resistance to depurination-induced strand scission under extremely acidic conditions. Notably, two FANA aptamers selected to target HIV-1 reverse transcriptase16 and integrase17 respectively, have been demonstrated with picomolar (pM) binding affinities and functioned in inhibiting the biological activity of their targets. This suggests that FANA aptamers tend to fold into compact structures, expanding their hydrophobic binding surface to mimic the interface of protein–protein interactions. Due to enhanced target RNA strand invasion capabilities conferred by thermodynamic stability and boosted biostability, XNAzymes comprising entirely FANA18 or partially FANA19,20 in the substrate-binding arms have shown dramatically improved efficiency in cleaving long, structured RNAs under physiological conditions. Moreover, they have enabled long-lasting allele-specific gene silencing intracellularly. Collectively, these superior functional properties underscore the vast potential FANA holds across a broad spectrum of biological and therapeutic applications.
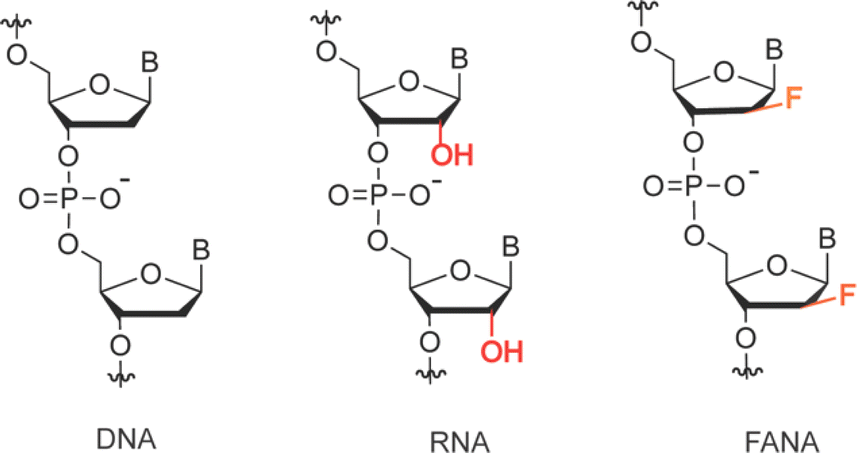 | ||
| Scheme 1 Constitutional structure for the linearized DNA, RNA, and FANA. FANA is an unnatural genetic polymer composed of repeating 2′-fluoro arabinofuranosyl units that are connected by 5,3′-phosphodiester bonds. | ||
Structurally analogous to DNA, various naturally occurring family B DNA polymerases exhibit DNA-templated FANA transcription activity, while family A DNA polymerase Bst demonstrates the ability to utilize FANA strands as templates to synthesize cDNA. In functional FANA molecule SELEX, the combined use of family B Tgo polymerase and Bst led to the isolation of the first RNA-cleaving FANAzyme, showcasing robust catalysis with Michaelis–Menten kinetics.15 Drawing inspiration from the remarkable efficiency and fidelity of naturally occurring DNA polymerases when interacting with FANA, our study herein explored a range of other nucleic acid processing enzymes routinely employed for phosphorylation, ligation, and amplification of DNA oligonucleotides (ONs) when FANA served as the substrate. We have demonstrated that T4 polynucleotide kinase (T4 PNK) efficiently phosphorylated the 5′-hydroxyl group of FANA using ATP as a phosphate donor. Subsequently, T4 DNA ligase effectively ligated the resulting phosphorylated FANA with an upstream FANA ON in the presence of a DNA splint ON, tethering the two FANA ONs in close proximity. This enabled the reconstruction of the RNA-cleaving FANAzyme 12-7 by ligating two FANA fragments amenable for solid-phase synthesis. Tgo (exo-) was capable of utilizing mixtures of dNTPs and fNTPs, containing up to two fNTPs, to perform polymerase chain reaction (PCR) and exponentially amplify DNA templates into DNA–FANA chimeras with high efficiency and fidelity. Furthermore, double-stranded DNA–fA chimeras containing T7 promoter sequences were recognized by T7 RNA polymerase (T7 RNAP) for templated RNA in vitro transcription (IVT), yielding long RNA transcripts with highly structured F30 motifs at the 3′ end. Our findings expand the toolkit for enzymatic manipulation of FANA, encompassing phosphorylation, ligation, chimeric amplification, and templating T7 RNAP-catalyzed RNA IVT. These advancements hold the potential to accelerate fundamental studies, therapeutic translations, and the functional evolution of FANA-based XNA agents.
Results and discussion
T4 PNK catalyzed 5′ phosphorylation of FANA ON1
Oligonucleotide 5′ phosphorylation reaction is the prerequisite step proceeding enzymatic ligation, and also is a critical strategy for 5′ radiolabeling of ON by using γ-32P-ATP. We therefore initiated our study by examining the T4 PNK-catalyzed 5′ phosphorylation reaction of FANA using FANA ON1 under typical T4 PNK reaction buffer conditions for phosphorylating DNA or RNA, with ATP supplementation as the phosphate donor. The molecular weight (MW) of the oligos was characterized using matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry both before (PNK-) and after 18 hours of incubation at 37 °C (PNK+) in the PNK labeling reaction. MALDI-TOF spectra revealed complete conversion of FANA ON1 by T4 PNK into a monophosphorylated FANA ON2 and a diphosphorylated FANA ON3 (Fig. 1A). This was evidenced by the disappearance of the peak corresponding to the MW of FANA ON1 and the appearance of peaks at +80 and +160 (Fig. 1B). Intrigued by the unexpected appearance of diphosphorylated FANA ON3, we further characterized DNA ON1 of the same sequence alongside FANA ON1 before and after the PNK labeling reaction for comparison. While both monophosphorylated DNA ON2 and diphosphorylated DNA ON3 were observed via MALDI-TOF, the relative peak intensity between DNA ON2 and DNA ON3 was significantly greater than that observed for FANA (Fig. S1, ESI†). Upon shortening the reaction time of FANA PNK labeling from 18 hours to 1 hour, we observed an obvious increase in the relative peak ratio between monophosphorylated FANA ON2 and diphosphorylated FANA ON3via MALDI-TOF (Fig. S2, ESI†). Although the peak intensity of the MALDI-TOF spectrum does not necessarily reflect the abundance of the analyte in the analyzed sample,21 we conducted PNK phosphorylation reactions of both FANA and DNA for 1 hour in preparation for subsequent oligonucleotide ligation studies.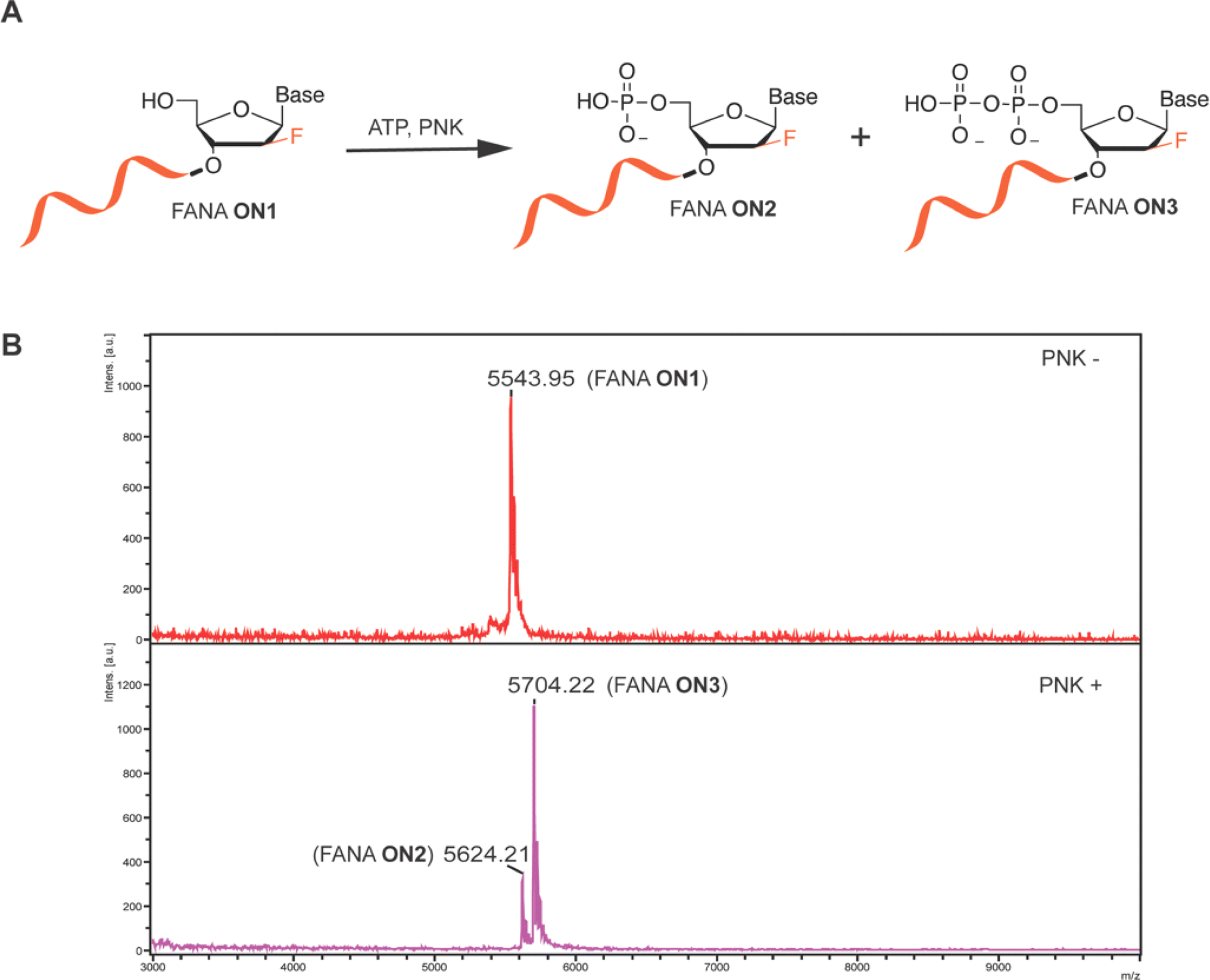 | ||
| Fig. 1 T4 PNK catalyzed 5′ phosphorylation of FANA ON. (A) Schematic representation of T4 PNK catalyzed 5′ phosphorylation reaction of FANA. (B) MALDI-TOF spectra of the FANA ON before and after PNK phosphorylation reaction, which suggested that both 5′ mono- (FANA ON2) and diphosphorylation (FANA ON3) products were generated. The reaction was performed in 1× T4 PNK reaction buffer containing 0.4 U μL−1 of T4 PNK supplemented with 1 mM ATP for 18 h at 37 °C. | ||
T4 DNA ligase catalyzed FANA ligation
Building upon the efficient phosphorylation of FANA by PNK, we proceeded to investigate the activity of T4 DNA ligase, a widely utilized ligase for DNA fragment ligation, in catalyzing the ligation of an upstream FANA acceptor with a downstream 5′ phosphorylated FANA donor in the presence of a DNA splint that brings the acceptor and donor into close proximity (Fig. 2A). Specifically, we designed the acceptor and donor sequences as depicted in Fig. 2B, aiming to reconstitute the RNA-cleaving FANAzyme 12-7 upon ligation.15 The time-dependent progression of ligation, visualized through the 5′-IR signal on the acceptor sequence, is depicted in Fig. 2C. Our findings revealed approximately 60% of the donor sequence was ligated with the acceptor within 180 minutes as quantified by the IR-labeled acceptor and calculated based on the equivalents of the ligation pair. Extending the ligation reaction time to 20 hours did not yield further improvement in overall ligation yield (see Fig. S3B, ESI†). We successfully recovered the ligation product, FANAzyme 12-7, from a preparative-scale ligation resolved by denaturing urea-PAGE (Fig. S3C, ESI†). This FANAzyme 12-7 demonstrated comparable activity in cleaving the matched RNA substrate under pseudo first-order conditions (Fig. 2D), affirming the fidelity of the ligation process. Our observation reinforces that the generation of 5′ diphosphorylated FANA ONs by PNK labeling does not impede subsequent ligation reactions, a fact further supported by the indiscernible difference in ligation yield observed between a FANA donor and a DNA donor with the same FANA acceptor sequence (Fig. S3C, ESI†). The combination of readily achievable T4 PNK-catalyzed 5′ phosphorylation with efficient T4 DNA ligase-catalyzed ligation facilitates access to long FANA oligonucleotides beyond the scope achievable by solid-phase chemical synthesis or enzymatic transcription. | ||
| Fig. 2 T4 DNA ligase catalyzed FANA ligation. (A) Schematic representation of ligation of a dye-labeled acceptor FANA ON with a 5′-phosphorylated donor FANA ON in the presence of DNA splint tethering the two FANA ONs together by forming reverse complementary duplex. (B) The sequence scheme of ONs depicted in (A) for T4 DNA ligase catalyzed ligation to generate the full-length FANAzyme 12-7. (C) Representative PAGE showing the time-dependent progression of ligation reaction. The reaction was performed in 1× T4 DNA ligase reaction buffer containing 5 μM of donor, 10 μM of acceptor, 20 μM of splint, and 20 U μL−1 of T4 DNA ligase for 18 h at 24 °C. (D) Representative PAGE showing the catalytic RNA cleavage activity of FANAzyme 12-7 obtained by ligation under single-turnover conditions in buffer (pH 8.5) containing 25 mM MgCl2 and 200 mM NaCl at 24 °C ([FANAzyme] = 2.5 μM, [S] = 0.5 μM). | ||
FANA substitutional polymerase chain reaction (PCR) using Tgo (exo-)
Several family B DNA polymerases, such as Thermococcus gorgonarius (Tgo), Thermococcus kodakarensis (Kod), Thermococcus sp. 9°N (9°N), and Pyrococcus sp. Deep vent (DV), deficient in exonuclease activity, have been demonstrated to transcribe FANA from DNA templates through primer extension reactions with high fidelity and sufficient efficiency under isothermal conditions. However, polymerase chain reaction (PCR) using FANA triphosphates (fNTPs) has not been reported, likely due to the inhibitory effect of highly stable FANA–FANA duplexes on strand separation and primer binding in PCR cycles. Our recent discovery that FANAzyme 12-7 retains a large extent of activity even when the G and C nucleosides are substituted by DNA congeners (data not shown) has sparked interest in exploring DNA–FANA chimeric aptamers or catalysts. This approach allows for the introduction of external functionalities onto DNA nucleosides via established nucleic acid modification chemistry to compensate for functions, while the FANA composition provides conformational and biological stability. This integration offers a practical means to simultaneously leverage expanded functionality and the advantages of an XNA backbone.22To investigate the possibility of constructing DNA–FANA chimeric sequences via exponential amplification, we first compared the rate of DNA-templated single nucleotide incorporation of fNTPs and dNTPs using Tgo polymerase deficient in 3′-5′ exonuclease activity [Tgo (exo-)]. When nucleotide concentrations were set at 300 and 600 nM, spanning routine dNTP concentrations in PCR, Tgo (exo-) displayed an incorporation rate for fATP and fGTP about 1.1–2 fold that of their DNA counterparts, similar incorporation rates for fCTP and dCTP, and a 2 to 4-fold reduction in the incorporation rates of fTTP and fUTP relative to their respective DNA counterparts (Fig. 3A and S4 in ESI†). These incorporation rate differences between fNTPs and dNTPs were within the same range as those observed among different dNTPs. Additionally, a full-length product of 79 nt was generated in 1 minute through DNA template-directed primer extension reactions using triphosphate mixtures containing a single fNTP substitution (Fig. S5, ESI†). Together, these results suggest that substituting dNTPs with fNTPs in PCR reactions may be feasible.
 | ||
| Fig. 3 Single-nucleotide incorporation kinetics and FANA substitutional polymerase chain reaction (PCR) using Tgo (exo-). (A) Single-nucleotide incorporation kinetic profiles of fNTPs in comparison to dNTPs. dNTPs are color coded in black, fA, G, C, UTPs are in orange, with fTTP in blue. All reactions were performed in 1× ThermoPol buffer containing 50 nM of template-primer duplex, 300 or 600 nM tested dNTP or fNTP and 60 nM of Tgo (exo-) at 55 °C. Values of kobs were calculated by fitting the percentage of incorporation and reaction time (min) to the first-order decay equation using Prism 8, GraphPad. (B) Schematic representation of PCR using dNTPs or a mixture of dNTPs and fNTPs wherein single or double dNTPs were substituted by fNTP counterparts. (C) Representative agarose gel electrophoresis showing the PCR amplicons (denoted by the asterisk) of different dNTP and fNTP combinations. All single fNTP substitutions led to increases of PCR efficiency as reflected by the intensity of amplicon band, and only the combination of fA and fG was able to generate amplicon of similar yield with control using all dNTPs. All reactions were performed in 1× ThermoPol buffer supplemented with 1 μM of each primers (forward and reverse), 0.25 mM of each dNTP or fNTP, 1 mM MgSO4, and 40 nM of Tgo (exo-). PCR protocol: step 1: 95 °C for 5 min; step 2: 25 cycles of (98 °C for 30 s; 55 °C for 30 s; 72 °C for 60 s); step 3: 72 °C for 5 min. | ||
Then we proceeded to perform Tgo (exo-) catalyzed DNA–FANA chimeric PCR following standard PCR protocols. Specifically, we systematically substituted individual or multiple dNTPs with their FANA counterparts in PCR reactions using a DNA template with 54% GC content to generate chimeric amplicons of 93 bp (see Fig. 3B). Remarkably, all four single fNTP substitutions (fATP, fGTP, fCTP, or fUTP) led to increased PCR yield for this specific template, resulting in amplicon bands with approximately twice the intensity compared to those produced using dNTPs. Notably, no correct amplification occurred when the tested fNTP was omitted from the reactions. Instead, a truncational band appeared, presumably due to suboptimal template-primer designs, which was also observed to varying extents in the all-dNTP control and some other combinations. When two fNTPs were present in the PCR reactions, only the combination of fA and fG yielded amplicons of similar yield to controls using all dNTPs (Fig. 3B). To further investigate the generalizability of FANA substitutional PCR, we amplified a DNA template coding for the spinach aptamer, which has a higher GC content of 65%, to generate 117 bp amplicons. Fortuitously, amplicons of the correct size containing single substitutions of all five fNTPs, including fTTP, were generated without the truncational products observed in Fig. 3C. This reinforces the notion that truncational products are likely related to suboptimal template-primer designs. The use of a highly structured DNA template with high GC content did result in a decreased yield of PCR amplicons, particularly in the cases of fGTP and fCTP substitutions. As expected, double-fNTP substitutional PCR failed to generate detectable amplicons for this highly structured GC-rich template (Fig. S6, ESI†). It is noteworthy that, even without extreme denaturation conditions to promote strand separation, amplicons up to 1000 bp were obtained for single FANA substitutional PCR containing either fA, fG, or fC. However, the yield was substantially compromised compared to the control using all dNTPs (Fig. S7, ESI†).
We further quantitatively assessed the fidelity of Tgo (exo-) mediated FANA substitutional PCR for all four single fNTP substitutions and the combined fA and fG substitution. To achieve this, we designed primers and a synthetic DNA template enabling recombination of the resulting chimeric amplicon (93 bp) into a plasmid expressing UV-excitable green fluorescent protein (GFPuv) using Gibson assembly. A forward primer containing a 5′ overhang sequence region was employed to distinguish the synthetic DNA template from the PCR amplicon (Fig. 3A). After transforming the assembled plasmids into E. coli, recombinants harboring the inserted amplicon were screened on antibiotic-containing LB-agar plates. Green colonies were swiftly identified under a UV transilluminator for subsequent Sanger sequencing (Fig. 4A). Alignment of the obtained sequences revealed that among the 10 sequenced colonies for each substitutional PCR, 8 to 9 contained the inserted amplicon of the correct size, indicating a high level of recombinational success (Fig. S8, ESI†). Notably, no errors were observed for the single fC substitution (Fig. 4D), and only one G-to-T transversion was noted for the combined fA and fG substitution, corresponding to an error rate of ∼1 × 10−3 errors per base (Fig. 4F). The overall error rates for the other three single FANA substitutions were calculated to be in the range of ∼3.6 × 10−3 to 4.6 × 10−3 errors per base (Fig. 4B, C and E), with 2 deletions and 1 insertion observed only for fU substitution (Fig. S8, ESI†). These values are consistent with those reported for Kod (exo-), which exhibits error rates of ∼3.5 × 10−3 errors per base when amplifying DNA under the same number of PCR cycles.23,24
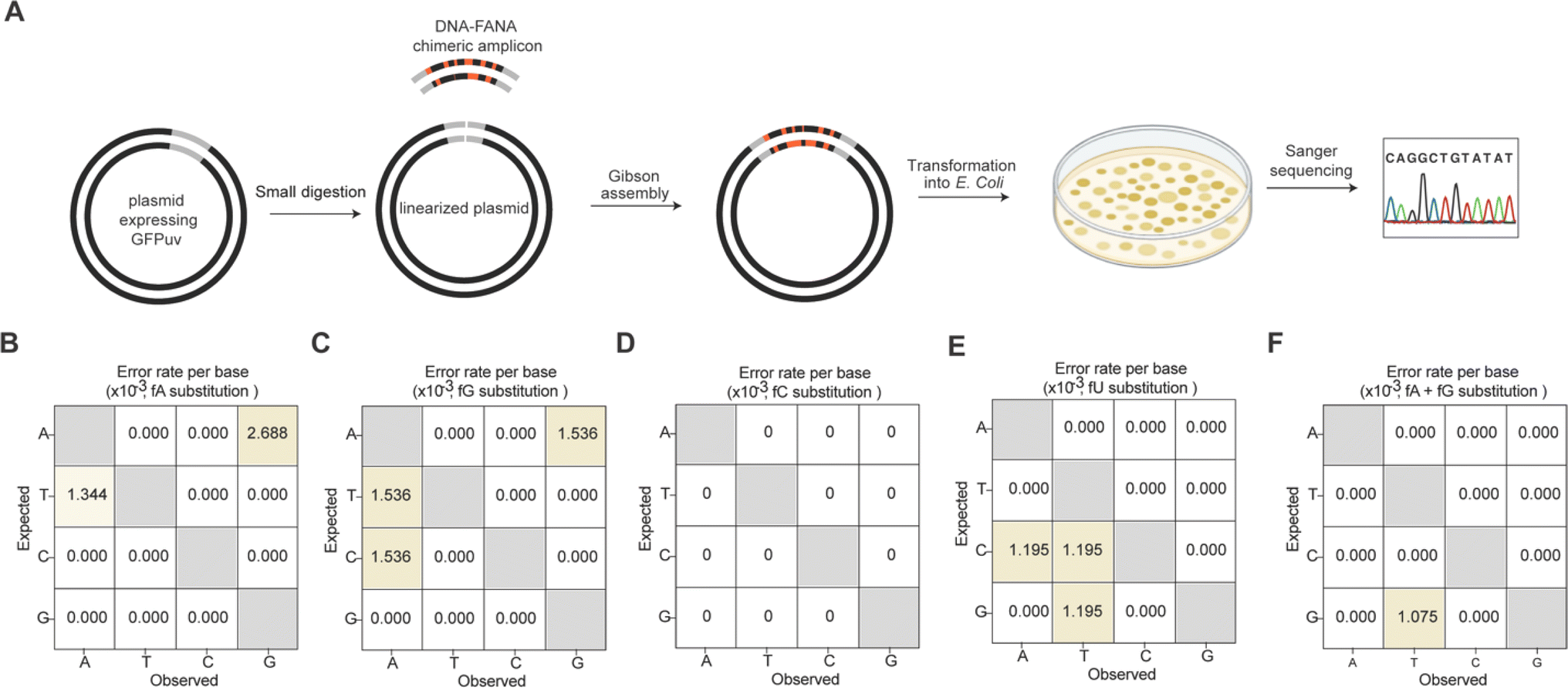 | ||
| Fig. 4 The replicational fidelity of FANA substitutional PCR using Tgo (exo-). (A) Schematic representation of fidelity measurement by Sanger sequencing of E. coli colonies transformed with GFPuv expressing plasmids recombined through Gibson assembly to contain the FANA substitutional PCR amplicon. (B–F) Fidelity profiles observed for fA (B), fG (C), fC (D), fU (E), and fA and fG (F) substitutional PCR, respectively. | ||
T7 RNA polymerase catalyzed in vitro RNA transcription (IVT) of using DNA–FANA chimeric amplicon as template
The insertion of DNA–FANA chimeric sequences into a linearized vector via Gibson assembly resulted in the generation of plasmids containing “alien” genetic material for the replicative system of E. coli. The remarkable replicational capability of E. coli with these alien plasmids was evidenced by both the regularly grown colonies expressing strong GFPuv signal and the faithful conservation of sequence information of the inserted chimera. This observation marked the first demonstration that FANA can be recognized by the E. coli replication system and converted into DNA in vivo, a phenomenon also known as genetic transliteration.25,26 While being transliterated into pure DNA plasmids to template RNA transcription was the primary fate of the transformed FANA-containing plasmid in E. coli, whether the small amount of chimeric plasmid could be directly recognized by certain RNA polymerases to directly template RNA transcription in vivo had not yet been explored (Fig. S9†). To investigate this possibility, we amplified a synthetic DNA template encoding the fluorescent spinach RNA aptamer, with one of the primers containing the T7 promoter sequence, through single fNTP substitutional PCR. The resulting chimeric amplicon served as a template for T7 RNA polymerase (T7 RNAP)-catalyzed in vitro RNA transcription. We chose T7 RNAP due to its wide application in in vitro mRNA production.27–29 The spinach RNA transcript was readily detected and quantified by the addition of 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI), which induced folding of aptamer and activated the fluorescence of DFHBI30,31 (Fig. 5A). Excitingly, T7 RNAP recognized the DNA–fA chimera and transcribed RNA with approximately 2-fold efficiency compared to using DNA template, as evidenced by the stronger fluorescent signal associated with DNA–fA-templated IVT reaction (Fig. 5B and C). However, only a faint fluorescent signal was detected for the IVT reaction using DNA–fG template, and no detectable signal was generated when DNA–fC or DNA–fU was used as a template. We further analyzed the fluorescent sample on an agarose gel, which resolved discrete RNA transcript bands with relative yields consistent with those quantified by fluorescent signal readings (Fig. 5D).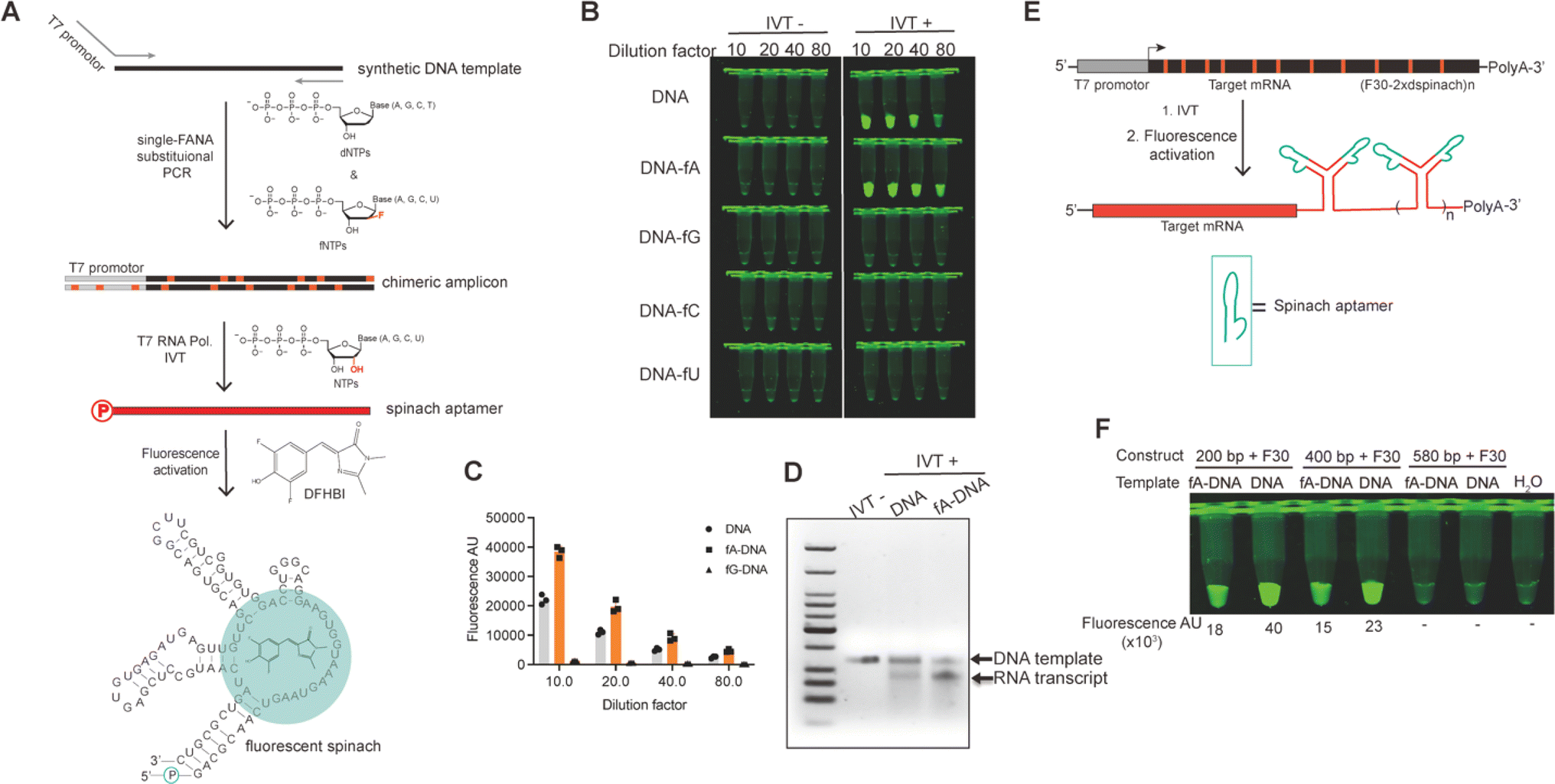 | ||
| Fig. 5 T7 RNA polymerase catalyzed in vitro RNA transcription (IVT) of the fluorescent spinach aptamer using DNA–FANA chimeric amplicon as template. (A) Schematic representation of the experimental protocol of single-fNTP substitutional PCR using one of the primers containing the T7 promoter sequence to obtain DNA–FANA chimeric amplicons as template for the IVT of spinach aptamer RNA, to which the addition of DFHBI activates the fluorescence of the aptamer-DFHBI complex. (B) Fluorescent image of DFHBI activated IVT reactions using different DNA–FANA chimeric amplicons as templates. (C) Quantitative analysis of the fluorescent signal intensities of different IVT reactions in panel (B). (D) Representative agarose gel resolving the IVT reaction templated by DNA and DNA–fA. (E) Schematic representation of the IVT of long RNA transcripts tagged with a 3′ structural motif of two spinach aptamers positioned by the F30 scaffold using DNA–fA templates. (F) Fluorescent image of DFHBI activated IVT reactions using different DNA–fA chimeric amplicons measuring different length as templates. | ||
Prompted by the doubled yield of spinach RNA IVT using T7 RNAP and DNA–fA template, we further challenged the system with the IVT of long RNA transcripts measuring 449, 649, and 829 nt, respectively. These RNA transcripts comprised a fragment of the ORF of GFP mRNA ranging from 200 to 580 nt, and a structural motif of two spinach aptamers positioned by the F30 scaffold32 (F30-2xdspinach, 249 nt) as illustrated in Fig. 5E. Fortuitously, the 449 and 649 nt transcripts were fully transcribed by T7 RNAP using the corresponding DNA–fA templates, albeit with relatively lower yield compared to the DNA controls, as indicated by the fluorescent signals of each individual IVT reaction (Fig. 5F). Visualization of the IVT reaction on agarose gel suggested that the relatively low yield of RNA transcripts using DNA–fA as template was associated with a lower concentration of template compared to that using DNA as template (Fig. S10†). Therefore, an improved RNA IVT yield using DNA–fA as a template could be expected by optimizing the IVT conditions. In contrast, no fluorescent signal was observed for the transcription of the 829 nt RNA using either fA–DNA or DNA template. This may be attributed to IVT reaction conditions that were suboptimal for transcribing such a long RNA transcript tagged with the 3′ highly structured F30 motif (Fig. 5F). However, the optimization of these conditions is beyond the scope of the current research.
Efficient strategies to increase the yield of RNA IVT would significantly benefit RNA-based biomedicine and biotechnology, representing a technical challenge in the mRNA vaccine industry. Our findings suggest that DNA–fA chimeras can be recognized by T7 RNAP and serve as templates for RNA in vitro transcription with even higher yields, providing an alternative RNA transcription system that utilizes biologically more stable XNA-containing chimeric templates.
Conclusions
We have demonstrated that, akin to DNA, a series of naturally occurring DNA processing enzymes can recognize and function with comparable activity on FANA. T4 PNK efficiently phosphorylated the 5′-hydroxyl group of FANA ON using ATP as a phosphate donor. The resulting phosphorylated FANA was successfully ligated by T4 DNA ligase with an upstream FANA ON, facilitated by a DNA splint ON tethering the two FANA ONs in close proximity. This ligation reaction enabled the successful reconstruction of the RNA-cleaving FANAzyme 12-7 by ligating two FANA fragments amenable for solid-phase synthesis. By tandem ligation of multiple FANA fragments, it should be possible to produce long FANA oligonucleotides with complex functions. Tgo (exo-) exhibited the capability to utilize dNTP and fNTP mixtures to perform PCR, exponentially amplifying DNA templates into DNA–FANA chimeras with sufficiently high efficiency and fidelity. To the best of our knowledge, this is the first report of the exponential amplification of extensively FANA-decorated amplicons using standard PCR procedures. This breakthrough paves the way for the scalable preparation of biologically more stable and conformationally diversified chimeric nucleic acids. Additionally, T7 RNAP recognized double-stranded DNA–fA chimeras containing the T7 promoter sequence, facilitating the templating of IVT of RNA transcripts up to 649 nt with a 3′ highly structured F30 motif. The ability to use biologically more stable DNA–fA chimera to template RNA IVT with higher yield not only provides a feasible platform for more efficient RNA transcription but also inspires the exploration of other non-natural nucleic acid-based systems for templating RNA synthesis. Our results significantly expand the toolkit for enzymatic manipulation of FANA to include phosphorylation, ligation, chimeric amplification, and templating RNA IVT, using correspondingly naturally occurring enzymes. The ready availability of these enzymatic tools holds promise for accelerating the fundamental study, functional evolution, and therapeutic translation of FANA-based XNA agents and technologies.Data availability
All other data supporting this article, including all experimental procedures, characterization details, and copies of MALDI-TOF spectra, have been included as part of the ESI.†Author contributions
Y. W. conceived of the project and designed the experiments. Y. L., J. W. and Yashu Wu performed all the experiments. Y. L., J. W., and Y. W. wrote the manuscript. All authors reviewed and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by start-up funds from Hangzhou Institute of Medicine, Chinese Academy of Sciences and Soochow University to Y. W., and the Young Scientists Fund of National Natural Science Foundation of China to Y. W. (22107083). We graciously thank Prof. John Chaput for providing the plasmid of the Tgo (exo-) polymerase and some of the FANA oligonucleotides, Prof. Chun Kit Kwok and Prof. Ying Li for their generous gift of FANA triphosphates. We thank Dr Yufeng Pei for the helpful discussion. We also thank members of the Wang laboratory for helpful discussions and critical reading of the manuscript.References
- J. C. Chaput and P. Herdewijn, Angew. Chem., Int. Ed., 2019, 58, 11570–11572 CrossRef CAS PubMed
.
- J. C. Chaput, Acc. Chem. Res., 2021, 54, 1056–1065 CrossRef CAS PubMed
- M. J. Lange, D. H. Burke and J. C. Chaput, Nucleic Acid Ther., 2019, 29, 51–59 CrossRef CAS PubMed
- F. Wang, L. S. Liu, C. H. Lau, T. J. H. Chang, D. Y. Tam, H. M. Leung, C. Tin and P. K. Lo, ACS Appl. Mater. Interfaces, 2019, 11, 38510–38518 CrossRef CAS PubMed
- L. S. Liu, H. M. Leung, D. Y. Tam, T. W. Lo, S. W. Wong and P. K. Lo, ACS Appl. Mater. Interfaces, 2018, 10, 9736–9743 CrossRef CAS PubMed
- V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S.-Y. Peak-Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Science, 2012, 336, 341–344 CrossRef CAS PubMed
- A. I. Taylor, V. B. Pinheiro, M. J. Smola, A. S. Morgunov, S. Peak-Chew, C. Cozens, K. M. Weeks, P. Herdewijn and P. Holliger, Nature, 2014, 518, 427–430 CrossRef PubMed
- Y. Wang, Y. Wang, D. Song, X. Sun, Z. Zhang, X. Li, Z. Li and H. Yu, J. Am. Chem. Soc., 2021, 143, 8154–8163 CrossRef CAS PubMed
- Y. Wang, Y. Wang, D. Song, X. Sun, Z. Li, J.-Y. Chen and H. Yu, Nat. Chem., 2021, 1–10 Search PubMed
- K.-U. Schöning, P. Scholz, S. Guntha, X. Wu, R. Krishnamurthy and A. Eschenmoser, Science, 2000, 290, 1347–1351 CrossRef PubMed
- A. Eschenmoser, Science, 1999, 284, 2118–2124 CrossRef CAS PubMed
- A. I. Taylor, G. Houlihan and P. Holliger, Cold Spring Harbor Perspect. Biol., 2019, 11, a032490 CrossRef CAS PubMed
- M. Y. Anzahaee, G. F. Deleavey, P. U. Le, J. Fakhoury, K. Petrecca and M. J. Damha, Nucleic Acid Ther., 2014, 24, 336–343 CrossRef CAS PubMed
- T. Dowler, D. Bergeron, A.-L. Tedeschi, L. Paquet, N. Ferrari and M. J. Damha, Nucleic Acids Res., 2006, 34, 1669–1675 CrossRef CAS PubMed
- Y. Wang, A. K. Ngor, A. Nikoomanzar and J. C. Chaput, Nat. Commun., 2018, 9, 5067 CrossRef PubMed
- I. A. Ferreira-Bravo, C. Cozens, P. Holliger and J. J. DeStefano, Nucleic Acids Res., 2015, 43, 9587–9599 CAS
- K. M. Rose, I. A. Ferreira-Bravo, M. Li, R. Craigie, M. A. Ditzler, P. Holliger and J. J. DeStefano, ACS Chem. Biol., 2019, 14, 2166–2175 CAS
- A. I. Taylor, C. J. K. Wan, M. J. Donde, S.-Y. Peak-Chew and P. Holliger, Nat. Chem., 2022, 14, 1295–1305 CrossRef CAS PubMed
- Y. Wang, K. Nguyen, R. C. Spitale and J. C. Chaput, Nat. Chem., 2021, 13, 319–326 CrossRef CAS PubMed
- K. Nguyen, Y. Wang, W. E. England, J. C. Chaput and R. C. Spitale, J. Am. Chem. Soc., 2021, 143, 4519–4523 CrossRef CAS PubMed
- W. Timm, A. Scherbart, S. Böcker, O. Kohlbacher and T. W. Nattkemper, BMC Bioinf., 2008, 9, 443 CrossRef PubMed
- K. B. Wu, C. J. A. Skrodzki, Q. Su, J. Lin and J. Niu, Chem. Sci., 2022, 13, 6873–6881 RSC
- A. Bębenek and I. Ziuzia-Graczyk, Curr. Genet., 2018, 64, 985–996 CrossRef PubMed
- M. Takagi, M. Nishioka, H. Kakihara, M. Kitabayashi, H. Inoue, B. Kawakami, M. Oka and T. Imanaka, Appl. Environ. Microbiol., 1997, 63, 4504–4510 CrossRef CAS PubMed
- M. Luo, E. Groaz, M. Froeyen, V. Pezo, F. Jaziri, P. Leonczak, G. Schepers, J. Rozenski, P. Marlière and P. Herdewijn, J. Am. Chem. Soc., 2019, 141, 10844–10851 CrossRef CAS PubMed
- C. Liu, C. Cozens, F. Jaziri, J. Rozenski, A. Maréchal, S. Dumbre, V. Pezo, P. Marlière, V. B. Pinheiro, E. Groaz and P. Herdewijn, J. Am. Chem. Soc., 2018, 140, 6690–6699 CrossRef CAS PubMed
- A. Ishaqat and A. Herrmann, J. Am. Chem. Soc., 2021, 143, 20529–20545 CrossRef CAS PubMed
- Z. J. Kartje, H. I. Janis, S. Mukhopadhyay and K. T. Gagnon, J. Biol. Chem., 2021, 296, 100175 CrossRef CAS PubMed
- A. Dousis, K. Ravichandran, E. M. Hobert, M. J. Moore and A. E. Rabideau, Nat. Biotechnol., 2022, 1–9 Search PubMed
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642–646 CrossRef CAS PubMed
- J. Wu, S. Zaccara, D. Khuperkar, H. Kim, M. E. Tanenbaum and S. R. Jaffrey, Nat. Methods, 2019, 16, 862–865 CrossRef CAS PubMed
- G. S. Filonov, C. W. Kam, W. Song and S. R. Jaffrey, Chem. Biol., 2015, 22, 649–660 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02904f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |